

Abstract
Corneal limbal epithelial stem cells (LESCs) are responsible for continuously renewing the corneal epithelium, and thus maintaining corneal homeostasis and visual clarity. Human pluripotent stem cell (hPSC)-derived LESCs provide a promising cell source for corneal cell replacement therapy. Undefined, xenogeneic culture and differentiation conditions cause variation in research results and impede the clinical translation of hPSC-derived therapeutics. This protocol provides a reproducible and efficient method for hPSC-LESC differentiation under xeno- and feeder cell-free conditions. Firstly, monolayer culture of undifferentiated hPSC on recombinant laminin-521 (LN-521) and defined hPSC medium serves as a foundation for robust production of high-quality starting material for differentiations. Secondly, a rapid and simple hPSC-LESC differentiation method yields LESC populations in only 24 days. This method includes a four-day surface ectodermal induction in suspension with small molecules, followed by adherent culture phase on LN-521/collagen IV combination matrix in defined corneal epithelial differentiation medium. Cryostoring and extended differentiation further purifies the cell population and enables banking of the cells in large quantities for cell therapy products. The resulting high-quality hPSC-LESCs provide a potential novel treatment strategy for corneal surface reconstruction to treat limbal stem cell deficiency (LSCD).
Keywords: Developmental Biology, Issue 140, Human embryonic stem cells, human induced pluripotent stem cells, corneal limbal epithelial stem cells, xeno-free, feeder-free, small-molecule induction, directed corneal differentiation
Introduction
The transparent cornea at the ocular surface allows light to enter the retina and provides the majority of the eye's refractive power. The outermost layer, the stratified corneal epithelium, is continuously regenerated by limbal epithelial stem cells (LESCs). The LESCs reside in the basal layer of the limbal niches at the corneoscleral junction1,2. LESCs lack specific and unique markers, so their identification requires a more extensive analysis of a set of putative markers. Epithelial transcription factor p63, and especially N-terminally truncated transcript of the alpha isoform of p63 (ΔNp63α), has been proposed as a relevant positive LESC marker3,4. Asymmetric division of LESCs allows them to self-renew, but also produce progeny that migrate centripetally and anteriorly. As the cells progress toward the corneal surface they gradually lose their stemness and finally terminally differentiate to superficial squamous cells that are continuously lost from the corneal surface.
Damage to any of the corneal layers can lead to severe visual impairment, and corneal defects are thus one of the leading causes of vision loss worldwide. In limbal stem cell deficiency (LSCD), the limbus is destroyed by disease or trauma which leads to conjunctivalization and opacification of the corneal surface and subsequent loss of vision5,6. Cell replacement therapy using autologous or allogeneic limbal grafts offers a treatment strategy for patients with LSCD4,7,8,9. However, harvesting autologous grafts bears a risk of complications to the healthy eye, and donor tissue is in short supply. Human pluripotent stem cells (hPSCs), specifically human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs), can serve as an unlimited source of clinically relevant cell types, including corneal epithelial cells. Therefore, hPSC-derived LESCs (hPSC-LESCs) represent an attractive new cell source for ocular cell replacement therapy.
Traditionally, both the undifferentiated hPSC culture methods and their differentiation protocols to LESCs have relied on the use of undefined feeder cells, animal sera, conditioned media, or amniotic membranes10,11,12,13,14,15. Recently, efforts toward safer cell therapy products have prompted the search for more standardized and xeno-free culture and differentiation protocols. As a result, several defined and xeno-free methods for long-term culture of undifferentiated hPSCs are now commercially available16,17,18. As a continuum, directed differentiation protocols relying on molecular cues to guide hPSCs to corneal epithelial fate have been recently introduced19,20,21,22,23. Yet many of these protocols used either undefined, feeder based hPSCs as starting material, or complex, xenogeneic growth factor cocktails for differentiation.
The purpose of this protocol is to provide a robust, optimized, xeno-and feeder-free hPSC culture method and subsequent differentiation to corneal LESCs. Monolayer culture of pluripotent hPSCs on laminin-521 (LN-521) matrix in defined, albumin-free hPSC medium (specifically Essential 8 Flex) allows rapid production of homogeneous starting material for differentiations. Thereafter a simple, two-step differentiation strategy guides hPSCs toward surface ectodermal fate in suspension, followed by adherent differentiation to LESCs. A cell population where > 65% express ΔNp63α is obtained within 24 days. The xeno- and feeder-free protocol has been tested with several hPSC lines (both hESCs and hiPSCs), without any requirement for cell line specific optimization. The protocols for weekend-free maintenance, passaging, cryostoring and hPSC-LESC phenotyping described here enable production of large batches of high-quality LESCs for clinical or research purposes.
Protocol
University of Tampere has the approval of the National Authority for Medicolegal affairs Finland (Dnro 1426/32/300/05) to conduct research on human embryos. The institute also has supportive statements of the Ethical Committee of the Pirkanmaa Hospital District to derive, culture, and differentiate hESC lines (Skottman/R05116) and to use hiPSC lines in ophthalmic research (Skottman/R14023). No new cell lines were derived for this study.
NOTE: The protocol described is based on specific, commercially available hPSC and corneal epithelium differentiation media. Please refer to the Table of Materials for manufacturer/supplier information and catalog numbers.
1. Establishing Xeno- and Feeder-free hPSC Culture
- Preparations
- Coat 24-well plates with human recombinant laminin-521 (LN-521). For the first feeder-free (FF) passage, use LN-521 at a concentration of 1.09 µg/cm2, and at 0.55 µg/cm2 for the following passages. The suggested LN-521 concentrations serve as a starting point for successful FF culture but can be lowered.
- Thaw LN-521 vial slowly at 4 °C as instructed by the manufacturer. NOTE: Appropriately handled LN-521 solution may be stored at 4 °C for up to 3 months after thawing.
- To prepare the coating solution, dilute appropriate amount of LN-521 stock solution with 1x Dulbecco’s Phosphate-Buffered Saline (DPBS) containing Ca2+ and Mg2+ to a total volume of 300 µL per well.
- Pipet the coating solution to the wells, seal the well plate with parafilm, and incubate overnight at 4 °C. Coated plates may be stored at 4 °C for up to 2 weeks. (Optional): Alternatively, for a rapid coating protocol, incubate well plates with coating solution for 2 h at 37 °C, 5% CO2.
- Prepare hPSC culture medium (specifically Essential 8 Flex) by supplementing basal medium with provided supplement, as instructed by the manufacturer. Add 50 U/mL penicillin-streptomycin. Note that the formulation is sensitive to light and high temperatures. Thaw the supplement and warm the media at room temperature (RT), protected from light. Use the supplemented hPSC medium within two weeks from the supplementation date.
- Transferring the hPSC to FF culture on LN-521
- Pre-warm all the needed materials and reagents to RT in the laminar flow hood.
- Passage hPSCs from standard culture system on feeder layers (e.g. inactivated human foreskin (hFF) or mouse embryonic fibroblasts), or other FF culture systems using standard methodology. For example, hPSC colonies cultured on hFF feeder cells can be dissected to small pieces with a scalpel and the pieces then detached with a needle tip. Human PSCs cultured using FF culture systems can be transferred using cluster passaging method with or without prior enzyme treatment.
- Remove the LN-521 coating solution from the 24-wells and add 1 mL pre-warmed hPSC medium per well. NOTE: Do not allow the wells to dry as LN-521 will inactivate upon drying.
- Transfer the colony pieces/cell clusters to LN-521 coated 24-wells in hPSC medium with a pipette. Transfer 20–30 colony pieces per well, avoiding overcrowding the wells.
- Replace the medium with 1 mL of hPSC medium the day after transfer and every other day thereafter. The cultures are ready for passaging 3–4 days later as hPSCs have grown out to colonies with smooth, undifferentiated morphology. For reference, see Figure 1B (first image). For the first FF passage, the colonies should not be allowed to grow to a fully confluent monolayer, but the cultures should be passaged when colonies reach an approximate size of 1 mm.
- Passaging and maintenance of FF hPSC culture
- Pre-warm all the needed materials and reagents to RT in the laminar flow hood.
- From the second FF passage onwards, passage the FF hPSCs when the culture has reached 80-100% confluency. Passage FF hPSCs to new LN-521 coated 24-well plates using single cell passaging twice a week (on Mondays and Thursdays) to maintain high quality cultures with undifferentiated morphology and to achieve weekend-free feeding regimen. For details, please refer to18,24,25.
- Rinse the FF hPSCs twice with 1 mL of 1x DPBS without Ca2+ and Mg2+.
- Detach FF hPSCs with xeno-free trypsin-EDTA (specifically TrypLE Select Enzyme) by incubating 500 µL per well at 37 °C, 5% CO2. For optimal incubation time, allow the cells to round up but not to detach. This usually takes 3 min at 37 °C, 5% CO2 (do not exceed 5 min).
- Remove xeno-free trypsin-EDTA and immediately add 500 µL per well of defined trypsin inhibitor.
- Detach FF hPSCs by careful, yet thorough pipetting to obtain a single cell suspension. Transfer the FF hPSC suspension through a 40 µm cell strainer into a 15 mL conical centrifuge tube containing pre-warmed hPSC medium.
- Wash the wells with 1 mL of hPSC medium and add washing medium to the 15 mL conical centrifuge tube.
- Centrifuge the single cell suspension for 5 min at 300 x g, aspirate the cell pellet, and resuspend in 1 mL pre-warmed hPSC medium.
- Count the cells with hemocytometer or automated cell counter.
- Remove the LN-521 coating solution (0.55 µg/cm2) from the 24-wells and add hPSC medium as in 1.2.3.
- Plate FF hPSCs onto 0.55 µg/cm2 LN-521 coated 24-wells at a cell density of 40,000 – 50,000 cells/cm2.
- Replace medium with fresh hPSC medium the day after passaging, and every other day thereafter excluding Sundays.
- The cells are ready to be used for differentiation 3-4 days after passaging, when the culture has reached >85% confluency. For ensuring the high quality of the hPSCs, refer to characterization methods described in detail in previous works18,24,25. It is recommended to only culture hPSCs up to passage level 15 in the FF system using single cell passaging to avoid karyotypic changes. Only use high-quality, undifferentiated hPSCs as starting material for differentiations.
2. Directed Differentiation and Cryopreservation of hPSC-derived LESCs
- Preparations
- Prepare xeno-free basal induction medium (basal induction medium): Supplement Dulbecco’s modified Eagle’s medium (specifically KnockOut DMEM) with 15% xeno-free serum replacement (spesifically CTS KnockOut SR XenoFree), 2 mM L-glutamine, 0.1 mM 2-mercaptoethanol, 1% non-essential amino acids, and 50 U/mL penicillin-streptomycin. Use the basal induction medium within two weeks.
- Prepare media for corneal induction in suspension culture.
- Day 1: Supplement basal induction medium with 5 μM blebbistatin.
- Day 2: Supplement basal induction medium with 10 µM SB-505124 and 50 ng/mL human basic fibroblast growth factor (bFGF).
- Day 3-4: Supplement basal induction medium with 25 ng/mL bone morphogenetic protein 4 (BMP-4).
- Prepare corneal epithelium differentiation medium (differentiation medium, specifically CnT-30) for adherent culture: Add supplements to basal medium according to the manufacturer’s instructions and add 50 U/mL penicillin-streptomycin. NOTE: Differentiation medium formulation is sensitive to light. Use the supplemented differentiation medium within 6 weeks of the supplementation date.
- Coat 100 mm tissue culture dishes for adherent differentiation (see step 2.3) with a mixture of 5 µg/cm2 human placental collagen type IV (col IV) and 0.5 µg/cm2 LN-521 diluted in 1x DPBS containing Ca2+ and Mg2+, in a total coating volume of 5 mL per dish. Prepare and store the coatings with col IV and LN-521 as described in 1.1.1.3.
- Step I: Corneal induction in suspension culture
- Pre-warm all the needed materials and reagents to RT in the laminar flow hood.
- Detach FF hPSCs to single cell suspension with xeno-free trypsin-EDTA as instructed in steps 1.3.2.1–1.3.2.6. Count the cells, and distribute 2-3x106 cells per low attachment 6-well plate well, in total volume of 3 mL of basal induction medium supplemented with 5 μM blebbistatin to induce EB formation overnight at 37 °C, 5% CO2 (Day 1).
- On the following day (Day 2), remove the medium and replace with 3 mL of basal induction medium supplemented with 10 µM SB-505124 and 50 ng/mL bFGF.
- On the following two days (Days 3–4), remove the medium and replace with 3 mL of basal induction medium supplemented with 25 ng/mL BMP-4.
- Step II: Corneal differentiation in adherent culture
- Pre-warm all the needed materials and reagents to RT in the laminar flow hood.
- On day 5, plate the EBs down onto 100 mm tissue culture dishes coated with 5 µg/cm2 col IV and 0.5 µg/cm2 LN-521.
- Remove the coating solution from 100 mm tissue culture dishes and add 10 mL of pre-warmed differentiation medium per dish. NOTE: Do not allow the dishes to dry as LN-521 will inactivate upon drying.
- Transfer the EBs from one 6-well plate well to two to three 100 mm tissue culture dishes (approximately 50 EBs per cm2) by pipetting. Distribute the EBs evenly by gentle shaking.
- Maintain the cells in adherent culture at 37 °C, 5% CO2, replacing the medium with 10 mL of fresh differentiation medium three times per week (on Monday, Wednesday and Friday) for the next 2.5–3 weeks. Check the cells regularly for the emergence of correct epithelial morphology using phase contrast microscope.
- Step III: Cryo-banking hPSC-derived LESCs
- Pre-warm all the needed materials and reagents to RT in the laminar flow hood, except for the cryopreservation medium that should be pre-chilled.
- Detach hPSC-derived LESCs with xeno-free trypsin-EDTA and count the cells, as instructed for FF hPSCs in steps 1.3.2.1–1.3.2.6, but using differentiation medium. NOTE: For hPSC-derived LESCs, optimal incubation time with xeno-free trypsin-EDTA is longer (about 5 min). Use 3 mL of xeno-free trypsin-EDTA and defined trypsin inhibitor per 100 mm dish.
- After counting the cells, repeat centrifugation for 5 min at 300 x g, aspirate medium and resuspend the cell pellet in pre-chilled, xeno-free hPSC cryopreservation medium. Pipet the single cell suspension into cryotubes so that each cryotube contains 0.5 to 1 x 106 cells in 1 mL cryopreservation medium.
- Place the tubes in a freezing container and transfer immediately (within 5 min) to -80 °C overnight.
- On the following day, transfer the tubes to liquid nitrogen for long-term storage.
- Step IV: Thawing the cryopreserved hPSC-LESCs
- Prior to thawing, coat the dishes/well plates with 5 µg/cm2 col IV and 0.5 µg/cm2 LN-521.
- Pre-warm all the needed materials and reagents to RT in the laminar flow hood.
- Remove the coating solution from dishes/wells and add appropriate volume of pre-warmed differentiation medium. NOTE: Do not allow the dishes to dry as LN-521 will inactivate upon drying.
- Thaw the cells quickly at RT and immediately transfer the cell suspension to a 15 mL conical centrifuge tube containing 5 mL of pre-warmed differentiation medium.
- Centrifuge the cell suspension for 5 min at 300 x g, aspirate, and resuspend the pellet in differentiation medium to remove any cryopreservation medium.
- Plate the cells onto dishes/wells coated with 5 µg/cm2 col IV and 0.5 µg/cm2 LN-521, in differentiation medium at a density of 40,000 – 50,000 cells/cm2. Maintain the cells at 37 °C, 5% CO2, replacing the differentiation medium three times a week.
3. Phenotyping of hPSC-derived LESCs
- Qualitative immunofluorescence analysis
- For immunofluorescence, coat 24- or 12- well plate wells with 5 µg/cm2 col IV and 0.5 µg/cm2 LN-521 and plate/thaw hPSC-LESCs in differentiation medium at a density of 40 000–50 000 cells/cm2.
- When the cultures have reached confluency, fix the cells with 4% paraformaldehyde (PFA): Wash the wells twice with 1x DPBS without Ca2+ and Mg2+and incubate 15-20 min with 4% PFA at RT. Thereafter, wash twice with 1x DPBS to remove any PFA residues. Use 0.5-1 mL of solutions per well. NOTE: Fixed cells may be stored in 1x DPBS at 4 °C for up to one week prior to staining. Caution: PFA is toxic and corrosive. Handle PFA in a fume hood and wear protective clothing, eye protection, and gloves.
- Aspirate 1x DPBS and permeabilize cell membranes by incubating for 10-15 min with 0.1% Triton X-100 in 1x DPBS.
- Aspirate 0.1% Triton X-100 and block nonspecific antibody binding sites by incubating for 1 h with 3% bovine serum albumin (BSA) in 1x DPBS at RT. Prepare primary antibody dilutions in 0.5% BSA in 1x DPBS. NOTE: See Table 1 for recommended primary antibodies.
- Aspirate 3% BSA and incubate with appropriately diluted primary antibody overnight at 4 °C.
- Wash the wells 3x for 5 min with 1x DPBS. Prepare secondary antibody dilutions in 0.5% BSA in 1x DPBS. NOTE: See Table 1 for recommended secondary antibodies.
- Aspirate 1x DPBS and incubate with suitable, appropriately diluted secondary antibody for 1 h at RT, protected from light. NOTE: From this step on, keep the samples protected from light in order to prevent fading of the fluorescent dyes.
- Wash the wells 3x for 5 min with 1x DPBS and finally counterstain nuclei with 4',6-diamidino-2-phenylindole (DAPI) and mount with fluorescence mounting media. DAPI can be used separately according to manufacturer’s instructions or included in the mounting medium. Place round coverslips (diameter 19 mm and 13 mm for 12- and 24-well plates, respectively) in each well. Follow the manufacturer’s instructions regarding drying and storage of the mounted samples.
- Image the stained cells with a fluorescent microscope.
- Quantitative immunofluorescence analysis
- Prepare cytospin samples of hPSC-derived LESCs on object glasses.
- Detach hPSC-derived LESCs with xeno-free trypsin-EDTA and count the cells, as instructed in step 2.4.2.
- After counting, add pre-chilled 1x DPBS and centrifuge for 5 min at 300 x g. Adjust the sample volume and cell concentration according to the manufacturer’s instructions of the given cytocentrifuge, e.g. 50,000 – 100,000 cells in a sample volume of 150 µL.
- Spin cells down to object glasses with a cytocentrifuge e.g. 5 min at 28 x g and fix immediately for 15-20 min with 4% PFA in 1x DPBS at RT. For the recommended spinning time and speed, refer to the instruction manual of the given cytocentrifuge.
- Proceed to staining the cytospin samples as described in steps 3.1.3–3.1.8 Use liquid blocker pen to surround the samples with a hydrophobic circle and conduct the staining in a droplet for economical practice. Washes may be performed in containers with slide holders, in order to ensure efficient removal of excess antibodies and minimal background staining. Counterstain nuclei with DAPI and mount the stained samples with fluorescence mounting media, covering with coverslips. Follow manufacturer’s instructions regarding drying and storage of the mounted samples.
- Capture 5–10 images per sample from randomly selected locations using e.g. 10X magnification of a fluorescence microscope.
- Estimate the percentage of positively stained cells in relation to total (DAPI positive) cells, e.g. with ImageJ Image Processing and Analysis software (https://imagej.nih.gov/ij/) tools. Preferably analyze >500 cells per sample.
- Open the image to be analyzed in ImageJ software. Duplicate the image and filter with default Gaussian blur to remove noise.
- Create a threshold. Adjust the threshold values to optimal selection of positively stained cell nuclei, and apply. The duplicated image is now converted to a binary view.
- Process the binary image with binary processing tools “Fill holes” and “Watershed”, which will automatically separate merged areas representing single nuclei.
- Use the “Analyze particles” tool to automatically list regions of interests (ROIs) to the ROI manager window, which will open upon applying the command. Close the binary image.
- Visualize the ROIs in the original image by choosing “Show all” in the ROI Manager. Confirm correct selection of stained cell nuclei, and if needed, manually remove and add individual selections.
- Flow cytometry analysis
- To confirm the p63α expression levels, stain the cells for flow cytometry.
- Detach hPSC-derived LESCs with xeno-free trypsin-EDTA and count the cells, as instructed in step 2.4.2.
- Wash the cells twice with 1 mL pre-chilled 1x DPBS and centrifuge for 5 min at 300 x g. Fix and permeabilize with ready-to-use fixation/permeabilization solution for 20 min at 4 °C. Thereafter wash the cells twice with 1 mL pre-chilled 1x permeabilizing wash buffer. NOTE: From this step on, keep the cells at 4 °C or on ice, unless indicated otherwise.
- CAUTION: Fixation/permeabilization solution contains 4.2% formaldehyde. Handle the hazardous solution in a fume hood and wear protective clothing, eye protection, and gloves.
- Divide samples into 5 mL polypropylene tubes. Each sample should contain 100,000–200,000 cells and the sample volume should be adjusted to approximately 100 µL of 1x wash buffer.
- Add 2 µL of fluorochrome conjugated p63-α FACS antibody (for recommended antibody, see Table of Equipment and Materials) into the sample tubes. Leave one sample unstained to serve as negative control. Vortex the samples and incubate for 1 h at RT, protected from light.
- Wash the cells twice with 1 mL of pre-chilled 1x wash buffer and lastly, resuspend the pellets with 300 µL of buffer. Store the tubes on ice, protected from light.
- Analyze the samples with a flow cytometer. Use the unstained negative control sample for gating of the correct cell population, and for excluding the fluorescent background signal. Analyze a minimum of 10,000 p63-α -stained cells. For detailed technical implementation, please refer to the user manual of the given flow cytometer.
Representative Results
From hPSCs to hPSC-LESCs
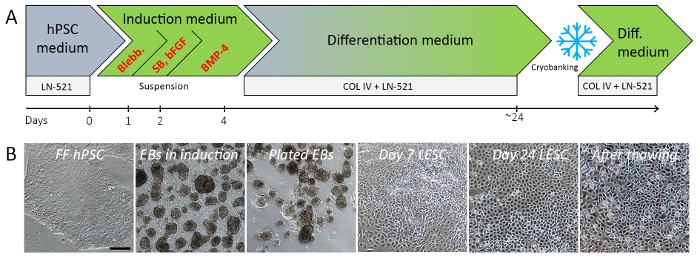
The entire process from inducing differentiation of FF hPSCs to cryostoring hPSC-LESCs takes around 3.5 weeks. Schematic overview of the differentiation method highlighting its key steps is presented in Figure 1A. Figure 1B shows typical morphologies of cell populations in different phases of the protocol. The data presented are obtained with Regea08/017 hESC line and UTA.04607.WT hiPSC line, both derived and characterized at the University of Tampere, Finland, as described previously26,27,28.
On LN-521 in hPSC medium, the undifferentiated, high quality FF hPSCs first form distinct colonies with sharp edges, which merge to homogeneous monolayers upon confluence (Figure 1B, first image). Several individually derived and genetically distinct hESC and hiPSC lines were successfully adapted and cultured with this system. The FF hPSC populations multiply approximately 3-fold within each passage, providing robust means to generate xeno-and feeder-free starting material for differentiations25. The 24 h induction in EB medium typically produces a suspension of tight, regular EBs of varying sizes (Figure 1B, second image). During the surface ectodermal induction in suspension (day 2–4), the EB morphology should not change dramatically. Colonial outgrowth appears soon after the EBs are plated onto col IV/LN-521 combination matrix in differentiation medium (Figure 1B, third and fourth images), and within 21–25 days of differentiation the cells form confluent homogeneous layers with polygonal morphology typical to epithelial cells (Figure 1B, fifth image). The cells may then be cryostored for later use. Viability and morphology are well preserved after thawing the hPSC-LESCs (Figure 1B, last image).
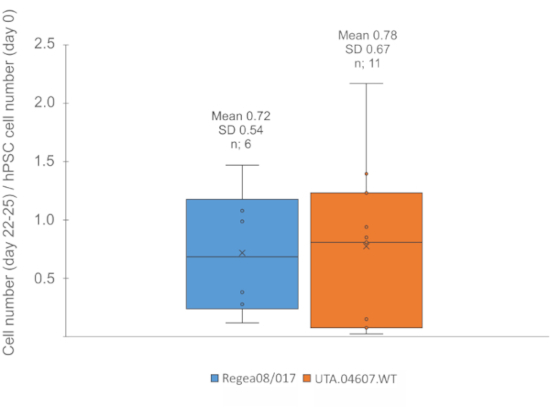
Typically, 3 x 106 FF hPSCs plated to a single 6-well plate yield enough EBs to be plated for adherent culture in 2–3 cell culture dishes (100 mm). From each 100 mm dish, 1 to 1.5 x 106 cells may be harvested for cryobanking by day 22–25 of differentiation. On average, each undifferentiated FF hPSC generates 0.7 cells by day 25 (Figure 2).
Validation of hPSC-derived LESCs
In the absence of specific LESC marker proteins, the correct cell phenotype is confirmed with a set of markers that demonstrate the decrease in expression of the pluripotency associated proteins and increased expression of acknowledged LESC markers. At day 24 of differentiation, the vast majority of the hPSC-derived LESCs express Paired box protein PAX6 (PAX6), the key regulator of eye development, as well as p63α, the widely recognized LESC marker. The truncated p63-isoform ΔNp63 is co-expressed in most of the p63α-positive cells, confirming the most cornea-specific ΔNp63α-positive cell phenotype. Basal epithelial markers and putative LESC markers cytokeratin (CK)-15 and CK-14 are expressed in part, whereas pluripotent stem cell marker OCT3/4 and mature corneal epithelial marker CK-12 are undetectable at this point. This indicates differentiation of FF hPSCs toward the unipotent limbal epithelial progenitors, but not yet terminal differentiation into mature corneal epithelial cells. (Figure 3A) The hPSC-LESCs successfully retain their phenotype after recovery from cryostorage (Data comparable to Figure 3A).
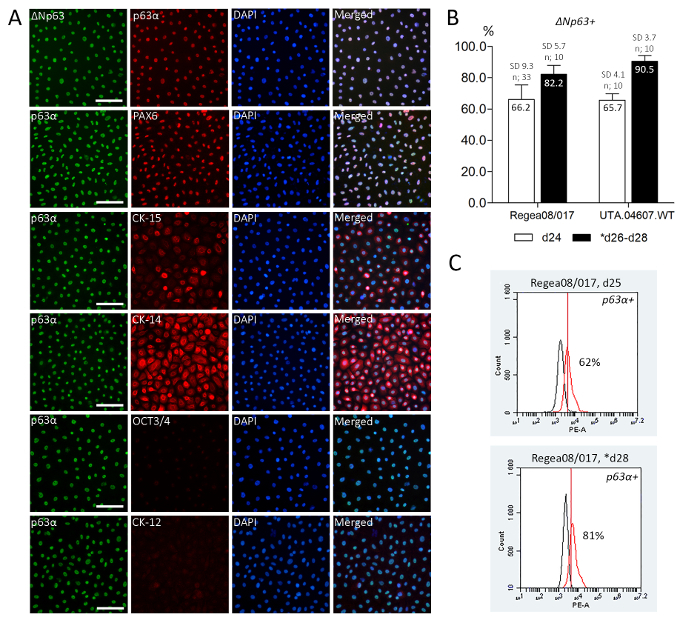
After 24 days of differentiation, ΔNp63 was expressed in 66.2% (n=33, SD 9.3%) of Regea08/017 hESC-LESCs, and in 65.7% (n=10, SD 4.1%) UTA.04607.WT hiPSC-LESCs (quantified from cytospin samples, Figure 3B). After recovery from cryostorage, 82.2% (n=10, SD 5.7%) of hESC-LESCs and 90.5% (n=10, SD=3.7%) of hiPSC-LESCs expressed ΔNp63 (quantified from well plates on day 26–28, Figure 3B).
The p63α expression was further confirmed with flow cytometry analysis for Regea08/017 hESC-LESCs. At day 25 of differentiation, 62% of the freshly differentiated hPSC-LESCs were positive for p63α. Two days after recovery from cryostorage (day 28 in total), 81% of the cells were p63α-positive (Figure 3C).
Figure 1: Schematic illustration of the hPSC-LESC differentiation protocol (A) and typical cell morphologies observed in different phases of the process (B). EBs are formed from a single cell suspension of high-quality, undifferentiated hPSCs during the first 24 h. Three-day small molecule induction toward surface ectoderm in suspension is followed by adherent differentiation phase. By day 24 of differentiation, the majority of the cells show typical LESC-like morphology. Representative images shown for Regea08/017 hESC line before and during differentiation. Black scale bar = 200 µm, valid for all images in the panel. Abbreviations: Blebb.: blebbistatin, SB: SB-505124 small molecule inhibitor, bFGF: basic fibroblast growth factor, BMP-4: bone morphogenetic protein 4, LN-521: human recombinant laminin 521, col IV: human placental collagen type IV, hPSC: human pluripotent stem cell, EB: embryoid body, LESC: limbal epithelial stem cells. Please click here to view a larger version of this figure.
Figure 2: Expected cell yield. Boxplots showing the number of cells obtained for cryostoring on day 22-25 of differentiation, divided by number of undifferentiated hPSCs plated for EB formation step on day 0. On average 0.72 (SD 0.4) cells were produced from each pluripotent Regea08/017 hESC, while 0.78 (SD 0.67) cells were produced from each UTA.04607.WT hiPSC. n: number of differentiation experiments. Please click here to view a larger version of this figure.
Figure 3. Expected phenotype of hPSC-derived LESCs. Immunofluorescence antibody labeling (A) showing uniform expression of eye development regulator PAX6 and acknowledged LESC markers p63α and its ΔNp63 isoform, as well as two other suggested LESC markers - CK15 and 14. Pluripotency marker OCT3/4 and mature corneal epithelial marker CK-12 are negative. Representative IF images shown for Regea08/017 hESC-LESCs at day 24 of differentiation. Scale bars = 100 µm. (B) ΔNp63 cell counting from freshly differentiated and cryostored hPSC-LESCs demonstrates that the cells do not only retain, but show increased ΔNp63 expression after cryostorage. Cell counting results show >65% of the freshly differentiated hPSC-LESCs stained positive for ΔNp63 before the cryobanking procedure, and >80% after successful recovery. n = number of separate differentiation experiments (minimum of 600 cells counted per sample and >1600 cells per time point) (C) Flow cytometry analysis showing 62% of the freshly differentiated hPSC-LESCs and 81% of the thawed cells positive for p63α, confirming the cell counting results for line Regea08/017. * in B-C indicate thawed cells. Red histogram for positive sample and black for negative (unstained) sample. Please click here to view a larger version of this figure.
| Antibody | Host | Dilution |
| Primary antibodies for IF | ||
| PAX6 | rabbit | 1:200 |
| p63α | mouse | 1:200 |
| ΔNp63α | rabbit | 1:200 |
| CK-15 | mouse | 1:200 |
| CK-14 | mouse | 1:200 |
| CK-12 | goat | 1:200 |
| OCT3/4 | goat | 1:200 |
| Secondary antibodies for IF | ||
| Alexa Fluor 568 anti-goat Ig | donkey | 1:800 |
| Alexa Fluor 568 anti-mouse Ig | donkey | 1:800 |
| Alexa Fluor 488 anti-rabbit Ig | donkey | 1:800 |
| Alexa Fluor 488 anti-mouse Ig | donkey | 1:800 |
Table 1: Recommended primary and secondary antibodies used for immunofluorescence (IF) labeling of hPSC-derived LESCs. See Table of Materials for manufacturers.
Discussion
The expected result of this protocol is the successful and robust generation of LESCs from a single cell suspension of FF hPSC within approximately 3.5 weeks. As corneal epithelium develops from surface ectoderm29, the first step of the protocol aims at steering hPSCs towards this lineage. A short 24 h induction with transforming growth factor beta (TGF-β) antagonist SB-505124, and bFGF are used to induce ectodermal differentiation, followed by 48 h mesodermal BMP-4 cue to push the cells towards surface ectoderm. The following adherent differentiation step on col IV/LN-521 combination matrix together with chemically defined differentiation medium is used to further guide differentiation towards LESCs.
High quality of the starting material (the FF hPSCs) is critical for successful differentiation. Only FF hPSC cultures with near confluence and close to 100% of undifferentiated phenotype should be used. Regular hPSC karyotyping is recommended, as single cell passaging can predispose hPSCs to karyotypic abnormalities that lead to growth and differentiation advantages30. Prolonged exposure to trypsin can cause inadequate EB formation or hPSC death. The protocol was tested with five individually derived and genetically distinct hPSC lines, both hESC and hiPSC lines. There was no need for cell line specific modifications to the small molecule concentrations or induction times. However, during the initial optimization of the protocol, excessive cell death or appearance of cells with fibroblastic, neuronal or other morphology indicated differentiation to undesired lineages. In such case, the method might require fine-tuning. The culture vessel formats provide a starting point and can be upscaled from the recommended sizes.
The entire protocol from hPSC culture to LESC differentiation and cryopreservation is defined, allowing easy transition to Good Manufacturing Practice (GMP) for production of cell therapy products. As the commercial media and reagents undergo robust development, manufacture and quality control procedures, they provide a consistent, uniform quality platform for hPSC culture and differentiations.The defined conditions minimize batch-to-batch variation, which offers an advantage over existing hPSC-LESC differentiation protocols10,11,12,13,14,20,22,23. The fast and relatively simple protocol also provides an advantage over three-dimensional corneal organoids which are difficult to standardize across cell lines and laboratories, and require robust methods to purify desired cell types31,32. Additionally, the highly efficient protocol provides robust means to produce LESCs for research purposes, e.g. disease modeling, genetic engineering, drug screening, and toxicological testing. Moreover, the platform can be easily fine-tuned for differentiation of other ocular epithelial cell types such as retinal pigment epithelial cells (RPE cells)25.
Successful limbal cell replacement therapy requires only a few thousand p63-positive LESCs4, per eye, but quality assurance requires additional cell populations and therefore large scale production. Cryobanking allows preparation of readily available cell stocks for transplantation, as well as for quality and safety testing. Further, the hPSC-LESC purity improves after cryostoring, and further after passaging and prolonged culture25, as suggested by increased ΔNp63 expression.
In summary, this robust method generates ΔNp63α-positive cells from hPSCs within 3.5 weeks under xeno- and feeder cell-free culture conditions. The cell culture methods presented here enable production of high-quality LESCs applicable to ocular cell therapy use.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The study was supported by the Academy of Finland (grant number 297886), the Human spare parts program of Tekes, the Finnish Funding Agency for Technology and Innovation, the Finnish Eye and Tissue Bank Foundation and the Finnish Cultural Foundation. The authors thank the biomedical laboratory technicians Outi Melin, Hanna Pekkanen, Emma Vikstedt, and Outi Heikkilä for excellent technical assistance and contribution to cell culture. Professor Katriina Aalto-Setälä is acknowledged for providing the hiPSC line used and BioMediTech Imaging Core facility for providing equipment for fluorescence imaging.
References
- Dua HS, Shanmuganathan VA, Powell-Richards AO, Tighe PJ, Joseph A. Limbal epithelial crypts: a novel anatomical structure and a putative limbal stem cell niche. The British Journal of Ophthalmology. 2005;89(5):529–532. doi: 10.1136/bjo.2004.049742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdanpanah G, Jabbehdari S, Djalilian AR. Limbal and corneal epithelial homeostasis. Current Opinion in Ophthalmology. 2017;28(4):348–354. doi: 10.1097/ICU.0000000000000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Iorio E, Barbaro V, Ruzza A, Ponzin D, Pellegrini G, De Luca M. Isoforms of DeltaNp63 and the migration of ocular limbal cells in human corneal regeneration. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9523–9528. doi: 10.1073/pnas.0503437102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rama P, Matuska S, Paganoni G, Spinelli A, De Luca M, Pellegrini G. Limbal stem-cell therapy and long-term corneal regeneration. The New England Journal of Medicine. 2010;363(2):147–155. doi: 10.1056/NEJMoa0905955. [DOI] [PubMed] [Google Scholar]
- Notara M, et al. In sickness and in health: Corneal epithelial stem cell biology, pathology and therapy. Experimental Eye Research. 2010;90(2):188–195. doi: 10.1016/j.exer.2009.09.023. [DOI] [PubMed] [Google Scholar]
- Osei-Bempong C, Figueiredo FC, Lako M. The limbal epithelium of the eye--a review of limbal stem cell biology, disease and treatment. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology. 2013;35(3):211–219. doi: 10.1002/bies.201200086. [DOI] [PubMed] [Google Scholar]
- Kolli S, Ahmad S, Lako M, Figueiredo F. Successful clinical implementation of corneal epithelial stem cell therapy for treatment of unilateral limbal stem cell deficiency. Stem Cells. 2010;28(3):597–610. doi: 10.1002/stem.276. [DOI] [PubMed] [Google Scholar]
- Sangwan VS, et al. Clinical outcomes of xeno-free autologous cultivated limbal epithelial transplantation: a 10-year study. The British Journal of Ophthalmology. 2011;95(11):1525–1529. doi: 10.1136/bjophthalmol-2011-300352. [DOI] [PubMed] [Google Scholar]
- Basu S, Mohan S, Bhalekar S, Singh V, Sangwan V. Simple limbal epithelial transplantation (SLET) in failed cultivated limbal epithelial transplantation (CLET) for unilateral chronic ocular burns. The British Journal of Ophthalmology. 2018. [DOI] [PubMed]
- Ahmad S, et al. Differentiation of human embryonic stem cells into corneal epithelial-like cells by in vitro replication of the corneal epithelial stem cell niche. Stem Cells. 2007;25(5):1145–1155. doi: 10.1634/stemcells.2006-0516. [DOI] [PubMed] [Google Scholar]
- Hanson C, et al. Transplantation of human embryonic stem cells onto a partially wounded human cornea in vitro. Acta Ophthalmologica. 2013;91(2):127–130. doi: 10.1111/j.1755-3768.2011.02358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi R, et al. Generation of corneal epithelial cells from induced pluripotent stem cells derived from human dermal fibroblast and corneal limbal epithelium. PLoS ONE. 2012;7(9):e45435. doi: 10.1371/journal.pone.0045435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt KJ, Shamis Y, Carlson MW, Aberdam E, Aberdam D, Garlick JA. Three-dimensional epithelial tissues generated from human embryonic stem cells. Tissue Engineering. Part A. 2009;15(11):3417–3426. doi: 10.1089/ten.tea.2009.0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalom-Feuerstein R, et al. Pluripotent stem cell model reveals essential roles for miR-450b-5p and miR-184 in embryonic corneal lineage specification. Stem Cells. 2012;30(5):898–909. doi: 10.1002/stem.1068. [DOI] [PubMed] [Google Scholar]
- Cieslar-Pobuda A, et al. Human induced pluripotent stem cell differentiation and direct transdifferentiation into corneal epithelial-like cells. Oncotarget. 2016;7(27):42314–42329. doi: 10.18632/oncotarget.9791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig TE, et al. Derivation of human embryonic stem cells in defined conditions. Nature Biotechnology. 2006;24(2):185–187. doi: 10.1038/nbt1177. [DOI] [PubMed] [Google Scholar]
- Chen G, et al. Chemically defined conditions for human iPSC derivation and culture. Nature Methods. 2011;8(5):424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodin S, Antonsson L, Hovatta O, Tryggvason K. Monolayer culturing and cloning of human pluripotent stem cells on laminin-521-based matrices under xeno-free and chemically defined conditions. Nature Protocols. 2014;9(10):2354–2368. doi: 10.1038/nprot.2014.159. [DOI] [PubMed] [Google Scholar]
- Mikhailova A, Ilmarinen T, Uusitalo H, Skottman H. Small-molecule induction promotes corneal epithelial cell differentiation from human induced pluripotent stem cells. Stem Cell Reports. 2014;2(2):219–231. doi: 10.1016/j.stemcr.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Garcia de la Torre RA, Nieto-Nicolau N, Morales-Pastor A, Casaroli-Marano RP. Determination of the Culture Time Point to Induce Corneal Epithelial Differentiation in Induced Pluripotent Stem Cells. Transplantation Proceedings. 2017;49(10):2292–2295. doi: 10.1016/j.transproceed.2017.09.047. [DOI] [PubMed] [Google Scholar]
- Zhang C, Du L, Pang K, Wu X. Differentiation of human embryonic stem cells into corneal epithelial progenitor cells under defined conditions. PLoS One. 2017;12(8):e0183303. doi: 10.1371/journal.pone.0183303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aberdam E, Petit I, Sangari L, Aberdam D. Induced pluripotent stem cell-derived limbal epithelial cells (LiPSC) as a cellular alternative for in vitro ocular toxicity testing. PLoS One. 2017;12(6):e0179913. doi: 10.1371/journal.pone.0179913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamarudin TA, et al. Differences in the Activity of Endogenous Bone Morphogenetic Protein Signaling Impact on the Ability of Induced Pluripotent Stem Cells to Differentiate to Corneal Epithelial-Like Cells. Stem Cells. 2018;36(3):337–348. doi: 10.1002/stem.2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodin S, et al. Clonal culturing of human embryonic stem cells on laminin-521/E-cadherin matrix in defined and xeno-free environment. Nature Communications. 2014;5:3195. doi: 10.1038/ncomms4195. [DOI] [PubMed] [Google Scholar]
- Hongisto H, Ilmarinen T, Vattulainen M, Mikhailova A, Skottman H. Xeno- and feeder-free differentiation of human pluripotent stem cells to two distinct ocular epithelial cell types using simple modifications of one method. Stem Cell Research & Therapy. 2017;8(1):4. doi: 10.1186/s13287-017-0738-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skottman H. Derivation and characterization of three new human embryonic stem cell lines in Finland. In vitro Cellular & Developmental Biology. Animal. 2010;46(3-4):206–209. doi: 10.1007/s11626-010-9286-2. [DOI] [PubMed] [Google Scholar]
- Ahola A, Kiviaho AL, Larsson K, Honkanen M, Aalto-Setälä K, Hyttinen J. Video image-based analysis of single human induced pluripotent stem cell derived cardiomyocyte beating dynamics using digital image correlation. Biomedical Engineering Online. 2014;13:39. doi: 10.1186/1475-925X-13-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala M, et al. Mutation-Specific Phenotypes in hiPSC-Derived Cardiomyocytes Carrying Either Myosin-Binding Protein C Or α-Tropomyosin Mutation for Hypertrophic Cardiomyopathy. Stem Cells International. 2016;2016:1684792. doi: 10.1155/2016/1684792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali RR, Sowden JC. Regenerative medicine: DIY eye. Nature. 2011;472(7341):42–43. doi: 10.1038/472042a. [DOI] [PubMed] [Google Scholar]
- International Stem Cell Initiative , et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nature Biotechnology. 2011;29(12):1132–1144. doi: 10.1038/nbt.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JW, Wahlin K, Adams SM, Birk DE, Zack DJ, Chakravarti S. Cornea organoids from human induced pluripotent stem cells. Scientific Reports. 2017;7:41286. doi: 10.1038/srep41286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susaimanickam PJ, et al. Generating minicorneal organoids from human induced pluripotent stem cells. Development. 2017;144(13):2338–2351. doi: 10.1242/dev.143040. [DOI] [PubMed] [Google Scholar]