

Abstract
Genome-wide association studies of childhood acute lymphoblastic leukemia (ALL) have identified regions of association at PIP4K2A and upstream of BMI1 at chromosome 10p12.31-12.2. The contribution of both loci to ALL risk and underlying functional variants remain to be elucidated. We carried out single nucleotide polymorphism (SNP) imputation across chromosome 10p12.31-12.2 in Latino and non-Latino white ALL cases and controls from two independent California childhood leukemia studies, and additional Genetic Epidemiology Research on Aging study controls. Ethnicity-stratified association analyses were performed using logistic regression, with meta-analysis including 3133 cases (1949 Latino, 1184 non-Latino white) and 12,135 controls (8584 Latino, 3551 non-Latino white). SNP associations were identified at both BMI1 and PIP4K2A. After adjusting for the lead PIP4K2A SNP, genome-wide significant associations remained at BMI1, and vice-versa (Pmeta<10−10), supporting independent effects. Lead SNPs differed by ethnicity at both peaks. We sought functional variants in tight linkage disequilibrium with both the lead Latino SNP among Admixed Americans and lead non-Latino white SNP among Europeans. This pinpointed rs11591377 (Pmeta=2.1×10−10) upstream of BMI1, residing within a hematopoietic stem cell enhancer of BMI1, and which showed significant preferential binding of the risk allele to MYBL2 (P=1.73×10−5) and p300 (P=1.55×10−3) transcription factors using binomial tests on ChIP-Seq data from a SNP heterozygote. At PIP4K2A, we identified rs4748812 (Pmeta=1.3×10−15), which alters a RUNX1 binding motif and demonstrated chromosomal looping to the PIP4K2A promoter. Fine-mapping chromosome 10p12 in a multi-ethnic ALL GWAS confirmed independent associations and identified putative functional variants upstream of BMI1 and at PIP4K2A.
Keywords: childhood acute lymphoblastic leukemia, genome-wide association study, fine-mapping, BMI1, enhancer element
Introduction
Acute lymphoblastic leukemia (ALL), which comprises approximately one third of childhood cancer diagnoses, is driven by disruption of hematopoietic regulatory pathways leading to the proliferation of immature lymphocytes. Although the mutational burden of ALL is relatively low, the disease is characterized by somatic alterations in B-cell differentiation genes, as well as those involved in the Ras pathway and in cell-cycle control 1, 2.
Heritable variation in genes associated with these same functional pathways is a risk factor for childhood ALL. Genome-wide association studies (GWAS) of ALL have identified SNP associations in genes involved in hematopoiesis and B-cell development, including ARID5B, IKZF1, CEBPE, and GATA3, as well as the cell-cycle control gene CDKN2A 3–6. Association has also been detected at the chromosome 10p12.31-12.2 region, with neighboring peaks located at PIP4K2A and upstream of the proto-oncogene BMI1 7, the latter region at which genome-wide significance has yet to be confirmed. The chromosome 10p12 SNPs were found to be specifically associated with the high hyperdiploid subtype of ALL 8, however a functional role for these loci in leukemogenesis has yet to be determined.
In a recent California-based GWAS of childhood ALL (using the same study population as current analyses), we identified novel SNP associations at chromosome 17q12-q21.1, which harbors the hematopoietic transcription factor IKZF3, and at chromosome 8q24, within a gene desert that appears to interact with the MYC oncogene via long-range chromatin interactions 9. We also replicated previously reported loci, including genome-wide significant associations at the chromosome 10p12 region containing PIP4K2A and BMI1. Here, we aimed to discover functional variants underlying the chromosome 10p12 associations and to identify their likely gene targets. To this end, imputation-based fine-mapping was carried out across the two 10p12 association peaks in Latino and non-Latino white ALL cases and controls from two independent California-based childhood leukemia studies, the California Cancer Records Linkage Project (CCRLP) and the California Childhood Leukemia Study (CCLS), with additional controls from the Genetic Epidemiology Research on Aging (GERA) study. Capitalizing on ethnic differences in haplotype structure, investigation of SNPs in strong linkage disequilibrium (LD) with both the lead SNP in Latinos and the lead SNP in non-Latino whites in corresponding populations was performed to pinpoint likely causal variants at both peaks.
Materials and Methods
Study Subjects
The study protocol was approved by the Institutional Review Boards at the California Health and Human Services Agency, University of California (San Francisco and Berkeley), Yale University, and of all participating hospitals. Sample acquisition for the California-based Childhood Cancer Record Linkage Project (CCRLP) GWAS of ALL is described in detail in Wiemels et al. 9. In brief, newborn dried bloodspots (DBS) for cases and controls were obtained from the California Biobank Program, California Department of Public Health (CDPH), Genetic Disease Screening Program. Childhood ALL cases were identified through linkage between the CDPH statewide birth records (years 1982-2009) and the California Cancer Registry (CCR, diagnosis years 1988-2011), with controls randomly selected and matched on year and month of birth, sex, and race/ethnicity (non-Latino white, non-Latino black, Latino (any race), Asian/Pacific Islander, other). Additional controls were included from the Genetic Epidemiology Research on Aging (GERA) study 10. In this study, analyses were limited to Latino and non-Latino white subjects.
ALL subtype-stratified analyses were carried out in the California Childhood Leukemia Study (CCLS), an independent case-control study of childhood leukemia that is described in detail elsewhere 11. In brief, patients with childhood leukemia under 15 years of age were identified and rapidly ascertained into the CCLS from California-based pediatric hospitals from 1995 to 2015. One or two controls were matched to each case on child’s date of birth, sex, Latino ethnicity, and maternal race as indicated on the birth certificate record. DNA samples for cases and controls were obtained from newborn DBS from the California Biobank Program Genetic Disease Screening Program, or from saliva or buccal swab samples. In the current study, we limited analyses to Latino and non-Latino white subjects with available genome-wide SNP array data (n=927 cases and 750 controls), and these were largely representative of the CCLS population as a whole.
Genome-wide SNP array data in the CCRLP
DNA was extracted from newborn DBS and genotyped on genome-wide SNP arrays as previously described 9. Briefly, CCRLP ALL cases and controls were genotyped using Affymetrix Axiom World LAT arrays. Following quality control filtering, 757,935 polymorphic autosomal SNPs were included in analyses. Genotype data for additional GERA controls, genotyped on the same Affymetrix arrays, were downloaded from dbGAP (Study Accession: phs000788.v1.p2). In total, 3133 childhood ALL cases (1949 Latino and 1184 non-Latino white) and 12,135 healthy controls (8584 Latino and 3551 non-Latino white) were included in association analyses. Case demographic data are included in Table S1. Case-control analyses were stratified by ethnicity, with SNP associations calculated using logistic regression and adjusted for the first ten ancestry-informative principal components, calculated using Eigenstrat 12, to control for population stratification. After adjusting for the first ten principal components, genomic inflation factors were calculated for the Latino and non-Latino white association analyses, which revealed minimal inflation of test statistics (λLatinos = 1.034; λNon-Latino whites = 1.004). A fixed-effects meta-analysis was used to combine results from the ethnicity stratified analyses.
Fine-mapping across chromosome 10p12
Imputation was carried out across the chromosome 10p12 locus encompassing the two association peaks upstream of BMI1 (“BMI1 peak”) and at PIP4K2A (“PIP4K2A peak”). BMI1 is centered between the two association peaks, therefore we took coordinates 500Kb upstream and downstream of this gene. Imputation of this 1Mb region was performed using the Impute2 v2.3.1 software and its standard Markov chain Monte Carlo algorithm, with default settings for targeted imputation 13 and using 1,000 Genomes Phase 3 haplotypes for the imputation reference panel 14. Poorly imputed SNPs were removed, i.e. those with imputation quality (info) scores <0.60 or posterior probabilities <0.90. Association statistics for imputed and directly-genotyped SNPs were calculated using logistic regression in SNPTESTv2, using an allelic additive model and probabilistic genotype dosages 15. The effect of individual SNPs on ALL risk was calculated while adjusting for the first 10 principal components from Eigenstrat. Analyses were carried out separately in Latino cases and controls and in non-Latino white subjects. Meta-analysis was carried out using the program META 16.
Following identification of two physically separate association peaks at chromosome 10p12.31 and 10p12.2, we investigated whether they were independently associations with ALL or whether this was due to linkage disequilibrium (LD) between SNPs in the two peaks. Thus, association analysis across 10p12 was repeated using the same logistic regression model adjusted for 10 principal components and also adjusting for additive effects of the lead PIP4K2A peak SNP rs10741006, and then again adjusting for the lead BMI1 peak SNP rs12769953.
LD structure across 10p12 was assessed separately in Latinos and non-Latino whites using Haploview v4.2. Haplotypes were constructed using all SNPs with minor allele frequencies >0.05 among control subjects, and blocks were plotted using the default block definition of Gabriel, et al. 17.
Epistasis analysis
Pairwise interaction analyses were carried out between three BMI1 peak SNPs and three PIP4K2A peak SNPs for a total of 9 interaction analyses. The three BMI1 peak SNPs included the most significant SNP in Latinos (rs12769953), the most significant SNP in non-Latino whites (rs4397732), and the candidate functional SNP rs11591377 that is in high LD with the former two SNPs (r2=0.93 and 0.97 respectively). The three PIP4K2A peak SNPs included the most significant SNP in Latinos (rs10741006), the most significant SNP in non-Latino whites (rs7912551), and the candidate functional SNP rs4748812 that is in LD with the former two SNPs (r2=0.82 and 0.80 respectively). Each interaction model contained the main effects for each SNP and the interaction term between each pair of SNPs, as well as the first 10 principal components. Analyses were performed separately in Latino subjects and non-Latino white subjects. P-values are reported for the interaction term of each logistic regression model performed using the glm command in the R statistical environment.
Multi-ethnic linkage disequilibrium correlation analysis
To identify putatively causal variants in the BMI1 peak and the PIP4K2A peak, at each locus we sought SNPs that demonstrated strong LD with both the lead SNP in Latinos and the lead SNP in non-Latino whites in the corresponding reference populations, i.e. in Admixed Americans (AMR) and Europeans, respectively. Thus, for the BMI1 peak we looked for SNPs in strong LD with rs12769953 in AMR and with rs4397732 in European populations, and for the PIP4K2A peak we sought SNPs in strong LD with rs10741006 in AMR and rs7912551 in Europeans. The LDlink “LDproxy” tool, a publicly available application for SNP LD analysis in 1000Genomes Project Phase 3 genotype data 18, was used to calculate LD values (r2 and D’) for all SNPs within +/−500Kb of queried variants. For AMR populations, we included the “MXL” (Mexican ancestry from Los Angeles), “PUR” (Puerto Ricans from Puerto Rico), “CLM” (Colombians from Medellin, Colombia), and “PEL” (Peruvians from Lima, Peru) datasets. For European populations we included “CEU” (Utah residents from North and West Europe), “TSI” (Toscani in Italia), “FIN” (Finnish in Finland), “GBR” (British in England and Scotland), and “IBS” (Iberian population in Spain).
Scatter plots were generated for the BMI1 peak and PIP4K2A peak SNPs, plotting the r2 of SNPs with the lead Latino SNP in AMR populations against r2 of the same SNPs with the lead non-Latino white SNP in Europeans. Triallelic SNPs were excluded. We included only those SNPs with P < 5.0 × 10−6 in the adjusted meta-analyses results for both the BMI1 peak and PIP4K2A peak.
Enhancer element analysis
The Roadmap Epigenome Browser 19 was used to identify enhancer elements overlapping the 10p12.31 and 10p12.2 association peaks. We assessed tracks for DNase I hypersensitive sites (DHS), histone 3 lysine 4 monomethylation (H3K4me1), and H3K27 acetylation (H3K27ac) in HSCs (“Mobilized CD34 primary cells”), lymphoblastoid cell line (LCL) GM12878, and in the HSC-like myelogenous leukemia cell line K562. Predicted gene targets of enhancer elements were investigated using EnhancerAtlas (www.enhanceratlas.org), a recently developed database containing a consensus of human genomic enhancers derived from data on 76 cell lines and 29 different tissue types 20. In brief, enhancers are predicted based on at least three independent experimental tracks (e.g. DNase hypersensitivity, transcription factor binding, and histone modification), and enhancer gene targets are predicted using the Integrated Method for Predicting Enhancer Targets (IM-PET) algorithm.
H3k27ac HiChIP data analysis
H3K27ac HiChIP is a recently developed protein-targeting chromatin conformation method for generating high-resolution contact maps between active enhancers and their target genes 21. H3K27ac HiChIP data was downloaded for GM12878 cells and for the K562 cell line (Gene Expression Omnibus, GEO dataset GSE101498), and uploaded into Juicebox software for visualization of 3D chromatin interactions across the chr10p12.31-p12.2 region. Knight-Ruiz (balanced) normalization was used to remove Hi-C matrix biases as recommended 22. Tracks for the same data were also uploaded into the Roadmap Epigenome Browser to assess locus interactions.
MethylC-Seq Analysis
Methylation levels at CpGs across the BMI1 peak enhancer locus (overlapping rs11591377) and the PIP4K2A peak enhancer locus (overlapping rs4748812) were assessed using whole genome shotgun bisulfite sequencing (MethylC-Seq) data tracks in the Epigenome Browser 23. MethylC-Seq data was assessed for HSCs (Mobilized CD34 primary cells) and a selection of different tissue types including fetal thymus, spleen, brain hippocampus, small intestine, liver, lung, gastric, adipose tissue, and ovary. At the rs11591377 (BMI1) enhancer, we assessed the proportion of methylated cytosine at 21 CpGs spanning a 2,215bp region, excluding analysis of CpGs with <10 sequenced reads in any tissue types. At the rs4748812 (PIP4K2A) enhancer, we assessed proportion of methylated cytosines at 7 CpGs spanning a 422bp region (again excluding CpGs with <10 reads).
Transcription factor binding analysis
UCSC Genome Browser was used to identify TFs found to bind to the BMI1 peak and PIP4K2A peak regulatory elements in ENCODE ChIP-seq analyses. To assess whether candidate SNPs altered any TF binding sites, we used the “TFBIND” software (http://tfbind.hgc.jp) 24, inputting an 11bp sequence that included the SNP itself (either risk or protective allele) plus 5bp up- and 5bp downstream sequence.
ENCODE ChIP-Seq analysis of K562 cells
For TFs predicted to bind to the BMI1 peak locus (at rs11591377), we downloaded available BAM files for corresponding ENCODE ChIP-seq experiments in K562 cells (from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeSydhTfbs/), including for ARID3A, BHLHE40, CEBPB, CTCF, c-Jun, c-Myc, p300, GATA2, JunD, MAFF, MAFK, MAX, TAL1, and ZNF143. BAM files for MYBL2 ChIP-seq experiments in K562 cells were downloaded separately (from https://www.encodeproject.org/experiments/ENCSR162IEM/), as these data were only released in August 2017. For each TF, separate BAM files were downloaded for each of two ChIP-seq biological replicates. BAM files were imported into the SNP & Variation Suite (SVS) software (Golden Helix), and visualized using the GenomeBrowse tool. For each TF, the number of sequenced reads was counted for the rs11591377 risk allele (G) and protective allele (A) in each biological replicate, and a binomial significance test used to calculate P values for the distribution of risk vs. protective allele reads compared with the expected distribution of 1:1. Fisher’s method was used for meta-analysis of P-values, using the R statistical package ‘metap’. To confirm that K562 cells are heterozygous for SNP rs11591377, we extracted DNA from an aliquot of cells (provided by Dr. Neil Shah, UCSF) using the Qiagen DNA Blood and Mini Kit, and genotyped using a Taqman assay for rs11591377 (Assay ID: C_____85910_10) on a Droplet Digital PCR machine (BioRad).
Replication and cytogenetic subtype analysis in the CCLS
Cytogenetic subtype information was not available for CCRLP cases. Therefore, to assess subtype-specific associations of the lead chromosome 10p12 SNPs, we utilized data from the CCLS, an independent case-control study of childhood leukemia 11. SNP genotype data was available for 927 ALL cases (589 Latino, 338 non-Latino white, Table S1) including 238 with high hyperdiploidy (HD) (156 Latino, 82 non-Latino white), as confirmed by FISH or G-banding 25, and 750 controls (506 Latino, 244 non-Latino white). DNA was extracted from neonatal DBS, saliva, or buccal cells, and genotyped on Illumina HumanOmniExpress or HumanOmniExpressExome genome-wide SNP arrays 26. SNP imputation was performed using Impute2, as described earlier for analysis in CCRLP. Multidimensional scaling (MDS) components were calculated using PLINK1.9 27 to control for population stratification. For this analysis, we included six SNPs in total: the lead Latino and non-Latino white SNPs and putative functional variants at both the BMI1 and PIP4K2A peaks (i.e. rs12769953, rs4397732, rs11591377, rs10741006, rs7912551, and rs4748812). Association of each SNP with ALL risk (overall and with HD-ALL subtype) was tested separately in Latinos and non-Latino whites using logistic regressions assuming an allelic additive model, adjusting for the first seven MDS components. A fixed-effects meta-analysis was used to combine results from the ethnicity stratified analyses, separately in overall ALL cases versus controls, in HD cases versus controls, and in non-HD cases versus controls. Cochran’s Q-test was used to test for heterogeneity between Latino and non-Latino white studies.
Results
SNP genotype imputation across chromosome 10p12 in Latino and non-Latino white ALL cases and controls followed by meta-analysis of ethnicity stratified case-control analyses confirmed two ALL association peaks, with multiple SNPs at each peak reaching genome-wide significance (P < 5.0 × 10−8). The strongest association was at a region encompassing the 3’ end of PIP4K2A at chr10p12.2 (hereafter the “PIP4K2A peak”). The lead PIP4K2A peak SNP in the multi-ethnic meta-analysis was rs10741006, with Pmeta = 5.78 × 10−19 and an odds ratio (OR) of 1.36 (95% confidence intervals, CI: 1.30-1.43). In ethnicity-stratified analyses this was also the most significant variant in Latinos (P = 3.19 × 10−12) and was strongly associated in non-Latino whites (P = 7.35 × 10−9); however, the lead SNP in non-Latino whites was rs7912551 (P = 7.41 × 10−10) (Table S2).
The second association peak was located ~ 450Kb upstream of PIP4K2A, between genes DNAJC1 and BMI1 at chr10p12.31 (hereafter the “BMI1 peak”). The lead SNP was rs12769953, with Pmeta = 1.33 × 10−13 and OR = 1.32 (95% CI: 1.25-1.39). This was also the most significant variant in Latinos (P = 2.99 × 10−9) and was associated in non-Latino whites (P = 3.56 × 10−6) (Table S2). The lead SNP in non-Latino whites was rs4397732 (P = 2.05 × 10−6), which was in very strong LD with the lead Latino SNP (rs12769953) in European reference populations (r2 = 0.98, D’ = 0.99) and in relatively strong LD in AMR reference populations from the 1000 Genomes project (r2 = 0.82, D’ = 0.93).
Assessment of LD structure across the chromosome 10p12 loci in Latinos and non-Latino whites revealed slight differences in haplotype structure, in particular showing shorter haplotype structure in Latinos at the PIP4K2A peak (Figure S1).
Two chromosome 10p12 association peaks associated independently with ALL
After adjusting for the lead PIP4K2A SNP (rs10741006), associations at the BMI1 peak remained genome-wide significant for several SNPs, including the lead Latino and lead non-Latino white SNPs (rs12769953 and rs4397732, respectively) (Table 1, Figure 1). Similarly, after adjusting for the lead SNP in the BMI1 peak (rs12769953), rs10741006 in the PIP4K2A peak retained genome-wide statistical significance (P = 2.21 × 10−16), thus confirming independent associations between these two loci and ALL risk (Table 1, Figure 1).
Table 1.
Lead SNPs at the independent chromosome 10p12 association peaks upstream of BMI1 and at PIP4K2A, in analyses of acute lymphoblastic leukemia cases and controls from the Childhood Cancer Record Linkage Project (CCRLP), and additional controls from the Genetic Epidemiology Research on Aging (GERA) study. Results adjusted for the lead SNP at the alternate peak (i.e. BMI1 SNPs adjusted for lead PIP4K2A SNP, and vice-versa).
| Peak | SNP | Chromosome 10 position (hg19) | CCRLP Latinos | CCLRP Whites | CCRLP Meta-analysis | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| RAF controls a | P-value b | OR (95% CI) b | RAF controls a | P-value b | OR (95% CI) b | P-value | OR (95% CI) | |||
| BMI1 | rs12769953 | 22407656 | 0.768 | 3.41 × 10−8 | 1.29 (1.20-1.39) | 0.777 | 2.11 × 10−4 | 1.25 (1.13-1.38) | 6.13 × 10−11 | 1.28 (1.21-1.35) |
| BMI1 | rs4397732 | 22435841 | 0.770 | 6.64 × 10−8 | 1.29 (1.20-1.38) | 0.776 | 1.14 × 10−4 | 1.27 (1.15-1.39) | 6.28 × 10−11 | 1.28 (1.21-1.36) |
| BMI1 | rs11591377 c | 22423302 | 0.778 | 6.89 × 10−8 | 1.29 (1.20-1.39) | 0.78 | 3.65 × 10−4 | 1.24 (1.12-1.37) | 2.07 × 10−10 | 1.27 (1.20-1.35) |
| PIP4K2A | rs10741006 | 22856019 | 0.7 | 3.78 × 10−11 | 1.37 (1.28-1.47) | 0.59 | 2.94 × 10−7 | 1.296 (1.20-1.40) | 2.21 × 10−16 | 1.33 (1.27-1.40) |
| PIP4K2A | rs7912551 | 22818337 | 0.716 | 5.0 × 10−9 | 1.32 (1.23-1.41) | 0.632 | 7.15 × 10−8 | 1.327 (1.22-1.43) | 4.38 × 10−15 | 1.32 (1.25-1.39) |
| PIP4K2A | rs4748812 c | 22839083 | 0.684 | 5.91 × 10−10 | 1.32 (1.24-1.41) | 0.593 | 1.85 × 10−7 | 1.30 (1.20-1.40) | 1.3 × 10−15 | 1.31 (1.25-1.38) |
Risk allele frequency (RAF) in Latino and non-Latino white control subjects
P-values and odds ratios (ORs) adjusted for the first ten principal components. In addition, P-values and ORs for BMI1 peak SNPs adjusted for lead PIP4K2A SNP rs10741006, and P-values and ORs for PIP4K2A peak SNPs adjusted for lead BMI1 SNP rs12769953
Candidate causal variant
Figure 1. Chromosome 10p12 loci independently associated with childhood ALL.
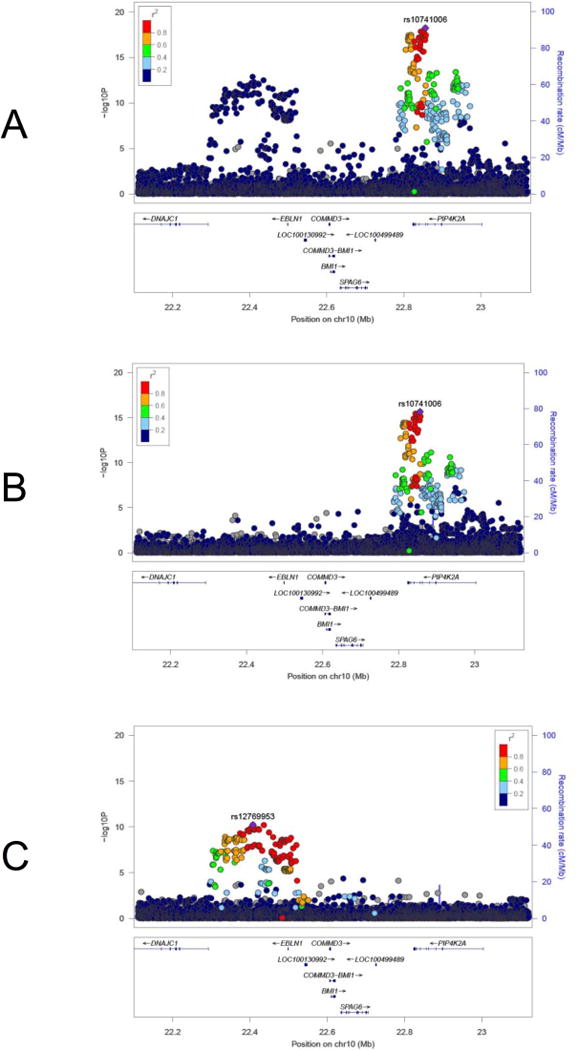
A: unadjusted meta-analysis of Latino and non-Latino white GWAS revealing two association peaks, upstream of BMI1 and at PIP4K2A; B: meta-analysis adjusted for the lead BMI1 peak SNP rs12769953; C: meta-analysis adjusted for the lead PIP4K2A peak SNP rs10741006. SNPs at both loci remain genome-wide significant (P<5×10−8) after adjustment. Manhattan plots generated using LocusZoom.
Epistasis analyses between the lead SNPs in the two association peaks revealed no evidence of synergistic or antagonistic interaction between SNPs (P>0.5 for all interactions tested in both Latinos and non-Latino whites).
Given the independence of association and lack of epistasis, we hypothesized that associated variants in the two peaks impart ALL risk via discrete mechanisms and possibly by affecting different gene targets. To explore this hypothesis, we capitalized on our multi-ethnic study design and that the lead SNP at both association peaks differed by ethnicity to investigate putative causal variants underlying the association at each peak. We predicted that underlying causal variant(s) would be in strong LD with lead tag SNPs in both Latino and non-Latino white populations.
rs11591377 is a putative BMI1 enhancer variant
At the BMI1 peak, there were 6 successfully imputed SNPs in tight LD (r2 >0.90) with both the lead Latino SNP (rs12769953) in AMR reference populations and the lead non-Latino white SNP (rs4397732) in Europeans (Figure 2a). All 6 SNPs were strongly associated with ALL (Pmeta < 5 × 10−10) after adjusting for the lead PIP4K2A SNP. Three SNPs – rs11591377, rs12773841, and rs11599410 – were in perfect LD (r2 =1) in AMR and in perfect or near-perfect LD (r2 >0.99) in Europeans (Table S3). None of the BMI1 peak SNPs were expression quantitative trait loci (eQTLs) for expression of BMI1 or any other genes in the Genotype-Tissue Expression (GTEx) project 28 or in the Genetic European Variation in Health and Disease (GEUVADIS) project RNA sequencing data (in which no significant eQTLs were detected for BMI1).
Figure 2. Multi-ethnic LD correlation analysis pinpoints BMI1 enhancer element SNP rs11591377.
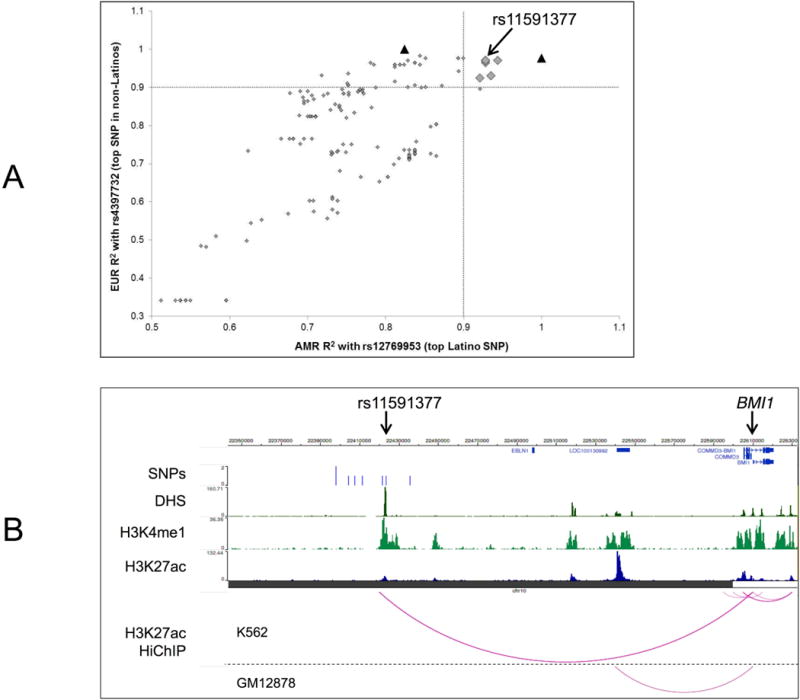
A: Scatter plot showing correlation between the LD (r2) of SNPs with the lead Latino BMI1 peak SNP (rs12769953) in Admixed American (AMR) populations with the LD (r2) of the same SNPs with the lead non-Latino white SNP (rs4397732) in European (EUR) populations. Plot includes data for 164 biallelic SNPs with P<5.0 × 10−6 in adjusted meta-analysis. Black triangles denote the lead Latino and lead non-Latino white SNPs. SNP rs11591377 is one of only 6 SNPs (grey diamonds) with a high LD (r2>0.9) with both the lead Latino and lead non-Latino white SNP in respective populations. LD values calculated using 1000 Genomes Project Phase 3 data in the LDlink tool “LDproxy”.
B: Epigenome Browser screenshot showing positions, indicated by vertical blue lines, of the lead Latino and lead non-Latino white SNPs along with the 6 additional SNPs in strong LD with both. SNP rs11591377 (in strong LD with both) overlaps a predicted enhancer element for BMI1, as supported by strong peaks for DNase I hypersensitivity (DHS), H3K4me1, and H3K27ac in hematopoietic stem cells (Mobilized CD34 cells). H3K27ac HiChIP data show strong support (score = 203.96) for chromosomal looping between rs11591377 locus and BMI1 gene in the hematopoietic stem cell-like K562 cell line, which was not found in GM12878 LCLs.
We next explored the potential regulatory function of candidate SNPs. Both rs11591377 and rs12773841 overlapped a ~2kb region containing multiple transcription factor (TF) binding sites in ENCODE 29 (Figure S2). A DNase I hypersensitivity (DHS) cluster was identified at this locus in 22 cell types in ENCODE, of which CD34+ Mobilized cells (i.e. hematopoietic progenitor cells) had by far the highest signal. In EnhancerAtlas 20, this DHS cluster lies within a designated enhancer for BMI1 in 7 different tissues/cell lines, including: CD34+ cells, GM12878 LCL, fetal thymus, and the leukemia cell lines K562 and HL-60. SNP rs11591377 is positioned at the maximum height of the enhancer consensus score, whereas rs12773841 lies ~200bp upstream of the 5’ end of the enhancer locus (Figure S3). This locus is the only predicted enhancer for BMI1 located in the BMI1 peak region.
We then used the Roadmap Epigenome browser 19 to assess signals for DHS, which represents open chromatin regions, and for the histone modifications H3K4me1 and H3K27ac, which are known markers of enhancer elements. In support of the rs11591377 locus as an enhancer, we found strong peaks for DHS, H3K4me1, and H3K27ac in hematopoietic stem cells (HSCs) (Figure 2b) and in K562 cells, with smaller peaks in GM12878 LCLs. Analysis of H3K27ac HiChIP chromatin interaction data supported this locus as an enhancer for BMI1 in K562 cells, with a very strong signal for looping between the rs11591377 SNP locus (chr10:22,420,000-22,425,000) and BMI1 (chr10:22,605,000-22,610,000), with score = 203.96 (Figure 2b). This interaction was detected in GM12878 LCLs with a much lower score (score = 5.84), and no looping was detected in T-cells.
Methylation of cytosines at CpGs is frequently associated with transcriptional silencing at promoters and enhancer regions. Thus, we assessed methylation levels at CpG loci across the BMI1 enhancer region, using whole genome bisulfite sequencing data in different tissue types in the Epigenome Browser. This revealed variation across tissues, with HSCs almost entirely unmethylated across 9 CpGs in a region spanning 758bp (mean methylation = 0.56%) (Figure S4), whereas average methylation levels at other tissues ranged from 6.7% (ovary) to 65.7% (brain hippocampus) (Figure S5). Assessment of other BMI1 enhancers revealed these to be largely unmethylated across different tissues (data not shown), suggesting that the BMI1 enhancer spanning SNP rs11591377 is unique in its specificity to HSCs.
rs11591377 associated with differential transcription factor binding
Several transcription factors (TFs), including known hematopoietic regulators, were predicted to bind to this enhancer locus and overlap rs11591377 (Table S4). Analysis of ENCODE ChIP-seq data revealed that the K562 cell line was heterozygous for SNP rs11591377, and revealed significant preferential binding of p300 (P=1.55×10−3) and JunD (P=2.61×10−3) to the ALL risk allele (G), with trends in the same direction for c-Myc (P=0.077), JunB (P=0.194), MAFK (P=0.069), SPI1 (P=0.134) and TAL1 (P=0.14) (Figures S6 and S7, Table S5). Additional TFs did not have available data in K562 cells or showed minimal binding at the rs11591377 enhancer. We assessed whether rs11591377 altered any TF binding motifs using TFBIND software 24, and found the highest motif score (0.88) for the MYB transcription factor when including the risk allele G, and no predicted MYB binding for the non-risk allele A (Figure 3). MYB-like 2 (MYBL2), a MYB paralog with a similar binding motif sequence, showed a high level of ChIP-seq coverage at the enhancer locus and with highly significant preferential binding of the risk allele in K562 cells (P=1.73×10−5) (Figure 3). Genotyping of the rs11591377 SNP in genomic DNA from K562 cells using droplet digital PCR (ddPCR) confirmed a 1:1 ratio of allele G:A, supporting that the bias in the ChIP-seq data was not due to allelic copy number differences.
Figure 3.
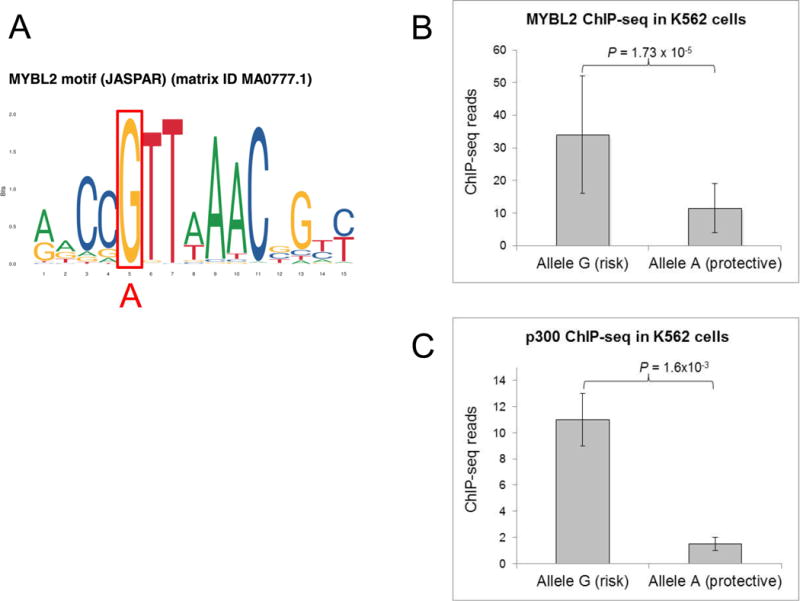
rs11591377 disrupts MYBL2 binding at BMI1 enhancer
A: MYBL2 binding motif showing high conservation of the rs11591377 risk allele G (red box), with no preference for the non-risk allele A (in red below). Binding motif downloaded from the JASPAR database (http://jaspar.genereg.net/).
B: MYBL2 ChIP-Seq data in K562 cells heterozygous for rs11591377 demonstrate significant preferential binding of the risk allele G (P = 1.73 × 10−5). The mean proportion of allele G out of total sequenced reads was 0.766. Bars show mean read depth of two biological replicates and error bars represent the standard error of the mean.
C: p300 ChIP-Seq data in K562 cells heterozygous for rs11591377 demonstrate significant preferential binding of the risk allele G (P = 1.6 × 10−3). The mean proportion of allele G out of total sequenced reads was 0.883. Bars show mean read depth of two biological replicates and error bars represent the standard error of the mean.
rs4748812 generates RUNX1 binding site in PIP4K2A enhancer
At the PIP4K2A peak, there were no variants with r2>0.90 but there were 4 SNPs with r2>0.75 with both the lead Latino SNP rs10741006 and the lead non-Latino white SNP rs7912551 in corresponding populations (Figure S8). All 4 SNPs – rs4747443, rs4748812, rs12146350, and rs746203 – were associated with ALL at Pmeta<5×10−14 after adjusting for the lead BMI1 peak SNP (rs12769953), and were significant eQTLs for PIP4K2A expression in GTEx with ALL risk alleles associated with increased expression (Table S6).
We next sought to determine whether any of these SNPs overlapped putative regulatory regions in PIP4K2A, based on DHS, H3K4me1, and H3K27ac peaks in relevant tissues in the Epigenome Browser. Within a ~38Kb region encompassing the lead Latino SNP, the lead non-Latino white SNP, and the 4 SNPs listed above, only rs4748812 overlapped a region with strong evidence of regulatory function, with a large DHS peak found at chr10: 22,839,000-22,840,000 (Figure S9). SNP rs4748812 was also the only variant found to overlap a DHS cluster locus in the UCSC Genome Browser (Table S6).
H3K27ac HiChIP data analysis in the Epigenome Browser revealed evidence of chromatin looping between the locus containing rs4748812 (chr10:22,835,000-22,840,000) and another PIP4K2A regulatory region ~100kb downstream (chr10:22,935,000-22,940,000) in GM12878 cells, with score = 11.2 (Figure S9). HiChIP analysis in Juicebox also supported interaction between these loci in GM12878 cells (observed/expected score = 1.49), and even stronger interaction between the rs4748812 locus and the PIP4K2A promoter region at chr10:23,000,000-23,005,000 (O/E score = 2.22) (Figure S10).
Inspection of ENCODE ChIP-seq data revealed multiple TFs binding at the rs4748812 locus in LCLs and in K562 cells, including TAL1, CTCF, p300, and ARID3A. Analysis of MethylC-seq data across tissues revealed that demethylation at the rs4748812 locus is largely specific to HSCs (Figures S11 and S12). Investigation of potential TF binding motifs revealed that the rs4748812 risk allele T creates a RUNX1 (AML1) binding site (TFbind score = 0.911), which is not predicted with the protective allele C (Figure S13).
Cytogenetic subtype analysis of BMI1 and PIP4K2A SNPs
Association analyses of lead chromosome 10p12 SNPs in the CCLS revealed that BMI1 SNP rs11591377 was more strongly associated with high hyperdiploid (HD) ALL (OR = 1.56; 95% CI: 1.07-2.27) than with overall ALL risk (OR = 1.22; 95% CI: 1.01-1.49), with no association in non-HD ALL (OR = 1.06; 95% CI: 0.87-1.29) (Table S7, Figure S14). The HD-ALL association with rs11591377 appeared to be stronger in Latinos (OR = 1.71; 95% CI: 1.20-2.50), although the inter-ethnic study difference was not significant (P-value for heterogeneity, Phet = 0.58, Figure S14). The PIP4K2A putative causal SNP rs4748812 also appeared to be more strongly associated with HD-ALL than with overall ALL risk in Latinos. This pattern was not seen in non-Latino whites, although the difference between ethnic groups was again not significant (Phet = 0.32) (Table S7, Figure S14).
Discussion
Our group and others have previously carried out fine-mapping across childhood ALL association loci, identifying both causal coding variants (CDKN2A) and causal regulatory variants (CEBPE and ARID5B) impacting ALL risk 30–32. Here, fine-mapping across chromosome 10p12 confirmed that two nearby association peaks are independently associated with childhood ALL predisposition, and likely mediate ALL risk through the effects of distinct genes, namely BMI1 and PIP4K2A. In the first reported fine-mapping of the chromosome 10p12.31 association signal, we pinpointed a putatively causal variant rs11591377 located in a predicted enhancer element for BMI1, and that demonstrated preferential transcription factor binding of the risk allele in ENCODE ChIP-Seq data. At chromosome 10p12.2, we identified SNP rs4748812 that lies within a regulatory element in PIP4K2A and is predicted to alter binding of the RUNX1 transcription factor.
The proto-oncogene BMI1 is a member of the polycomb repressive complex 1, and is a negative regulator of the cell-cycle checkpoint proteins p16 and p14ARF 33, both of which are encoded by CDKN2A, the most frequently deleted gene in ALL tumors in both Latinos and non-Latino whites 34. Hyperactivation of BMI1 promotes oncogenesis through increased cell proliferation and cell lifespan 33, and BMI1 overexpression is commonly detected in childhood ALL 35, in addition to several other hematological malignancies including AML 36 and B-cell non-Hodgkins lymphoma 37. Overexpression of BMI1 is associated with poorer prognosis in childhood ALL 35 and in AML 36. Although patient outcome data were not available in our study, it would be intriguing to investigate whether rs11591377 is associated with relapse rates in ALL.
The function of BMI1 in leukemogenesis is supported by its essential role in the maintenance of leukemic stem cells 41, and the reduction of apoptosis in CD34+ HSCs that overexpress BMI1 42. Our analyses support that BMI1 is the gene target of an enhancer element ~180Kb upstream containing SNP rs11591377, with data from recent H3K27ac HiChIP experiments 21, as well as MethylC-seq patterns of CpG methylation across tissues, suggesting this enhancer is specific to HSCs and early B-cell progenitors. Enhancers are often tissue or cell-type specific (reviewed in Heinz et al. 43), which may explain the lack of association with BMI1 expression levels in LCLs. Indeed, BMI1 was previously found to be expressed almost exclusively in CD34+ progenitor cells and not in differentiated cells 44.
The rs11591377 protective allele disrupts a MYB binding motif, and the risk allele demonstrated significant preferential binding to MYB-like 2 (MYBL2) transcription factor, in addition to several other TFs involved in hematopoiesis including p300 and c-Myc. MYBL2 is a regulator of cell cycle progression and cell survival, and is required for proliferation of hematopoietic cells 45 and maintenance of HSCs 46. Binding of the histone acetyltransferase p300 is a strong predictor of enhancer function 49. Moreover, c-Myc is a known positive regulator of BMI1 expression 50. Therefore, we posit that the rs11591377 risk allele upregulates BMI1 through binding of MYBL2 and recruitment of p300 and c-Myc, which enables leukemia cells to persist via increased cell proliferation and reduction of apoptosis.
The BMI1 locus may also have general effects on hematopoiesis. A recent GWAS of blood cell traits identified a genome-wide significant SNP rs3011641 in near-perfect LD with rs11591377 (r2=0.94 in Europeans and r2=0.99 in Latinos), in which the major allele was associated with a higher percentage of myeloid cells that are granulocytes 51, and which in our study was associated with ALL risk at Pmeta = 2.4 × 10−10. Mirroring this finding, the novel 8q24 locus identified in our recent GWAS of ALL displays chromosomal contact with a region overlapping several GWAS SNPs associated with blood cell traits, including granulocyte percentage of myeloid cells 9, 51.
We also determined the likely functional variant underlying the ALL association peak at PIP4K2A. The sequence including the rs4748812 risk allele matches a RUNX1 binding motif, TGAGGT, which is the second most frequent RUNX1 binding site in B-cells 52, thus supporting a broader role for RUNX1 beyond ETV6-RUNX1 fusion ALL. RUNX1, a crucial transcription factor in hematopoiesis, is located on chromosome 21, which is always triploid or tetraploid in HD-ALL. The effects of rs4748812 on PIP4K2A expression may, therefore, be more pronounced in hyperdiploid leukemia. PIP4K2A is a member of the phosphatidylinositol-4-phosphate 5-kinase family, and is involved in the synthesis of PIP2 (Phosphatidylinositol 4,5-bisphosphate). Activation of PIP2 is known to suppress apoptosis 53, and overexpression of a PIP4K2A paralog, PIP5K1A, has been shown to reduce stress-induced apoptosis 54. The overexpression of PIP4K2A associated with ALL risk alleles may thus lead to suppression of apoptosis via increased PIP2 synthesis.
Both BMI1 and PIP4K2A play a role in regulation of apoptosis and cell survival. High hyperdiploid leukemia cells, characterized by gross chromosomal aneuploidies, are inherently unstable and hence prone to apoptosis 55. Chromosome aneuploidies cause oxidative stress 56, and BMI1 expression may protect HSCs from oxidative stress-induced apoptosis 57. Survival of a high hyperdiploid leukemia cell likely relies more heavily on suppression of apoptosis than leukemia subtypes with more stable karyotypes, such as ETV6-RUNX1 fusion. Indeed, this may explain the stronger association of BMI1 and PIP4K2A variants with HD-ALL, demonstrated previously 8 and as suggested in our results, in particular for Latinos.
Previous GWAS identified genome-wide significant SNPs across chromosome 10p12.31-p12.2 6–8, however the functional variants were not determined. In this study, we have pinpointed putative causal SNPs at both association peaks upstream of BMI1 and at PIP4K2A, providing strong evidence of the function of both loci, in particular for BMI1 peak SNP rs11591377. Our results were based on a refined analysis of previously generated datasets, and additional experiments will be required to elucidate the precise leukemogenic effects of both loci. In this study, we capitalized on the finding of different lead SNPs in Latinos versus non-Latino whites, and on ethnic differences in haplotype structure, to reduce the list of associated SNPs to a handful of strong candidates. This LD correlation analysis should prove a useful strategy for causal variant discovery in other multi-ethnic GWAS datasets.
Supplementary Material
Novelty and Impact.
We report the first fine-mapping analysis of a chromosome 10p12.31 region associated with childhood acute lymphoblastic leukemia (ALL). In a large California-based genome-wide association study of childhood ALL, we confirm independent genome-wide significant associations at nearby peaks upstream of BMI1 and at PIP4K2A. Using a multi-ethnic linkage disequilibrium correlation approach, we pinpoint putative causal variants at both loci, including SNP rs11591377 that lies within an enhancer for BMI1 and showed differential transcription factor binding.
Acknowledgments
This work was supported by research grants from the National Institutes of Health (R01 CA155461 to J.L.W. and X.M., R01 CA175737 to J.L.W. and X.M., R01 ES009137 to C.M., R24 ES028524 to C.M., and P01 ES018172 to C.M.) and the Environmental Protection Agency (RD83451101 to C.M.), United States. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health and the EPA. AJD is supported by an Emerging Investigator Fellowship Grant award from the Pediatric Cancer Research Foundation, and AJD and KMW are supported by ‘A’ Awards from Alex’s Lemonade Stand Foundation.
The collection of cancer incidence data used in this study was supported by the California Department of Public Health pursuant to California Health and Safety Code Section 103885; Centers for Disease Control and Prevention’s (CDC) National Program of Cancer Registries, under cooperative agreement 5NU58DP003862-04/DP003862; the National Cancer Institute’s Surveillance, Epidemiology and End Results Program under contract HHSN261201000140C awarded to the Cancer Prevention Institute of California, contract HHSN261201000035C awarded to the University of Southern California, and contract HHSN261201000034C awarded to the Public Health Institute. The ideas and opinions expressed herein are those of the author(s) and do not necessarily reflect the opinions of the State of California, Department of Public Health, the National Cancer Institute, and the Centers for Disease Control and Prevention or their Contractors and Subcontractors.
The biospecimens and/or data used in this study were obtained from the California Biobank Program, (SIS request # 26), Section 6555(b), 17 CCR. The California Department of Public Health is not responsible for the results or conclusions drawn by the authors of this publication.
This study makes use of data from the Research Program on Genes, Environment and Health (RPGEH) (dbGaP Study Accession: phs000788.v1.p2). Data came from a grant, the Resource for Genetic Epidemiology Research in Adult Health and Aging (RC2 AG033067; Schaefer and Risch, PIs) awarded to the Kaiser Permanente Research Program on Genes, Environment, and Health (RPGEH) and the UCSF Institute for Human Genetics. The RPGEH was supported by grants from the Robert Wood Johnson Foundation, the Wayne and Gladys Valley Foundation, the Ellison Medical Foundation, Kaiser Permanente Northern California, and the Kaiser Permanente National and Northern California Community Benefit Programs. The RPGEH and the Resource for Genetic Epidemiology Research in Adult Health and Aging are described here: https://divisionofresearch.kaiserpermanente.org/genetics/rpgeh/rpgehhome
For recruitment of subjects enrolled in the California Childhood Leukemia Study (CCLS) replication set, the authors gratefully acknowledge the clinical investigators at the following collaborating hospitals: University of California Davis Medical Center (Dr. Jonathan Ducore), University of California San Francisco (Drs. Mignon Loh and Katherine Matthay), Children’s Hospital of Central California (Dr. Vonda Crouse), Lucile Packard Children’s Hospital (Dr. Gary Dahl), Children’s Hospital Oakland (Drs. James Feusner and Carla Golden), Kaiser Permanente Roseville (formerly Sacramento) (Drs. Kent Jolly and Vincent Kiley), Kaiser Permanente Santa Clara (Drs. Carolyn Russo, Alan Wong, and Denah Taggart), Kaiser Permanente San Francisco (Dr. Kenneth Leung), Kaiser Permanente Oakland (Drs. Daniel Kronish and Stacy Month), California Pacific Medical Center (Dr. Louise Lo), Cedars-Sinai Medical Center (Dr. Fataneh Majlessipour), Children’s Hospital Los Angeles (Dr. Cecilia Fu), Children’s Hospital Orange County (Dr. Leonard Sender), Kaiser Permanente Los Angeles (Dr. Robert Cooper), Miller Children’s Hospital Long Beach (Dr. Amanda Termuhlen), University of California, San Diego Rady Children’s Hospital (Dr. William Roberts), and University of California, Los Angeles Mattel Children’s Hospital (Dr. Theodore Moore). The authors additionally thank the families for their participation in the CCLS (formerly known as the Northern California Childhood Leukemia Study). The authors also thank Dr. Neil Shah (UCSF) for providing K562 cell DNA for copy number analysis.
Abbreviations
- ALL
acute lymphoblastic leukemia
- AMR
Admixed American
- GWAS
genome-wide association study
- SNP
single nucleotide polymorphism
- LD
linkage disequilibrium
- CCRLP
Childhood Cancer Record Linkage Project
- GERA
Genetic Epidemiology Research on Aging
- CCLS
California Childhood Leukemia Study
- DBS
dried bloodspots
- HSC
hematopoietic stem cell
- ChIP-Seq
chromatin immunoprecipitation sequencing
- GTEx
Genotype-Tissue Expression
- LCL
lymphoblastoid cell line
- HD
high hyperdiploid
References
- 1.Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui CH, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–64. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 2.Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, Alexandrov LB, Van Loo P, Cooke SL, Marshall J, Martincorena I, Hinton J, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet. 2014;46:116–25. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trevino LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, Willman C, Neale G, Downing J, Raimondi SC, Pui CH, Evans WE, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41:1001–5. doi: 10.1038/ng.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, Kinsey SE, Lightfoot T, Roman E, Irving JA, Allan JM, Tomlinson IP, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41:1006–10. doi: 10.1038/ng.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sherborne AL, Hosking FJ, Prasad RB, Kumar R, Koehler R, Vijayakrishnan J, Papaemmanuil E, Bartram CR, Stanulla M, Schrappe M, Gast A, Dobbins SE, et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet. 2010;42:492–4. doi: 10.1038/ng.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Migliorini G, Fiege B, Hosking FJ, Ma Y, Kumar R, Sherborne AL, da Silva Filho MI, Vijayakrishnan J, Koehler R, Thomsen H, Irving JA, Allan JM, et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood. 2013;122:3298–307. doi: 10.1182/blood-2013-03-491316. [DOI] [PubMed] [Google Scholar]
- 7.Xu H, Yang W, Perez-Andreu V, Devidas M, Fan Y, Cheng C, Pei D, Scheet P, Burchard EG, Eng C, Huntsman S, Torgerson DG, et al. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst. 2013;105:733–42. doi: 10.1093/jnci/djt042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walsh KM, de Smith AJ, Chokkalingam AP, Metayer C, Dahl GV, Hsu LI, Barcellos LF, Wiemels JL, Buffler PA. Novel childhood ALL susceptibility locus BMI1-PIP4K2A is specifically associated with the hyperdiploid subtype. Blood. 2013;121:4808–9. doi: 10.1182/blood-2013-04-495390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiemels JL, Walsh KM, de Smith AJ, Metayer C, Gonseth S, Hansen HM, Francis SS, Ojha J, Smirnov I, Barcellos L, Xiao X, Morimoto L, et al. GWAS in childhood acute lymphoblastic leukemia reveals novel genetic associations at chromosomes 17q12 and 8q24.21. Nat Commun. 2018;9:286. doi: 10.1038/s41467-017-02596-9. 017-02596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banda Y, Kvale MN, Hoffmann TJ, Hesselson SE, Ranatunga D, Tang H, Sabatti C, Croen LA, Dispensa BP, Henderson M, Iribarren C, Jorgenson E, et al. Characterizing Race/Ethnicity and Genetic Ancestry for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics. 2015;200:1285–95. doi: 10.1534/genetics.115.178616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metayer C, Zhang L, Wiemels JL, Bartley K, Schiffman J, Ma X, Aldrich MC, Chang JS, Selvin S, Fu CH, Ducore J, Smith MT, et al. Tobacco smoke exposure and the risk of childhood acute lymphoblastic and myeloid leukemias by cytogenetic subtype. Cancer Epidemiol Biomarkers Prev. 2013;22:1600–11. doi: 10.1158/1055-9965.EPI-13-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 13.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.1000 Genomes Project Consortium. Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 16.Liu JZ, Tozzi F, Waterworth DM, Pillai SG, Muglia P, Middleton L, Berrettini W, Knouff CW, Yuan X, Waeber G, Vollenweider P, Preisig M, et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet. 2010;42:436–40. doi: 10.1038/ng.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–9. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 18.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–7. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou X, Li D, Zhang B, Lowdon RF, Rockweiler NB, Sears RL, Madden PA, Smirnov I, Costello JF, Wang T. Epigenomic annotation of genetic variants using the Roadmap Epigenome Browser. Nat Biotechnol. 2015;33:345–6. doi: 10.1038/nbt.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao T, He B, Liu S, Zhu H, Tan K, Qian J. EnhancerAtlas: a resource for enhancer annotation and analysis in 105 human cell/tissue types. Bioinformatics. 2016;32:3543–51. doi: 10.1093/bioinformatics/btw495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mumbach MR, Satpathy AT, Boyle EA, Dai C, Gowen BG, Cho SW, Nguyen ML, Rubin AJ, Granja JM, Kazane KR, Wei Y, Nguyen T, et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat Genet. 2017;49:1602–12. doi: 10.1038/ng.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016;3:99–101. doi: 10.1016/j.cels.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou X, Li D, Lowdon RF, Costello JF, Wang T. MethylC Track: visual integration of single-base resolution DNA methylation data on the WashU EpiGenome Browser. Bioinformatics. 2014;30:2206–7. doi: 10.1093/bioinformatics/btu191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsunoda T, Takagi T. Estimating transcription factor bindability on DNA. Bioinformatics. 1999;15:622–30. doi: 10.1093/bioinformatics/15.7.622. [DOI] [PubMed] [Google Scholar]
- 25.Aldrich MC, Zhang L, Wiemels JL, Ma X, Loh ML, Metayer C, Selvin S, Feusner J, Smith MT, Buffler PA. Cytogenetics of Hispanic and White children with acute lymphoblastic leukemia in California. Cancer Epidemiol Biomarkers Prev. 2006;15:578–81. doi: 10.1158/1055-9965.EPI-05-0833. [DOI] [PubMed] [Google Scholar]
- 26.Wallace AD, Francis SS, Shao X, de Smith AJ, Walsh KM, Mckean-Cowdin R, Ma X, Dahl G, Barcellos LF, Wiemels JL, Metayer C. A germ-line deletion of APOBEC3B does not contribute to subtype-specific childhood acute lymphoblastic leukemia etiology. Haematologica. 2018;103:e29–31. doi: 10.3324/haematol.2017.179317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–5. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walsh KM, de Smith AJ, Hansen HM, Smirnov IV, Gonseth S, Endicott AA, Xiao J, Rice T, Fu CH, McCoy LS, Lachance DH, Eckel-Passow JE, et al. A Heritable Missense Polymorphism in CDKN2A Confers Strong Risk of Childhood Acute Lymphoblastic Leukemia and Is Preferentially Selected during Clonal Evolution. Cancer Res. 2015;75:4884–94. doi: 10.1158/0008-5472.CAN-15-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wiemels JL, de Smith AJ, Xiao J, Lee ST, Muench MO, Fomin ME, Zhou M, Hansen HM, Termuhlen A, Metayer C, Walsh KM. A functional polymorphism in the CEBPE gene promoter influences acute lymphoblastic leukemia risk through interaction with the hematopoietic transcription factor Ikaros. Leukemia. 2016;30:1194–7. doi: 10.1038/leu.2015.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Studd JB, Vijayakrishnan J, Yang M, Migliorini G, Paulsson K, Houlston RS. Genetic and regulatory mechanism of susceptibility to high-hyperdiploid acute lymphoblastic leukaemia at 10p21.2. Nat Commun. 2017;8:14616. doi: 10.1038/ncomms14616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–8. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 34.de Smith AJ, Kaur M, Gonseth S, Endicott A, Selvin S, Zhang L, Roy R, Shao X, Hansen HM, Kang AY, Walsh KM, Dahl GV, et al. Correlates of Prenatal and Early-Life Tobacco Smoke Exposure and Frequency of Common Gene Deletions in Childhood Acute Lymphoblastic Leukemia. Cancer Res. 2017;77:1674–83. doi: 10.1158/0008-5472.CAN-16-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng HX, Liu XD, Luo ZY, Zhang XH, Luo XQ, Chen X, Jiang H, Xu L. Upregulation of the proto-oncogene Bmi-1 predicts a poor prognosis in pediatric acute lymphoblastic leukemia. BMC Cancer. 2017;17:76. doi: 10.1186/s12885-017-3049-3. 017-3049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chowdhury M, Mihara K, Yasunaga S, Ohtaki M, Takihara Y, Kimura A. Expression of Polycomb-group (PcG) protein BMI-1 predicts prognosis in patients with acute myeloid leukemia. Leukemia. 2007;21:1116–22. doi: 10.1038/sj.leu.2404623. [DOI] [PubMed] [Google Scholar]
- 37.van Kemenade FJ, Raaphorst FM, Blokzijl T, Fieret E, Hamer KM, Satijn DP, Otte AP, Meijer CJ. Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood. 2001;97:3896–901. doi: 10.1182/blood.v97.12.3896. [DOI] [PubMed] [Google Scholar]
- 38.Moorman AV, Ensor HM, Richards SM, Chilton L, Schwab C, Kinsey SE, Vora A, Mitchell CD, Harrison CJ. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010;11:429–38. doi: 10.1016/S1470-2045(10)70066-8. [DOI] [PubMed] [Google Scholar]
- 39.van der Lugt NM, Domen J, Linders K, van Roon M, Robanus-Maandag E, te Riele H, van der Valk M, Deschamps J, Sofroniew M, van Lohuizen M. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 1994;8:757–69. doi: 10.1101/gad.8.7.757. [DOI] [PubMed] [Google Scholar]
- 40.Iwama A, Oguro H, Negishi M, Kato Y, Morita Y, Tsukui H, Ema H, Kamijo T, Katoh-Fukui Y, Koseki H, van Lohuizen M, Nakauchi H. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 2004;21:843–51. doi: 10.1016/j.immuni.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 41.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–60. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 42.Rizo A, Dontje B, Vellenga E, de Haan G, Schuringa JJ. Long-term maintenance of human hematopoietic stem/progenitor cells by expression of BMI1. Blood. 2008;111:2621–30. doi: 10.1182/blood-2007-08-106666. [DOI] [PubMed] [Google Scholar]
- 43.Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–54. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lessard J, Baban S, Sauvageau G. Stage-specific expression of polycomb group genes in human bone marrow cells. Blood. 1998;91:1216–24. [PubMed] [Google Scholar]
- 45.Arsura M, Introna M, Passerini F, Mantovani A, Golay J. B-myb antisense oligonucleotides inhibit proliferation of human hematopoietic cell lines. Blood. 1992;79:2708–16. [PubMed] [Google Scholar]
- 46.Baker SJ, Ma’ayan A, Lieu YK, John P, Reddy MV, Chen EY, Duan Q, Snoeck HW, Reddy EP. B-myb is an essential regulator of hematopoietic stem cell and myeloid progenitor cell development. Proc Natl Acad Sci U S A. 2014;111:3122–7. doi: 10.1073/pnas.1315464111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mucenski ML, McLain K, Kier AB, Swerdlow SH, Schreiner CM, Miller TA, Pietryga DW, Scott WJ, Jr, Potter SS. A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell. 1991;65:677–89. doi: 10.1016/0092-8674(91)90099-k. [DOI] [PubMed] [Google Scholar]
- 48.Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, Loh ML, Hunger SP, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–7. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, Afzal V, Ren B, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–8. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang R, Cheung NK, Vider J, Cheung IY, Gerald WL, Tickoo SK, Holland EC, Blasberg RG. MYCN and MYC regulate tumor proliferation and tumorigenesis directly through BMI1 in human neuroblastomas. Faseb J. 2011;25:4138–49. doi: 10.1096/fj.11-185033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, Mead D, Bouman H, Riveros-Mckay F, Kostadima MA, Lambourne JJ, Sivapalaratnam S, et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell. 2016;167:1415–1429.e19. doi: 10.1016/j.cell.2016.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Niebuhr B, Kriebitzsch N, Fischer M, Behrens K, Gunther T, Alawi M, Bergholz U, Muller U, Roscher S, Ziegler M, Buchholz F, Grundhoff A, et al. Runx1 is essential at two stages of early murine B-cell development. Blood. 2013;122:413–23. doi: 10.1182/blood-2013-01-480244. [DOI] [PubMed] [Google Scholar]
- 53.Azuma T, Koths K, Flanagan L, Kwiatkowski D. Gelsolin in complex with phosphatidylinositol 4,5-bisphosphate inhibits caspase-3 and -9 to retard apoptotic progression. J Biol Chem. 2000;275:3761–6. doi: 10.1074/jbc.275.6.3761. [DOI] [PubMed] [Google Scholar]
- 54.Halstead JR, van Rheenen J, Snel MH, Meeuws S, Mohammed S, D’Santos CS, Heck AJ, Jalink K, Divecha N. A role for PtdIns(4,5)P2 and PIP5Kalpha in regulating stress-induced apoptosis. Curr Biol. 2006;16:1850–6. doi: 10.1016/j.cub.2006.07.066. [DOI] [PubMed] [Google Scholar]
- 55.Ito C, Kumagai M, Manabe A, Coustan-Smith E, Raimondi SC, Behm FG, Murti KG, Rubnitz JE, Pui CH, Campana D. Hyperdiploid acute lymphoblastic leukemia with 51 to 65 chromosomes: a distinct biological entity with a marked propensity to undergo apoptosis. Blood. 1999;93:315–20. [PubMed] [Google Scholar]
- 56.Santaguida S, Amon A. Aneuploidy triggers a TFEB-mediated lysosomal stress response. Autophagy. 2015;11:2383–4. doi: 10.1080/15548627.2015.1110670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rizo A, Olthof S, Han L, Vellenga E, de Haan G, Schuringa JJ. Repression of BMI1 in normal and leukemic human CD34(+) cells impairs self-renewal and induces apoptosis. Blood. 2009;114:1498–505. doi: 10.1182/blood-2009-03-209734. [DOI] [PubMed] [Google Scholar]
- 58.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, Ku M, Durham T, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.