

Abstract
Multi-omic insights into microbiome function and composition typically advance one study at a time. However, to understand relationships across studies, they must be aggregated into meta-analyses. This makes it possible to generate new hypotheses by finding features that are reproducible across biospecimens and data layers. Qiita dramatically accelerates such integration tasks in a web-based microbiome comparison platform, which we demonstrate with Human Microbiome Project and iHMP data.
Recent years have seen exponential growth in studies that generate large quantities of microbiome and metabolome data, enabled by advances in high-throughput techniques1. New bioinformatics tools allow us to put these samples in the context of other studies, revolutionizing our picture of microbial diversity2, and generating insights into dysbiotic states relevant to human health3. In principle, the vast increase in available data should enable broader and more accurate insights into the diversity and functional impacts of the microbial world. However, these tools require increasing investments of time and effort by highly trained individuals, and more facile meta-analysis of summary statistics is infeasible due to the inconsistency of methods applied by different analysts. Despite these challenges, meta-analyses of microbiomes have a rich history of success, identifying the major global drivers of diversity in microbial communities4, characterizing the evolution of the vertebrate gut microbiome5, and surveying specialized fields such as the built environment6. Meta-analyses also enable scientists to identify important biases such as DNA extraction, primers, or analytical pipelines7,8, which need to be controlled to generate biological discoveries.
To address these challenges, we developed Qiita (https://github.com/biocore/qiita and Supplementary Software), an open-source web-based platform that enables non-bioinformaticians to perform their own analyses and meta-analyses easily using standardized pipelines such as QIIME29 and GNPS10. Analyses are carried out within a simple graphical user interface, starting with primary data and ending with statistical analyses and publication-quality figures.
Meta-analyses typically involve tremendous effort, primarily due to three common issues. First, raw data (e.g., sequence data, spectra, study covariates) are frequently not open or completely accessible11. Second, common standards for sample metadata (i.e., study covariates), such as Minimum Information about any (x) Sequence (MIxS) standards12, are not enforced by the major sequence repositories, leading to varying degrees of use. Third, even when provided, processed data files rarely contain details about the processing itself. Differences in sample or data processing can lead to technical differences that obscure biological differences in the data7,13.
Qiita alleviates these issues using a number of strategies. First, it requires that new studies include a description of the work; relevant publications; collection and processing parameters for each sample; and relevant covariates, based on the MIxS standards12. Only administrator-reviewed standards-compliant metadata are made public (for an example, see Supplementary Table 1). Second, users must upload the rawest form of the data possible, typically multiplexed or demultiplexed FASTQ files. Qiita can thus re-access the raw data as new pipelines and databases are adopted. Third, users select from a constrained set of processing parameters, which are subsequently retained with the data. This tracking and standardization ensures that newly processed data can be immediately compared to hundreds of thousands of samples in the database, and enables automated data deposition into ENA-EBI (as has been performed now for 102,292 samples; Supplementary Fig. 1A). Finally, relevant samples can be discovered via search of study title, metadata values, or even sequence data through the redbiom plugin (https://github.com/biocore/redbiom), and quickly combined for analysis using a QIIME2-based analysis plugin. When more specialized analyses are required, combined feature tables, metadata, and analytical artifacts (e.g. distance matrices, filtered subsets of samples) can be downloaded for use in other pipelines.
By establishing an accessible path from annotated data to interoperable results, Qiita applies the “living data” concept10 of adding value to data by ongoing reprocessing and annotation. To date, this resource hosts over 50TB of omics data from over 460,000 samples originating from studies that span the world (Supplementary Fig. 1B). More than 168,000 of these samples, including the entire recently released Earth Microbiome Project (EMP)2 are public and immediately available for meta-analyses. As this collection grows, it will become increasingly important to improve the quality of associated metadata. “Gold” studies with exceptional metadata are highlighted to promote better practices in the community.
To demonstrate Qiita’s utility, we tested the reproducibility of a study that investigated how microbiomes of Inflammatory Bowel Disease (IBD) subtypes relate to those of healthy individuals3. We combined the 16S data from three studies of IBD-affected cohorts 3,14 and iHMP, with the HMP1 study of healthy individuals15 and a study of Clostridium difficile-affected patients that underwent Fecal Microbiota Transplants (FMT)16. Using the web interface, Principal Coordinates Analysis (PCoA) on Unweighted UniFrac17 computations shows the expected clustering by body site (Fig. 1A). However, examining only fecal samples (‘UBERON:feces’ category) reveals a pattern explained by sequencing platform as previously observed8 (Fig. 1B). Restricting analysis to samples processed using the same sequencing platform (all but the HMP1 study), produces spatial enrichment of the different IBD subtypes as previously reported3,14 (Fig. 1C). Employing the feces-only distance matrix generated via the Qiita interface, we used QIIME2 to calculate the distance from each sample to a “healthy plane”3, replicating the PCoA result across these independent studies. The C. difficile samples are also further from the healthy plane than the IBD subtype samples, yet are much closer to the healthy plane after restoration of the microbiome via FMT (Fig. 1D). This analysis took under 5 minutes of hands-on time, and did not require manual intervention between pipeline initiation and use of the files in a Jupyter Notebook (https://github.com/knightlab-analyses/qiita-paper).
Figure 1.
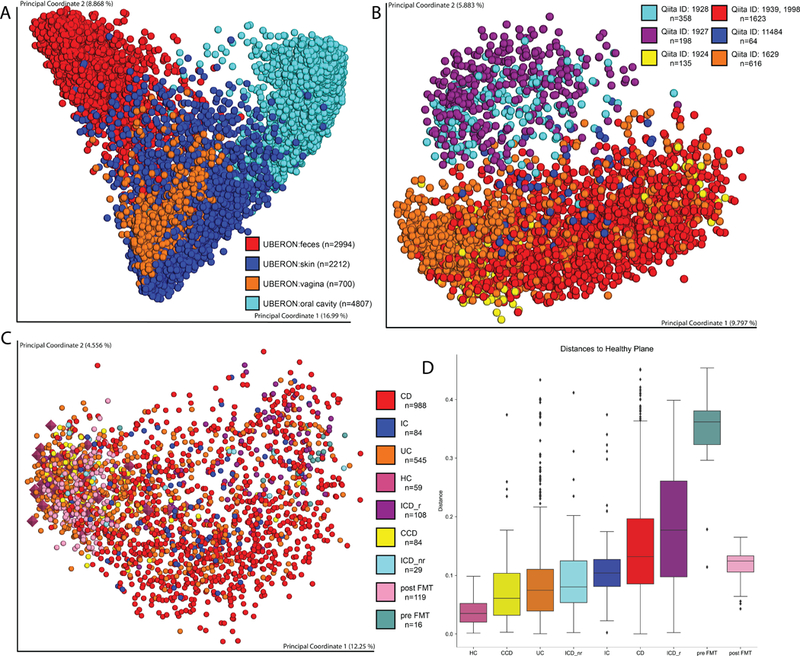
Example meta-analysis in Qiita. A. Unweighted UniFrac PCoA meta-analysis of three studies examining different IBD subtypes, C. difficile patients who underwent FMT, and the HMP1 and iHMP data. B. Only fecal samples from the same studies as in A. C. Fecal samples only from studies that used the same data-generation methods. D. Calculated distances from a healthy plane as described in Ref. 3. Box plots show the median value (center line), the upper and lower quartiles of the data (box edges), maxima and minima (whiskers), and outliers (individual data points). CD, Crohn’s disease; IC, ileal Crohn’s disease; UC, ulcerative colitis; HC, healthy cohort; CCD, colonic Crohn’s disease; ICD_r, ileal Crohn’s disease patients with previous ileocecal resection; ICD_nr, ileal Crohn’s disease patients with no previous ileocaecal resection; post-FMT, patients with C. difficile infection pre-FMT; pre-FMT, patients with C. difficile infection post-FMT.
Qiita provides a unique resource allowing researchers to contextualize their data, perform meta-analyses across hundreds of studies and thousands of samples, and seamlessly deposit data into standards-compliant databases. Custom instances of Qiita can also be easily set up on virtual or physical machines to host specific datasets (e.g. the iHMP IBDMDB, http://ihmp.ucsd.edu/). We expect that Qiita will assist researchers considerably in conducting microbiome analyses and meta-analyses.
Online Methods
Code design
Qiita is designed using a three layer pattern: storage, logic, and interface. We describe each layer individually.
The storage layer design is a combination of a PostgreSQL 9.3.17 database and a structured filesystem. This approach allows Qiita to maintain referential integrity within and between studies, sample metadata, the analysis pipeline(s), and the commands executed over the different data types. However, the data volume is such that it can encumber a relational database, so the data (e.g., sequence files, contingency tables etc.) are stored in standard formats (e.g FASTA, FASTQ, BIOM). The database maintains file path locations using indirection to allow files to reside on any number of filesystems. Additionally, this layer also stores the covariates (metadata) of each sample split in two main tables: a sample and a preparation information. The sample information are the covariates pertinent to the sample, while the preparation is how the sample was processed in the wet-lab and data generation (target gene sequencing, shotgun, metabolomic, etc).
The Qiita logic layer is written in Python using Object Oriented Programming, defining an object for each important element of the system. All data in Qiita are represented by an “artifact” object. An artifact represents a collection of files which reside on the filesystem, the logical types associated with each file, and a logical type of the artifact itself. Commands can specify which type of artifacts they accept as input and which type of artifacts they generate as output. The type of artifacts and the commands used to analyze artifacts are defined by Qiita plugins, which encapsulate the compute logic. Qiita defines two types of plugins: Qiita Type Plugins and Qiita Plugins. The Qiita Type Plugins define new artifact types, and is how data are imported into Qiita. A Qiita Type Plugin must define only two operations: “Validate” and “Generate HTML summary”. The “Validate” operation receives as input the set of files, and user associated types, for a new artifact and the preparation information and determines if the set of files defines a valid “artifact” for the given preparation. For example, in the case of a set of per-sample FASTQ files, the validator checks that each of the samples has a unique file, and that the names of these files match those in the run_prefix column in the preparation information. The “Generate HTML summary” obtains the contents of an artifact and generates an HTML file summarizing the contents of such artifact. This summary provides a user-interpretable overview of the artifact, usually helpful enough to determine if something went wrong with the processing of the artifact. In contrast, the Qiita Plugin represents a collection of logically related commands (e.g., methods for constructing distance matrices). Each command within a Qiita Plugin accepts one or more artifacts as input, runtime parameters, and produces one or more artifacts as output. Each command execution is logged in the Qiita relational database, specifically, Qiita stores the plugin used, the command executed within the plugin, the artifacts provided as inputs, the parameters specified, and the artifacts generated.
The motivation for a modular plugin system is separation of concerns and encapsulation as each plugin runs in its own discrete environment and communicates with Qiita through an internal communication layer. This approach allows the plugins to be written in any programming language, with plugin specific dependencies, without introducing dependency conflicts with other plugins in the system. These environments are managed using plugin-specific conda environments. To facilitate the development of new Qiita plugins by external developers, we have created a Qiita client library (https://github.com/qiita-spots/qiita_client) and two Cookiecutter (Qiita Type Plugin: https://github.com/qiita-spots/qtp-template-cookiecutter & Qiita Plugin: https://github.com/qiita-spots/qp-template-cookiecutter) templates that set up the boilerplate code needed for an initial plugin repository and communication with Qiita.
The interface layer is a web-based interface accessible via Google Chrome, and that is powered from the server side via Tornado 3.1.1 (http://www.tornadoweb.org/). The interface design and implementation has gone through multiple rounds of review, utilizing feedback kindly provided by users attending Qiita workshops.
The source code, and comprehensive test suite, for the Qiita package can be found in https://github.com/biocore/qiita. The source code for the officially supported Qiita plugins can be found under the qiita-spots GitHub organization at https://github.com/qiita-spots. All source code in the qiita repository and qiita-spots organization are BSD-licensed.
Data analysis
One of the most important items for a successful meta-analysis is consistency during the data processing. To achieve this consistency, Qiita processes all raw data with one of several standard parameter sets, based on the recommendations published in the literature. The parameters for demultiplexing and quality control the 16S rRNA gene sequences are based on the assessment performed Bokulich et al.18, while the parameters for OTU picking are based on the recommendations provided in Navas-Molina et al19. In addition to OTU picking, Qiita also permits sub-OTU sequence clustering with Deblur20. In the deblur manuscript, the authors used more stringent quality control parameters from those outlined by Bokulich et al.18.
Comparison to other resources
Qiita contains information from more samples than does MG-RAST (326,705 samples spanning 1.195 billion sequences) or the EBI Metagenomics Portal (113,805 samples, number of sequences not readily available), although the latter two resources likely contain more shotgun metagenomics datasets than does Qiita at present. Qiita uses a more up-to-date version of QIIME than does MG-RAST or the last QIIME-based version of the EBI metagenomics portal, and offers a choice of taxonomy databases (Greengenes, RDP and SILVA).
Statistics
Figures 2A, 2B, and 2C show the 3 first Principal Coordinates of a PCoA based on the unweighted UniFrac distances of the close reference picking independent samples rarefied at 1000 sequences per sample and visualized via Emperor21. The boxplots in Figure 2C follow the Seaborn22 defaults; in brief, each boxplot represent the quartiles of the data, the whiskers extend to show the rest of the distribution, except for outliers determined using a method that is a function of the inter-quartile range.
Supplementary Material
Acknowledgements
We are grateful to Justine Debelius, Janet Jansson, Dante Bazaldua, and Justin Kuczynski for their help improving Qiita via suggestion, code changes, contributed data sets, or during the preparation of this manuscript; and Jeff Gordon and his laboratory for helpful discussions. This work was supported in part by Alfred P. Sloan Foundation 2017–9838 & 2015–13933, NIH/NIDDK P01DK078669, NSF DBI-1565057 & 1565100, Office of Naval Research (ONR) N00014–15-1–2809, and U.S. ARMY CDMRP W81XWH-15–1-0653.
Footnotes
Competing Financial Interests Statement
The authors declare no competing financial interests.
Data availability
All data used is available via Qiita and EBI (where applicable). The Human Microbiome Project (HMP) and Integrative Human Microbiome Project (iHMP) data is available via the HMP Data Analysis and Coordination Center (DACC) https://hmpdacc.org/, Table 2. Analytical steps for this paper can be found in https://github.com/knightlab-analyses/qiita-paper. Additionally, the Qiita Analysis can be found here: https://qiita.ucsd.edu/analysis/description/15093/, you must be log in to Qiita to access it.
Code availability
All Qiita code is available in https://github.com/biocore/qiita and the public deploy is at http://qiita.microbio.me/.
References
- 1.Caporaso JG et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624, doi:10.1038/ismej.2012.8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson LR et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463, doi:10.1038/nature24621 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halfvarson J et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol 2, 17004, doi:10.1038/nmicrobiol.2017.4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lozupone CA & Knight R Global patterns in bacterial diversity. Proc Natl Acad Sci U S A 104, 11436–11440, doi:10.1073/pnas.0611525104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley RE, Lozupone CA, Hamady M, Knight R & Gordon JI Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol 6, 776–788, doi:10.1038/nrmicro1978 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adams RI, Bateman AC, Bik HM & Meadow JF Microbiota of the indoor environment: a meta-analysis. Microbiome 3, 49, doi:10.1186/s40168-015-0108-3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Debelius J et al. Tiny microbes, enormous impacts: what matters in gut microbiome studies? Genome Biol 17, 217, doi:10.1186/s13059-016-1086-x (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lozupone CA et al. Meta-analyses of studies of the human microbiota. Genome Res 23, 1704–1714, doi:10.1101/gr.151803.112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caporaso JG et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336, doi:10.1038/nmeth.f.303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat Biotechnol 34, 828–837, doi:10.1038/nbt.3597 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langille MGI, Ravel J & Fricke WF “Available upon request”: not good enough for microbiome data! Microbiome 6, 8, doi:10.1186/s40168-017-0394-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yilmaz P et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat Biotechnol 29, 415–420, doi:10.1038/nbt.1823 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sinha R et al. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat Biotechnol 35, 1077–1086, doi:10.1038/nbt.3981 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gevers D et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392, doi:10.1016/j.chom.2014.02.005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214, doi:10.1038/nature11234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weingarden A et al. Dynamic changes in short- and long-term bacterial composition following fecal microbiota transplantation for recurrent Clostridium difficile infection. Microbiome 3, 10, doi:10.1186/s40168-015-0070-0 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lozupone C & Knight R UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71, 8228–8235, doi:10.1128/AEM.71.12.8228-8235.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bokulich NA et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10, 57–59, doi:10.1038/nmeth.2276 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navas-Molina JA et al. Advancing our understanding of the human microbiome using QIIME. Methods Enzymol 531, 371–444, doi:10.1016/B978-0-12-407863-5.00019-8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amir A et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2, doi:10.1128/mSystems.00191-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods Only References
- 21.Vazquez-Baeza Y, Pirrung M, Gonzalez A & Knight R EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2, 16, doi:10.1186/2047-217X-2-16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waskom, M. Seaborn: statistical data visualization. (2012).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.