

Abstract
Background:
Rho-associated kinases (ROCK1 and ROCK2) are important regulators of the actin cytoskeleton and endothelial nitric oxide synthase (eNOS). Because the phosphorylation of eukaryotic elongation factor-1 A1 (eEF1 A1) by ROCK2 is critical for eNOS expression, we hypothesized that this molecular pathway may play a critical role in neuroprotection following focal cerebral ischemia.
Methods and Results:
Adult male wild-type (WT) and mutant ROCK2 and eNOS−/− mice were subjected to middle cerebral artery occlusion (MCAO), and cerebral infarct size, neurological deficit and absolute cerebral blood flow were measured. In addition, aortic endothelium-dependent response to acetylcholine, NG-nitro-L-arginine methyl ester (L-NAME) and sodium nitroprusside were assessed ex vivo. Endothelial cells from mouse brain or heart were used to measure eNOS and eEF1A activity, as well as NO production and eNOS mRNA half-life. In global hemizygous ROCK2+/− and endothelial-specific EC-ROCK2−/− mice, eNOS mRNA stability and eNOS expression were increased, which correlated with enhanced endothelium-dependent relaxation and neuroprotection following focal cerebral ischemia. Indeed, when ROCK2+/− mice were place on an eNOS−/− background, the neuroprotective effects observed in ROCK2+/− mice were abolished.
Conclusions:
These findings indicate that the phosphorylation of eEF1A1 by ROCK2 is physiologically important for eNOS expression and NO-mediated neuroprotection, and suggest that targeting endothelial ROCK2 and eEF1A may have therapeutic benefits in ischemic stroke and cardiovascular disease.
Keywords: Cerebral ischemia, Endothelial nitric oxide synthase, Eukaryotic elongation factor-1 A, mRNA stability, Rho kinase
Rho-associated coiled-coil-containing kinases (ROCK1 and ROCK2) are important regulators of cell contractility, motility and proliferation.1 In the vascular wall, ROCKs mediate smooth muscle contraction, and their abnormal activities could contribute to coronary and cerebral vasospasm,2 hypertension3 and pulmonary hypertension.4 ROCKs are also critical regulators of endothelial function. In particular, we have previously shown that ROCK1nhibitors (e.g., Y27632, Fasudil) or HMG-CoA reductase inhibitors (e.g., statins), improve endothelial function, increase cerebral blood flow (CBF), and protects against focal cerebral ischemia.5–8 Indeed, in patients with ischemic stroke, leukocyte ROCK activity is elevated within 48 h of stroke onset,9 and acute treatment with the ROCK1nhibitor, fasudil, leads to improvements in both neurological function and clinical outcome.10 Thus, the inhibition of ROCKs by statins may contribute to some of their cholesterol-independent or pleiotropic neuroprotective effects.11–13
One of the primary mechanisms underlying statin pleiot-ropy on endothelial function is due to the upregulation of endothelial nitric oxide synthase (eNOS). The increase in eNOS expression is due to the stabilization of eNOS mRNA.7,14 Furthermore, statins also increase eNOS phosphorylation and activity through the rapid activation of protein kinase Akt.15,16 Because statins can inhibit ROCK activity,13,17 ROCKs, therefore, may be important pathophysiological mediators of endothelial function through their inhibitory effects on both eNOS expression and activity. However, statins and ROCK1nhibitors such as fasudil and Y27632 are non-selective and cannot distinguish between the 2 ROCK1soforms. Furthermore, when given in vivo, they cannot distinguish ROCK1nhibition in a tissue-specific manner, and at higher concentrations, they could also inhibit other serine-threonine protein kinases such as PKA and PKC.8 Hence, a genetic approach with tissue-specific gene targeting of ROCK1soforms offers the greatest likelihood to dissect the role of ROCKs in pathophysiological processes. In this study, we used a genetic approach to determine the pathophysiological role of ROCKs in mediating endothelial function and cerebral ischemia.
Methods
Materials and methods information are available in the Supplementary Materials section (Supplementary File 1).
Results
R0CK2 Deficiency Protects Against Cerebral Injury Following Transient Focal Cerebral Ischemia
We have previously generated mice with global deficiency of ROCK118,19 or ROCK220 on a C57BL/6 background. However, embryonic lethality and developmental abnormalities were limiting factors in adult studies. In agreement with other studies, ROCK2−/− mice were embryonically lethal due to placental insufficiency,21,22 while ROCK1−/− mice died soon after birth from the development of large omphalocoele.23 However, global hemizygous ROCK1+/− and ROCK2+/− mice were normal in terms of growth, appearance, and fertility, with approximately 50% reduction in protein levels of the corresponding ROCK isoform in mouse brain endothelial cells (MBEC)8,20,21 (Figure 1A,B, n=3). To determine the pathophysiological relevance of ROCK deficiency on stroke protection, we induced transient focal cerebral ischemia by subjecting ROCK1+/− and ROCK2+/− mice to intra-filament transient middle cerebral artery occlusion (MCAO). Following reperfusion, cerebral infarct volume was substantially smaller in ROCK2+/− (13.6±1.8%, n=16) compared to that of wild-type (WT) (22±1.9%, n=16, P<0.01) or ROCK1+/− (20.5±1.7%, n=17, P<0.01) mice (Figure 1C). There was no difference in cerebral infarct volume between WT and ROCK1+/− mice (P=NS). This reduction in the severity of cerebral injury was correlated with a decreased neurological deficit score (NDS) in ROCK2+/− mice (1.9±0.1) compared to that of WT (2.4±0.2) and ROCK1+/− (2.2±0.1) mice (P<0.05 for both) (Figure 1D). Absolute CBF was higher in ROCK2+/− mice (168±15mL/100g per min, n=11) compared to that of ROCK1+/− mice (133±11 mL/l00g per min, n = 11) and WT mice (120±19mL/100g per min, n=9) (P<0.05 for both) (Figure 1E).
Figure 1.
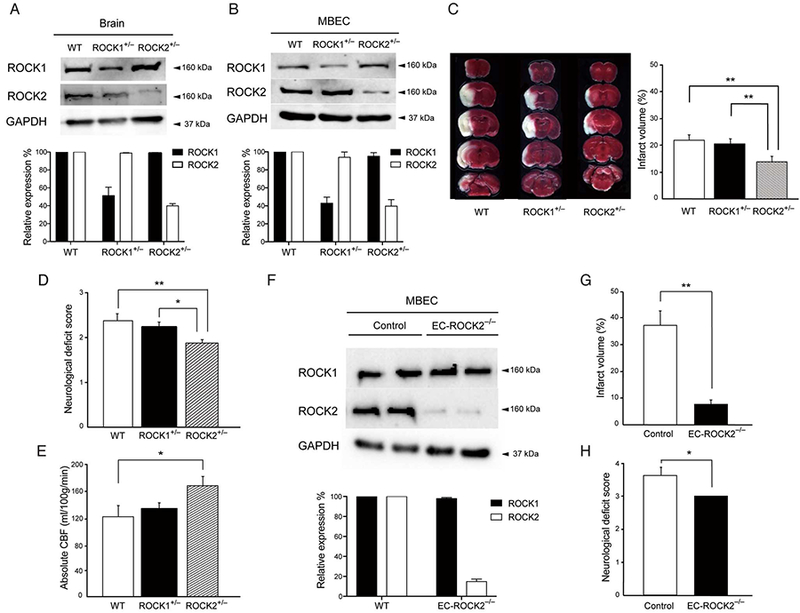
Effect of Rho-associated kinase (ROCK) deficiency on cerebral injury following transient middle cerebral artery occlusion (MCAO). (A) Expression of ROCK1, ROCK2 and GAPDH in brain tissues and (B) mouse brain endothelial cells (MBEC) of wild-type (WT), hemizygous ROCK1+/−and hemizygous ROCK2+/− mice, as assessed by Western blotting (n=3). (C) Coronal brain sections (Top to Bottom, Rostral to Caudal) of WT, ROCK1+/− and ROCK2+/− mice stained with 2,3,5 triphenyltetrazolium chloride (Left panel). Cerebral infarct volume following MCAO in WT, ROCK1+/− and ROCK2+/− mice (Right panel, n=16–17). (D) Neurological deficit score following MCAO in WT, ROCK1+/− and ROCK2+/− mice (n=16–17). (E) Absolute cerebral blood flow, as measured by an indicator fractionation technique using radiolabeled [14C] iodoamphetamine in WT, ROCK1+/− and ROCK2*/_ mice (n=9–11). (F) Expression of ROCK1, ROCK2 and GAPDH in MBECs isolated from control and endothelial cell-specific ROCK2-KO (EC-ROCK2−/−) mice (n=3). (G) Cerebral infarct volume following MCAO in control and EC-ROCK2−/− mice (n=5–6). (H) Neurological deficit score following MCAO in control and EC-ROCK2−/− mice (n=5–6). *P<0.05, **P<0.01. All data are expressed as mean±SEM.
Neuroprotection in Mice With Endothelial Cell (EC)-Targeted R0CK2 Deficiency
To circumvent the lethality associated with global homozygous ROCK2 deficiency and to exclude the role of ROCK2 in non-ECs that could contribute to neuroprotection, we generated EC-specific ROCK2 deletion using Tie2-Cre mice and conditional ROCK2 “floxed” mice.24 At 10 weeks of age, ROCK2 expression in MBECs from EC-ROCK2−/− mice was substantially reduced compared to that of Tie2-Cre control mice (Figure 1F, n=3). Compared to controls, the EC-ROCK2−/− mice displayed no substantial differences in body weight (control, 40.3±3.3g; EC-ROCK2−/−, 43.8±3.4g, n=5), heart rate (control, 525±35 beats/min; EC-ROCK2−/−, 566±34beats/min, n=5), or systolic blood pressure (control, 101±2mmHg; EC-ROCK2−/−, 105±2mmHg, n=5) (P=NS for all). However, similar to global hemizygous ROCK2+/− mice, EC-ROCK2−/− mice developed smaller cerebral infarct volume (7.5±1.8% vs. 36.9±5.5%, n=5–6, P<0.01) and lower NDS (3.0±0.0 vs. 3.6±0.3, n=5–6, P<0.05) following MCAO (Figure 1G,H). These results indicate that endothelial-specific ROCK2 deletion is associated with greater neuroprotection following focal cerebral ischemia.
R0CK2 Deficiency Enhances Endothelium-Dependent Vasomotor Response
To determine whether ROCK2 expression affects vasomotor response, we measured isometric tension in aortic rings isolated from WT (n=7), ROCK1+/− (n=6), ROCK+/− (n=5) and EC-ROCK2−/− (n=6) mice. The endothelium-dependent vasodilatory response to acetylcholine (ACh) was greater in ROCK2+/− mice than WT mice (P<0.05), whereas no differences were observed between ROCK1+/− and WT mice (P=NS) (Figure 2A). Similarly, the vasoconstrictive response to the eNOS inhibitor, L-NAME, was greater in ROCK2+/− mice, and to a lesser extent in ROCK1+/− mice, compared to that of WT mice (P<0.01 and P<0.05, respectively; Figure 2B). Indeed, the vasodilatory response of aorta from EC-ROCK2−/− (n=6) mice was augmented to ACh (P<0.05), but not to sodium nitro-prusside (SNP; P=NS) compared to that of controls (n=5) (Figure 2D,E). These findings suggest that global or EC-specific ROCK2-deficient mice may have greater aortic basal NO release and enhanced endothelium-dependent relaxation.
Figure 2.
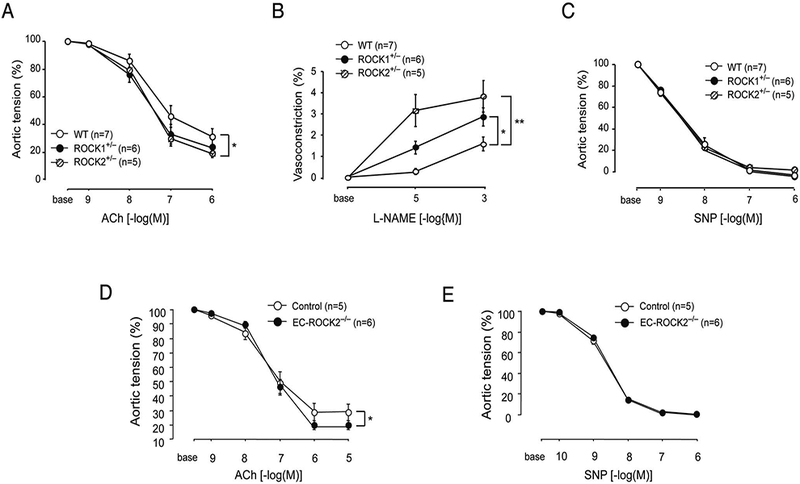
Aortic vasomotor responses to acetylcholine (ACh), NG-nitro-L-arginine methylester (L-NAME), and sodium nitroprusside (SNP). Vascular reactivity of aortic rings from wild-type (WT; n=7), ROCK1+/− (n=6) or ROCK2+/− (n=5) mice to (A) ACh, (B) L-NAME, and (C) SNP. (D,E) Aortic vasodilatory response to ACh and SNP from control (n=5) and endothelial cell-specific ROCK2-KO (EC-ROCK2−/−) (n=6) mice. *P<0.05, **P<0.01. All data are expressed as mean±SEM. ROCK, Rho-associated kinase.
R0CK2 Deficiency Leads to Increased eNOS Expression and NO Production
To investigate the effect of ROCK deletion on eNOS expression, we performed Western blotting of brain tissue and MBECs from WT, ROCK1+/−, ROCK2+/− and EC-ROCK2−/− mice. The expression of eNOS was increased by 2-fold in ROCK2+/− mice compared with that of WT and ROCK1+/− mice. Although phospho-Ser1177-eNOS tended to be higher in ROCK2+/− mice, the ration of p-eNOS to total eNOS was not different when standardized to total eNOS expression. Furthermore, the Thr853 phosphorylation of the myosin-binding subunit (p-MBS) of myosin light chain phosphatase was more than four-fold decreased in ROCK2+/− mice compared to that of WT and ROCK1+/− mice (Figure 3A,B, n=3, P<0.01 and P<0.0001, respectively). Similar to global hemizygous ROCK2+/− mice, MBECs isolated from EC-ROCK2−/− mice exhibited an even higher increase in eNOS expression and a further decrease in phosphorylation of MBS compared to cells from control mice (Figure 3C, n=3, P<0.001 and P<0.0001, respectively). Taken together, our findings indicate that ROCK2 deficiency in ECs leads to higher eNOS expression compared to that of WT or ROCK1 deficiency.
Figure 3.
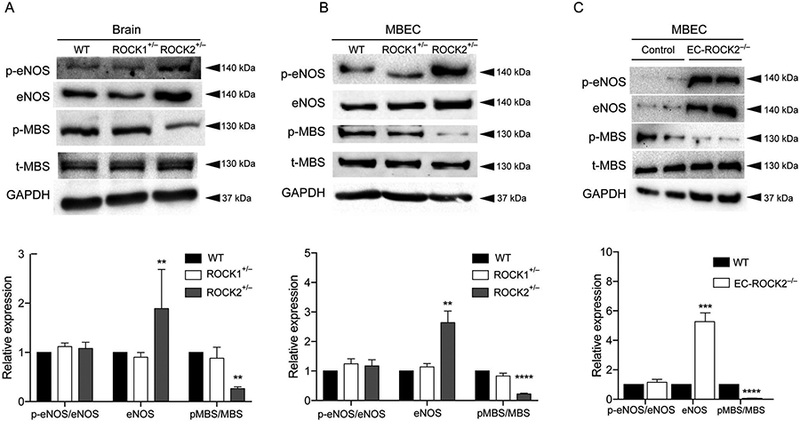
Increased endothelial nitric oxide synthase (eNOS) expression with Rho-associated kinase (ROCK)2 deficiency. Expression of eNOS, p-eENOS, p-MBS, t-MBS, and GAPDH in (A) brain tissues and (B) mouse brain endothelial cells (MBECs) of wild-type (WT), hemizygous ROCK1+/−, hemizygous ROCK2+/− and (C) endothelial cell-specific ROCK2-KO (EC-ROCK2−/−) mice, as assessed by Western blotting (n=3). **P<0.01, ****P<0.0001. All data are expressed as mean±SEM.
To determine whether increased eNOS expression correlated with higher NO production, we measured nitrite (NO2) accumulation in the culture medium from mouse heart ECs (MHECs) isolated from WT, ROCK1+/−, and ROCK2+/− mice. MHECs were isolated from the same animals that were used for isolation of MBECs. We found that MHECs show similar eNOS expression and MBS phosphorylation as MBECs (Figure 4A, n=3, P<0.0001). The NO2 levels were substantially higher in MHECs from ROCK2+/−mice compared to MHECs from ROCK1+/− or WT mice (Figure 4B, n=4, P<0.01 for both). To determine whether the increased eNOS expression in ROCK2+/− mice was due to increased eNOS mRNA stability, we treated MHECs with the RNA polymerase inhibitor, 5,6-dichlo-robenzimidazole riboside (DRB), and evaluated the half-life of eNOS mRNA (Figure 4C, n=4 in each group). The half-life of eNOS mRNA in MHECs from ROCK2+/− mice was greater than that of ROCK1+/− mice (55.7 vs. 31.5 h, P<0.05). The eNOS mRNA half-life in ROCK1+/− mice was also slightly greater than that of WT mice (31.5 vs. 20.1 h, P<0.05). These findings indicate that ROCK2, and to a lesser extent, ROCK1, decreases eNOS mRNA stability.
Figure 4.
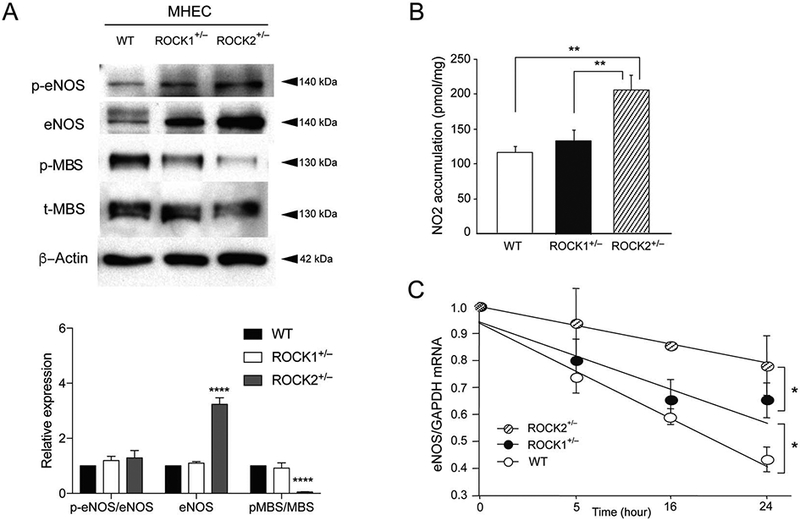
Nitric oxide (NO) production and endothelial NO synthase (eNOS) mRNA stability in Rho-associated kinase (ROCK)2-deficient mice. (A) Expression of eNOS, p-eENOS, p-MBS, t-MBS, and β-actin in mouse heart endothelial cells (MHEC, n=3) of wild-type (WT), hemizygous ROCK1+/−, and hemizygous ROCK2+/− mice. (B) NO production in the culture supernatants of MHECs isolated from WT, ROCK1+/− and ROCK2+/− mice (n=4 in each group). (C) Effect of ROCK1 or ROCK2 hemizygous deletion on eNOS mRNA stability in cultured MHECs treated with 5,6-dichlorobenzimidazole riboside (5×10−5M) for the indicated time periods (n=4 in each group). *P<0.05, **P<0.01, ****P<0.0001. All data are expressed as mean±SEM.
eNOS Mediates the Neuroprotective Effects of R0CK2 Deficiency
We assessed potential differences in eNOS expression and NO production in brains from WT, ROCK1+/− and ROCK2+/− mice following MCAO using Western blotting and DAF-FM diacetate, respectively. Brains from ROCK2+/− mice exhibited higher eNOS expression in the penumbra region compared with that of WT and ROCK1+/− mice (Figure 5A,B, n=3, P<0.05). Furthermore, NO production in the ipsilateral brain hemisphere of ROCK2+/− mice was higher 24 h after MCAO (Figure 5C, n=3). These findings indicate that ROCK2+/− mice have higher eNOS expression and NO production in the ipsilateral brain hemisphere compared with that of WT and ROCK1+/− mice, 24h after transient cerebral ischemia.
Figure 5.
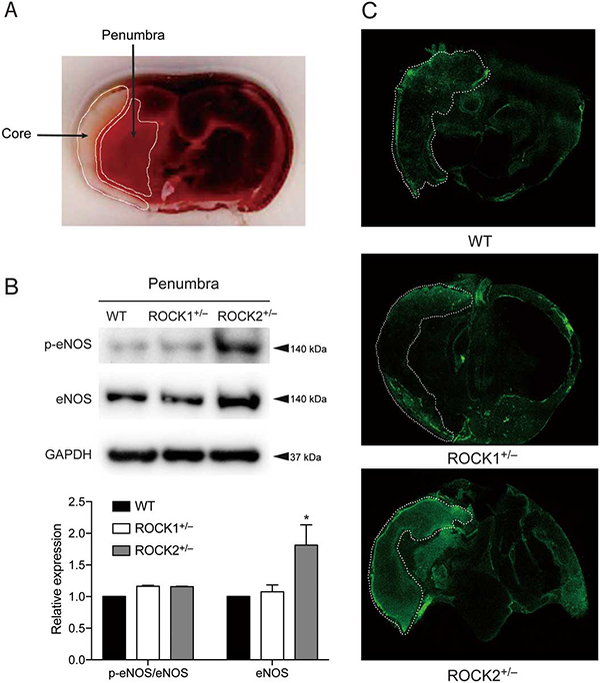
Rho-associated kinase (ROCK)2 deficiency leads to increased endothelial nitric oxide synthase (eNOS) expression and NO production in brain tissue after middle cerebral artery occlusion (MCAO). Mouse brain tissues were analyzed for eNOS expression and NO production with Western blot and DAF-FM diacetate staining, respectively, 24h after MCAO. (A) Coronal brain sections were stained with TTC, 24 h after MCAO with indicated infarct core and penumbra of ipsilateral hemisphere. (B) eNOS expression in brain penumbra of wild-type (WT), ROCK1+/− and ROCK2+/− mice, 24 h after MCAO (n=3). (C) DAF-FM diacetate staining for NO levels in brain of WT, ROCK1+/− and ROCK2+/− mice, 24 h after MCAO with indicated ischemic core (n=3). *P<0.05. All data are expressed as mean±SEM.
Endothelium-derived NO is an important regulator of CBF, and global deletion of eNOS leads to increased cerebral infarct size following focal cerebral ischemia.6,25 To determine whether endothelium-derived NO mediates the neuroprotective effects observed in ROCK2+/− mice, we generated double mutant mice by placing global hemizygous ROCK1+/− and ROCK2+/− mice on eNOS−/− background. In both eNOS−/− ROCK1+/− and eNOS3−/− ROCK2+/− mice, the protein expression of each ROCK isoform was approximately half that of WT mice, with minimal compensatory changes in the other ROCK isoform (Figure 6A). Furthermore, at 10 weeks of age, there were no differences in body weight, heart rate or systolic blood pressure in the double mutant mice compared to eNOS−/− mice (data not shown). Following MCAO, there were no differences in cerebral infarct volumes and NDS, respectively, between eNOS−/− (27.2±2.2% and 3.0±0.0, n=10), eNOS−/−ROCK1+/− (25.6±1.9% and 3.0±0.0, n=14), and eNOS−/−ROCK2+/− (30.0±4.3% and 2.8±0.2, n=12) mice (P>0.05 for inter-group comparisons) (Figure 6B,C). Furthermore, the mortality rate was not different between eNOS−/− (37.5%, n=16), eNOS−/− ROCK1+/− (26.3%, n=19), and eNOS−/−ROCK2+/− (26.7%, n=15) mice (P>0.05 for inter-group comparisons) (Figure 6D). These findings indicate that the neuroprotective effects observed in global hemizygous ROCK2+/− mice are completely abolished in the absence of eNOS, suggesting that eNOS mediates the neuroprotective effects of ROCK2 deficiency
Figure 6.
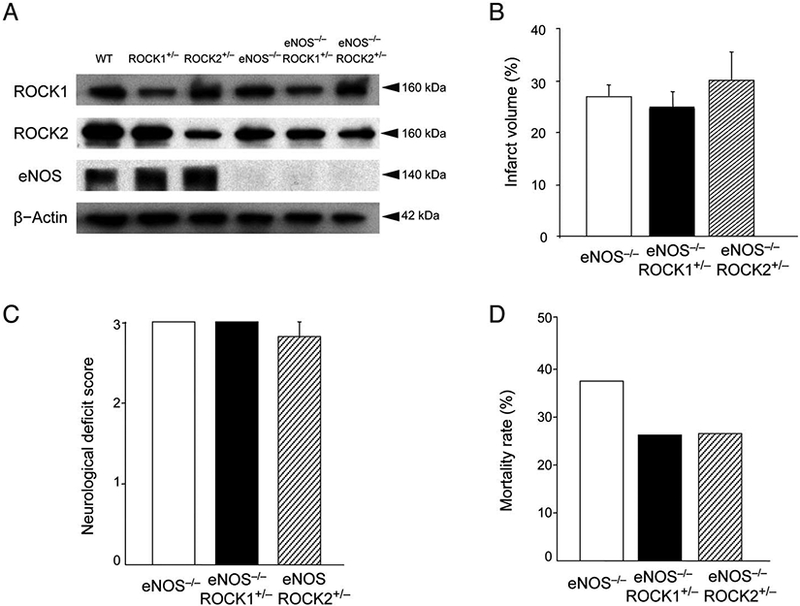
Effects of endothelial nitric oxide synthase (eNOS) deletion on the neuroprotective effects of Rho-associated kinase (ROCK)1 and ROCK2 deficiency. (A) Expression of ROCK1, ROCK2, eNOS, and β-actin in brain tissues of wild-type (WT), ROCK1+/− ROCK2+/−, eNOS−/−, eNOS−/−ROCK1+/−, and eNOS−/−ROCK2+/− mice. (B,C) Cerebral infarct volume (n=10–14), neurological deficit score, and (D) mortality rate following middle cerebral artery occlusion (MCAO) (n=15–19) in eNOS−/−, eNOS−/−ROCK1+/−, and eNOS−/−ROCK2+/− mice. P>0.05 for all inter-group comparisons.
Phosphorylation of eEF1A1 Thr432 by R0CK2 Increases Its Interaction With eNOS mRNA 3’-UTR
eEF1A1 is an abundant and highly conserved protein in eukaryotic cells involved in protein translation machinery with strong RNA binding activity. Phosphorylation of Thr432 in Domain III of eEF1A1 by ROCK2 regulates its competitive reaction with F-actin and eNOS mRNA. To analyze eEF1A1 phosphorylation on Thr432, we developed a phospho-specific eEF1A1 rabbit polyclonal antibody that was generated against the synthetic peptide-containing phosphorylated Thi432 (425AVRDMRQ26VAVGV437-amide). First, we assessed the Thr432 phosphorylation state of eEF1A1 in brain sections and brain ECs in ROCK1+/−, ROCK2+/− and EC-ROCK2−/− mice using Western blotting. For both tissues, eEF1A1 Thr432 phosphorylation was lower in ROCK2+/− and EC-ROCK2 mice compared to that of WT or ROCK1+/− mice, with higher differences observed in MBEC from EC-ROCK2−/− mice (Figure 7A, n=3, P<0.0001). Total eEF1A1 expression was not different between the groups.
Figure 7.
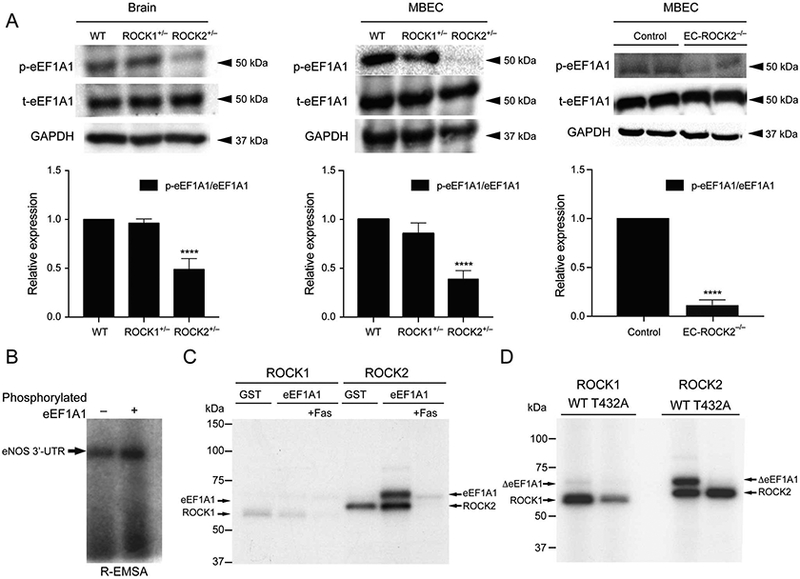
Phosphorylation of eukaryotic elongation factor-1A1 (eEF1A1) by Rho-associated kinase (ROCK)2. (A) Expression of P-EF1A1, t-eEF1A1 and GAPDH in mouse brain endothelial cells (MBEC) from wild-type (WT), ROCK1+/−, ROCK2+/− and EC-ROCK2−/− (n=3). (B) Increased affinity of phosphorylated eEF1A1 to endothelial nitric oxide synthase (eNOS) mRNA 3’-UTR. GST-eEF1A1 phosphorylated by ROCK2 increased its affinity to eNOS mRNA 3’-UTR (n=5). (C) Phosphorylation of eEF1A1 by ROCK1 and ROCK2 in vitro. Purified ROCK2 protein robustly phosphorylated GST-eEF1A1, whereas purified ROCK1 did so very weakly. The upper band represents phosphorylated GST-eEF1A1 (78 kDa), and the lower band auto-phosphorylated ROCK (ROCK1: 61.4kDa; ROCK2: 63.3kDa). GST, GST only; eEF1A1, GST-tagged eEF1A1; Fas, fasudil (100μmol/L), (n=5). (D) Phosphorylation of eEF1 A1 at Thr432 by ROCK2. Mutagenesis of Thr432Ala (ΔT432A) abolished the phosphorylation of eEF1A1 by ROCK2. The upper band represents phosphorylated GST-eEF1A1 Thr432 and the lower bands the auto-phosphorylated ROCK proteins (caROCK1: 61.4kDa; caROCK2: 63.3kDa). ****P<0.0001. All data are expressed as mean±SEM.
To determine whether the ROCK2-dependent phosphorylation of eEF1A1 affects its interaction with eNOS mRNA 3’-UTR, we examined eEF1A1-eNOS mRNA binding using RNA electrophoretic mobility shift assay (R-EMSA). Phosphorylation of eEF1A1 by ROCK2 enhanced eEF1A1 binding to eNOS mRNA (Figure 7B), which is consistent with eEF1A1’s ability to bind and destabilize eNOS mRNA.27
Because ROCKs are serine-threonine protein kinases, we also tested whether both ROCK isoforms could phos-phorylate eEF1A1. We performed in vitro kinase assays using GST-eEF1A1 protein and constitutive-active ROCK1 or ROCK2 proteins. ROCK2 strongly phosphorylated eEF1A1, while ROCK1 did so very weakly (Figure 7C). This phosphorylation was reduced in the presence of the ROCK inhibitor, fasudil (Fas, 100μmol/L). To pinpoint the site of ROCK2 phosphorylation on eEF1A1 and to confirm previous findings, we introduced a phospho-negative mutation, T432A, into eEF1A1. The ΔeEF1A1 T432A protein was unable to be phosphorylated by either ROCK1 or ROCK2 (Figure 7D), confirming that Thr432 is the ROCK phosphorylation site. Taken together, these findings indicate that phosphorylation of eEF1A1 at Thr432 by ROCK2 is critical for binding and destabilization of eNOS mRNA.
Inhibition of eEF1A1 Thr432 Phosphorylation by Statin and ROCK inhibitor
To determine whether statins or ROCK inhibitors could reduce eEF1A1 Thr432 phosphorylation, human brain microvascular ECs (HBMECs) were treated with atorvastatin (10μmol/L) and fasudil (10μmol/L) for varying durations, followed by measurement of phospho-eEF1A1 and total eEF1A1 by Western blotting. The phosphorylation of a myosin-binding subunit (Thr853 of MBS or MYPT1) of myosin light chain phosphatase served as a marker for ROCK activity.28 The phosphorylation of eNOS and total eNOS was used to assess eNOS activity. Treatment with atorvastatin (10μmol/L, 24h) led to the greatest reduction in ROCK activity, which coincided with the greatest reduction of eEF1A1 phosphorylation (P<0.0001 for both). Maximum phosphorylation of eNOS was detected at 6h (P<0.0001, n=5; Figure 8A). In addition, treatment with fasudil (10μmol/L) for 24h was also sufficient to inhibit ROCK activity and eEF1A1 Thr432 phosphorylation, with maximum effect observed at 24 h (P<0.0001). Maximum level of eNOS phosphorylation was observed at 12h after treatment (P<0.0001, n=5; Figure 8B). These findings indicate that statins and fasudil can inhibit eEF1A1 Thr432 phosphorylation and increase eNOS phosphorylation, which may contribute to some of their neuroprotective effects.
Figure 8.
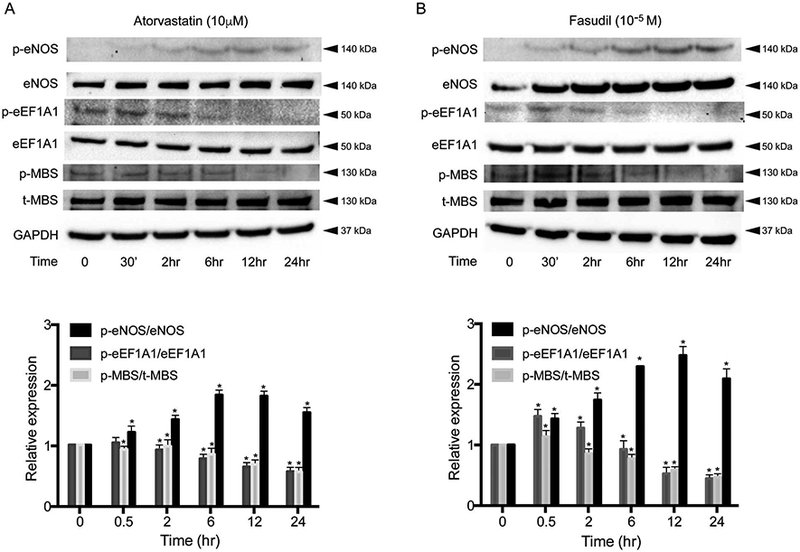
Inhibition of Rho-associated kinase (ROCK) activity and eEF1A1 phosphorylation by statin and ROCK1nhibitor. Human brain microvascular endothelial cells (HBMECs) were grown to confluence, serum-starved overnight, and treated with atorvastatin (10μmol/L) or fasudil (10−5mol/L) for 24h. Cells were then washed, lysed and analyzed by Western blot. (A) Atorvastatin inhibited ROCK activity (pMBS/tMBS), decreased eEF1A1 Thr432 phosphorylation, and increased endothelial nitric oxide synthase (eNOS) activity (p-eNOS/t-eNOS) in a time-dependent manner (n=5, P<0.05). (B) Time-dependent effects of the ROCK1nhibitor, fasudil, on ROCK activity, eEF1 A1 Thr432 phosphorylation and eNOS activity (n=5). *P<0.05. All data are expressed as mean±SEM.
Discussion
We have shown that the loss of endothelial ROCK2, and to a lesser extent ROCK1, leads to enhanced endothelial function, higher CBF, and greater neuroprotection through upregulation of eNOS. This was associated with decreased Thr432 phosphorylation of eEF1A1, a protein that is directly phosphorylated by ROCK2.29 This mechanism probably also contributes to the NO-dependent neuroprotective effects of statins and ROCK inhibitors, as statins could also inhibit ROCK activity (p-MBS/t-MBS) and Thr432 phosphorylation of eEF1A1 (p-eEF1A1/t-eEF1A1) while upregulating eNOS expression and activity (p-eNOS/t-eNOS). Indeed, concomitant loss of eNOS abrogated the neuroprotective effects of global ROCK2 hemizygous deletion, indicating that the upregulation of eNOS is the underlying mechanism of neuroprotection in ROCK2-deficient mice. Interestingly, ROCK1 hemizygous mice exhibited lesser eNOS upregulation, which was not sufficient to reduce stroke severity.
ROCK2 is the main ROCK isoform in the brain, with significant expression in many cell types of the neurovascular unit that is relevant to stroke severity.30–32 With availability of global ROCK1+/− and ROCK2+/− hemizygous mice, we have shown that ROCK2 haploinsufficiency has a neuroprotective effect in a mouse model of transient ischemic stroke. Similar to our results, a recently published study using the selective ROCK2 inhibitor, KD025, has shown beneficial effects after focal cerebral ischemia.33 However, the precise mechanism contributing to the neuroprotective effects of KD025 is not known. To determine the tissue specificity in the global ROCK2+/− hemizygous mice, we generated endothelial-specific ROCK2 deletion using Tie2-Cre mice and conditional ROCK2 “floxed” mice.24 This model has some limitations as Tie2 is also expressed in hematopoietic cells, which could affect stroke severity. Indeed, when the Tie2-Cre mouse strain was crossed to EGFP or eYFP reporter mouse strain, Cre expression was observed in 80–90% of hematopoietic cells.34 Thus, it is possible that ROCK2 deletion in hematopoietic cells could contribute to some of the neuroprotective effects observed in our Tie2-Cre/ROCK2f/f mice. However, because the neuroprotective effects of Tie2-Cre/ROCK2f/f mice was absent when these mice were placed on an eNOS-KO background, most, if not all, of the neuroprotection by Tie2-mediated Cre deletion of ROCK2 appears to be due to ROCK2 in ECs. Furthermore, vasomotor response of aortic rings also suggests that ROCK2-deficient mice have higher basal NO release. Indeed, vasomotor responses were observed only after endothelial-dependent agents (i.e., ACh and L-NAME) treatment, but not after non-endothelial-dependent stimulation with SNP.
Our findings also demonstrate that the neuroprotective effect of ROCK2 deletion occurs through a reduction in eEF1A1 phosphorylation in vivo, and an increase in eNOS mRNA stability and expression in vitro. Although eNOS expression can be regulated at the transcriptional level by various conditions and factors such as shear stress and TGF-β1, other important mediators of ischemic stroke such as hypoxia and TNF-α could also affect eNOS expression through modulating eNOS mRNA stability.35 For example, upon TNF-α stimulation, eEF1A1 binds to the 3’-untrans-lated region (3’-UTR) of eNOS mRNA, leading to decreased eNOS mRNA stability and expression.27 Similar to previous studies, we found that eEF1A1 is a substrate of ROCK2, and that ROCK2 can alter F-actin dynamics by directly phosphorylating eEF1A1 on Thr432, thereby allowing the formation of actomyosin bundles.29,36 Our results, therefore, confirm and extend the physiological importance of eEF1A1 phosphorylation by ROCK2 on eNOS expression in ECs. This may have important implications for the treatment of cerebral ischemia.
Statins have been shown to improve endothelial function by upregulating eNOS expression and activity, in part, through a cholesterol-independent or “pleiotropic” mechanism.7,14,37,38 Our findings suggest that some of this pleiotropic mechanism of statins may be due to Rho/ROCK inhibition, reduction of eEF1A1 phosphorylation, and the subsequent upregulation of eNOS expression. Indeed, similar to statins, the ROCK inhibitor, fasudil, has been shown to inhibit eEF1A1 phosphorylation, upregulate eNOS, and is neuroprotective.8 In addition to eNOS expression, statins can increase phosphatidylinositol 3-kinase/protein kinase Akt and eNOS activities,15,39 and co-treatment with PI3K and eNOS inhibitors have been shown to block the vascular protective effects of fasudil.5,15 Other potential pleiotropic mechanisms of statin could also be due to decreasing activation of reactive oxygen species, reduction of abundance of caveolin-1, or increasing of Hsp90, which serves as a molecular chaperone or scaffolding for the activation of eNOS.7,40,41
The expression of eEF1A1 is elevated in diabetic patients and animal models of diabetes, which is reversed by treatment with insulin.42 Likewise, patients with metabolic syndrome or diabetes have elevated ROCK activity, which correlated with endothelial dysfunction in these patients. Therefore, ROCK2-mediated eEF1A1 phosphorylation may be an important central mechanism leading to endothelial dysfunction in diabetic patients.
In summary, we have identified eEF1A1 and eNOS as physiologically important ROCK2-dependent targets for ischemic stroke and other endothelium-dependent vascular diseases. Improving endothelial function through the inhibition of endothelial ROCK2 and eEF1A1 phosphorylation, therefore, may have therapeutic benefits in cardiovascular disease, and could contribute to some of the cholesterol-independent or “pleiotropic” effects of statin therapy. Further studies are need, however, to determine how the binding of phosphorylated eEF1A1 to eNOS mRNA leads to eNOS mRNA degradation, and whether this mechanism could extend to other eukaryotic mRNAs.
Supplementary Material
Acknowledgments
We appreciate the editorial assistance of Phetcharat Chen and Christine Blaumueller.
Funding Sources
This work was supported by grants from the National Institute of Health (NIH) (HL052233, HL080187, NS070001) and the American Heart Association (Bugher Foundation Award) to J.K.L. Both N.S. and C.E.T. were recipients of the American Heart Association Research Fellowship Award. Y.H. and R.O. were recipients of the Uehara Memorial Fellowship. And both K.N. and N.O. were recipients of the Japan Heart Foundation/Bayer-Yakuhin Research Grant Abroad.
Footnotes
Disclosures
J.K.L. is a consultant for Asahi-Kasei Pharmaceuticals, Inc.
Supplementary Files
Supplementary File 1
Supplementary Methods
Please find supplementary file(s); http://dx.doi.org/10.1253/circj.CJ-17-0732
References
- 1.Riento K, Ridley AJ. Rocks: Multifunctional kinases in cell behaviour. Nat Rev Mol CelI Biol 2003; 4: 446–456. [DOI] [PubMed] [Google Scholar]
- 2.Masumoto A, Mohri M, Shimokawa H, Urakami L, Usui M, Takeshita A. Suppression of coronary artery spasm by the Rho-kinase inhibitor fasudil in patients with vasospastic angina. Circulation 2002; 105: 1545–1547. [DOI] [PubMed] [Google Scholar]
- 3.Masumoto A, Hirooka Y, Shimokawa H, Hironaga K, Setoguchi S, Takeshita A. Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension 2001; 38:1307–1310. [DOI] [PubMed] [Google Scholar]
- 4.Abe K, Shimokawa H, Morikawa K, Uwatoku T, Oi K, Matsumoto Y, et al. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ Res 2004; 94: 385–393. [DOI] [PubMed] [Google Scholar]
- 5.Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, et al. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci USA 1998; 95: 8880–8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laufs U, Endres M, Stagliano N, Amin-Hanjani S, Chui DS, Yang SX, et al. Neuroprotection mediated by changes in the endothelial actin cytoskeleton. J Clin Invest 2000; 106: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 1998; 97: 1129–1135. [DOI] [PubMed] [Google Scholar]
- 8.Rikitake Y, Kim HH, Huang Z, Seto M, Yano K, Asano T, et al. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke 2005; 36:2251–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feske SK, Sorond FA, Henderson GV, Seto M, Hitomi A, Kawasaki K, et al. Increased leukocyte ROCK activity in patients after acute ischemic stroke. Brain Res 2009; 1257: 8993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shibuya M, Hirai S, Seto M, Satoh S, Ohtomo E; Fasudil Ischemic Stroke Study Group. Effects of fasudil in acute ischemic stroke: Results of a prospective placebo-controlled double-blind trial. J Neurol Sci 2005; 238: 31–39. [DOI] [PubMed] [Google Scholar]
- 11.Liao JK. Isoprenoids as mediators of the biological effects of statins. J Clin Invest 2002; 110: 285–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol 2005; 45: 89–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu PY, Liu YW, Lin LJ, Chen JH, Liao JK. Evidence for statin pleiotropy in humans: Differential effects of statins and ezetimibe on rho-associated coiled-coil containing protein kinase activity, endothelial function, and inflammation. Circulation 2009; 119: 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem 1998; 273: 24266–24271. [DOI] [PubMed] [Google Scholar]
- 15.Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, et al. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vase Biol 2004; 24: 1842–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolfrum S, Grimm M, Heidbreder M, Dendorfer A, Katus HA, Liao JK, et al. Acute reduction of myocardial infarct size by a hydroxymethyl glutaryl coenzyme A reductase inhibitor is mediated by endothelial nitric oxide synthase. J Cardiovasc Pharmacol 2003; 41:474–480. [DOI] [PubMed] [Google Scholar]
- 17.Nohria A, Prsic A, Liu PY, Okamoto R, Creager MA, Selwyn A, et al. Statins inhibit Rho kinase activity in patients with atherosclerosis. Atherosclerosis 2009; 205: 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rikitake Y, Liao JK. Rho-kinase mediates hyperglycemia-induced plasminogen activator inhibitor-1 expression in vascular endothelial cells. Circulation 2005; 111: 3261–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rikitake Y, Oyama N, Wang CY, Noma K, Satoh M, Kim HH, et al. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/− haploinsufficient mice. Circulation 2005; 112: 2959–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noma K, Rikitake Y, Oyama N, Yan G, Alcaide P, Liu PY, et al. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J Clin Invest 2008; 118: 1632–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thumkeo D, Shimizu Y, Sakamoto S, Yamada S, Narumiya S. ROCK-I and ROCK-II cooperatively regulate closure of eyelid and ventral body wall in mouse embryo. Genes Cells 2005; 10: 825–834. [DOI] [PubMed] [Google Scholar]
- 22.Thumkeo D, Keel J, Ishizaki T, Hirose M, Nonomura K, Oshima H, et al. Targeted disruption of the mouse rho-associated kinase 2 gene results in intrauterine growth retardation and fetal death. Mol Cell Biol 2003; 23: 5043–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimizu Y, Thumkeo D, Keel J, Ishizaki T, Oshima H, Oshima M, et al. ROCK-I regulates closure of the eyelids and ventral body wall by inducing assembly of actomyosin bundles. J Cell Biol 2005; 168: 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okamoto R, Li Y, Noma K, Hiroi Y, Liu PY, Taniguchi M, et al. FHL2 prevents cardiac hypertrophy in mice with cardiac-specific deletion of ROCK2. FASEB J 2013; 27: 1439–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science 1994; 265: 1883–1885. [DOI] [PubMed] [Google Scholar]
- 26.Biswas PS, Gupta S, Chang E, Song L, Stirzaker RA, Liao JK, et al. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J Clin Invest 2010; 120: 3280–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan G, You B, Chen SP, Liao JK, Sun J. Tumor necrosis factor-alpha downregulates endothelial nitric oxide synthase mRNA stability via translation elongation factor 1-alpha 1. Circ Res 2008;103:591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu PY, Chen JH, Lin LJ, Liao JK. Increased Rho kinase activity in a Taiwanese population with metabolic syndrome. J Am Coll Cardiol 2007; 49: 1619–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Couzens AL, Gill RM, Scheid MP. Characterization of a modified ROCK2 protein that allows use of N6-ATP analogs for the identification of novel substrates. BMC Biotechnol 2014; 14: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iizuka M, Kimura K, Wang S, Kato K, Amano M, Kaibuchi K, et al. Distinct distribution and localization of Rho-kinase in mouse epithelial, muscle and neural tissues. Cell Struct Fund 2012; 37: 155–175. [DOI] [PubMed] [Google Scholar]
- 31.Duffy P, Schmandke A, Schmandke A, Sigworth J, Narumiya S, Cafferty WB, et al. Rho-associated kinase II (ROCKII) limits axonal growth after trauma within the adult mouse spinal cord. J Neurosci 2009; 29: 15266–15276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hashimoto R, Nakamura Y, Kosako H, Amano M, Kaibuchi K, Inagaki M, et al. Distribution of Rho-kinase in the bovine brain. Biochem Biophys Res Commun 1999; 263: 575.-. [DOI] [PubMed] [Google Scholar]
- 33.Lee JH, Zheng Y, von Bomstadt D, Wei Y, Balcioglu A, Daneshmand A, et al. Selective ROCK2 inhibition in focal cerebral ischemia. Ann Clin Transi Neurol 2014; 1: 2–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang Y, Harrington A, Yang X, Friesel RE, Liaw L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis 2010; 48: 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshizumi M, Perrella MA, Burnett JC Jr, Lee ME. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ Res 1993; 73: 205–209. [DOI] [PubMed] [Google Scholar]
- 36.Izawa T, Fukata Y, Kimura T, Iwamatsu A, Dohi K, Kaibuchi K. Elongation factor-1 alpha is a novel substrate of rho-associated kinase. Biochem Biophys Res Commun 2000; 278: 72–78. [DOI] [PubMed] [Google Scholar]
- 37.Laufs U, Liao JK. Direct vascular effects of HMG-CoA reductase inhibitors. Trends Cardiovasc Med 2000; 10: 143–148. [DOI] [PubMed] [Google Scholar]
- 38.Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methyl-glutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. J Biol Chem 1997; 272:31725–31729. [DOI] [PubMed] [Google Scholar]
- 39.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholes-terolemic animals. Nat Med 2000; 6: 1004–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feron O, Dessy C, Desager JP, Balligand JL. Hydroxy-methyl-glutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation 2001; 103: 113–118. [DOI] [PubMed] [Google Scholar]
- 41.Wassmann S, Laufs U, Baumer AT, Muller K, Ahlbory K, Linz W, et al. HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension 2001; 37: 1450–1457. [DOI] [PubMed] [Google Scholar]
- 42.Reynet C, Kahn CR. Unbalanced expression of the different subunits of elongation factor 1 in diabetic skeletal muscle. Proc Natl Acad Sci USA 2001; 98: 3422–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.