

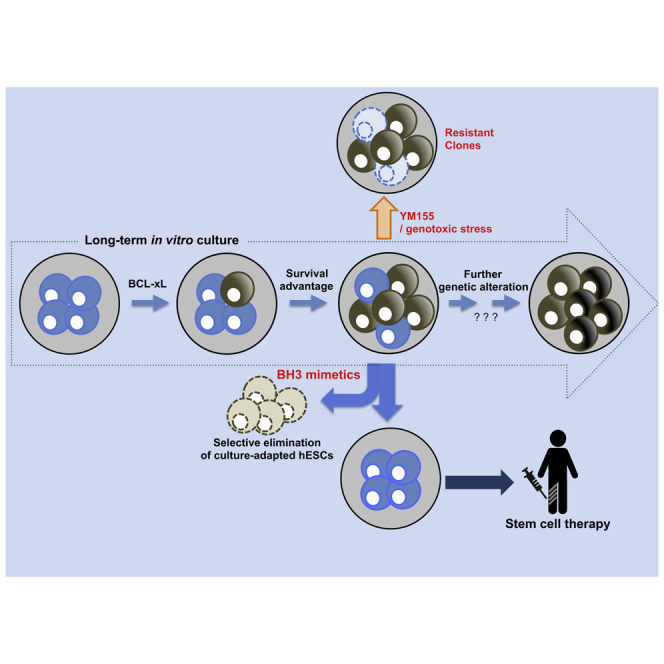
Summary
The selective survival advantage of culture-adapted human embryonic stem cells (hESCs) is a serious safety concern for their clinical application. With a set of hESCs with various passage numbers, we observed that a subpopulation of hESCs at late passage numbers was highly resistant to various cell death stimuli, such as YM155, a survivin inhibitor. Transcriptome analysis from YM155-sensitive (YM155S) and YM155-resistant (YM155R) hESCs demonstrated that BCL2L1 was highly expressed in YM155R hESCs. By matching the gene signature of YM155R hESCs with the Cancer Therapeutics Response Portal dataset, BH3 mimetics were predicted to selectively ablate these cells. Indeed, short-course treatment with a sub-optimal dose of BH3 mimetics induced the spontaneous death of YM155R, but not YM155S hESCs by disrupting the mitochondrial membrane potential. YM155S hESCs remained pluripotent following BH3 mimetics treatment. Therefore, the use of BH3 mimetics is a promising strategy to specifically eliminate hESCs with a selective survival advantage.
Keywords: culture adaptation, survival advantage, BCL2L1, BH3 mimetics, YM155, CTRP, BCL-xL
Graphical Abstract
Highlights
-
•
Culture-adapted hESCs against YM155/genotoxic agents mediated by high BCL-xL expression
-
•
Selective cell death of culture-adapted hPSCs by BH3 mimetics
-
•
Pluripotency maintenance of normal hESCs after exposure to BH3 mimetics
In this article, Cha and colleagues demonstrate that in culture-adapted hESCs, survival advantage toward several genotoxic stresses as well as YM155, a stemtoxic chemical, results from increased Bcl-xL expression. BH3 mimetics, predicted by a bioinformatics tool based on the gene expression signature of culture-adapted hESCs, induces selective cell death while normal hESCs remain functionally intact even after BH3 mimetics exposure.
Introduction
Genetic aberrations have been reported to arise in human embryonic stem cells (hESCs) during in vitro culture (Baker et al., 2007, Draper et al., 2004, Spits et al., 2008). These alterations are an important safety issue because their cause and biological significance are uncertain. Although cells differentiated from aneuploid hESCs give rise to tumors in vivo (Moon et al., 2011, Werbowetski-Ogilvie et al., 2009), the safety margins are unclear because genetic alterations frequently occur in many chromosomal loci not only of serially passaged human pluripotent stem cells (hPSCs) (Andrews et al., 2017, International Stem Cell Initiative et al., 2011) but also of human induced pluripotent stem cells (hiPSCs) at early passage numbers (Martins-Taylor et al., 2011). Genetic alterations that arise during repeated in vitro culture of hPSCs are frequently associated with gain of BCL2L1, a typical anti-apoptotic gene (International Stem Cell Initiative et al., 2011), and/or loss of TP53, a well-known tumor suppressor gene (Merkle et al., 2017). Both these changes are responsible for the selective survival advantage of hPSCs (Andrews et al., 2017, Avery et al., 2013), and such cells outnumber normal hPSCs upon serial passage in standard culture conditions (Andrews et al., 2017).
The genetic integrity of hESCs, which is crucial to produce normal progenies, is maintained by efficient DNA damage repair and high susceptibility to genotoxic stimuli (Stambrook, 2007, Weissbein et al., 2014). In particular, high sensitivity to genotoxic stress, leading to cell death (or “high mitochondrial priming;” Liu et al., 2013) via mitochondrial translocation of p53 (Kim et al., 2016, Lee et al., 2013) and Golgi-localized active BCL2-associated X (BAX) (Dumitru et al., 2012), was proposed to help maintain genetic stability by ablating genetically altered hESCs (Liu et al., 2014). A subpopulation of hESCs that escape from high mitochondrial priming due to a selective survival advantage may acquire further mutations that are undetected by the aforementioned surveillance system. Hence, it is important to selectively remove or sequester the subpopulation of hESCs with a selective survival advantage to prevent the acquisition of additional harmful genetic aberrations. In this regard, CD30 was identified as a surface marker of transformed hPSCs (Herszfeld et al., 2006), although this has since been disputed (Harrison et al., 2009). A recent study demonstrated that hESCs with trisomy 12 (T12) are more susceptible to several cytotoxic DNA replication inhibitors (Ben-David et al., 2014). In addition, treatment with ABT-263, a BCL-xL inhibitor, reduces the clonogenicity of hESCs that have acquired a selective survival advantage due to genetic aberrations (such as a 20q11.21 copy number variation) (Avery et al., 2013). The risk of residual undifferentiated hPSCs forming teratomas has been resolved by selectively inducing their death using small molecules (Ben-David et al., 2013) such as YM155, a survivin inhibitor (Lee et al., 2013), recombinant proteins (Tateno et al., 2015), suicide genes (Cho et al., 2015), and other approaches (Cho et al., 2016) (briefly summarized in a recent review; Jeong et al., 2017). However, no study has attempted to selectively remove hESCs with genetic alterations, with the exception of a report that screened a library of anti-cancer drugs (Ben-David et al., 2014).
In this study, we used hESCs at various passage numbers in which the size of the YM155-resistant (YM155R) subpopulation differed. Transcriptome analysis identified BCL2L1 as an important factor for the selective survival advantage of YM155R hESCs. In silico analysis based on the Cancer Therapeutics Response Portal (CTRP) predicted that BH3 mimetics would selectively induce the death of YM155R hESCs. Importantly, treatment with BH3 mimetics efficiently induced the death of YM155R, but not YM155-sensitive (YM155S), hESCs lines. YM155S hESCs remained pluripotent after treatment with BH3 mimetics. These findings suggest that the use of BCL-xL inhibitors is a promising strategy to prevent genetic variation in hESCs.
Results
hESCs at Late Passage Numbers Are Resistant to YM155
We and others have reported that treatment with YM155, a survivin inhibitor, selectively ablates undifferentiated hPSCs and inhibits teratoma formation (Bedel et al., 2017, Lee et al., 2013). However, surprisingly, a few hESC colonies occasionally survived following treatment with a relatively high concentration of YM155 (data not shown), while others were eliminated as previously reported (Kim et al., 2017, Lee et al., 2013). Since we observed the complete elimination of the hESCs line with YM155 (Lee et al., 2013), the same hESC clone has been serially passaged for several years. Thus, we speculated that the sensitivity of hESCs to YM155 might differ according to the passage number. To investigate this, we used hESCs (H9) at various passage numbers (passage number 40s, P1; passage number 100s, P2; passage number 200s, P3; and passage number 300s, P4) (Figure S1A), which expressed similar levels of POU5F1, SOX2, and NANOG (Figure S1B). The subpopulation that survived (Annexin− and 7AAD−) after YM155 treatment was dramatically larger in P3 and P4 hESCs than in P1 and P2 hESCs (Figure 1A). Similar results were obtained by examining alkaline phosphatase activity after YM155 treatment (Figures 1B and S1C). The difference in sensitivity to YM155 between P1 and P4 hESCs was confirmed by immunoblotting (Figure S1D) and live-cell imaging of YM155-treated GFP-expressing P1 (EGFP-P1) hESCs and P4 hESCs (Figures S1E–S1G, Videos S1, S2, and S3). P4 hESCs exhibited T12 (Figure S1H), one of the most frequent genomic aberrations in cultured hESCs (Baker et al., 2007, Ben-David et al., 2014, Draper et al., 2004, Lamm et al., 2016, Moon et al., 2011), and both P1 and P4 hESCs formed teratomas (Figure S1I). However, consistent with the previous finding that the number of OCT-4+ cells is high in teratomas formed by T12 hESCs (Ben-David et al., 2014), the population of OCT-4+ cells was larger in teratomas formed by P4 hESCs than in teratomas formed by P1 hESCs (Figure S1J). hESCs adapt to in vitro culture by acquiring genetic alterations in a passage-number-dependent manner (Baker et al., 2007), and this adaptation is highly associated with cell growth (International Stem Cell Initiative et al., 2011) or a selective survival advantage (Avery et al., 2013). The growth rates of YM155R P3 and P4 hESCs were similar to that of YM155S P1 hESCs (Figure S1K); therefore, we speculated that P3 and P4 hESCs gain a survival advantage. The gene signatures of P1 and P2 hESCs (YM155S group) clearly differed from those of P3 and P4 hESCs (YM155R group) (Figures 1C–1E). To examine significantly altered pathways in P4 hESCs, we performed gene set enrichment analysis (GSEA) with P4 hESC signature genes. In GSEA with GO biological process (GOBP) gene sets, it appeared that the various apoptotic signaling pathways were the gene sets that P4 signature genes highly enriched (Figures 1F and 1G). Among 719 gene sets that P4 signatures highly enriched, apoptotic signaling pathways were significantly concentrated (21/50, p = 0.0185, total = 4,436 GOBP gene sets, data not shown). Through gene network analysis of the apoptosis hallmark gene set (see Supplemental Information), we found that BCL2L1, ANAXA1, and PLAT were highly upregulated in the apoptosis hallmark gene set network (Figure 1H). This suggested that the apoptosis process and related gene regulation significantly differed between P1 and P4 hESCs.
Figure 1.

hESCs at Late Passage Numbers Are Resistant to YM155
(A) P1, P2, P3, and P4 hESCs were exposed to 5 nM YM155 for 24 hr and stained with 7-AAD and FITC-Annexin-V antibody. The live and dead cell populations were quantified through fluorescence-activated cell sorting analysis.
(B) The P1, P2, P3, and P4 hESCs were treated with 10 nM and 40 nM YM155 for 24 hr. The cells were stained with alkaline phosphatase (AP) stain in order to visually show the live hESC population after YM155 treatment (n = 3 independent experiment; mean ± SEM).
(C) Cluster heatmap of a sample cluster showed that P1, P2 hESCs were clustered as YM155S and P3, P4 hESCs as YM155R.
(D) Principal component analysis was performed with P1, P2, P3, and P4 hESCs. P1 and P2 hESCs were clustered into the YM155S group while P3 and P4 hESCs were clustered into the YM155R group.
(E) Gene distribution matrix analysis was performed and clustered to P1, P2 hESCs as one group and P3, P4 hESCs as the other group.
(F) GSEA of P4 hESCs signatures with hallmark gene sets in MSigDB. Only significantly enriched gene sets are represented as a heatmap. Sky blue indicates the significance threshold (p = 0.05), and dark red indicates the dose of enrichment beyond the significance threshold.
(G) Gene sets for hallmark of apoptosis and the apoptotic signaling pathway (left) and heatmap of differentially expressed genes of the apoptosis-related gene set (right).
(H) Undirected network of the apoptosis hallmark gene set with P4 hESC gene expression. To represent the change of gene expression, the fold-change (P4/P1, logged) level has been added in red (upregulation) or green (downregulation) to the only genes (nodes) that altered by at least 1.5-fold.
YM155R hESCs Are Resistant to Other Stresses
Due to the clear differences in apoptosis-related gene sets between P1 and P4 hESCs (Figures 1F–1H), we next examined whether YM155R hESCs were resistant to other DNA-damaging stimuli, such as bleomycin (BNM) and ionizing radiation (IR). P4 hESCs survived upon BNM and IR treatment, while all P1 hESCs died (Figures 2A and S2A). Given that hESCs are normally highly sensitive to genotoxic stress (Liu et al., 2013, Smith et al., 2012), this result suggests that P4 hESCs acquired a strong survival advantage. In addition to genotoxic stimuli, P4 hESCs were also resistant to dissociation-induced apoptosis (Ohgushi et al., 2010) (Figures S2B and S2C). Such a strong selective survival advantage of YM155R hESCs would be problematic because a clone resistant to various stresses due to genetic alterations would readily become dominant upon serial passage. To confirm this with a competitive proliferation assay (Cha et al., 2017), we mixed equal numbers of EGFP-P1 and P4 hESCs and cultured them under standard conditions. P4 hESCs outnumbered EGFP-P1 hESCs even after one passage (Figure 2B), despite these cells having similar growth rates (Figure S1K). Consistently, treatment with Y-27632, a chemical inhibitor of Rho-associated protein kinase (ROCK), which inhibits dissociation-induced apoptosis (Watanabe et al., 2007), significantly prevented the outnumbering of EGFP-P1 hESCs by P4 hESCs upon serial passage (Figure 2C). This suggests that a difference in susceptibility to dissociation-induced apoptosis would be one of factors leading to outnumbering of P4 hESCs.
Figure 2.

YM155R hESCs Are Resistant to Other Stresses
(A) Quantified amounts of live and dead cell populations were analyzed through fluorescence-activated cell sorting (FACS) after treatment with 10 eg/μL bleomycin (BNM) and 1 Gy of ionizing radiation (IR) on P1 and P4 hESCs. Cells were stained with both Annexin-V and 7-AAD (n = 3 independent experiments; mean ± SEM; Student's t test; ∗∗∗p < 0.001).
(B) Competition assay was performed with EGFP-P1 and P4. Each population was quantified through FACS before and after passaging.
(C) EGFP-P1 and P4 cells were mixed and treated with 10 μM Y-27632 at every passage. Population of both EGFP-P1 and P4 hESCs was quantified for every passage by FACS analysis.
(D) P1 and P4 hESCs were treated with etoposide (ETO, 20, 50, and 100 nM) for 24 hr and the live-cell population was quantified through an Annexin-V apoptosis detection assay. Quantified amounts of live-cell population are shown (n = 3 independent experiments; mean ± SEM).
(E) JC-1 MMP assay was performed with P1 and P4 hESCs after etoposide treatment. Each population was quantified through FACS analysis.
DNA replication inhibitors, including etoposide, selectively induce the death of hESCs with T12 (Ben-David et al., 2014). Hence, we investigated whether etoposide could abrogate the selective survival advantage of YM155R hESCs (especially P4 hESCs with T12). Unexpectedly, P4 and P3 hESCs were resistant to etoposide, unlike P1 hESCs (Figures 2D and S2D). Chemotherapeutic agents that induce DNA damage, including etoposide, BNM, and IR, trigger apoptosis by disrupting the mitochondrial membrane potential (MMP) (Galluzzi et al., 2006), which can be stabilized by the BCL2 family of proteins (Youle and Strasser, 2008). Consistent with the resistance of YM155R hESCs to etoposide, BNM, and IR (Figures S2A, 2D, and 2E), the MMP, as determined by JC-1 staining, remained stable upon etoposide treatment in P3 hESCs but not in P1 hESCs (Figure S2E), suggesting that high resistance toward chemotherapeutics and YM155 in YM155R hESCs results from stabilization of MMP.
BCL2L1 Expression Is Induced in YM155R hESCs
To explain the resistance of YM155R hESCs to cell death stimuli, we performed gene network analysis from the gene sets related to “DNA-damaging drug metabolism and resistance” of differentially expressed genes in P4 hESCs and found that BCL2L1 was upregulated in such gene networks (Figure S3), suggesting that BCL2L1 may be involved in the resistance to DNA-damaging drugs and YM155 in P4 hESCs. To confirm such prediction, a PCR array was performed comparing the expression levels of pro- and anti-apoptotic genes between P1 and P4 hESCs. Consistently, expression of BCL2L1, which encodes the anti-apoptotic protein BCL-xL, was significantly induced in P4 hESCs, whereas expression of several pro-apoptotic genes was significantly attenuated (Figure 3A). Given that BCL2L1 has already been demonstrated to contribute to the selective survival advantage (Avery et al., 2013) or culture adaptation (International Stem Cell Initiative et al., 2011) of hESCs, we compared its expression level between YM155S and YM155R hESCs. Consistently, expression of BCL2L1 (Figure 3B) and BCL-xL (Figure 3C) was significantly upregulated in YM155R hESCs. Furthermore, expression of BCL-xL in the surviving subpopulation of P3 hESCs significantly increased as the concentration of YM155 increased, implying that high expression of BCL-xL underlies the resistance of YM155R hESCs (Figure 3D).
Figure 3.

BCL2L1 Expression Is Induced in YM155R hESCs
(A) PCR array based on human apoptosis-related genes was performed with P1 and P4 hESCs. Anti-apoptotic and pro-apoptotic genes were sorted, and each gene profile was determined between P1 and P4 hESCs.
(B) RT-qPCR analysis was performed with P1, P2, P3, and P4 hESCs in order to compare the BCL2L1 mRNA expression (n = 4 independent experiments; mean ± SEM, ∗p < 0.05, ∗∗p < 0.01).
(C) Immunoblotting assay showed the relative Bcl-xL protein expression between P1, P2, P3, and P4 hESCs. β-actin was used as a protein loading control.
(D) P3 hESCs were exposed to the indicated concentrations of YM155 (0, 25, and 50 nM) for 24 hr and the surviving population was stained with Bcl-xL antibody in order to quantify the relative Bcl-xL expression level in the YM155-surviving population.
Prediction of Compounds that Selectively Target YM155R hESCs Using the CTRP
A set of compounds was recently found to selectively induce the death of therapy-resistant cancer cells by matching the gene signature of the target cells with the CTRP database (https://portals.broadinstitute.org/ctrp/) (Viswanathan et al., 2017). Similarly, we applied the gene signatures of YM155S and YM155R hESCs to the CTRP dataset in an attempt to identify compounds that target these cells. Using an algorithm that we developed (see Supplemental Experimental Procedures, Figure 4A), BH3 mimetics, such as ABT-737, ABT-263, and a mixture thereof, which target the BCL2 family of proteins, were predicted to specifically target YM155R hESCs (Figure 4B). Although histone deacetylase and microtubule inhibitors were also listed as candidate drugs, they were similarly predicted to target randomly selected cells (data not shown). DNA damage inducers, such as DNA alkylators and topoisomerase inhibitors, which were not effective against YM155R hESCs (Figure 2), were predicted to be rather resistant to YM155R hESCs (Figure 4B) as shown in Figure 2.
Figure 4.

Prediction of Compounds that Selectively Target YM155R hESCs Using the CTRP
(A) The scheme of the drug prediction. Pearson correlation coefficients were used for the pattern matching of signature gene expression and the CTRP dataset.
(B) The correlation scores of all drugs for YM155R-hESC effective compound prediction (boxplot). Partial BH3 mimetics (red circles) showed significantly high correlation scores. In contrast, DNA-damaging drug classes (blue and dark blue circles) were poorly correlated.
Targeting BCL-xL with Chemical Inhibitors Selectively Induces the Death of YM155R hESCs
When the survival of cancer cells is completely dependent on a certain highly expressed survival factor (referred to as oncogenic addiction), this factor is an optimal druggable target to selectively induce the death of these cells (referred to as oncogenic shock) (Sharma and Settleman, 2007).
Because P4 hESCs were predicted to be sensitive to BH3 mimetics (Figure 4), we hypothesized that interference in the anti-apoptotic function of BCL-xL, which may be closely associated with the selective survival advantage of YM155R hESCs, by BH3 mimetics would selectively trigger the death of these cells. To test this, we determined the sensitivities of YM155R and YM155S hESCs to ABT-263 and ABT-737, which are BH3 mimetic inhibitors of BCL-2 and BCL-xL (Tse et al., 2008). As predicted (Figure 4), treatment with ABT-263 (Figures 5A and 5B) or ABT-737 (Figures S4A–S4C) induced the death of P3 and P4 hESCs in a dose-dependent manner, whereas P2 hESCs were less sensitive. Similarly, treatment with ABT-263 induced the death of P4, but not EGFP-P1, hESCs in a time-dependent (Figure 5C and Videos S4 and S5) and dose-dependent (Figure 5D) manner.
Figure 5.

Targeting BCL-xL with Chemical Inhibitors Selectively Induces the Death of YM155R hESCs
(A) Annexin-V, 7-AAD staining was applied after ABT-263 treatment of P2, P3, and P4 hESCs with at designated concentrations (0, 10, 20, 50, and 100 nM). Annexin-V-negative, 7-AAD-negative populations were quantified and plotted.
(B) Immunoblotting assay was performed for P2, P3, and P4 hESCs after ABT-263 treatment for 48 hr. Cleaved caspase-3 (Ccasp-3) was blotted as a representative cell death marker protein, α-tubulin was used as a loading control.
(C) Time-lapse images were taken every 10 min in order to monitor morphological cell death in EGFP-P1 and P4 hESCs. Representative images were selected.
(D) EGFP-P1 and P4 hESCs were treated with ABT-263 with designated concentrations for 48 hr. Annexin-V staining was applied after ABT-263 treatment, and the viable cell population was quantified with FACS analysis (n = 3 independent experiments; mean ± SEM).
(E) JC-1 MMP analysis was performed after each drug treatment (ABT-263 50 nM, ABT-737 50 nM, etoposide 50 nM). MMP change was quantified with FACS analysis after JC-1 staining.
(F) P1 and P4 hESCs were treated with designated concentrations of WEHI-539 and propidium iodide (PI) staining was applied in order to quantify cell viability after drug treatment. The live-cell population was quantified through FACS (n = 3 independent experiments; mean ± SEM).
(G) Phase contrast images of CHA3- and CHA3-T12 hESCs after YM155 treatment for 24 hr (scale bar, 200 μm). The live-cell population was quantified with the Annexin-V apoptosis assay (n = 3 independent experiments; mean ± SEM).
(H) Cell viability of CHA3 and CHA3-T12 hESCs after ABT-263 treatment was quantified through Annexin-V assay. PI co-staining was used to indicate the cell death population (n = 3 independent experiments; mean ± SEM).
High mitochondrial priming (i.e., prompt cell death via the mitochondrial pathway partly due to constitutively active BAX localized at the Golgi complex, Dumitru et al., 2012; or mitochondrial translocation of p53, Lee et al., 2013) upon exposure to genotoxic stimuli, a typical feature of hPSCs compared with differentiated cells (Liu et al., 2013), was remarkably attenuated in YM155R hESCs (Figure 2F). This may be due to dramatic induction of the anti-apoptotic protein BCL-xL (Figure 3). Hence, inhibition of BCL-xL via treatment with a sub-optimal dose of BH3 mimetics was predicted to restore high mitochondrial priming because a similar level of active BAX remained localized at the Golgi in P1 and P3 hESCs (Figures S4D and S4E). As expected, treatment with BH3 mimetics clearly destabilized the MMP in P3, but not in P1, hESCs, while etoposide treatment destabilized the MMP in P1 hESCs (Figure 5E). ABT-263 and ABT-737 do not specifically target BCL-xL. Therefore, we next used WEHI-539, a selective BCL-xL inhibitor (Lessene et al., 2013), to confirm that YM155R hESCs were highly sensitive to BCL-xL inhibition. P3 hESCs were more sensitive to WEHI-539 than P1 hESCs (Figure 5F). To confirm these findings in another set of hESCs, we used CHA3-hESCs (Moon et al., 2011) at an early passage number (P67: CHA3), which have a normal karyotype, and those at a late passage number (P328: CHA3-T12), which have T12 (Figure S4F) (Moon et al., 2011). Consistently, CHA3-T12 hESCs were resistant to YM155, whereas CHA3 hESCs were not (Figure 5G). In addition, CHA3-T12 hESCs showed higher BCL2L1 expression than that of CHA3 hESCs (Figure S4G). Treatment with a relatively high concentration of ABT-263 selectively induced the death of CHA3-T12 hESCs (Figures 5H and S4H). For further confirmation, we used a pair of BJ-iPSCs, with different passage number (passage 35, P35; passage 156, P156). As expected, long-term BJ-iPSCs (P165) showed trisomy in chromosome 12 (hereafter BJ-iPSC-T12) (Figure S4I), resistance to YM155 treatment (Figure S4J), and high BCL2L1 expression (Figure S4K). BJ-iPSC-T12 with “survival advantage” (or resistance to YM155) (Figures 1 and 2), similar as those of YM155R hESCs and CHA3-T12 (Figure 5G), were more sensitive to ABT-263 than normal control (Figure S4L). In order to next examine whether high sensitivity to ABT-263 occurs in other chromosome alterations, we took advantage of H9 hESCs with duplication mutations in chromosome 1 between 1q.11 and 1q.44 loci (H9-D1 [or D1], Figure S5A). As D1 was highly sensitive to YM155, similar to P1 hESCs but unlike P4 hESCs (Figures S5B and S5C), we assumed that duplication mutation in chromosome 1 did not confer survival advantage. D1 hESCs were not sensitive to ABT-263 unlike P4 hESCs (Figures S5D and S5E), suggesting that high sensitivity to ABT-263 is associated with the survival advantage acquired during culture adaptation.
A BCL-xL Inhibitor Selectively Ablates YM155R hESCs in Co-culture with YM155S hESCs
The high sensitivity of YM155R hESCs to BCL-xL inhibitors may enable these cells to be selectively removed without markedly damaging YM155S hESCs. To test this, P4 hESCs that had been pre-stained with CDy1, a selective fluorescent probe for hESCs (Cho et al., 2016, Im et al., 2010), were mixed with P2 hESCs, and then the mixed culture was exposed to a sub-optimal dose of ABT-263. Active caspase-3 was detected in CDy1+ P4 hESCs, but not in CDy1- P2 hESCs, implying that ABT-263 selectively induced the death of P4 hESCs in a mixed culture (Figure 6A). To exclude the possibility that CDy1 had an off-target effect on the sensitivity of P4 hESCs to ABT-263, unlabeled P4 hESCs were mixed with EGFP-P1 hESCs, and then the mixed culture was treated with a sub-optimal dose of ABT-263. P4 hESCs rapidly died, while the majority of EGFP-P1 hESCs survived (Figure 6B and Video S6). Thus, in a mixed culture of P1 and P4 hESCs, the former cells became dominant after treatment with a sub-optimal dose of ABT-263 (Figure 6C), while the latter cells were dominant in standard culture conditions (Figure 2C). Similarly, WEHI-539 treatment triggered the death of P4, but not EGFP-P1, hESCs (Figures 6D and 6E) and eventually increased the population of P1 hESCs in the mixed culture (Figure 6F).
Figure 6.

A BCL-xL Inhibitor Selectively Ablates YM155R hESCs in Co-culture with YM155S hESCs
(A) Immunofluorescence cytochemistry (IFC) was performed on P2 and P4 co-culture conditions after 37.5 nM of ABT-263 treatment for 48 hr. CDy1 staining was used in order to indicate P4 hESCs, cleaved caspase-3 (Ccasp-3) was stained as a representative cell death marker protein (scale bars, 20 μm).
(B) Time-lapse images were taken every 10 min under EGFP-P1 and P4 co-culture conditions after ABT-263 treatment (37.5 nM, 48 hr).
(C) The EGFP-P1 and P4 cell population was quantified before and after ABT-263 treatment. Each cell was exposed to 37.5 nM ABT-263 for 48 hr, and the surviving population recovered without ABT-263 for 5 days. Each cell population was quantified with FACS analysis (n = 3 independent experiments; mean ± SEM).
(D) WEHI-539 treatment was applied at 0, 0.5, 0.75, 1.0, and 2.0 μM for 40 hr and Annexin-V staining was used in order to quantify the cell viability in EGFP-P1 and P4 mixed culture conditions. Annexin-V-positive populations (Annexin+) were plotted (right; n = 3 independent experiments; mean ± SEM).
(E) Time-lapse image was taken every 10 min in EGFP-P1 and P4 mixed culture conditions after 1 μM WEHI-539 for 48 hr.
(F) The cell population was quantified before and after WEHI-539 treatment in EGFP-P1 and P4 mixed culture conditions. The cells were treated with 1 μM WEHI-539 for 48 hr, and surviving cells recovered for 5 days. Each population was quantified with FACS analysis (n = 4 independent experiments; mean ± SEM, ∗p < 0.05).
(G) Treatment with 37.5 nM ABT-263 was applied to a co-culture of EGFP-P1 and P4 hESCs, and surviving cells recovered without drug for 5 days. Thereafter, the cells were passaged with or without Y-27632 (10 μM). Each cell population was quantified with FACS analysis.
Despite the high susceptibility of YM155R hESCs to BCL-xL inhibitors (Figures 5 and 6), it was technically challenging to completely eliminate these cells without damaging YM155S hESCs because long-term exposure to even a sub-optimal dose of BCL-xL inhibitors eventually induced the death of YM155S hESCs (data not shown). Hence, even after treatment with a sub-optimal dose of ABT-263 to significantly reduce the population of YM155R hESCs, residual YM155R hESCs eventually outnumbered YM155S hESCs due to their high resistance to dissociation-induced apoptosis. Consequently, treatment with Y-27632 delayed this outnumbering (Figure 6G).
P1 hESCs Remain Pluripotent after Exposure to ABT-263
After treatment with BCL-xL inhibitors to selectively reduce the population of YM155R hESCs, surviving YM155S hESCs must remain pluripotent for further applications. To investigate this, P1 hESCs that survived after exposure to ABT-263 were characterized. These cells expressed OCT4 and Lin-28A (Figure 7A) as well as POU5F1 and NANOG (Figure 7B), similar to non-treated P1 hESCs, and exhibited alkaline phosphatase activity (Figure 7C). Similarly, POU5F1 and SOX2 mRNA expression was maintained in normal BJ-iPSCs after ABT-263 treatment (Figure S6). These data imply that ABT-263 treatment does not affect the pluripotency of hESCs. The induction of ectoderm (NESTIN), mesoderm (MSX1), and endoderm (AFP and SOX17) marker genes upon spontaneous differentiation (Figure 7D) and the formation of teratomas comprising all three-germ layers (Figure 7E) clearly indicated that P1 hESCs remained pluripotent upon ABT-263 treatment.
Figure 7.

P1 hESCs Remain Pluripotent after Exposure to ABT-263
(A) P1 and ABT-P1 hESCs were immuno-stained with pluripotency marker proteins (OCT4 and Lin-28A). Insets show the magnified images. DAPI co-staining was used to indicate the nucleus (scale bars, 100 μm).
(B) RT-qPCR analysis was performed for human dermal fibroblasts (hDFs), P1 hESCs, and ABT-P1 hESCs in order to compare the expression of pluripotency marker genes (POU5F1 and NANOG) (n = 3 independent experiments; mean ± SEM).
(C) Alkaline phosphatase (AP) staining assay was performed in order to check pluripotency between hDFs, P1 hESCs, and ABT-P1 hESCs (scale bars, 500 μm).
(D) Three-germ-layer-specific marker gene expression was analyzed through RT-qPCR after spontaneous differentiation of P1 and ABT-P1 hESCs (MSX1 for mesoderm, NESTIN for ectoderm, and SOX17 and AFP for endoderm marker genes; n = 3 independent experiments; mean ± SEM).
(E) Teratoma formation assay was performed with ABT-P1 hESCs and representative three-germ layer tissues are shown. Teratoma tissues were histologically analyzed after H&E staining, Masson's trichrome staining, Alcian blue staining (scale bars, 50 μm).
Discussion
While the risk of tumorigenicity from residual undifferentiated hPSCs has been resolved by a variety of approaches (Jeong et al., 2017), the safety of hPSCs with genetic aberrations remains a serious concern (Cell Stem Cell Editorial Team, 2016). While unexpected genetic mutations in iPSCs recently stopped a clinical trial for macular degeneration (Mandai et al., 2017), the biological effects of hPSCs with genetic alterations remain hugely uncertain (Andrews et al., 2017). However, a selective survival advantage due to gain of BCL2L1 (Avery et al., 2013) and/or loss of TP53 (Merkle et al., 2017) allows these cells to escape cell death from genotoxic stresses. Thus, hPSCs with a selective survival advantage must be ablated to prevent the acquisition of further genetic abnormalities. In this line, CD30 was suggested to be a surface marker of transformed hPSCs (Herszfeld et al., 2006), and DNA replication inhibitors, including etoposide, were shown to selectively induce the death of hPSCs with T12 (Ben-David et al., 2014). However, YM155R hESCs in this study did not highly express CD30 (data not shown), consistent with a previous report (Harrison et al., 2009), and were not sensitive to etoposide or other genotoxic stimuli (Figure 2E).
Instead, we predicted that BH3 mimetics, which were previously shown to induce cell death in various hPSC models (Avery et al., 2013, Garcia et al., 2016, Lee et al., 2013), would effectively ablate YM155R hESCs based on their gene signatures and the CTRP dataset. In particular, survival advantage from culturally adapted hESCs due to gain of 20q11.21 to induce BCL2L1 gene expression, was also abrogated by BH3 mimetics (Avery et al., 2013). However, short-course treatment with a sub-optimal dose of BH3 mimetics selectively induced the death of three different hPSCs with survival advantage (e.g., high resistance to YM155 treatment) (Figures 5 and S4) by BCL2L1 gene expression (Figures 3, S4G, and S4K). On the contrary, hESCs without survival advantage (Figures S5B and S5C) were not sensitive to BH3 mimetics (Figure S5E) despite genetic aberration on chromosome 1 (Figure S5A). These results suggest that hPSCs with survival advantage acquired by BCL2L1 expression would be more sensitive to BH3 mimetics. High mitochondrial priming normally occurs in hPSCs exposed to genotoxic stimuli (Liu et al., 2013) via activation of the pro-apoptotic machinery (Dumitru et al., 2012, Lee et al., 2013, Liu et al., 2013), which is important to remove hESCs with damaged DNA and thereby maintain genome integrity (Weissbein et al., 2014). Inhibition of any anti-apoptotic factors in the mitochondria, which are critical for the balance between cell death and survival, perturbs this fine equilibrium and leads to cell death via the mitochondrial pathway (Lee et al., 2013, Liu et al., 2014). In this regard, hPSCs with induction of BCL2L1, a frequent genetic aberration in long-term cultured hPSCs (International Stem Cell Initiative et al., 2011) that was also observed in culturally adapted hPSCs (or hPSCs with survival advantage), readily escape high mitochondrial priming and become resistant to genotoxic stimuli. Thus, inhibition of BCL-xL by treatment with a sub-optimal dose of BH3 mimetics selectively induced the death of YM155R hESCs (Figures 5 and 6). Unfortunately, as BH3 mimetics did not completely remove YM155R hESCs, residual YM155R hESCs soon outnumbered YM155S hESCs once BH3 mimetics are withdrawn in the culture. Treatment with Y-27632 to inhibit dissociation-induced apoptosis of YM155S hESCs merely delayed this outnumbering (Figure 6G). Thus, identification of surface markers of normal or culture-adapted hESCs (if any) would be a more practical approach to minimize the risk of acquisition of further genetic abnormalities as previously suggested (Herszfeld et al., 2006). Nevertheless, this is an important proof of concept that will assist the development of a chemical approach to selectively enrich normal hPSCs.
Experimental Procedures
All animal care and experimental procedures were performed under the approval of the animal care committees in Konkuk University (IACUC # KU16203).
CTRP Drug Prediction Based on Transcriptional Signature
Drug sensitivity raw data were downloaded from CTRPv2 (https://portals.broadinstitute.org/ctrp/). Drug sensitivity raw data were processed for fitting growth inhibition curves using the nplr package in R. The profiles with poor data quality and the profiles of compounds annotated as screening hits were removed. Drug sensitivity was represented as the area under the curve of the fitted response curve. For gene expression data of CTRP cell lines, RNA-sequencing data were downloaded from Genomic Data Commons (GDC, https://gdc.cancer.gov/) and gene-wise standardization was performed by median centering for drug prediction. Drug prediction was performed based on pattern matching analysis of drug sensitivity and signature genes expression as described previously with some modifications (Viswanathan et al., 2017). We introduced the expression signature score (ESS) to represent expression values of signature genes as a single score; then we calculated the correlations between the drug sensitivity of CTRP cell lines and ESS of matched cells. We ranked the drugs that showed negative correlations by correlation p values because the cell numbers tested varied for each compound. The enrichment of the drug class of candidate drugs was tested by the hypergeometric test.
Fluorescence-Based Competitive Proliferation Assay
Fluorescence-based competitive proliferation assay was performed as described previously (Cha et al., 2017). Briefly, the same number of GFP-expressing hESCs (EGFP-P1) were mixed with P4 hESCs and maintained under standard culture conditions with 10 μM Y-27632. At each passage, the proportion of GFP+/GFP cells was measured by flow cytometry.
JC-1 Mitochondrial Membrane Potential Assay
In order to monitor the MMP hESCs before and after the drug treatment, JC-1 dye was stained after each small-molecule treatment. After staining with JC-1 dye, cells were analyzed and quantified through fluorescence-activated cell sorting.
Apoptosis-Related Gene PCR Array
RNA from P1 and P4 hESCs was isolated and cDNA was synthesized through reverse transcription following the manufacturer's instructions (RT2 profiler PCR array system, Human Apoptosis PCR Array, QIAGEN). After the gDNA elimination step, qPCR was performed and quantified using the online analysis program.
Karyotyping
H9 (P1, P2, P3, and P4) and CHA3 hESCs (P67, CHA3; P328, CHA3-T12) were incubated with 100 nM colcemid for 10 hr and were then collected. The karyotypes were determined using a standard G-banding procedure. A total of 20 (for CHA3) to 50 (for H9) metaphase cells for each type of cell line were examined. Chromosome analyses followed the Standard and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics.
Statistical Analysis
The quantitative data are expressed as the mean values ± standard error of the mean (SEM). Student's paired t tests or one-way ANOVAs were performed to analyze the statistical significance of each response variable. Pre-specified comparisons between groups were conducted (when appropriate) by Tukey’s post hoc test using the SPSS program (Statistical Package for the Social Sciences, version 17). p values less than 0.05 were considered statistically significant.
Author Contributions
S.-J.C., K.-T.K., H.-C.J, J.-C.P., and Y.-H.S. performed hESC cell death experiments. O.-S.K., J.-G.S., M.-O.L., and H.D.S. contributed to production and analysis of microarrays. S.K. and W.K. performed CTRP dataset analysis for drug prediction. S.-H.M. conceived the study. H.-J.C. conceived the study and wrote the manuscript.
Acknowledgments
This work was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare (grant number HI14C3365), and by a grant from the National Research Foundation of Korea (NRF) (2017M3A9B3061843 and 2017R1A2A2A05000766).
Published: October 4, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and six videos and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.09.002.
Contributor Information
Sung-Hwan Moon, Email: sunghwanmoon@kku.ac.kr.
Hyuk-Jin Cha, Email: hjcha93@snu.ac.kr.
Accession Numbers
The microarray dataset in this study was deposited in Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo) with accession number GEO: GSE119386.
Supplemental Information
References
- Andrews P.W., Ben-David U., Benvenisty N., Coffey P., Eggan K., Knowles B.B., Nagy A., Pera M., Reubinoff B., Rugg-Gunn P.J. Assessing the safety of human pluripotent stem cells and their derivatives for clinical applications. Stem Cell Reports. 2017;9:1–4. doi: 10.1016/j.stemcr.2017.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery S., Hirst A.J., Baker D., Lim C.Y., Alagaratnam S., Skotheim R.I., Lothe R.A., Pera M.F., Colman A., Robson P. BCL-XL mediates the strong selective advantage of a 20q11.21 amplification commonly found in human embryonic stem cell cultures. Stem Cell Reports. 2013;1:379–386. doi: 10.1016/j.stemcr.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D.E., Harrison N.J., Maltby E., Smith K., Moore H.D., Shaw P.J., Heath P.R., Holden H., Andrews P.W. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat. Biotechnol. 2007;25:207–215. doi: 10.1038/nbt1285. [DOI] [PubMed] [Google Scholar]
- Bedel A., Beliveau F., Lamrissi-Garcia I., Rousseau B., Moranvillier I., Rucheton B., Guyonnet-Duperat V., Cardinaud B., de Verneuil H., Moreau-Gaudry F. Preventing pluripotent cell teratoma in regenerative medicine applied to hematology disorders. Stem Cells Transl. Med. 2017;6:382–393. doi: 10.5966/sctm.2016-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David U., Arad G., Weissbein U., Mandefro B., Maimon A., Golan-Lev T., Narwani K., Clark A.T., Andrews P.W., Benvenisty N. Aneuploidy induces profound changes in gene expression, proliferation and tumorigenicity of human pluripotent stem cells. Nat. Commun. 2014;5:4825. doi: 10.1038/ncomms5825. [DOI] [PubMed] [Google Scholar]
- Ben-David U., Gan Q.F., Golan-Lev T., Arora P., Yanuka O., Oren Y.S., Leikin-Frenkel A., Graf M., Garippa R., Boehringer M. Selective elimination of human pluripotent stem cells by an oleate synthesis inhibitor discovered in a high-throughput screen. Cell Stem Cell. 2013;12:167–179. doi: 10.1016/j.stem.2012.11.015. [DOI] [PubMed] [Google Scholar]
- Cell Stem Cell Editorial Team 10 questions: clinical outlook for iPSCs. Cell Stem Cell. 2016;18:170–173. doi: 10.1016/j.stem.2016.01.023. [DOI] [PubMed] [Google Scholar]
- Cha Y., Han M.J., Cha H.J., Zoldan J., Burkart A., Jung J.H., Jang Y., Kim C.H., Jeong H.C., Kim B.G. Metabolic control of primed human pluripotent stem cell fate and function by the miR-200c-SIRT2 axis. Nat. Cell Biol. 2017;19:445–456. doi: 10.1038/ncb3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.J., Kim S.Y., Jeong H.C., Cheong H., Kim D., Park S.J., Choi J.J., Kim H., Chung H.M., Moon S.H. Repair of ischemic injury by pluripotent stem cell based cell therapy without teratoma through selective photosensitivity. Stem Cell Reports. 2015;5:1067–1080. doi: 10.1016/j.stemcr.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.J., Kim S.Y., Park S.J., Song N., Kwon H.Y., Kang N.Y., Moon S.H., Chang Y.T., Cha H.J. Photodynamic approach for teratoma-free pluripotent stem cell therapy using CDy1 and visible light. ACS Cent. Sci. 2016;2:604–607. doi: 10.1021/acscentsci.6b00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper J.S., Smith K., Gokhale P., Moore H.D., Maltby E., Johnson J., Meisner L., Zwaka T.P., Thomson J.A., Andrews P.W. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat. Biotechnol. 2004;22:53–54. doi: 10.1038/nbt922. [DOI] [PubMed] [Google Scholar]
- Dumitru R., Gama V., Fagan B.M., Bower J.J., Swahari V., Pevny L.H., Deshmukh M. Human embryonic stem cells have constitutively active bax at the Golgi and are primed to undergo rapid apoptosis. Mol. Cell. 2012;46:573–583. doi: 10.1016/j.molcel.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L., Larochette N., Zamzami N., Kroemer G. Mitochondria as therapeutic targets for cancer chemotherapy. Oncogene. 2006;25:4812–4830. doi: 10.1038/sj.onc.1209598. [DOI] [PubMed] [Google Scholar]
- Garcia C.P., Videla Richardson G.A., Dimopoulos N.A., Fernandez Espinosa D.D., Miriuka S.G., Sevlever G.E., Romorini L., Sassi M.E. Human pluripotent stem cells and derived neuroprogenitors display differential degrees of susceptibility to BH3 mimetics ABT-263, WEHI-539 and ABT-199. PLoS One. 2016;11:e0152607. doi: 10.1371/journal.pone.0152607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison N.J., Barnes J., Jones M., Baker D., Gokhale P.J., Andrews P.W. CD30 expression reveals that culture adaptation of human embryonic stem cells can occur through differing routes. Stem Cells. 2009;27:1057–1065. doi: 10.1002/stem.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herszfeld D., Wolvetang E., Langton-Bunker E., Chung T.L., Filipczyk A.A., Houssami S., Jamshidi P., Koh K., Laslett A.L., Michalska A. CD30 is a survival factor and a biomarker for transformed human pluripotent stem cells. Nat. Biotechnol. 2006;24:351–357. doi: 10.1038/nbt1197. [DOI] [PubMed] [Google Scholar]
- Im C.N., Kang N.Y., Ha H.H., Bi X., Lee J.J., Park S.J., Lee S.Y., Vendrell M., Kim Y.K., Lee J.S. A fluorescent rosamine compound selectively stains pluripotent stem cells. Angew. Chem. Int. Ed. 2010;49:7497–7500. doi: 10.1002/anie.201002463. [DOI] [PubMed] [Google Scholar]
- International Stem Cell Initiative. Amps K., Andrews P.W., Anyfantis G., Armstrong L., Avery S., Baharvand H., Baker J., Baker D., Munoz M.B. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011;29:1132–1144. doi: 10.1038/nbt.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H.C., Cho S.J., Lee M.O., Cha H.J. Technical approaches to induce selective cell death of pluripotent stem cells. Cell. Mol. Life Sci. 2017;74:2601–2611. doi: 10.1007/s00018-017-2486-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.T., Jeong H.C., Kim C.Y., Kim E.Y., Heo S.H., Cho S.J., Hong K.S., Cha H.J. Intact wound repair activity of human mesenchymal stem cells after YM155 mediated selective ablation of undifferentiated human embryonic stem cells. J. Dermatol. Sci. 2017;86:123–131. doi: 10.1016/j.jdermsci.2017.01.011. [DOI] [PubMed] [Google Scholar]
- Kim S.Y., Jeong H.C., Hong S.K., Lee M.O., Cho S.J., Cha H.J. Quercetin induced ROS production triggers mitochondrial cell death of human embryonic stem cells. Oncotarget. 2016;8:64964–64973. doi: 10.18632/oncotarget.11070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamm N., Ben-David U., Golan-Lev T., Storchova Z., Benvenisty N., Kerem B. Genomic instability in human pluripotent stem cells arises from replicative stress and chromosome condensation defects. Cell Stem Cell. 2016;18:253–261. doi: 10.1016/j.stem.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Lee M.O., Moon S.H., Jeong H.C., Yi J.Y., Lee T.H., Shim S.H., Rhee Y.H., Lee S.H., Oh S.J., Lee M.Y. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl. Acad. Sci. USA. 2013;110:E3281–E3290. doi: 10.1073/pnas.1303669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessene G., Czabotar P.E., Sleebs B.E., Zobel K., Lowes K.N., Adams J.M., Baell J.B., Colman P.M., Deshayes K., Fairbrother W.J. Structure-guided design of a selective BCL-X(L) inhibitor. Nat. Chem. Biol. 2013;9:390–397. doi: 10.1038/nchembio.1246. [DOI] [PubMed] [Google Scholar]
- Liu J.C., Guan X., Ryan J.A., Rivera A.G., Mock C., Agrawal V., Letai A., Lerou P.H., Lahav G. High mitochondrial priming sensitizes hESCs to DNA-damage-induced apoptosis. Cell Stem Cell. 2013;13:483–491. doi: 10.1016/j.stem.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.C., Lerou P.H., Lahav G. Stem cells: balancing resistance and sensitivity to DNA damage. Trends Cell Biol. 2014;24:268–274. doi: 10.1016/j.tcb.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandai M., Watanabe A., Kurimoto Y., Hirami Y., Morinaga C., Daimon T., Fujihara M., Akimaru H., Sakai N., Shibata Y. Autologous induced stem-cell-derived retinal cells for macular degeneration. N. Engl. J. Med. 2017;376:1038–1046. doi: 10.1056/NEJMoa1608368. [DOI] [PubMed] [Google Scholar]
- Martins-Taylor K., Nisler B.S., Taapken S.M., Compton T., Crandall L., Montgomery K.D., Lalande M., Xu R.H. Recurrent copy number variations in human induced pluripotent stem cells. Nat. Biotechnol. 2011;29:488–491. doi: 10.1038/nbt.1890. [DOI] [PubMed] [Google Scholar]
- Merkle F.T., Ghosh S., Kamitaki N., Mitchell J., Avior Y., Mello C., Kashin S., Mekhoubad S., Ilic D., Charlton M. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature. 2017;545:229–233. doi: 10.1038/nature22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon S.H., Kim J.S., Park S.J., Lim J.J., Lee H.J., Lee S.M., Chung H.M. Effect of chromosome instability on the maintenance and differentiation of human embryonic stem cells in vitro and in vivo. Stem Cell Res. 2011;6:50–59. doi: 10.1016/j.scr.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Ohgushi M., Matsumura M., Eiraku M., Murakami K., Aramaki T., Nishiyama A., Muguruma K., Nakano T., Suga H., Ueno M. Molecular pathway and cell state responsible for dissociation-induced apoptosis in human pluripotent stem cells. Cell Stem Cell. 2010;7:225–239. doi: 10.1016/j.stem.2010.06.018. [DOI] [PubMed] [Google Scholar]
- Sharma S.V., Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007;21:3214–3231. doi: 10.1101/gad.1609907. [DOI] [PubMed] [Google Scholar]
- Smith A.J., Nelson N.G., Oommen S., Hartjes K.A., Folmes C.D., Terzic A., Nelson T.J. Apoptotic susceptibility to DNA damage of pluripotent stem cells facilitates pharmacologic purging of teratoma risk. Stem Cells Transl. Med. 2012;1:709–718. doi: 10.5966/sctm.2012-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits C., Mateizel I., Geens M., Mertzanidou A., Staessen C., Vandeskelde Y., Van der Elst J., Liebaers I., Sermon K. Recurrent chromosomal abnormalities in human embryonic stem cells. Nat. Biotechnol. 2008;26:1361–1363. doi: 10.1038/nbt.1510. [DOI] [PubMed] [Google Scholar]
- Stambrook P.J. An ageing question: do embryonic stem cells protect their genomes? Mech. Ageing Dev. 2007;128:31–35. doi: 10.1016/j.mad.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Tateno H., Onuma Y., Ito Y., Minoshima F., Saito S., Shimizu M., Aiki Y., Asashima M., Hirabayashi J. Elimination of tumorigenic human pluripotent stem cells by a recombinant lectin-toxin fusion protein. Stem Cell Reports. 2015;4:811–820. doi: 10.1016/j.stemcr.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse C., Shoemaker A.R., Adickes J., Anderson M.G., Chen J., Jin S., Johnson E.F., Marsh K.C., Mitten M.J., Nimmer P. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Viswanathan V.S., Ryan M.J., Dhruv H.D., Gill S., Eichhoff O.M., Seashore-Ludlow B., Kaffenberger S.D., Eaton J.K., Shimada K., Aguirre A.J. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–457. doi: 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K., Ueno M., Kamiya D., Nishiyama A., Matsumura M., Wataya T., Takahashi J.B., Nishikawa S., Nishikawa S., Muguruma K. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 2007;25:681–686. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]
- Weissbein U., Benvenisty N., Ben-David U. Quality control: genome maintenance in pluripotent stem cells. J. Cell Biol. 2014;204:153–163. doi: 10.1083/jcb.201310135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werbowetski-Ogilvie T.E., Bosse M., Stewart M., Schnerch A., Ramos-Mejia V., Rouleau A., Wynder T., Smith M.J., Dingwall S., Carter T. Characterization of human embryonic stem cells with features of neoplastic progression. Nat. Biotechnol. 2009;27:91–97. doi: 10.1038/nbt.1516. [DOI] [PubMed] [Google Scholar]
- Youle R.J., Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.