

Abstract
Controlled activation is a critical component in prodrug development. Herein we report a concentration-sensitive platform approach for bioorthogonal prodrug activation by taking advantage of reaction kinetics. Using two “click and release” systems, we demonstrate enrichment and prodrug activation specifically in mitochondria to demonstrate the principle of this approach. In both cases, the payload (doxorubicin or carbon monoxide) was released inside the mitochondrial matrix upon the enrichment-initiated click reaction. Furthermore, mitochondria-targeted delivery yielded substantial augmentation of functional biological and therapeutic effects in vitro and in vivo, as compared to controls that did not result in enrichment. This method is thus a platform for targeted drug delivery amenable to conjugation with a variety of molecules and not limited to cell-surface delivery. Taken together, these two click and release pairs clearly demonstrate the concept of enrichment-triggered drug release and critical feasibility of treating clinically relevant diseases such as acute liver injury and cancer.
Graphical Abstract
Introduction
Prodrug strategies have been widely used to address delivery problems with pharmaceuticals.1 Essentially all such approaches have one goal, i.e. to deliver the drug to the desired location at a sufficiently high concentration. Prodrug efforts focused on improving physicochemical properties allow for enhanced permeability and solubility. Site-selective activations allow for targeting based on environmental factors such as pH,2 unique redox chemistry including levels of H2O2,3 glutathione4 and other thiol species,5 and elevated levels of enzymes such as esterases,6, 7 proteases,8 and phosphatases.9 In addition, gut bacteria also present unique redox chemistry and an active enzymatic environment for site-selective targeting.10 In recent years, targeted drug delivery has quickly gained attention with some remarkable success, especially in the field of cancer with the goal being minimizing toxicity.11 For example, antibody-drug conjugates (ADC) allow for targeted delivery of drugs to the desired site.12, 13 There are many other targeting molecules that can be used to hone in on biomarkers such as the high affinity folate receptor,14 carbohydrate biomarkers,15 and prostate-specific membrane antigen (PSMA).16 Such targeted approaches work very well in enriching the concentration of the drug conjugates and prodrugs at the desired site. However, targeted release after enrichment is still a significant challenge.
Herein we describe a novel concentration-sensitive platform approach to prodrug activation based on controls by reaction kinetics using bioorthogonal click chemistry, which has been applied in prodrug preparation with excellent success.12, 17, 18, 19, 20, 21, 22, 23, 24, 25 Ideally, one would like to use linker chemistry that tethers the active drug to the targeting molecule in a stable fashion, and allow for selective cleavage at the desired site of action.20, 26, 27, 28, 29 Through the use of click chemistry and the concept of co-localization,30, 31 the linker can be very stable until enrichment-triggered release (ETR). The applicability of the approach is not limited to cell surface as in the case of antibody-mediated delivery (or ADC) because of the use of small molecules for both targeting and cleavage.
Results and Discussion
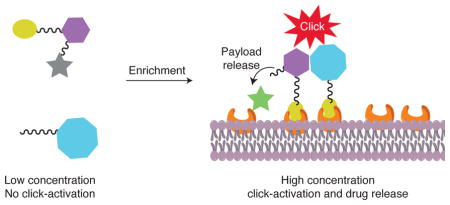
To establish the proof of concept, we designed two pairs of prodrug-release trigger partners that can undergo a bioorthogonal reaction with tunable release rates, which should set the stage for subsequent release of a drug moiety. As for the drug moiety, we chose two biologically and clinically relevant agents, the chemotherapeutic genotoxin doxorubicin (Dox) and the gasotransmitter carbon monoxide (CO), as model drugs. These two examples represent drugs of distinctly different properties and delivery challenges. Doxorubicin and CO have IC50 values generally in the range of low micromolar to high nanomolar concentrations depending on the cell lines used and drug exposure time. In the first pair of prodrug-trigger partners, we used the tetrazine-cyclooctyne chemistry as the prodrug-trigger pair for the click reaction,32, 33 a lactonization reaction of the cycloaddition intermediate for drug release,34 and mitochondria-targeting as a way to achieve enrichment, which is discussed later.35 Specifically, tetrazines are known to react with transcyclooctene36, 37 and strained cyclooctyne32, 33 with second order rate constants ranging from 0.0001 M−1s−1 to more than 1000 M−1s−1. As a proof of principle study, we contemplated a scenario where the prodrug could be used at a concentration of 10 μM in cell culture without cytotoxicity until triggered release is achieved based on control by reaction kinetics. For this design, we desire the second order rate constant to be below 0.25 M−1s−1, which would result in the first half-life being over 100 h without enrichment at 10 μM. Triphenylphosphine conjugation is known to enrich its “payload” to about 500 μM or higher in mitochondria,35, 38 which would lead to a decrease in its first half-life to about 2.2 h. What this means is that the prodrug would be non-toxic to cells until enrichment-triggered release. The general idea is presented in Figure 1, which depicts a trigger-prodrug pair going through targeted enrichment, reaction kinetics-controlled click reaction, and spontaneous cyclization-based release. For the second pair of prodrug-trigger partners, we used cyclopentadienone-strained alkyne chemistry for the controlled release rates and mitochondria-targeting as a way to achieve enrichment. The reaction kinetics issues are the same as for the first one. Below, we separately described these two systems and in vivo demonstration of proof of principle.
Figure 1. Schematic illustration of the concept of enrichment-triggered prodrug activation.

The two components would not click with each other during circulation at low concentrations. Upon enrichment in the targeted area, the significant increase in local concentrations leads to enhanced reaction rate, and thereby the drug is released in the targeted area.
The click, cyclization and release (CCR) system
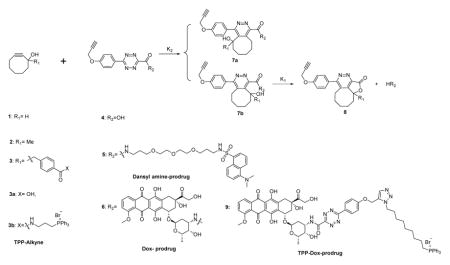
First, we studied the chemical feasibility of the first system using Dox as a model drug. Thus, we synthesized the two components of this drug release system: alkyne triggers (AT, 1–3) with an appropriately positioned hydroxyl group to initiate lactonization, model tetrazine-prodrugs (TP, 4 and 5), and tetrazine-linked prodrugs of Dox (6 and 9; Table 1). The reason that we also prepared a tetrazine conjugate (5) with dansyl amine was to take advantage of the strong fluorescence of dansyl amine as a marker to easily monitor and also check feasibility of the click, cyclization and release (CCR) sequence of events for amine containing prodrugs.
Table 1.
Chemical structure of CCR systems and evaluation of the reaction kinetics of the CCR system. All reactions were conducted in PBS containing 10% DMSO.
| |||||
|---|---|---|---|---|---|
| Tetrazine | Alkyne | k2 at r.t (M−1s−1) | k2 at 37 °C (M−1s−1) | k1 at r.t (h−1) | Dox Peak % within 48 h, at r.t (%) |
| 5 | 1 | 0.25± 0.06 | 1.9± 0.4 | 0.029± 0.006 | 60± 6 (48 h) |
| 2 | 0.0075± 0.0009 | 0.021± 0.005 | * | 90± 5 (20 h) | |
| 3b | 0.042± 0.012 | 0.14± 0.04 | 0.16±0.04 | 90 ±3 (16 h) | |
| 6 | 1 | 0.36± 0.07 | 2.1 ±0.4 | Δ | 20±5 (48 h) |
| 2 | 0.0078±0.0009 | 0.025±0.006 | * | 85±5 (21 h) | |
| 3b | 0.065±0.013 | 0.19±0.04 | 0.20±0.04 | 80±5 (18 h) | |
| 3a | 0.061±0.013 | 0.17±0.05 | 0.21±0.04 | 80±5 (19 h) | |
| 9 | 3b | 0.091±0.013 | 0.25±0.05 | 0.22±0.05 | 78±6 (18 h) |
not detectable because of the slow second order reaction and fast lactonization reaction. No intermediates were observed.
not detectable because of Dox decomposition in PBS. (n = 3, p= 0.95).
As the first step, we studied the basic chemical feasibility of the proposed approach. Thus, we treated 25 μM of the prodrugs 5 or 6 with alkynes 1, 2 or 3 at different concentrations at room temperature (r.t.) or 37 °C respectively, and monitored prodrug consumption and the release of the parent drug by HPLC (Table 1). There are three issues in this study: i.) cycloaddition regiochemistry, ii.) substituent effect at the R1 position, and iii.) the effect of the “drug” on the reaction rates and regiochemistry.
On the issue of regiochemistry, one can see from Table 1 that the cycloaddition chemistry has two possible regioisomeric products, but only one would lead to lactonization and drug release. For the strategy to work, it is critical that the correct regioisomer (7b) is the predominant product. Fortuitously, it was found that more than 80% Dox or dansyl amine was released within 48 h of the prodrug treatment with alkyne 2 or 3. Such results indicate that the regiochemistry of the Inverse Electron Demand Diels-Alder reaction (DAinv) is such that it favors the reaction leading to 7b with the hydroxyl group on the cyclooctyne positioned on the same side of the amide group linked to the parent drug. This allows for subsequent lactonization and drug release. To acquire initial insight into the observed regiochemistry, we performed theoretical calculations. Specifically, we used reported methods to calculate possible transition state(s) and activation energies for two regio-isomers (See supplementary Fig. 3).32 Schematic representations of the energy profiles for the tetrazine alkyne reactions are shown in supplementary Fig. 3. To simplify the calculation process, we used tetrazine 10 and alkyne 2 as models.32 The results showed that the activation energy for the first step of the DAinv between 10 and 2 via transition state TS1b is 17.1 kcal·mol−1 as compared to 19.0 kcal·mol−1 for the reaction via transition state TS1a. The qualitative difference in activation energy correlates well with our observed regiochemistry outcome.
There are several reasons that we needed to understand the effect of the R1 group on the alkyne trigger for this project. First, we are interested in understanding factors that would allow us to tune the reaction rate. It is easy to intuitively understand that the R1 group would have an effect on both the DAinv reaction and the subsequent lactonization. Second, for eventual applications, we need to conjugate both the tetrazine and alkyne to a targeting moiety. This means that we need a “handle” for such conjugation chemistry. For the tetrazine part, this can be easily accomplished through the phenolic hydroxyl group. For the cyclooctyne, we need to introduce an additional “handle” for such conjugations. For this reason, the R1 group is essential as a handle for further conjugation. Thus, we need to understand the substituent effect at this position. It was found that a methyl group or a benzyl group significantly reduced the rate of the DAinv reaction. For example, when R1 was a hydrogen, the second order rate constant was 0.25 M−1s−1 at r.t and 1.9 M−1s−1 at 37 °C when dansyl amine-prodrug 5 was used. In comparison, the second order rate constant was 0.0075 M−1s−1 and 0.021 M−1s−1 at r.t and 37 °C, respectively, for alkyne 2 with a methyl substituent; and 0.042 M−1s−1 and 0.14 M−1s−1 at r.t and 37 °C, respectively, for alkyne 3 with a phenyl substituent. Such results indicate that the DAinv reaction rates can easily be tuned over a range of more than 30-fold by using different R1 groups on the alkyne. Additionally, varying the temperature from r.t to 37 °C can afford another 3- to 10-fold of reaction rate variations. Besides, the reaction rate (0.25 M−1s−1) between 9 and 3b is similar to that for the reaction between 6 and 3a (0.17 M−1 s−1), confirming that the introduction of the TPP moiety would not significantly affect the reaction kinetics. As discussed earlier, the second order rate constant was targeted to be below 0.25 M−1s−1. The range of second order rate constants that can be tuned falls exactly where needed. It should also be noted that the R1 substituent accelerated the rate of the lactonization, as expected due to added conformational constraints. Therefore, all the reaction studies thus far clearly demonstrate one thing, i.e., the designed CCR system can be used for prodrug release for the intended purpose.
Regarding the third issue of the effect of the “drug” moiety on the reaction outcomes, it was found that dansylamine-prodrug 5 and Dox-prodrug 6 did not show statistically significant differences in terms of reaction rates. For example, the second order rate constant for the reaction of alkyne 1 with tetrazine 5 or 6 at 37 °C is 1.9 or 2.1 M−1s−1, respectively. Similarly, the second order rate constant for the reaction of alkyne 3b with tetrazine 5 or 6 at 37 °C is 0.14 or 0.19 M−1s−1, respectively. Such results are expected since the “drug” portion is positioned away from all reaction centers, and thus would not be expected to have a major effect on reaction rates. The only issue is that if the lactonization rate is slow, it may only lead to partial drug release within a reasonable period of time. This is the case with alkyne trigger 1. For example, only 60% dansylamine and 20% Dox was detected after treating prodrugs with alkyne 1 (1 mM) for 48 h. This low percentage Dox recovery was likely because of the slow lactonization rate of 7b (t1/2 = 24 h) when R1 was hydrogen, and decomposition of Dox in PBS (t1/2= 50 h).39 In contrast, with alkyne 2, the lactonization rate was so fast that the HPLC method did not detect the accumulation of the intermediate (CI). This also led to 85–90% release of the active drug at the 48-h point. For alkyne 3, the situation was similar with 80–90% drug release. All such results indicate the importance of the R1 group not only for tethering a targeting molecule, but also for an improved lactonization rate.
In addition to the three chemical feasibility issues discussed above, we were also interested in understanding the stability of the tetrazine-linked prodrugs in the presence of high concentrations of thiol species, due to the electron deficient nature of the tetrazine moiety. It is important to note that we did not encounter stability problems (Supplementary Table 4).
In vitro assessment of the CCR system
Next, we were interested in testing the drug delivery system in a cellular environment. We first chose the alkyne 1 and 3b. HeLa cells were incubated with different concentrations of Dox-prodrug and alkynes, or doxorubicin, for 48 hours at 37 °C. The IC50 values are shown in Table 2. After modification, the Dox-prodrug 6 lost its activity with IC50 well over 100 μM, while the IC50 of the parent drug Dox was about 1.0 μM. Such results were expected and desirable because the activity of the prodrug means that it would not be cytotoxic before Dox is released through the CCR process. As designed, the IC50 of Dox-prodrug 6 is about 1.5 μM in the presence of 50 μM alkyne 1 and 2.0 μM in the presence of 50 μM alkyne 3b. Such results indicate efficient release of the parent drug, Dox, by alkynes 1 and 3b.
Table 2.
IC50 in Hela cell line (n = 3, p = 0.95)
| Compounds | IC50 (μM) |
|---|---|
| Dox-prodrug 6 | >100 |
| Alkyne 1 | >100 |
| Alkyne 3b | >50 |
| Dox | 1.0±0.2 |
| Dox-prodrug 6 + 50 μM 1 | 1.5±0.3 |
| Dox-prodrug 6 + 100 μM 1 | 1.3±0.2 |
| Dox-prodrug 6 + 50 μM 3b | 2.3±0.4 |
| Tetrazine 4+ 100 μM 1 | >100 |
Having established a well-defined release system, we next examined the feasibility of using reaction kinetics to control drug release. Our goal was thus to target and enrich the prodrug at a particular location within the mammalian cell. It is well-known that folic acid, Arg-Asp-Asp (RGD), and many other molecules can target cancer, and be used for drug delivery.1 We chose mitochondrion-targeting through the use of a well-known triphenylphosphonium (TPP) moiety, which is known to enrich in mitochondria by approximately 100 to 500 fold.38 We elected to use mitochondria targeting because this can easily be assessed in a cell culture system. Thus, we conjugated the tetrazine prodrug and the alkyne trigger with triphenylphosphine to give 9 and 3b, respectively. HeLa cells were treated with 10 μM or 20 μM TPP-alkyne 3b and various concentrations of TPP-Dox-prodrug 9 (0, 1.2, 2.5, 5 and 10 μM) for 48 hours. It is clear that the IC50 (~1.5 μM) for TPP-Dox 9 in the presence of 20 μM of TPP-alkyne 3b is much lower than that of its non-TPP conjugated counterpart Dox prodrug 6 (~10 μM) in the presence of 20 μM alkyne 3a (Figure 2a). We see a similar trend when the alkyne concentration was decreased to 10 μM. For example, the IC50 for TPP-Dox prodrug 9 was about 2 μM in the presence of 3b, while the IC50 for Dox prodrug 6 was well over 10 μM in the presence of 10 μM of the alkyne trigger 3a (Figure 2b). As controls, both the IC50 values of 9 and 3b alone are higher than 40 μM, and the treatment of 20 μM of alkyne 3a or 3b alone had no effect on cell viability. Such dose-dependent results strongly support the underlying hypothesis that conjugation with TPP (3b and 9) allows for the enhanced release of Dox through enrichment of the prodrug in mitochondria, which subsequently led to an enhanced bimolecular reaction rate and facilitated CCR-mediated activation of the prodrug. Without such enrichment, the reaction is expected to be slow, resulting in a lower level of active Dox being released. Specifically, the second order rate constant of the reaction between the Dox-prodrug 6 and alkyne 3a was determined to be 0.17 M−1s−1, with the first half-life at 10 μM concentration of 6 and 10 μM concentration of 3a being about 163 h. Such results also mean that only 22% of the drug is released at the 48-h time point. If the concentrations of both the tetrazine prodrug (9) and alkyne (3b) are enriched to 500 μM in the mitochondria, the first half-life of the reaction would be 2.0 h (confirmed by experiments), which means almost complete Dox release at the 48-h time point. As a consequence, major differences in the apparent potency of the two prodrugs were observed with and without conjugation to TPP. Therefore, we conclude that enrichment in the mitochondria indeed led to triggered release of the drug, as designed. The same concept is applicable to the delivery of drugs through other targeting approaches such as receptor-mediated drug delivery.
Figure 2. The cytotoxicity of click and release pairs in Hela cells.

The click pairs (9 + 3b) with TPP conjugation showed more pronounced cytotoxicity as compared to the ones without TPP conjugation (6 + 3a) as designed. The Hela cells were incubated with different concentrations of Dox-prodrug 6 and alkyne trigger 3a or TPP -Dox-prodrug 9 and TPP- alkyne trigger 3b for 48h. a) Prodrugs with 10 μM the respective trigger; b) prodrugs with 20 μM of the respective trigger; The cytotoxicity was evaluated by a crystal violet assay. Results represent mean±SD of n =4. *p<0.05, **p<0.001 vs without TPP conjugation.
The CO delivery system via click chemistry
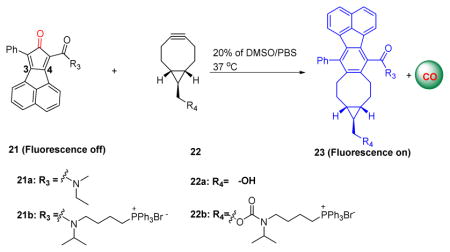
In order to demonstrate the general applicability of the “enrichment to release” concept for prodrug activation, we designed a second system, which uses different chemistry and a different “drug” component. Specifically, we designed a bioorthogonal reaction “cyclopentadienone-alkyne” pair for the targeted delivery of carbon monoxide (CO) to mitochondria (Table 3). CO has been widely accepted as an endogenous signaling molecule with potent physiologic and therapeutic importance on par with that of nitric oxide.40, 41, 42, 43, 44, 45 Mitochondria have been proposed to be a major site for action for CO.46 Indeed, Motterlini and Foresti have developed metal-based CO-releasing molecules (CORMs) that release CO extracellularly, rapidly diffuse into the cell, and influence mitochondrial function in part by binding to their heme-containing oxidases on the inner membrane.46, 47 Later on, organic CO prodrugs were reported with the ability to release CO extracellularly.17, 23, 48 Thus, targeting mitochondria would allow for CO release at the site of action. Given its high diffusivity, CO should be able to subsequently traverse out of the mitochondria into the cytosol to modulate additional functional effects. Previously, we successfully developed metal-free CO prodrugs by employing the click reaction between cyclopentadienone and a strained alkyne (e.g. BCN).18 For mitochondrial targeted delivery, we conjugated both components of the Diels-Alder reactants with TPP as the targeting moiety for mitochondria for ETR (Table 3).49 Additionally, in order to facilitate visualization of intracellular click reaction and CO release, a naphthalene group was attached to the 3, 4 position of the dienone ring to enable the fluorescence “turn-on” upon clicking these two components.23 Therefore, compounds 21 and 22 were readily synthesized (Table 3). With those compounds in hand, we initially confirmed CO release from the click reaction between 21 and 22 using the widely-accepted CO-myoglobin assay (Supplementary Fig. 8). We next tested their second order rate constants by monitoring the blue fluorescence of the click products. Much like the previous CCR system, the ligation of TPP did not significantly affect the reaction kinetics between the alkyne and dienone compound (Table 3).
Table 3.
The structure of TPP-conjugated cyclopentadienone-alkyne pairs and the kinetics data.
| ||
|---|---|---|
| Dienone | Alkyne | k2 (M−1s−1) |
| 21a | 22a | 0.14 ± 0.02 |
| 21b | 22b | 0.20 ± 0.03 |
We next probed whether compound 21b and 22b could be employed for targeted delivery of CO to mitochondria. In this case, the “enrichment and release” process can be conveniently monitored by fluorescence “turn-on” upon the click reaction.17, 23 To test this, RAW264.7 macrophages were co-treated with 21b and 22b simultaneously at different concentrations at 1 and 5 μM for 3 h and imaged under the DAPI channel (absorption at 358 nm and emission at 461 nm). RAW264.7 cells co-treated with 21a and 22a as controls.
As shown in Figure 3, RAW 264.7 cells co-treated with 21a and 22a showed no fluorescence signal at either 1 μM or 5 μM indicating that intracellular CO release did not occur or was undetectable by this method. In contrast, RAW cells treated with 21b and 22b showed strong blue fluorescence in a dose-dependent fashion (Figure 3c–d), indicating CO release. The cells treated with either 21b or 22b alone showed no fluorescence (data not shown). In the case of the reaction between 21a and 22a, the first half-life was roughly calculated to be around 397 h with an initial concentration of 5 μM. Therefore, it was not surprisingly that cells treated with 21a and 22a for 3 h did not show any fluorescence. However, after conjugation with TPP, compounds 22b and 21b can be enriched in the mitochondria, and hence the local concentration for 22b and 21b is substantially increased. Assuming that the concentration of compound 21b and 22b was enriched to 400 μM, we determined the half-life for the click reaction to be around 2 h. Consequently, one would expect that cells treated with 21b and 22b to show strong blue fluorescence. The observed results are consistent with such analysis. In order to further confirm that CO release primarily occurred in mitochondria, RAW 264.7 cells were co-treated with 22b (5 μM) and 21b (2.5 μM) for 4 h, followed by addition of the mitochondrial tracker MT-deep red for 20 minutes and then imaged by confocal microscopy. As shown in Figure 3e–h, the blue fluorescence (adjusted to green for ease of visualization) resulted from the click reaction between 21b and 22b clearly co-localized with the MT-deep red mitochondrial tracker. Collectively, the results are consistent with CO release from the enrichment of 21b and 22b within mitochondria.
Figure 3. Fluorescent imaging studies of the click reaction between 21 and 22 in Raw264.7 cells.

The click reaction between 21b and 22b resulted in dose-dependent increase in fluorescent intensity, which tracked that of a fluorescent mitochondrion-tracker, while the control click reaction between 21a and 22a only showed negligible fluorescence. Images a, b, c and d show the results from the click reaction between compound 21 and 22. a) RAW 264.7 cells treated with 22a (1 μM) and 21a (1 μM); b) RAW 264.7 cells treated with 22a (5 μM) and 21a (5 μM); c) RAW 264.7 cells treated with 22b (1 μM) and 21b (1 μM); d) RAW 264.7 cells treated with 22b (5 μM) and 21b (5 μM). Images e, f, g and h are confocal images of RAW 264.7 cells treated with compound 21b (5 μM), 22b (2.5 μM) and MT-deep red (50 nM). e) Bright field; f) Red channel; g) DAPI channel; h) Merged images of e), f) and g). Images represent 3 fields of view from 3 independent experiments in triplicate.
Assessment of the CO-prodrug’s ability to exert anti-inflammatory effects in vitro
Having confirmed that compounds 21b and 22b can be employed for selective targeting of CO to mitochondria through an “enrichment and release” process, we next examined if such a strategy would be effective in recapitulating CO-associated biological responses.17, 18, 23 Therefore, RAW 264.7 macrophages were pretreated with compounds 21b and 22b at different concentrations for 4 h, and then challenged with bacterial lipopolysaccharide (LPS; 1 μg/mL) for 1 h. No decrease in cell viability was observed throughout the experiment. Cell supernatants were collected for TNF expression determination by ELISA. The cells treated with 21a and 22a, or either compound 21b or 22b alone were used as negative controls. Unlike co-treatment with 21a and 22a, co-treatment of 21b and 22b dose-dependently suppressed LPS-induced TNF expression. Importantly, no effects on TNF production was observed for RAW 264.7 cells treated with 21a and 22a (without TPP conjugation) (Supplementary Fig. 11). These data make clear that the observed TNF suppression was associated with the presence of CO generated upon clicking 21b with 22b in the mitochondria. The inhibition of TNF by this mechanism of CO exposure mimicked that observed with gaseous CO as reported in numerous earlier literature reports.17, 23, 48, 50, 51 Furthermore, the remarkably low IC50 value (~5 μM) is the lowest we have seen of all the organic CO-prodrugs tested under the same experimental protocols.17, 18 For example, with organic CO-prodrugs, we have seen IC50 values in the 25–100 μM range depending on the release half-life (ranging from 2 min-2 h). Of particular importance is the nearly complete suppression of TNF production at 10 μM (Supplementary Fig. 11), which has not been observed with any previously tested organic CO-prodrugs.17, 18 Such results further suggest the power of directly delivering CO to mitochondria.
Assessing the utility of the enrichment-triggered release system in vivo
To assess whether the concept of enrichment–triggered release would work in vivo and result in the intended pharmacological effect, we tested the CO-prodrug system in a well-characterized in vivo model of acetaminophen (APAP)-induced acute liver injury where TNF is known to be involved and where inhaled CO gas has been shown to impart salutary effects when given after APAP and at a time when liver injury has already begun.52, 53, 54 In this model, fasted male CD-1 mice were administered an overdose of APAP, which leads to rapid pro-inflammatory sequelae ultimately resulting in massive hepatocyte cell death (Figure 4). We reasoned that the described CO-prodrug should match the hepatoprotection observed with inhaled CO. APAP injury occurs in two phases, the first is a rapid radical-mediated hepatocyte cell death that releases local inflammatory mediators. The second phase that ultimately leads to liver failure is a cytokine storm that results in massive hepatocyte death and has systemic implications.55, 56 It is well-known that CO’s targets are largely in the mitochondria.57 Thus, we hypothesized that targeting CO release to mitochondria would lead to significant anti-inflammatory and anti-apoptotic effects. Therefore, we next compared administering a combination of the TPP-conjugated CO-prodrugs (21b+22b) to their counterparts without the TPP unit and ability to enrich (21a + 22a). Mice were treated with 21a and 22a or 21b and 22b. Controls received equivalent volumes of DMSO as the vehicle. Mice were administered APAP followed 4 h later with 21b followed 3 min later by 22b at 4 mg/kg, i.v. or 0.4 mg/kg, i.v. in separate groups. These animals were compared to those that received 21a plus 22a using identical treatment regimens. Serum and liver tissue were collected 24 h later and analyzed for serum alanine aminotransferase (ALT) as a measure of liver injury and immunostaining of liver sections for indices of cell death (TUNEL) and architectural changes [Hemotoxylin and Eosin (H&E)]. Both 21a plus 22a or 21b plus 22b showed significantly lower serum ALT levels relative to DMSO vehicle-treated animals in response to APAP (Figure 4), which is consistent with literature reported protective effects of CO.53 At the higher 4 mg/kg dose the reduction in ALT achieved with 21a and 22a was 58%. In contrast those animals treated with the TTP-conjugates 21b and 22b known to enrich in the mitochondria, the reduction in serum ALT levels was >95%. At a 10-fold lower dose (0.4 mg/kg), treatment with the TPP conjugates (21b and 22b) resulted in 55% reduction in serum ALT levels, compared to the non-TPP conjugated compounds (21a and 22a) which had no effect on APAP-induced ALT levels. It should also be noted that treatment with 21b and 22b increased carboxyhemoglobin levels, which is an important pharmacokinetic indicator for CO presence in the circulation (Figure 4b). Finally, histological staining analyses of the livers from these mice corroborated the serum ALT data showing less cell death as measured by TUNEL and H&E in those animals treated wtih the 21b and 22b pair (Figure 4c–h). Collectively, these results clearly show that TPP conjugation enhances local delivery of CO in a dose-dependent fashion that provides potent heaptoprotection. It should be noted that the molecular weights of the TPP conjugates (21b, 22b) are nearly twice that of the parent compounds (21a, 22a). Thus, the same dose based on weight is half the dose when considering the molarity for the TPP conjugates. Taken together, these results demonstrate in part the proof of principle of the “enrich and release” concept in vivo for effective delivery of CO to impart tissue cytoprotection.
Figure 4. CO prodrugs protect against acute liver failure.

a. The effect of CO prodrugs on APAP induced liver injury. Mice were treated with APAP (300 mg/kg, i.p.) followed by administration of compounds 21a plus 22a or the 21b and 22b (4 (high) or 0.4 (low) mg/kg, i.v.) or DMSO. Three minutes separated the first and second injections. Serum ALT levels were determined 24h after APAP. Results represent mean±SD of n = 6–8 animals/group. **p<0.03, *p<0.001 vs DMSO. b. COHb levels over time after administration of 21b plus 22b (4 mg/kg, i.v.). Results represent mean±SD of 3 mice/time point. *p<0.005 vs baseline. c–e. Hemotoxylin and Eosin staining of liver sections from mice treated as in a with 21b and 22b at 4 mg/kg, i.v. Dotted sections designate dead/dying tissue. f–h. TUNEL staining of liver sections from the same animals in c–e. Brown staining indicates cell death. All images are representative 4–5 fields of view from at least 3 animals/group.
Conclusions
In conclusion, in targeted therapy, there is a strong need for the development of new chemistry to achieve selective delivery to complement existing ones. In this study, we developed and tested a new concept and platform approach, which relies on concentration-sensitive chemistry to allow for selective drug release upon enrichment. This is achieved by taking advantage of reaction kinetics, that allows for accelerated cleavage at an elevated concentration. Such an approach will may prove very useful in delivering small molecules through targeting including the use of antibody drug conjugates and receptor-mediated drug delivery. As a result, the delivery is not limited to the cell surface and can be used for sub-cellular delivery, with enriched concentrations, which could not be achieved by ADC based delivery. Collectively, we describe a method that could be leveraged for biologic and medicinal efforts in treating disease. These compounds may be very useful therapeutics for the treatment of cancer and acute liver injury.
Data availability
Additional details on methods and compound characterizations are available in the Supplementary Information. Supplementary figures include 1H NMR spectra of 21a at different temperatures (Supplementary Fig. 1), schematic representations of the reaction between tetrazine 10 and alkyne 2 (Supplementary Fig. 3), studies of reaction kinetics (Supplementary Figs. 4–6), CO-myoglobin assay (Supplementary Fig. 8), cytotoxicity assay (Supplementary Figs. 9 and 10), Elisa assay (Supplementary Fig. 11) and NMR spectra of all synthesized compounds. Supplementary tables include results of studies on reaction kinetics (Supplementary Tables 1–3), stability tests (Supplementary Table 4) and computational studies (Supplementary Table 5) data.
Supplementary Material
Acknowledgments
We would like to express our gratitude to Dr. Zhaoyu Liu at the Center for Molecular and Translational Medicine at Georgia State University for taking the confocal images. We also thank the GSU Molecular Basis of Disease (ZP and MC), Brains & Behaviors (BY) and University 2CI (LKDLC) fellowships for their support. Department of Defense Peer Reviewed Medical Research Program, W81XWH-16-0464 and NIH R44 DK111260-02 to LEO. Leo is a scientific consultant for Hillhurst Biopharmaceuticals and an Adjunct Professor of Aston University, United Kingdom.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Wang B, Hu L, Siahaan T. Wiley Series in Drug Discovery and Development. 2. John Wiley and Sons; 2016. Drug Delivery: Principles and Applications. [Google Scholar]
- 2.He X, Li J, An S, Jiang C. pH-sensitive drug-delivery systems for tumor targeting. Ther Deliv. 2013;4:1499–1510. doi: 10.4155/tde.13.120. [DOI] [PubMed] [Google Scholar]
- 3.Saravanakumar G, Kim J, Kim WJ. Reactive-Oxygen-Species-Responsive Drug Delivery Systems: Promises and Challenges. Adv Sci. 2017;4:1600124. doi: 10.1002/advs.201600124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quinn JF, Whittaker MR, Davis TP. Glutathione responsive polymers and their application in drug delivery systems. Polym Chem. 2017;8:97–126. [Google Scholar]
- 5.Al-Nahain A, et al. Triggered pH/redox responsive release of doxorubicin from prepared highly stable graphene with thiol grafted Pluronic. Int J Pharm. 2013;450:208–217. doi: 10.1016/j.ijpharm.2013.04.053. [DOI] [PubMed] [Google Scholar]
- 6.Perez C, Daniel KB, Cohen SM. Evaluating Prodrug Strategies for Esterase-Triggered Release of Alcohols. ChemMedChem. 2013;8:1662–1667. doi: 10.1002/cmdc.201300255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng Y, et al. An Esterase-Sensitive Prodrug Approach for Controllable Delivery of Persulfide Species. Angew Chem Int Ed. 2017;56:11749–11753. doi: 10.1002/anie.201704117. [DOI] [PubMed] [Google Scholar]
- 8.Hamilton JR, Trejo J. Challenges and Opportunities in Protease-Activated Receptor Drug Development. Annu Rev Pharmacol Toxicol. 2017;57:349–373. doi: 10.1146/annurev-pharmtox-011613-140016. [DOI] [PubMed] [Google Scholar]
- 9.Wiemer AJ, Wiemer DF. Prodrugs of phosphonates and phosphates: crossing the membrane barrier. Top Curr Chem. 2015;360:115–160. doi: 10.1007/128_2014_561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Philip AK, Philip B. Colon Targeted Drug Delivery Systems: A Review on Primary and Novel Approaches. Oman Med J. 2010;25:79–87. doi: 10.5001/omj.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agarwal P, Bertozzi CR. Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjug Chem. 2015;26:176–192. doi: 10.1021/bc5004982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kern JC, et al. Discovery of Pyrophosphate Diesters as Tunable, Soluble, and Bioorthogonal Linkers for Site-Specific Antibody–Drug Conjugates. J Am Chem Soc. 2016;138:1430–1445. doi: 10.1021/jacs.5b12547. [DOI] [PubMed] [Google Scholar]
- 13.Chudasama V, Maruani A, Caddick S. Recent advances in the construction of antibody-drug conjugates. Nat Chem. 2016;8:114–119. doi: 10.1038/nchem.2415. [DOI] [PubMed] [Google Scholar]
- 14.Zhao X, Li H, Lee RJ. Targeted drug delivery via folate receptors. Expert Opin Drug Deliv. 2008;5:309–319. doi: 10.1517/17425247.5.3.309. [DOI] [PubMed] [Google Scholar]
- 15.Ahmed M, Narain R. Carbohydrate-based materials for targeted delivery of drugs and genes to the liver. Nanomedicine (Lond) 2015;10:2263–2288. doi: 10.2217/nnm.15.58. [DOI] [PubMed] [Google Scholar]
- 16.Jin W, et al. Discovery of PSMA-specific peptide ligands for targeted drug delivery. Int J Pharm. 2016;513:138–147. doi: 10.1016/j.ijpharm.2016.08.048. [DOI] [PubMed] [Google Scholar]
- 17.Ji X, et al. Click and Release: A Chemical Strategy toward Developing Gasotransmitter Prodrugs by Using an Intramolecular Diels-Alder Reaction. Angew Chem Int Ed. 2016;55:15846–15851. doi: 10.1002/anie.201608732. [DOI] [PubMed] [Google Scholar]
- 18.Wang D, et al. A click-and-release approach to CO prodrugs. Chem Commun (Camb) 2014;50:15890–15893. doi: 10.1039/c4cc07748b. [DOI] [PubMed] [Google Scholar]
- 19.Rossin R, et al. Triggered Drug Release from an Antibody-Drug Conjugate Using Fast “Click-to-Release” Chemistry in Mice. Bioconjug Chem. 2016;27:1697–1706. doi: 10.1021/acs.bioconjchem.6b00231. [DOI] [PubMed] [Google Scholar]
- 20.Versteegen RM, et al. Click to release: instantaneous doxorubicin elimination upon tetrazine ligation. Angew Chem Int Ed. 2013;52:14112–14116. doi: 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- 21.Matikonda SS, et al. Bioorthogonal prodrug activation driven by a strain-promoted 1,3-dipolar cycloaddition. Chem Sci. 2015;6:1212–1218. doi: 10.1039/c4sc02574a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ji X, et al. Click and Release: SO2 Prodrugs with Tunable Release Rates. Org Lett. 2017;19:818–821. doi: 10.1021/acs.orglett.6b03805. [DOI] [PubMed] [Google Scholar]
- 23.Ji X, et al. Click and Fluoresce: A Bioorthogonally Activated Smart Probe for Wash-Free Fluorescent Labeling of Biomolecules. J Org Chem. 2017;82:1471–1476. doi: 10.1021/acs.joc.6b02654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jimenez-Moreno E, et al. Vinyl Ether/Tetrazine Pair for the Traceless Release of Alcohols in Cells. Angew Chem Int Ed. 2017;56:243–247. doi: 10.1002/anie.201609607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steiger AK, Yang Y, Royzen M, Pluth MD. Bio-orthogonal “click-and-release” donation of caged carbonyl sulfide (COS) and hydrogen sulfide (H2S) Chem Commun. 2017;53:1378–1380. doi: 10.1039/c6cc09547j. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Jia S, Chen PR. Diels-Alder reaction-triggered bioorthogonal protein decaging in living cells. Nat Chem Biol. 2014;10:1003–1005. doi: 10.1038/nchembio.1656. [DOI] [PubMed] [Google Scholar]
- 27.Eising S, Lelivelt F, Bonger KM. Vinylboronic Acids as Fast Reacting, Synthetically Accessible, and Stable Bioorthogonal Reactants in the Carboni-Lindsey Reaction. Angew Chem Int Ed. 2016;55:12243–12247. doi: 10.1002/anie.201605271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo J, Liu Q, Morihiro K, Deiters A. Small-molecule control of protein function through Staudinger reduction. Nat Chem. 2016;8:1027–1034. doi: 10.1038/nchem.2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anseth KS, Klok HA. Click Chemistry in Biomaterials, Nanomedicine, and Drug Delivery. Biomacromolecules. 2016;17:1–3. doi: 10.1021/acs.biomac.5b01660. [DOI] [PubMed] [Google Scholar]
- 30.Kang J, Hilmersson G, Santamaría J, Rebek J. Diels–Alder Reactions through Reversible Encapsulation. J Am Chem Soc. 1998;120:3650–3656. [Google Scholar]
- 31.Murase T, Horiuchi S, Fujita M. Naphthalene Diels–Alder in a Self-Assembled Molecular Flask. J Am Chem Soc. 2010;132:2866–2867. doi: 10.1021/ja9107275. [DOI] [PubMed] [Google Scholar]
- 32.Chen W, et al. Clicking 1,2,4,5-Tetrazine and Cyclooctynes with Tunable Reaction Rates. Chem Commun. 2012:1736–1738. doi: 10.1039/c2cc16716f. [DOI] [PubMed] [Google Scholar]
- 33.Wang D, et al. 3,6-Substituted-1,2,4,5-tetrazines: Tuning Reaction Rates for Staged Labeling Applications. Org Biomol Chem. 2014;12:3950–3955. doi: 10.1039/c4ob00280f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shan D, Nicolaou MG, Borchardt RT, Wang B. Prodrug Strategies Based on Intramolecular Cyclization Reactions. J Pharm Sci. 1997;86:765–767. doi: 10.1021/js970069d. [DOI] [PubMed] [Google Scholar]
- 35.Murphy MP, Smith RAJ. Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Ann Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 36.Blackman ML, Royzen M, Fox JM. Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels– Alder Reactivity. J Am Chem Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Devaraj NK, Weissleder R, Hilderbrand SA. Tetrazine-based cycloadditions: application to pretargeted live cell imaging. Bioconjug Chem. 2008;19:2297–2299. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith RAJ, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci USA. 2003;100:5407–5412. doi: 10.1073/pnas.0931245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janssen MJH, Crommelin DJA, Storm G, Hulshoff A. Doxorubicin decomposition on storage. Effect of pH, type of buffer and liposome encapsulation. Int J Pharm. 1985;23:1–11. [Google Scholar]
- 40.Otterbein LE, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 41.Otterbein LE, et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med. 2003;9:183–190. doi: 10.1038/nm817. [DOI] [PubMed] [Google Scholar]
- 42.Klinger-Strobel M, et al. Bactericidal Effect of a Photoresponsive Carbon Monoxide-Releasing Nonwoven against Staphylococcus aureus Biofilms. Antimicrob Agents Chemother. 2016;60:4037–4046. doi: 10.1128/AAC.00703-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clark JE, et al. Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ Res. 2003;93:e2–8. doi: 10.1161/01.RES.0000084381.86567.08. [DOI] [PubMed] [Google Scholar]
- 44.Nagel C, et al. Introducing [Mn(CO)3(tpa-kappa(3)N)](+) as a novel photoactivatable CO-releasing molecule with well-defined iCORM intermediates - synthesis, spectroscopy, and antibacterial activity. Dalton Trans. 2014;43:9986–9997. doi: 10.1039/c3dt51848e. [DOI] [PubMed] [Google Scholar]
- 45.Kuramitsu K, et al. Carbon monoxide enhances early liver regeneration in mice after hepatectomy. Hepatology. 2011;53:2016–2026. doi: 10.1002/hep.24317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Motterlini R, Foresti R. Biological signaling by carbon monoxide and carbon monoxide-releasing molecules. Am J Physiol Cell Physiol. 2017;312:C302–c313. doi: 10.1152/ajpcell.00360.2016. [DOI] [PubMed] [Google Scholar]
- 47.Motterlini R, et al. Carbon monoxide-releasing molecules: characterization of biochemical and vascular activities. Circ Res. 2002;90:E17–24. doi: 10.1161/hh0202.104530. [DOI] [PubMed] [Google Scholar]
- 48.Pan Z, et al. Organic CO Prodrugs: Structure-CO-Release Rate Relationship Studies. Chem Eur J. 2017;23:9838–9845. doi: 10.1002/chem.201700936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hernandez-Aguilera A, et al. Mitochondrial dysfunction: a basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediators Inflamm. 2013;2013:135698. doi: 10.1155/2013/135698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sawle P, et al. Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW264.7 murine macrophages. Br J Pharmacol. 2005;145:800–810. doi: 10.1038/sj.bjp.0706241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schallner N, Otterbein LE. Friend or foe? Carbon monoxide and the mitochondria. Front Physiol. 2015;6:17. doi: 10.3389/fphys.2015.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marques AR, et al. Generation of Carbon Monoxide Releasing Molecules (CO-RMs) as Drug Candidates for the Treatment of Acute Liver Injury: Targeting of CO-RMs to the Liver. Organometallics. 2012;31:5810–5822. [Google Scholar]
- 53.Zuckerbraun BS, et al. Carbon monoxide protects against liver failure through nitric oxide-induced heme oxygenase 1. J Exp Med. 2003;198:1707–1716. doi: 10.1084/jem.20031003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov. 2010;9:728–743. doi: 10.1038/nrd3228. [DOI] [PubMed] [Google Scholar]
- 55.Robinson MW, Harmon C, O’Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. 2016;13:267–276. doi: 10.1038/cmi.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bradley MJ, et al. Host responses to concurrent combined injuries in non-human primates. J Inflamm (Lond) 2017;14:23. doi: 10.1186/s12950-017-0170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Queiroga CSF, Almeida AS, Vieira HLA. Carbon Monoxide Targeting Mitochondria. Biochem Res Int. 2012;2012:9. doi: 10.1155/2012/749845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Additional details on methods and compound characterizations are available in the Supplementary Information. Supplementary figures include 1H NMR spectra of 21a at different temperatures (Supplementary Fig. 1), schematic representations of the reaction between tetrazine 10 and alkyne 2 (Supplementary Fig. 3), studies of reaction kinetics (Supplementary Figs. 4–6), CO-myoglobin assay (Supplementary Fig. 8), cytotoxicity assay (Supplementary Figs. 9 and 10), Elisa assay (Supplementary Fig. 11) and NMR spectra of all synthesized compounds. Supplementary tables include results of studies on reaction kinetics (Supplementary Tables 1–3), stability tests (Supplementary Table 4) and computational studies (Supplementary Table 5) data.