

Abstract
Tandem mass spectrometry (MS/MS) is the primary method for discovering, identifying, and localizing post-translational modifications (PTMs) in proteins. However, conventional positive ion mode collision induced dissociation (CID)-based MS/MS often fails to yield site-specific information for labile and acidic modifications due to low ionization efficiency in positive ion mode and/or preferential PTM loss. While a number of alternative methods have been developed to address this issue, most require specialized instrumentation or indirect detection. In this work, we present an amine-reactive TEMPO-based free radical initiated peptide sequencing (FRIPS) approach for negative ion mode analysis of phosphorylated and sulfated peptides. FRIPS-based fragmentation generates sequence informative ions for both phosphorylated and sulfated peptides with no significant PTM loss. Furthermore, FRIPS is compared to positive ion mode CID, electron transfer dissociation (ETD) as well as negative ion mode electron capture dissociation (niECD) and CID, both in terms of sequence coverage and fragmentation efficiency for phospho- and sulfopeptides. Because FRIPS-based fragmentation has no particular instrumentation requirements and shows limited PTM loss, we propose this approach as a promising alternative to current techniques for analysis of labile and acidic PTMs.
Graphical Abstract
While mass spectrometry (MS) remains the primary method for annotating protein post-translational modifications (PTMs),1–3 acidic PTMs such as sulfation and phosphorylation are often not detected or are challenging to localize with conventional methods. In fact, it has been suggested that only 20–40% of phosphopeptides are detected with standard MS techniques4 and sulfopeptides are notoriously difficult to observe under standard conditions.5–8 Given the critical biological relevance of these acidic modifications, it is a priority to develop robust analytical techniques for their direct analysis, accessible without the need for costly, specialized instrumentation.3,8
Phosphorylation and sulfation are challenging to analyze with conventional MS techniques for two major reasons: 1) their acidic nature, and 2) their gas-phase lability. Ionization in positive ion mode electrospray-based experiments requires protonation of the analyte. Acidic PTMs resist protonation, thus interfering with ionization and limiting their detection. Once ionized, the site of a PTM can be identified if the peptide backbone is cleaved by tandem mass spectrometry (MS/MS) without loss of the modification. MS/MS is typically performed with collision induced dissociation (CID), which preferentially cleaves the lowest energy bonds. While CID is the most widely available activation method for MS/MS, collisional activation of la-bile PTM-modified peptides preferentially cleaves PTM-linked bonds, limiting sequence informative fragmentation and precluding PTM site localization.5,9 Alternative activation techniques involving UV photons or electrons have shown promise in positive ion mode analyses of phosphopeptides and metal adducted sulfopeptides, but are hindered by the reduced ion abundance of these species.10–14 Furthermore, in the absence of metal ligands the extreme proton-mediated lability of the sulfate group triggers loss during the positive ion mode ionization process and even under the gentlest activation techniques.4,7,15
Conversely, negative ion mode electrospray ionization improves ion abundance of both phospho- and sulfopeptides.5 Negative ion mode also adequately increases sulfopeptide stability to remain mostly intact through the ionization process, but not sufficiently to survive collisional activation.7 Ultraviolet photodissociation (UVPD) presents a promising approach to overcome these issues. In UVPD, sulfopeptide anions are irradiated with 193 nm photons to promote backbone dissociation with little PTM loss.10,16 Our laboratory and others also previously achieved peptide anion dissociation through the introduction or removal of an electron, initiating radical-driven backbone cleavages while retaining the PTM. Metastable atom-activated dissociation (MAD), electron-detachment dissociation (EDD), negative-ion electron capture dissociation (niECD), and negative electron transfer dissociation (NETD) are some of the techniques that utilize electron-based fragmentation and differ by the manner in which the electron is introduced or removed. Each technique has demonstrated success in the site specific localization of acidic PTMs.17–22 However; many of these techniques, while successful, require advanced instrumentation. Moreover, for many of these techniques, the reaction times required for sufficient product ion signal-to-noise ratios are not ideal for standard LC/MS proteomics workflows.
Free radical initiated peptide sequencing (FRIPS) is an alternative radical-driven MS/MS technique in which an odd-electron species is generated through homolytic bond cleavage of a stable free radical initiator, added to free amines via N-hydroxy-succinimide (NHS) chemistry.23,24 Radical initiation is accomplished through collisional activation, initiating a series of radical propagated reactions to achieve peptide backbone cleavage (Scheme 1). Unlike externally driven electron-based reactions, FRIPS and related techniques can be applied to peptides regardless of charge state with duty cycles approaching those of standard CID methods.23–32 Despite the growing number of reports on FRIPS applications, to date, we are unaware of FRIPS to localize labile acidic modifications in negative ion mode.29,33
Scheme 1.

Here we demonstrate that negative ion mode TEMPO-based FRIPS30 generates a virtually complete set of sequence ions for site-specific assignment of sulfation and phosphorylation in peptides with little neutral PTM loss. We also demonstrate that FRIPS can outperform other MS/MS techniques in both sequence coverage and fragmentation efficiency.
Assignment of Phosphorylation Sites with Negative Ion Mode FRIPS.
Because radical initiation in FRIPS is achieved through a low energy collisionally-induced homolytic cleavage, the initiation step is in direct competition with other low energy CID processes. Thus, for FRIPS efficiency to be high, the targeted chemical bond in the radical initiator must be highly la-bile. While the phosphodiester bond is considerably more stable than the sulfodiester bond,34 both phosphate and sulfate groups are labile upon CID and could thus effectively compete with FRIPS radical initiation. Accordingly, we first examined the FRIPS applicability towards labile modifications with the following phosphorylated peptides: tyrosine phosphopeptide (TyPP, TSTEPQyPGEN) and serine phosphopeptide (SePP, RRAsVA).
Following collisional activation, the MS/MS spectrum of doubly deprotonated, underivatized TyPP is dominated by products lacking phosphate or phosphoric acid. The two product ions, b8 and b10, that retained the PTM, are not sufficient to localize the modification to a single residue (Figure 1A). After conjugation with o-TEMPO-Bz, however, equivalent collisional activation of doubly deprotonated TyPP generates the truncated, radical methyl-Bz containing species (the subscript “r” denotes this additional mass, when present).30 Subsequent isolation and further collisional activation yields significant peptide backbone cleavage with little neutral loss of the phospho group (Figure 1B). FRIPS-based fragmentation resulted in a virtually complete set of product ions and enabled the precise assignment of the phospho group to Tyrosine 7. The formation of predominately cʹ-, z•-, a•-, and ʹx-type ions are indicative of radical-mediated dissociation (Figure 1B).
Figure 1.

CID MS/MS spectra of doubly deprotonated tyrosine phosphopeptide (TyPP, A) and singly deprotonated serine phosphopeptide (SePP, C). MS3 spectra of the same peptides conjugated to o-TEMPO-Bz and subjected to FRIPS-based dissociation (B, D). r subscript: product ions that retained the truncated methyl-Bz tag. *: phosphorylated residue.
When applied to the singly deprotonated, TEMPO-tagged species of the phosphorylated peptide SePP, FRIPS-mediated dissociation once again outperforms collisional activation. While CID was able to properly assign the PTM to Serine 4 (Figure 1C), FRIPS generated a complete set of product ions, again providing increased confidence in the assignment (Figure 1D). Additionally, radical-mediated FRIPS dissociation generates significant neutral side chain loss (Figures 1B, 2B, and 2D).29 These peaks are annotated in each spectrum according to the system proposed by Julian and co-workers and can be used to improve the confidence of a peptide assignment.28,32
Figure 2.

CID MS2 spectra of doubly deprotonated cholecystokinin sulfate (CCKS, A) and hirudin (C). MS3 spectra of the same peptides conjugated to o-TEMPO-Bz, and subjected to FRIPS-based dissociation (B, D). r subscripts: product ions that retained the truncated tag. *: sulfated residue.
Assignment of Sulfation Sites with Negative Ion Mode FRIPS.
The above observed homolytic cleavage of the o-TEMPO-Bz moiety prior to loss of the phosphate group suggests that this desired cleavage within the radical initiator requires less energy than phosphodiester bond cleavage. To examine whether the energetics of the o-TEMPO-Bz cleavage pathway are sufficiently low to also outcompete the elimination of a sulfo group, we applied FRIPS to the sulfopeptides hirudin (DFEEIPEEY*LQ) and cholecystokinin (CCKS, DY*MGWMDF-NH2).
Similarly to phosphopeptides, when doubly deprotonated CCKS is collisionally activated the resulting MS/MS spectrum predominately shows PTM elimination products (Figure 2A). Only a y7 ion including the sulfate is generated, showing, as expected, that the CID inability to generate PTM-containing product ions is even more pronounced for sulfopetides compared with phosphopeptides. However, when conjugated with o-TEMPO-Bz and collisionally activated, homolytic o-TEMPOBz cleavage once again outcompetes PTM elimination. Isolation of the resulting radical-containing species and further collisional activation generates a near complete set of radically derived product ions (Figure 2B). The observed a-, c-, x-, and z-type ions enable unambiguous assignment of the sulfation site. The application of CID and FRIPS to doubly deprotonated hirudin yielded similar results as for CCKS: CID failed to produce sufficient sequence coverage to confirm the location of the sulfation and FRIPS generated significantly improved sequence coverage, enabling the precise assignment of the sulfation to Tyrosine 9 (Figures 2C and2D).
The application of FRIPS to sulfated peptides presented some insight into the mechanism of TEMPO-based FRIPS. The elimination of a sulfate group in limited proton mobility environments has been calculated to require between 150 and 240 kcal mol-1.35,36 The preferential dissociation of the o-TEMPO-Bz moiety in the presence of these PTMs suggests that the homo-lytic cleavage must require lower energies than elimination of either PTM and, indeed, we calculate its dissociation energy to be 49.3 kcal mol−1 with B3LYP level theory using a 6–31G* basis set.
Comparison of Negative Ion Mode FRIPS to other MS/MS Methods.
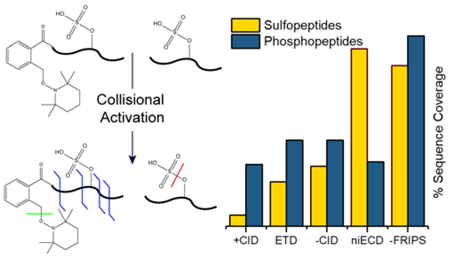
To assess the potential of negative ion mode FRIPS for analysis of acidic PTM-containing peptides, we also analyzed each peptide with ETD, positive ion mode CID, and niECD, a technique that has previously shown superior performance for sulfation analysis. Negative ion mode FRIPS, on average, outperforms all other dissociation techniques at generating product ions and maximizing sequence coverage (Figure 3). As expected, positive ion mode analyses of sulfopeptides were particularly poor, showing low coverage of hirudin (approximately 10%), and the PTM failing to survive the ionization process for CCKS (Figure 3A, S1 and S2). While negative ion mode CID does demonstrate improved sequence coverage compared with the positive ion mode techniques, it still is inferior to niECD in most cases and to FRIPS in every example (Figures 3A and S3). For sulfated peptides, niECD is comparable to FRIPS in sequence coverage, even surpassing it for hirudin. When probing phosphorylated peptides, however, FRIPS eclipsed niECD. Sequence coverage is vital for the site specific localization of a PTM and, thus, an important measure of a technique’s applicability. Additionally, the number of generated product ions may improve peptide assignment confidence in a proteomics workflow.
Figure 3.

Observed sequence coverage for hirudin, cholecystokinin, serine- and tyrosine phosphopeptide following various MS/MS techniques (A). Abundance of sequence ions that retain the PTM (B). *: insufficient detection of intact peptide.
Another important factor in peptide scoring is product ion abundance. Typically, when CID or exogenous radical-mediated techniques are applied to phosphorylated and sulfated peptides, neutral losses or low fragmentation efficiencies suppress sequence informative product ion signals and thus yield lower peptide scores.37 FRIPS, however, on average generates sequence informative ions that retain the PTM at higher ion abundances than all of the other techniques (Figure 3B). The lone exception is positive ion mode CID of TyPP. This peptide contains two proline residues and the majority of the observed product ion abundance is derived from the cleavage of the associated labile peptide bonds.
The increased sequence coverage and product ion abundance, the charge state independence, and duty cycles approaching those of CID are all attributes that make FRIPS attractive for large scale labile-PTM profiling workflows. For peptide phosphorylation and sulfation analysis, negative ion mode FRIPS induces significant backbone cleavage while demonstrating limited PTM loss, enabling the localization of the PTM to a single residue. Given the universal implementation on any mass spectrometer capable of collisional activation, we predict that FRIPS can be a powerful tool for the analysis of these and other labile PTMs.
Supplementary Material
ACKNOWLEDGMENT
Financial support was provided by the National Science Foundation CHE 16–09840 (KH), the National Institutes of Health F31 EB019320 (CTMBT), DP2 GM114848 (BRM), and S10 OD021619 (acquisition of Thermo Scientific Orbitrap Fusion Lumos), and the University of Michigan. We thank Adam W. Gann for helpful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Additional ECD, ETD, and CID MS/MS spectra, Materials, o-TEMPO-Bz-NHS synthesis, and methods. These items can be found online free of charge at http://pubs.acs.org/.
REFERENCES
- (1).Witze ES; Old WM; Resing KA; Ahn NG Nat. Methods 2007, 4, 798–806. [DOI] [PubMed] [Google Scholar]
- (2).Olsen JV; Mann M Mol. Cell. Proteomics 2013, 12, 3444–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Analysis of Protein Post-Translational Modifications by Mass Spectrometry; Griffiths JR; Unwin RD, Eds.; 1st ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017. [Google Scholar]
- (4).Hsu C; Xue L; Arrington JV; Wang P; Paez JSP; Zhou Y; Zhu J; Tao WA J. Am. Soc. Mass Spectrom. 2017, 28, 1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Nemeth-Cawley JF; Karnik S; Rouse JC J Mass Spectrom 2001, 36, 1301–1311. [DOI] [PubMed] [Google Scholar]
- (6).Severs JC; Carnine M; Eguizabal H; Mock KK Rapid Commun. Mass Spectrom. 1999, 13, 1016–1023. [DOI] [PubMed] [Google Scholar]
- (7).Yagami T; Kitagawa K; Futaki S Rapid Commun. Mass Spectrom. 1995, 9, 1335–1341. [DOI] [PubMed] [Google Scholar]
- (8).Yu Y; Hoffhines AJ; Moore KL; Leary JA Nat. Methods 2007, 4, 583–588. [DOI] [PubMed] [Google Scholar]
- (9).Medzihradszky KF; Guan S; Maltby DA; Burlingame AL J. Am. Soc. Mass Spectrom. 2007, 18, 1617–1624. [DOI] [PubMed] [Google Scholar]
- (10).Robinson MR; Taliaferro JM; Dalby KN; Brodbelt JS J. Proteome Res. 2016, 15, 2739–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Halim MA; MacAleese L; Lemoine J; Antoine R; Dugourd P; Girod MJ Am. Soc. Mass Spectrom. 2018, 29, 270–283. [DOI] [PubMed] [Google Scholar]
- (12).Kim T; Reilly JP J. Am. Soc. Mass Spectrom. 2009, 20, 2334–2341. [DOI] [PubMed] [Google Scholar]
- (13).Cook SL; Zimmermann CM; Singer D; Fedorova M; Hoffmann R; Jackson GP J. Mass Spectrom. 2012, 47, 786–794. [DOI] [PubMed] [Google Scholar]
- (14).Liu H; Håkansson K Anal. Chem. 2006, 78, 7570–7576. [DOI] [PubMed] [Google Scholar]
- (15).Monigatti F; Hekking B; Steen H Biochim. Biophys. Acta -Proteins Proteomics 2006, 1764, 1904–1913. [DOI] [PubMed] [Google Scholar]
- (16).Robinson MR; Moore KL; Brodbelt JS J. Am. Soc. Mass Spectrom. 2014, 25, 1461–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Huzarska M; Ugalde I; Kaplan DA; Hartmer R; Easterling ML; Polfer NC Anal. Chem 2010, 82, 2873–2878. [DOI] [PubMed] [Google Scholar]
- (18).Coon JJ; Shabanowitz J; Hunt DF; Syka JE J. Am. Soc. Mass Spectrom. 2005, 16, 880–882. [DOI] [PubMed] [Google Scholar]
- (19).Hersberger KE; Håkansson K Anal. Chem 2012, 84, 6370–6377. [DOI] [PubMed] [Google Scholar]
- (20).Cook SL; Jackson GP J. Am. Soc. Mass Spectrom. 2011, 22, 1088–1099. [DOI] [PubMed] [Google Scholar]
- (21).Yoo HJ; Wang N; Zhuang S; Song H; Håkansson K J. Am. Chem. Soc 2011, 133, 16790–16793. [DOI] [PubMed] [Google Scholar]
- (22).Budnik BA; Haselmann KF; Zubarev RA Chem. Phys. Lett 2001, 342, 299–302. [Google Scholar]
- (23).Hodyss R; Cox HA; Beauchamp JL J. Am. Chem. Soc 2005, 127, 12436–12437. [DOI] [PubMed] [Google Scholar]
- (24).Oh HB; Moon B Mass Spectrom. Rev 2015, 34, 116–132. [DOI] [PubMed] [Google Scholar]
- (25).Chu IK; Rodriquez CF; Lau T; Hopkinson AC; Siu KWM J. Phys. Chem. B 2000, 104, 3393–3397. [Google Scholar]
- (26).Masterson DS; Yin H; Chacon A; Hachey DL; Norris JL; Porter NA J. Am. Chem. Soc 2004, 126, 720–721. [DOI] [PubMed] [Google Scholar]
- (27).Yin H; Chacon A; Porter NA; Masterson DS J. Am. Soc. Mass Spectrom. 2007, 18, 807–816. [DOI] [PubMed] [Google Scholar]
- (28).Sun Q; Nelson H; Ly T; Stoltz BM; Julian RR J. Proteome Res. 2009, 8, 958–966. [DOI] [PubMed] [Google Scholar]
- (29).Hage C; Ihling CH; Götze M; Schäfer M; Sinz A J. Am. Soc. Mass Spectrom. 2017, 28, 56–68. [DOI] [PubMed] [Google Scholar]
- (30).Lee M; Kang M; Moon B; Oh HB Analyst 2009, 134, 1706–1712. [DOI] [PubMed] [Google Scholar]
- (31).Laskin J; Yang Z; Lam C; Chu IK Int. J. Mass Spectrom. 2012, 316–318, 251–258. [Google Scholar]
- (32).Moore B; Sun Q; Hsu JC; Lee AH; Yoo GC; Ly T; Julian RR J. Am. Soc. Mass Spectrom. 2012, 23, 460–468. [DOI] [PubMed] [Google Scholar]
- (33).Lee J; Park H; Kwon H; Kwon G; Jeon A; Kim HI; Sung BJ; Moon B; Oh HB Anal. Chem 2013, 85, 7044–7051. [DOI] [PubMed] [Google Scholar]
- (34).Rappsilber J; Steen H; Mann M J. Mass Spectrom. 2001, 36, 832–833. [DOI] [PubMed] [Google Scholar]
- (35).Škulj S; Rožman M Int. J. Mass Spectrom. 2015, 391, 11–16. [Google Scholar]
- (36).Shi X; Huang Y; Mao Y; Naimy H; Zaia J J. Am. Soc. Mass Spectrom. 2012, 23, 1498–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Haas W; Faherty BK; Gerber SA; Elias JE; Beausoleil SA; Bakalarski CE; Li X; Villén J; Gygi SP Mol. Cell. Proteomics 2006, 5, 1326–1337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.