

Abstract
The misbehaving attitude of Ca2+ signaling pathways could be the probable reason in many muscular disorders such as myopathies, systemic disorders like hypoxia, sepsis, cachexia, sarcopenia, heart failure, and dystrophy. The present review throws light upon the calcium flux regulating signaling channels like ryanodine receptor complex (RyR1), SERCA (Sarco-endoplasmic Reticulum Calcium ATPase), DHPR (Dihydropyridine Receptor) or Cav1.1 and Na+/Ca2+ exchange pump in detail and how remodelling of these channels contribute towards disturbed calcium homeostasis. Understanding these pathways will further provide an insight for establishing new therapeutic approaches for the prevention and treatment of muscle atrophy under stress conditions, targeting calcium ion channels and associated regulatory proteins.
Keywords: Calcium, Ryanodine receptor, RyR1, Muscle, Atrophy
Introduction
Calcium ion (Ca2+) is the 5th of the most abundant elements in the earth’s crust and one of the universal intracellular messengers which controls a variety of cellular processes such as gene transcription, muscle contraction, cell proliferation, programmed cell death and neurotransmission (Berridge 2002; Bootman et al. 2001) and also considered as an important contributor in regulating skeletal muscle plasticity (Gehlert et al. 2015). In skeletal muscle fibers, Ca2+ has a crucial role in excitation-contraction coupling process which results into action potential of muscle fiber and also involved with innumerable functions such as myosin-actin cross bridging, protein synthesis, protein degradation, fiber type shifting, calcium-regulated proteases and transcription factors, mitochondrial adaptations, plasticity and respiration (Gehlert et al. 2015).
Abnormal cytosolic calcium ions [Ca2+] cyt or dysregulated calcium homeostasis caused by disturbances of Ca2+ channels, exchangers, calcium ion pumps, calcium ion transport channels and calcium ion binding proteins induce multiple pathologies (Missiaen et al. 1992). Recent evidences implicate Ca2+ dysregulation as a common underlying phenomenon in the pathophysiology of muscles such as hypoxia, sepsis, cachexia, sarcopenia, heart failure, and dystrophy (Fig. 1). Thus, the review summarizes the latest findings and emerging concepts in calcium ion signaling with a special focus on Ca2+ channels/receptors and how they are engaged with the muscle atrophic conditions. Laterally, clinical implication and novel therapeutic strategies will be discussed to provide a probable solution for muscular pathologies.
Fig. 1.

Schematic representation of the different diseases involved in causing muscle dystrophy and changes occur in muscle fibres which may finally lead to muscle degeneration
Excitation-contraction coupling
Calcium ions play a prime role in excitation-contraction coupling process. It starts with the binding of acetylcholine with receptors that lead to the opening of voltage-gated sodium channels, present on sarcolemma and down the t-tubule into the myofibers. The wave of depolarization leads to conformational change in L-type calcium channels (Cav1.1) which further governs the direct gating of ryanodine receptors (RyR) within the sarcoplasmic reticulum (SR) and this entire process allows a very large release of calcium (Wei and Dirksen 2010). The release of Ca2+ from SR via RyR1 channels facilitates a rapid and the enormous amount of cytoplasmic Ca2+ which make a binding with troponin C and this Ca2+-troponin C binding forms a cross-bridge between actin and myosin filaments. The actin-myosin cross bridging results into shortening of sarcomere that ultimately lead to muscle contraction and force generation (Fill and Copello 2002; Catterall 1991). This entire sequence of events is known as excitation-contraction coupling (Fig. 2).
Fig. 2.

Stress responses in skeletal muscle during E-C coupling. Depolarization of the T-tubule membrane activates Cav1.1, triggering SR Ca2+ release through RyR1 and leading to sarcomere contraction, a process known as E-C coupling. During pathological stress intracellular signaling pathways activated and affect RyR1 function and alter E-C coupling. Stress-induced RyR1 dysfunction can result in SR Ca2+ leak, which potentially activates numerous Ca2+-dependent cellular damage mechanisms. AC, adenylate cyclise; CSQ, Calsequestrin; SERCA1a, Sarcoplasmic Reticulum Calcium-ATPase; RyR1, Ryanodine receptor 1
Muscle relaxation involves SR calcium-ATPase (SERCA) pumps by which cytosolic calcium is pumped back into the sarcoplasm. Impaired skeletal muscle function due to altered E-C coupling is associated with many muscular stress conditions like running of the marathon, strenuous exercise or heart failure etc. (Reiken et al. 2003; Lunde et al. 2001). A defect in E-C coupling leads to decrease in Ca2+ ions released from the SR and impaired muscle contraction and force generation. While, consistent and continuing Ca2+ release is possibly due to impaired function of SR sarcoplasmic/endoplasmic reticulum calcium ATPase1 (SERCA1a), Ca2+ reuptake pump (Scherer and Deamer 1986; Bellinger et al. 2008). The SERCA pump plays a major role in muscle contraction as its decrement leads to aging and heart failure (Periasamy and Kalyanasundaram 2007).
Resting intracellular calcium ion concentration
Ca2+ concentration in the cytoplasm during unstimulated state is very low, nearly 0.1 M (Endo 2009), while under stimulated conditions, consequently mobilizes Ca2+ from its sources to cytoplasm which result in increase of local or global cytoplasmic Ca2+. Once the concentration of Ca2+ crosses the threshold level that could further conjure up cellular adverse responses. There could be two possible reasons for increased calcium concentration either extracellular or intracellular calcium pool or sometimes both. If both sources work together, the concentration of Ca2+ could be increased till millimolar level that is fourfold higher concentrations as compared to cytoplasmic calcium concentration. Stimulated conditions provoke the Ca2+ to be rapidly transported from the source to cytoplasm and this transportation furnish a large electrochemical potential gradient. A calcium channel is an ion channel which is selective for calcium ions and there is a number of calcium channels present in the cell.
Ca2+ entry across the sarcolemma of muscles can occur through a number of perceptible channels contributed by members of the transient receptor potential (TRP) family or ORAI family members, highly Ca2+ selective channels or Ca2+ release activated Ca2+ Channels (CRAC). The CRAC channel is the best classified store operated channel (SOC) with electrophysiological properties (Kiviluoto et al. 2011).
There are two types of cells: excitable and non-excitable cells. In non-excitable cells, the influx of Ca2+ from extracellular medium occurs via calcium channels such as receptor-operated channels, the second messenger operated channels, and store-operated calcium entry (SOCE). As discovered recently, the main participants of SOCE are plasma membrane channel Orai1 and transmembrane protein of the reticulum STIM1 (Avdonin 2012).
Basically, SOCs are plasma membrane calcium channels that are opened in reaction to decreased calcium concentration in the lumen of the SR (Parekh and Putney Jr 2005). The key fact which initiates the opening of SOC is the decrement in SRCa but not calcium released from SR. Members of the canonical transient receptor potential cation channels family (TRPC), especially TRPC1 are involved in SOCE in vascular smooth muscles (Leung et al. 2008). Another single membrane-spanning protein termed as STIM1 (stromal-interacting molecule 1) also plays a major role in activation of SOCs. STIM1 protein serves to provide information regarding stored Ca2+ (Roos et al. 2005). Orai 1 (Orai, the keepers of the gates of the Heaven in Greek mythology) is a pore subunit of the store-operated Ca2+ release-activated Ca2+ channels (Parekh and Putney Jr 2005).
Normally, a complex of STIM1-Orai incorporating TRPC proteins, advocated the mechanism controlling activation of SOC in smooth muscle cells (Roos et al. 2005). Experimental studies revealed that STIM1 interacts with TRPC channels through electrostatic interactions. STIM1 was shown to directly bind and regulate TRPC1, TRPC4, and TRPC5, while indirect actions of STIM1 on TRPC3 and TRPC6 has been proposed (Kiviluoto et al. 2011; Yuan et al. 2007; Zeng et al. 2008).
An entirely different story exists in excitable cells in which influx of calcium ion occurs via voltage-operated channels. There are various voltages-gated channels L-type (Cav1.1) channels; N, P/Q, R and T-type channels which have been described in Table 1.
Table 1.
Voltage dependent calcium channels
| S.No. | Ca2+ current type | Voltage | Most often found in | Principal physiological functions |
|---|---|---|---|---|
| 1 | L-type calcium channel (“Long-Lasting” AKA “DHP Receptor”) | HVA (high voltage activated) | Skeletal muscle, smooth muscle, bone (osteoblasts), ventricular myocytes (also termed DHP receptors), dendrites and dendritic spines of cortical neurones | Excitation-contraction coupling in cardiac and smooth muscle, regulation of transcription endocrine secretion, neuronal Ca2+ transients in cell bodies and dendrites, regulation of enzyme activity, cardiac pacemaking, neuronal, visual transduction |
| 2 | P-type calcium channel(“Purkinje”) /Q-type calcium channel | HVA (high voltage activated) | Purkinje neurons in the cerebellum / Cerebellar granule cells | Neurotransmitter release, Dendritic Ca2+ transients |
| 3 | N-type calcium channel(“Neural”/"Non-L”) | HVA (high-voltage-activated) | Throughout the brain and peripheral nervous system. | Neurotransmitter release, Dendritic Ca2+ transients |
| 4 | R-type calcium channel(“Residual”) | intermediate-voltage-activated | Cerebellar granule cells, other neurons | Neurotransmitter release, Dendritic Ca2+ transients |
| 5 | T-type calcium channel(“Transient”) | low-voltage-activated | Neurons, cells that have pacemaker activity, bone (osteocytes), thalamus (thalamus) | Pacemaking and repetitive firing |
A group of transmembrane ion channel proteins which allow transporting Na+, K+, Ca2+, or Cl− ions via binding of chemical messengers (ligands such as neurotransmitters) are known as ligand-gated ion channels (LGICS) (Table 2).
Table 2.
Ligand-gated ion channels
| Type | Gated by | Location | Function |
|---|---|---|---|
| IP3 receptor | IP3 | ER/SR | Releases calcium from ER/SR in response to IP3 by e.g. GPCRs (Bosanac et al. 2002) |
| Ryanodine receptor | Dihydropyridine receptors in T-tubules and increased intracellular calcium (Calcium Induced Calcium Release - CICR) | ER/SR | Calcium-induced calcium release in myocytes (Bosanac et al. 2002) |
Calcium ion storage in SR
The endoplasmic reticulum is the major intracellular Ca2+ storage site, contains thousands of times greater calcium concentration in the cytosol (Xu et al. 2005). Under resting conditions, a majority of Ca2+ ions are bound to Ca2+ binding proteins which include calreticulin, parvalbumin, calsequestrin (calretinin), calsequestrin-like proteins CLP-150, CLP-170, CLP-220 (Schreiber et al. 2004) and sarcalumenin (SAR) in the SR (Felix et al. 1997; Milner et al. 1992). In skeletal muscle and cardiac muscle, calsequestrin is found to be the main Ca2+ binding protein (Beard et al. 2004), while for other tissues, Ca2+ binds to calreticulin (Michalak et al. 2002) and other Ca2+-dependent chaperones or foldases like calnexin, 78-kDa glucose-regulated protein/ immunoglobulin heavy chain binding protein (GRP78/BiP), GRP94, and various protein disulfide isomerases (PDI) (Papp et al. 2003). All these proteins perform at least two of the following three properties for calcium ion signaling: Ca2+ binding, regulation of Ca2+ pumps or Ca2+-release channels, and chaperone function (Berridge 2002) which further emphasizes the close interrelation between the [Ca2+]ER and ER function.
Hypoxic conditions stimulate the mitochondria to generate excess ROS that further leads to activation of Ca2+ channels and protein folding enzymes to promote Ca2+ release from the ER and generate ER stress. Our recent study also made an agreement as hypoxic exposure leads to oxidative stress and disrupted intracellular calcium homeostasis (Agrawal et al. 2017). The increased calcium levels further led to activation of calcium-activated protease, calpain which was associated with protein degradation (Jain et al. 2013b; Jain et al. 2013a). Our findings also provided strong evidence that the elevated protein turnover rate leads to skeletal muscle atrophy under chronic hypobaric hypoxia exposure and this atrophy occurs via the upregulation of ubiquitin-proteasome pathway and calcium-activated protease, calpain, indicating the important role of calcium in muscle atrophy (Chaudhury et al. 2012).
Besides this, another Ca2+ binding protein is Calmodulin (CaM), ubiquitously expressed 17- kDa protein which regulates Ryanodine Receptors (RyR). CaM contains four calcium ion binding pockets (two in the carboxy-terminal domain and another two in the amino-terminal domain) and binds to one site per RyR subunit (Moore et al. 1999). Calmodulin (CaM) behaves in dual form: either apo-calmodulin (apoCaM) or Ca2+-calmodulin (Ca-CaM). If it is present in former form, it could not bind with calcium, which provokes RyR1 channel activity, but if it is present in the latter form, it shows channel inhibitor activity (Yamaguchi et al. 2001). One of the common properties between both forms is its binding property with skeletal Cav1.1. This common activity of calmodulin makes it special and coordinate the RyR1 or Cav1.1 interaction and controls skeletal muscle excitation-contraction coupling (Takeshima 1993). Whether Ca2+-CaM binds with Cav1.1 has now recently been questioned as a study presented a comparison of abilities of CaM to bind to the proximal C termini of two L-type Ca2+ channels, Cav1.1 (skeletal isoform) and Cav1.2 (cardiac isoform). The conclusion offered by the result that Ca2+-CaM is bound strongly to the proximal Cav1.2 C terminus, but not to that of Cav1.1 (Ohrtman et al. 2008). One of the reasons for weak binding of CaM with Cav1.1 is to provide access of this region to other proteins and these interactions might play a crucial role in excitation-contraction coupling. Sencer et al. 2001 also reported Cav1.1 and RyR1 interaction could be stabilized via binding of Ca2+-CaM with Cav1.1 and strong binding of CaM could be interfering with the stabilization interaction of RyR1 and Cav1.1.
In normal conditions, numerous mechanisms control Ca2+ overload or depletion in the ER. The cytosolic requirement of Ca2+ promotes the release of calcium from the ER, but it should not decrease the (Ca2+) ER to the level at which ER functions and Ca2+ signaling become compromised (Sammels et al. 2010). On the requirement, a mechanism is initiated that couple ER Ca2+ depletion to an increase of Ca2+ entry into the cell. This mechanism is known as “capacitative” (Putney Jr 1986) or “store-operated” Ca2+ entry.
In hypoxic or ischemic conditions, an increase in intracellular calcium levels is observed as a primary response (Seta et al. 2004). Hypoxia is also involved in modulating intracellular calcium levels in smooth muscle, cardiomyocytes, epithelial, neuronal and in non-excitable cells such as astrocytes (Aley et al. 2005, 2006; Chen et al. 2006). Although, the effect of hypoxia on the mobilization of calcium pools from skeletal muscle is vague. But few of the studies described that chronic hypoxia increases the levels of cytosolic Ca2+ specifically by boosting the release of calcium from the ER and potentiating Ca2+ influx via the L-type Ca2+ channels (Kanatous et al. 2009).
SR receptors control calcium ion homeostasis
In skeletal muscles, the following prime receptors are associated with maintaining calcium ion homeostasis during E-C coupling.
Ryanodine Receptor (RyR)
SERCA (Sarco-endoplasmic reticulum calcium ATPase)
Cav1.1 or Dihydropyridine Receptor (DHPR)
Na+/Ca2+ exchange pump
Calcium is considered as an important secondary messenger for signal transduction like excitation-contraction coupling (E-C coupling). Intracellular Ca2+ is mostly present in the sarcoplasmic reticulum (SR) in striated muscle and the endoplasmic reticulum (ER) in other cell types. Major Ca2+ release channels which are localized in the SR/ER are ryanodine receptors (RyRs) (Otsu et al. 1990) and inositol 1, 4, 5-triphosphate receptors (IP3Rs) (Nixon et al. 1994).
RyRs exist in multiple isoforms: RyR1 in skeletal muscle, RyR2 in myocardium (heart muscle) whereas RyR3 is expressed in brain (Hakamata et al. 1992). Vukcevic et al. (2010) reported that RyR1 is also expressed in B-lymphocytes. RyR2 is highly expressed in Purkinje cells of the cerebellum and cerebral cortex (Lai et al. 1992; Nakanishi et al. 1992; Furuichi et al. 1994) and very low levels in stomach, kidney, adrenal glands, ovaries, thymus, and lungs (Kuwajima et al. 1992; Giannini et al. 1995). RyR3 is expressed in brain regions such as hippocampal neurons, thalamus, Purkinje cells, corpus striatum (Hakamata et al. 1992; Lai et al. 1992; Furuichi et al. 1994), skeletal muscles (highest expression in the diaphragm) (Neylon et al. 1995; Marks et al. 1989), the smooth muscle cells of the coronary vasculature, lung, kidney, ileum, jejunum, spleen, stomach of mouse and aorta, uterus, ureter, urinary bladder, and esophagus of rabbit (Giannini et al. 1995; Ottini et al. 1996).
Ca2+ release channels (RyR1)
Mainly, ryanodine receptor 1 (RyR1) is abundant in skeletal muscle. The receptor is a tetramer structure which consists of four RyR1 (565 KD) polypeptide and four FK-506 binding proteins (FKBP1) (12KD). The RyR1 complex also consists of catalytic subunits, PKA (protein kinase A) and regulatory subunits, PP1 (protein phosphatase1) (Gehlert et al. 2015). One FKBP12 bind with one RyR1 subunit hence four FKBP12 binds with four RyR1 polypeptides. The opening probability (Po) depends on the binding of FKBP12 with the RyR1 polypeptide. The dissociation of FKBP12 from RyR1 polypeptide leads to increase open probability of the channel (Brillantes et al. 1994).
The cytoplasmic domain of the RyR1 channels include several other complexes like cAMP-dependent protein kinase (PKA), protein phosphatase 1 (PP1), and phosphodiesterase 4D3 (PDE4D3). Muscular A-kinase anchor protein (mAKAP) targets PKA and PDE4D3 to RyR1, whereas spinophilin targets PP1 to the channel (Marks et al. 1989; Zalk et al. 2007). Calmodulin also binds to RyR and the phosphorylation sites for CaMKII and PKA, including Ser2808 and Ser2030, in different subdomains within the clamp region of the channel (Lehnart et al. 2005; Marx et al. 2000). Muscular A-kinase anchoring protein (mAKAP) restraint to RyR1 at the residue of 3003 to 3039 via leucine zipper which tether PKA to come close to phosphorylation sites (Bers 2004). Another protein, sorcin also interacts with RyR1 via clamp domain. This binding further facilitates crosstalk between RyR1 and DHPRs and β-adrenergic receptors, surface protein membrane (Farrell et al. 2003; Melzer et al. 1995). During prolonged periods of stress condition, SNS get activated which leads to binding of catecholamine to a β-adrenergic receptor that resulted into the activation of adenyl cyclase and formation of cAMP, the secondary messenger via G-protein coupled receptor and further cAMP activates PKA.
PKA phosphorylation activates RyR1 and RyR2 by phosphorylating at serine2844and serine2843 respectively (Reiken et al. 2003; Bellinger et al. 2008). In skeletal muscles, Protein Kinase A (PKA) is phosphorylated at RyR1-S2844 which further, reduces the affinity of FKBP12 from the RyR1 channel (Reiken et al. 2003). This binding of FKBP12 to the RyR1 stabilizes the closed state of the channel and prevents a “leak” of calcium ion through channels, also facilitates coupled gating between neighboring channels that enhances the Ca2+ transients (Brillantes et al. 1994; Jayaraman et al. 1992). Several studies have also made an agreement that remodelling of RyR1 via nitrosylation, carbonylation and glutathionylation affects skeletal muscle function and Ca2+ signaling (Aracena-Parks et al. 2006; Barreiro and Hussain 2010; Hidalgo et al. 2006) (Fig. 3).
Fig. 3.

A model of “leaky” RYR1 channel in pathological skeletal muscles. a RyR1 from normal skeletal muscle is not “leaky” and the sequestration of calcium ion occurred due to RYR1 activation which triggers muscle contraction b In pathological state, ROS and RNS mediated remodelling of RYR1 channel and impaired calcium homeostasis which may lead to decreased muscle force and moreover, it may lead to muscular atrophy
A few recent studies reported the remodelling of the RyR1 macromolecular complex during chronic stress conditions. The remodelling of ryanodine receptor components include oxidation of phosphodiesterase PDE4D3 channel or depletion of Ca2+ release channel stabilizing protein FKBP12 or PKA hyperphosphorylation of the channel. Any conformation change in the component of ryanodine receptor complex leads to SR Ca2+ leak into the cytoplasm. Thus, the amount of Ca2+ released during each contraction of the muscle is reduced (Lehnart et al. 2005; Shan et al. 2010a; Shan et al. 2010b). Hence, these conformational changes under prolonged pathological stress could contribute to defective muscle function due to disturbances in Ca2+ signaling. Hence the elevated cytosolic calcium levels could be responsible for excessive muscle stress and muscle pathologies related to muscles.
Oxidative stress responsible for RyR1 channel modifications
RyR1 channel activity is highly sensitive to redox active reagents. ROS and NO have been shown to modify the RyR1 channel functions (Aracena et al. 2003; Favero et al. 1995; Stamler and Meissner 2001) and are potentially implicated in impaired Ca2+ signaling in heart failure and sarcopenia (Andersson et al. 2011). Oxidative stress is one of the major contributing factors which tend to increase ROS, RNS and the stress-induced protein oxidation (Jackson 2009; Muller et al. 2007). Recent studies reported that RyR1 activity is increased due to the presence of ROS and RNS, such as molecular oxygen (O2), superoxide anion (O2.-), hydrogen peroxide (H2O2), hydroxyl radical (OH−), nitric oxide (NO−), nitroxyl (HNO) species, glutathione disulfide (GSSG), and S-nitrosoglutathione (GSNO) (Stamler and Meissner 2001; Aghdasi et al. 1997; Eu et al. 2000; Feng et al. 2000; Oba et al. 2002; Xia et al. 2003; Cheong et al. 2005; Sun et al. 2001a). In contrast, the RyR1 activity gets decreased by intracellular reducing agent, glutathione (GSH) (Feng et al. 2000; Oba et al. 2002; Cheong et al. 2005; Sun et al. 2001b).
Skeletal muscle Ca2+ release channel RyR1 contains a large number of free thiol group whose oxidation or nitrosylation influences channel function (EU et al. 2000; Sun et al. 2001a; Salama et al. 2000). Nitric oxide involved to create S-nitrosylation of RyR1 that increases the same activity, on the other hand, calmodulin has an antagonist effect of nitric oxide (CaM 50 residues appeared to be in the reduced state of these, nearly 10–12). The RyR1 channel contains 100 cysteine residues (Takeshima 1993) which are highly susceptible to oxidation/modifications by exogenous sulfhydryl (SH) reagents (Sun et al. 2001a) (Table 3).
Table 3.
Modificatation of RyR1 on cystein residues sites
The modulation of RyR1, either S-nitrosylation or S-glutathionylation, both could reduce the affinity with calmodulin (Aracena et al. 2005). It has been reported that CaM, bound at a site of intersubunit contact, protects RyR1 from oxidation and possibly nitrosylation. However, during stress conditions and high concentrations of oxidants could lead to a loss of the interaction between RyR1 and CaM. Further this might contribute to altered muscle function or damage during periods of high oxidative stress, such as in fatigue (Moore et al. 1999).
SR Ca2+ leak is reported as aberrant calcium sparks in myofibres in numerous circumstances like after vigorous exercise, muscle dystrophy, a progression of heart failure, sarcopenia (Bellinger et al. 2008; Andersson et al. 2011). One of the general observations reported during these adverse conditions is disturbed Ca2+ homeostasis which could be associated with stress-induced remodelling of RyR1 channels.
Ryanodine receptors (RyRs) are highly susceptible to redox modifications due to the presence of hyperactive cysteine and serine residues. Hyperphosphorylation and redox modifications of RyRs perturbed calcium ion homeostasis. Reiken et al. (2003) reported hyperphosphorylation of RyR1 during heart failure (HF) in rats and humans, which lead to dissociation of FKBP12 from RyR1 and enhance its activity. During exhaustive exercise, remodelling of the RyR1 complex was noted that the resulted into limiting exercise capacity, decreased muscle contractility and calcium dysfunction. Remodelling of the RyR1 complex could be various types, including PKA-mediated phosphorylation of serine residues (Ser-2844), RyR1 S-nitrosylation, PDE4D3 depletion, and calstabin1 depletion (Bellinger et al. 2008). Some other in-vivo investigations also reported the role of S-nitrosylation of RyR1 in muscular atrophy, malignant hyperthermia and sarcopenia, which ultimately led to pathological calcium ion leak into the cytoplasm and reduced binding affinity of FKBP12 and other macromolecular complex to RyR1 (Bellinger et al. 2009; Durham et al. 2008; Andersson et al. 2011). Researchers also reported the role of RyR1 in arthritis-induced muscle weakness due to increased production of RNS and impaired calcium signaling (Yamada et al. 2015). RyR1 dysfunction due to excessive oxidation/nitrosylation was also observed during spinal cord injury (SCI) (Liu et al. 2016). A current report was also submitted on RyR1 mutations in Korean patients who suffered from congenital myopathy. The C-terminal dominant variant of RyR1 mutation were observed in core myopathy suffered patients (Jeong et al. 2018).
Besides RyR1 remodelling, few other Sarco (endo) plasmic reticulum (SR) receptors have also played an essential role in some diseases. Recently, Ravel-Chapuis et al. (2017) reported that SERCA1, SLN, and CSQ levels were ungoverned in myotonic dystrophy type 1 (DM1), suggested that the reduced capacity of pumping back calcium into SR which resulted into an aberrant release of calcium ion into the cytoplasm. Currently, Schartner et al. (2017) also highlighted the importance of Cav1.1 or DHPR in ECC and also revealed ten recessive or dominant mutations in CACNA1S (Cav1.1) in exome sequencing. Dominant CACNA1S mutations in Cav1.1 or DHPR of skeletal muscles associated with the muscle dysfunction and congenital myopathy (Schartner et al. 2017). These findings strengthened the importance of SR receptors in maintaining calcium ion homeostasis and ECC.
SERCA, a Sarco-Endoplasmic Reticulum Ca2+ATPase, an enzymatic pump that scavenges calcium from the cytosol and the transportation is coupled to ATP hydrolysis (MacLennan et al. 1997). This Ca2+ transport makes the cytosolic calcium three to four folds lower as compared to intra- SR/ER calcium concentration. But dystrophic mice or DMD patients show defects in calcium ion handling and uptake during relaxation (Divet and Huchet-Cadiou 2002; Goonasekera et al. 2011). Hence there is a possibility that skeletal muscle atrophy could be caused due to modulation of SERCA activity. Three homologous genes which encoded for SERCAs are SERCA1, SERCA2, and SERCA3 (Burk et al. 1989). Transcripts of these genes further endure for splicing and converted into isoforms which differ due to its C-terminal region (MacLennan et al. 1997). Fast-twitch (type 2) skeletal muscle encompass SERCA1a (adult form) and 1b (neonatal form). SERCA 2a is expressed in slow-twitch (type 1) skeletal and cardiac muscles, whereas SERCA 2b in smooth muscle and non-muscle tissues (Wuytack et al. 1992). Further, non-muscle tissues comprises of SERCA 3 at variable levels (Reuben et al. 1974). In skeletal muscle, SERCA activity can be modulated by two small regulatory proteins, sarcolipin and phospholamaban (Kranias and Hajjar 2012; MacLennan et al. 2003). Both are expressed in slow-twitch skeletal muscles and cardiac muscles and are an important regulator of muscle performance and cardiac diseases. Sarcolipin and phospholamban, both belong to the same family of proteins that bind to the same domain on SERCA2a (Kranias and Hajjar 2012; Tupling et al. 2011; Treves et al. 2016). Recently, Anderson et al. (2015) discovered the unrecognized functional open reading frames (ORFs) in RNA transcript encoding a conserved 46 amino acid micro peptide, named myoregulin. Myoregulin is a skeletal muscle-specific micro peptide and interacts with SERCA1 and decreases the SERCA-ATPase activity. Enhanced exercise capacity in myoregulin knock-out mice suggests its role in maintaining calcium homeostasis. It is reported that altered mRNA splicing or expressions might be impaired in DM1 (myotonic dystrophy Type 1) muscle and thus contribute to altered calcium homeostasis in skeletal muscle of DM1 patients. It is still questionable whether modulation of SERCA expression is responsible for muscular atrophy or remodelling of ryanodine receptors are responsible for the same as recent studies suggested the altered expression of SERCA in impaired DM1 (Kimura et al. 2005).
CaV1.1 or Dihydropyridine Receptor (DHPR) resides at the t-tubule region of the sarcolemma of skeletal muscles. Cav1.1 is a heteropentamer formed by the α1s, α2-δ1, β1a, and γ subunits (tetrads). Among all subunits, α1s is the principal subunit of CaV1.1 which involved in the L-type voltage-activated Ca2+ channel of EC coupling (Samso 2015).
During E-C coupling, four Cav1.1 formed a tetrad which are located on the alternate RyR1 in the case of a skeletal muscle while Cav1.2 tetrad not formed in cardiac muscle. Once an action potential reaches the t-tubule membrane, it leads to conformational changes of Cav1.1 which further induces direct protein-protein interaction of Cav1.1 and Ca2+ release channel, RyR1 to release calcium ion from the SR (Franzini-Armstrong et al. 1998). But in case of cardiac muscles, the mechanism is slightly different as the trigger for Ca2+ release through is RyR2. RyR2 is dependent on the influx of Ca2+ via Cav1.1. Once Ca2+ is entering via L-type calcium channel, it activates ryanodine receptor (RyR2) through CICR. However, the role of CICR in skeletal muscle is still questionable (Dulhunty et al. 2002). It is further reported that Cav1.1 is modulated by other protein components also such as Stac3, Rem, and JP45 (Mosca et al. 2016). Few latest studies mentioned about the novel component, Stac3 which is ostensible muscle specific adaptor protein. Stac3 binds to both Cav1.1 and RyR1 maintaining core protein complex and functional E-C coupling machinery (Dulhunty et al. 2017). Stac3 synchronizes the organization of Cav1.1 and RyR1s at triad junction of the t-tubules and SR. The amount of Cav1.1 and/or RyR1 at triads, regulated by Stac3, perhaps by modulating protein trafficking and/or stability of Cav1.1 and/or RyR1. Horstick et al. (2013) described the Stac3 in Zebrafish revealing, NAM (Native American Myopathy) mutation decreases E-C coupling. That Stac3 has been recognized only in the last 3–4 years highlight the interaction between RyR1 and Cav1.1 in skeletal muscles.
Calcium induced calcium release (CICR)
Calcium-induced calcium release (CICR) was first discovered and proposed in skeletal muscle (Endo et al. 1970; Endo 1975). It is a biological process in which calcium is able to activate calcium release from intracellular Ca2+ stores like endoplasmic reticulum or sarcoplasmic reticulum. During the excitation - coupling process, few calcium ions cross the sarcolemma but this limited amount is not sufficient to activate myofilaments hence this small amount calcium induce the sarcolemma to release more calcium and activate the EC process. Some of the studies reported that superficial calcium is required to encourage the release of sarcoplasmic calcium. This act is highly required to activate “depolarization-induced” release of calcium (Fabiato and Fabiato 1975). This phenomenon is called “Calcium-Induced Calcium Release” (CICR). CICR is also an important process for excitation-contraction coupling in cardiac muscle (Fabiato 1989). Both types of Ca2+ channels such as RyRs and IP3 receptors (IP3Rs) exhibit CICR behavior. But an important difference exists between these two receptors for CICR behavior that Ca2+ alone, without the help of any other agents or stimuli, can cause Ca2+ release via ryanodine receptors (Endo 1981; Smith et al. 1986) but in the case of IP3R, can cause Ca2+ release only in the presence of IP3 (Foskett et al. 2007). Due to these findings, CICR is an important activity of RyR, but not for IP3R. In cardiac muscle, the Ca2+ release is considered a prime physiological mechanism for contraction process. As per the contraction procedure, an influx of Ca2+ occurs via L-type voltage-dependent calcium channel which activates t-tubule membrane of myocytes and generates the action potential and Ca2+ release from SR (Bers 2001; Cannell and Soeller 1997).
CICR was first discovered in skeletal muscle, still the same was not considered as a primary mechanism of physiological Ca2+ release and whether it has secondary participates in skeletal muscle contraction is still controversial (Rios and Pizarro 1991; Schneider 1994).
Na+/Ca2+ exchange pump (NCX)
NCX is a membrane-associated protein that catalyzes the electronic exchange of three Na+ ions and one Ca2+ ion across the plasma membrane in a high capacity, and low Ca2+ affinity fashion depending on the electrochemical gradient of the substrate ion. Na+/Ca2+ exchange pump resides at the sarcolemma of the muscle cell.
In skeletal muscles, there is only small influx of extracellular Ca2+ during activity (Bianchi and Shanes 1959). Few of Ca2+ also influx through Cav1.1 but it is also reported that the primary function of Cav1.1 is the voltage sensor for excitation-contraction (EC) coupling, but not calcium channeling (Melzer et al. 1995). Although, reports also depicted that calcium influx through Cav1.1 is dispensable for EC coupling in skeletal muscles of most vertebrates (Armstrong et al. 1972). Now the question could arise if calcium influx through Cav1.1 is dispensable, then why the channel is required? Recent studies provided the answer that indicates voltage-dependent calcium influx is important for skeletal muscle differentiation, function, and health (Flucher and Tuluc 2017).
But in cardiac muscles, the event of contraction is accompanied by the large influx of Ca2+ from the extracellular medium through the voltage-sensitive Ca2+ channels of the plasma membrane (Cannell et al. 1995). In the case of cardiac muscle, Na+/Ca2+ exchange pump performed an important role as Ca2+ influx occurs via this channel it further induced cardiac excitation-contraction coupling (Bers 2002; Martonosi and Pikula 2003). However, the role of NCX is still controversial in skeletal muscles.
Calcium homeostasis and muscle atrophy
Intracellular calcium concentration controls numerous signaling mechanisms and biological processes. Calcium signaling regulates crucial processes such as gene transcription, signal transduction, contraction, and secretion, to the long-term regulation of fertilization, proliferation, migration, differentiation, apoptosis, and necrosis (Berridge et al. 2000). Specifically, a prolonged global concentration controls processes like fertilization and apoptosis, whereas a localized transient change in calcium concentration regulates cell migration and muscle contraction (Clapham 2007).
Calcium signal is required for cell survival, but it could be highly toxic if it exceeds the normal concentration. Continuous increase in calcium concentration augments production of reactive oxygen species and activates proteases (Batchelor and Winder 2006). Basically, calcium concentration is increased due to a rapid release from intracellular stores and slow entry from the extracellular pool to cells via membrane channels (Parekh 2003). Usually, the release of calcium is controlled by inositol-1,4,5-trisphosphate receptor (InsP3R) and ryanodine receptor1 (RyR1) in muscle cells, while its return regulated by Sarco-endoplasmic reticulum ATPase (SERCA) pumps. RyR- mediated calcium signaling is rapid (takes less than one second) where as IP3R- mediated calcium signaling is a delayed type (takes seconds to minutes) (Eltit et al. 2004).
Calcium homeostasis is an important phenomenon for maintaining an intracellular environment in cells and recent studies depicted its imbalance responsible for muscle atrophy. Few muscle disuse conditions such as spaceflight, hind limb unloading, bed rest, etc. contributed to intracellular Ca2+ overload and muscle atrophy. For eg. elevation of 246 and 215% in rats’ Soleus (SOL) muscle and gastrocnemius (GAS) muscle, respectively, was observed in intracellular resting Ca2+ concentration during 4 weeks of hindlimb immobilization (Booth and Giannetta 1973). Similarly, other studies also reported a 330% increase in intracellular resting Ca2+ concentration in the SOL muscle of rats after 14 days of hindlimb suspension (Wu et al. 2012).
Intracellular Ca2+ overload performs an interesting function in the mechanisms of muscle atrophy. Calpain, calcium-activated proteases activated by elevated intracellular Ca2+ concentration that plays a prime role in the degradation of filaments and initiation of most proteolytic pathways (e.g. the ubiquitin-proteasome pathway). Enhanced protein degradation due to calpain is believed as one of the crucial pathways of muscle atrophy (Ferreira et al. 2008). Our recent studies also made an agreement that activation of calpain leads to skeletal muscle atrophy (Agrawal et al. 2017; Chaudhury et al. 2012). Beside calpains, overloaded cytosolic Ca2+ also allied with mitochondrial apoptosis process (Fontana et al. 2015). Increase cytosolic Ca2+ leads to accumulation of the same to the mitochondria, which trigger mitochondrial depolarization once it reached the threshold level. Afterward, pro-apoptotic protein Bax was activated which leads to the formation and opening of the mitochondrial permeability transition pore (mPTP) via that cytochrome C, a mitochondria-resided apoptogenic factor released to cortisol which resultant into cell apoptosis (Adhihetty and Hood 2003) (Fig. 4).
Fig. 4.

Intracellular calcium regulates muscle atrophy via calpain regulated Ub-proteasome pathway and apoptosis
The opening of RyR channels is responsible for Ca2+ sparks. A single Ca2+ spark could produce a high level of local Ca2+ (10–100 μM or (0.1% of the cell volume), whereas global intracellular Ca2+ concentration was increased by 2 nM (Jaggar et al. 1998). In simple words, the term “Ca2+ sparks” refers to Ca2+ release through RyR channels (Jaggar et al. 2000). Ca2+ sparks or global Ca2+ depends on the following factors: (1) voltage-dependent Ca2+ channels (DHPR); (2) Communication between voltage-dependent Ca2+ channels and RyR channels; (3) positive- and negative-feedback regulation of global Ca2+; (4) frequency and amplitude modulation (FM and AM) of Ca2+ sparks (Fig. 5).
Fig. 5.
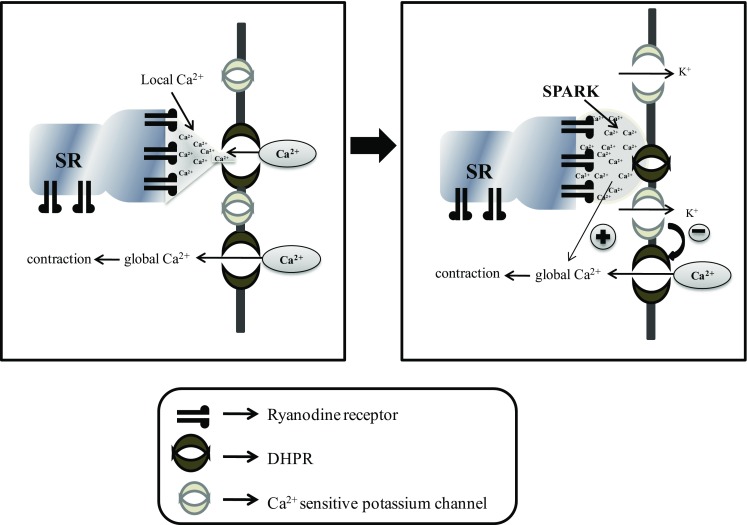
Ca2+ sparks in smooth muscle cells. Left: local control of Ca2+ sparks. Right. Ca2+ spark activate Ca2+-K+ channel to complete negative feedback loop () and also illustrated a positive-feedback () of Ca2+ release from RyR channels to contraction
Ca2+ sparks have been reported in cardiac (Cheng et al. 1993), skeletal (Klein et al. 1996) and smooth muscle cells (Nelson et al. 1995). A number of reports also evidenced to prove that Ca2+ sparks are due to the opening of RyR channels and increment in Ca2+ concentration around RyR are known as local Ca2+ which used to increase by a factor of 10 (Hoang-Trong et al. 2015). It is also assumed that RyR remodelling could play an important role in Ca2+ spark (Nelson et al. 1995; Tsugorka et al. 1995).
Calcium channels and related therapeutic implications/strategies
Leaky RyR1 suggested a potential role in muscular dystrophy (Andersson et al. 2012), muscular fatigue (Andersson et al. 2011) and ageing/sarcopenia (Bellinger et al. 2008) and this could be associated with cysteine nitrosylation or oxidation which further contributed to the weak binding of FKBP12 with RyR1 (Xia et al. 2000).
During stress conditions, ryanodine receptor (RyR1) could be remodeled which resulted into a production of PKA-hyperphosphorylation, S-nitrosylation, depletion of phosphodiesterase PDE4D3 and the RyR1 stabilizing subunit FKBP12 (FKBP12). The remodelling of RyR1 converted to a leaky RyR1 channel resultant into decrease exercise tolerance. Skeletal muscle-specific knockout of FKBP12 or PDE43 exhibited an impaired exercise capacity. Researchers tested the effect of a drug which prevents depletion of FKBP12 from the RyR1 complex on exercise capacity.
Derivatives of JTV519, a 1,4-benzodiazepine was screened that enhance the binding affinity of FKBP12 to PKA phosphorylated and/or S-nitrosylated RyR1. JTV519 is specific for RyR1 and have favorable drug-like properties (e.g., orally available, well absorbed, and stable). A small molecule (S107) met all the necessary criteria, was also used to enhance the binding of FKBP12 to RyR1 complex that improved force generation and exercise capacity and reduced SR Ca2+ leak, Ca2+ dependent neutral protease calpain activity and plasma creatine kinase levels (Bellinger et al. 2008). Also, treated aged mice with S107 enhances muscle strength without increasing the size of the muscle, at least during the 4 week period of treatment, this has been examined in their study (Andersson et al. 2011). Moreover, an improved cardiac function is seen through S107 treatment of heart failure (myocardial function) and in mdx mice (Shan et al. 2010a; Fauconnier et al. 2010). Sgcb−/− (Sarcoglycan deficient mice) mice treated with S107, displayed improved exercise capacity with improvement in RyR1-FKBP12 binding and the SR Ca2+ release during muscle contraction (Andersson et al. 2012).
Recent evidence also documented the therapeutics for few other calcium-related receptors, which basically involve L-type channels, SERCA–ATPase pump and the sodium-calcium pump (Na+/Ca2+ exchange pump). A clinical trial has been undertaken for L-type calcium channel inhibitors include diltiazem, verapamil, nifedipine and Flunarizine (Burr and Molkentin 2015). These inhibitors proven to be interesting targets for handling disrupted calcium ion homeostasis and they are already being clinically approved for human use. One study found that after a week of treatment of mdx mice with nifedipine, [Ca]i2+ was decreased and grip strength and swimming times were improved (Altamirano et al. 2013). Increased SERCA expression/activity could control the defects in SR calcium ion, a potent activator; BGP-15 increased SERCA activity and reduced muscle pathology in mdx mice (Gehrig et al. 2012). Ranolazine is the reverse mode NCX inhibitor reduces intracellular sodium ions (Burr et al. 2014). Many more inhibitors are yet to be tested for altered SR calcium ion release. Therefore, these are some probable therapeutics which could be used against muscular stress/atrophy; hence muscular problems could be treated effectively.
Conclusions
Ca2+ ions are the signaling molecules in muscles for excitation-contraction coupling and in the plasticity of skeletal muscles. Ca2+ also regulates various processes like myosin-actin cross bridging, protein synthesis, and protein degradation by calpain activation. Despite this, an introduction of Ca2+ leak channels during pathophysiological condition causes an increased cytosolic Ca2+ level and consequently activates calcium-dependent protease i.e., calpain which degrades myofilaments.
The present review provided a new insight into different pathophysiological conditions of muscular atrophy, which could be due to abnormal Ca2+ homeostasis and calcium related signaling pathways. Understanding these pathways, further offered some pharmacological interventions which could present beneficial effects under various muscular stress conditions like muscular fatigue, sarcopenia and in heart failure as mentioned above in therapeutic part of this review.
Acknowledgements
The authors are thankful to Dr. Bhuvnesh Kumar, Director, DIPAS for his constant support and encouragement. One of the authors, Ms. Akanksha Agrawal is thankful for obtaining Senior Research Fellowship from DRDO.
Compliance with ethical standards
Disclosures
No conflicts of interest, financial or otherwise, are declared by the author(s).
References
- Adhihetty PJ, Hood DA. Mechanisms of apoptosis in skeletal muscle. Basic Appl Myol. 2003;13:171–179. [Google Scholar]
- Aghdasi B, Reid MB, Hamilton SL. Nitric oxide protects the skeletal muscle Ca2+ Release Channel from oxidation induced activation. J Biol Chem. 1997;272:25462–25467. doi: 10.1074/jbc.272.41.25462. [DOI] [PubMed] [Google Scholar]
- Agrawal A, Rathor R, Suryakumar G. Oxidative protein modification alters proteostasis under acute hypobaric hypoxia in skeletal muscles: a comprehensive in vivo study. Cell Stress Chaper. 2017;22:429–443. doi: 10.1007/s12192-017-0795-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley PK, Porter KE, Boyle JP, Kemp PJ, Peers C. Hypoxic modulation of Ca2+signaling in human venous endothelial cells. Multiple roles for reactive oxygen species J Biol Chem. 2005;280:13349–13354. doi: 10.1074/jbc.M413674200. [DOI] [PubMed] [Google Scholar]
- Aley PK, Murray HJ, Boyle JP, Pearson HA, Peers C. Hypoxia stimulates Ca2+ release from intracellular stores in astrocytes via cyclic ADP ribose-mediated activation of ryanodine receptors. Cell Calcium. 2006;39:95–100. doi: 10.1016/j.ceca.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Altamirano F, Valladares D, Henríquez-Olguín C, Casas M, López JR, Allen PD, Jaimovich E. Nifedipine treatment reduces resting calcium concentration, oxidative and apoptotic gene expression, and improves muscle function in dystrophic mdx mice. PLoS One. 2013;8:e81222. doi: 10.1371/journal.pone.0081222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DM, Anderson KM, Chang CL, Makarewich CA, Nelson BR, McAnally JR, Kasaragod P, Shelton JM, Liou J, Bassel-Duby R, Olson EN. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 2015;160(4):595–606. doi: 10.1016/j.cell.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W, Shiomi T, Zalk R, Lacampagne A, Marks AR. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011;14:196–207. doi: 10.1016/j.cmet.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DC, Meli AC, Reiken S, Betzenhauser MJ, Umanskaya A, Shiomi T, D'Armiento J, Marks AR. Leaky ryanodine receptors in β-sarcoglycan deficient mice: a potential common defect in muscular dystrophy. Skelet Muscle. 2012;2(1):9. doi: 10.1186/2044-5040-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aracena P, Sanchez G, Donoso P, Hamilton SL, Hidalgo C. S-glutathionylation decreases Mg2+ inhibition and S-nitrosylation enhances Ca2+ activation of RyR1 channels. J Biol Chem. 2003;278:42927–42935. doi: 10.1074/jbc.M306969200. [DOI] [PubMed] [Google Scholar]
- Aracena P, Tang W, Hamilton S, Hidalgo C. Effects of S-glutathionylation and S-nitrosylation on calmodulin binding to triads and FKBP12 binding to type 1 calcium release channels. Antioxid Redox Signal. 2005;7:870–881. doi: 10.1089/ars.2005.7.870. [DOI] [PubMed] [Google Scholar]
- Aracena-Parks P, Goonasekera SA, Gilman CP, Dirksen RT, Hidalgo C, Hamilton SL. Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J Biol Chem. 2006;281:40354–40368. doi: 10.1074/jbc.M600876200. [DOI] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla FM, Horowicz P. Twitches in the presence of ethylene glycol bis (β-aminoethyl ether)-N,N -tetracetic acid. Biochim Biophys Acta. 1972;267:605–608. doi: 10.1016/0005-2728(72)90194-6. [DOI] [PubMed] [Google Scholar]
- Avdonin PV. Orai and TRP channels in skeletal muscle cells. Biochemistry (Moscow) Supplement Series A: Membrane and Cell Biol. 2012;6(2):159–168. [Google Scholar]
- Barreiro E, Hussain SN. Protein carbonylation in skeletal muscles: impact on function. Antioxid Redox Signal. 2010;12:417–429. doi: 10.1089/ars.2009.2808. [DOI] [PubMed] [Google Scholar]
- Batchelor CL, Winder SJ. Sparks, signals and shock absorbers: how dystrophin loss causes muscular dystrophy. Trends Cell Biol. 2006;16:198–205. doi: 10.1016/j.tcb.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Beard NA, Laver DR, Dulhunty AF. Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog Biophys Mol Biol. 2004;85:33–69. doi: 10.1016/j.pbiomolbio.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Dura M, Murphy PW, Deng SX, Landry DW, Nieman D, Lehnart SE, Samaru M, LaCampagne A, Marks AR. Remodelling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. PNAS. 2008;105:2198–2202. doi: 10.1073/pnas.0711074105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, Matecki S, Lacampagne A, Marks AR. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15:325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32:235–249. doi: 10.1016/s0143416002001823. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signaling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Bers DM. Excitation-contraction coupling and cardiac contractile force. Develop Cardiovas Med. 2001;237:978–994. [Google Scholar]
- Bers DM. Cardiac excitation–contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol. 2004;37:417–429. doi: 10.1016/j.yjmcc.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Bianchi CP, Shanes AM. Calcium influx in skeletal muscle at rest, during activity and during potassium contracture. J Gen Physiol. 1959;42:803–815. doi: 10.1085/jgp.42.4.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth FW, Giannetta CL. Effect of hindlimb immobilization upon skeleton muscle calcium in rat. Calcif Tissue Res. 1973;13:327–330. doi: 10.1007/BF02015423. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Lipp P, Berridge MJ. The organization and functions of local Ca2+ signals. J Cell Sci. 2001;114:2213–2222. doi: 10.1242/jcs.114.12.2213. [DOI] [PubMed] [Google Scholar]
- Bosanac Ivan, Alattia Jean-René, Mal Tapas K., Chan Jenny, Talarico Susanna, Tong Frances K., Tong Kit I., Yoshikawa Fumio, Furuichi Teiichi, Iwai Miwako, Michikawa Takayuki, Mikoshiba Katsuhiko, Ikura Mitsuhiko. Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature. 2002;420(6916):696–700. doi: 10.1038/nature01268. [DOI] [PubMed] [Google Scholar]
- Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasová E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Burk SE, Lytton J, MacLennan DH, Shull GE. cDNA cloning, functional expression and mRNA tissue distribution of a third organellar Ca2+ pump. J Biol Chem. 1989;264:18561–18568. [PubMed] [Google Scholar]
- Burr AR, Molkentin JD. Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ. 2015;22:1402–1412. doi: 10.1038/cdd.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burr AR, Millay DP, Goonasekera SA, Park KH, Sargent MA, Collins J, Altamirano F, Philipson KD, Allen PD, Ma J, López JR, Molkentin JD. Na+ dysregulation coupled with Ca2+ entry through NCX1 promotes muscular dystrophy in mice. Mol Cell Biol. 2014;34:1991–2002. doi: 10.1128/MCB.00339-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Soeller C. Numerical analysis of ryanodine receptor activation by L-type channel activity in the cardiac muscle diad. Biophys J. 1997;73:112–122. doi: 10.1016/S0006-3495(97)78052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–1049. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Excitation-contraction coupling in vertebrate skeletal muscle: a tale of two calcium channels. Cell. 1991;64:871–874. doi: 10.1016/0092-8674(91)90309-m. [DOI] [PubMed] [Google Scholar]
- Chaudhury P, Suryakumar G, Prasad R, Singh SM, Ali S, Ilavazhagan G. Chronic hypobaric hypoxia mediated skeletal muscle atrophy: role of ubiquitin–proteasome pathway and calpains. Mol Cell Biochem. 2012;364:101–113. doi: 10.1007/s11010-011-1210-x. [DOI] [PubMed] [Google Scholar]
- Chen L, Lu XY, Li J, Fu JD, Zhou ZN, Yang HT. Intermittent hypoxia protects cardiomyocytes against ischemia-reperfusion injury-induced alterations in Ca2+ homeostasis and contraction via the sarcoplasmic reticulum and Na+/Ca2+ exchange mechanisms. Am J Physiol Cell Physiol. 2006;290:C1221–C1229. doi: 10.1152/ajpcell.00526.2005. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Cheong E, Tumbev V, Abramson J, Salama G, Stoyanovsky DA. Nitroxyl triggers Ca2+ release from skeletal and cardiac sarcoplasmic reticulum by oxidizing ryanodine receptors. Cell Calcium. 2005;37:87–96. doi: 10.1016/j.ceca.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Divet A, Huchet-Cadiou C. Sarcoplasmic reticulum functions in slow- and fast-twitch skeletal muscles from mdx mice. Pflugers Arch. 2002;444:634–643. doi: 10.1007/s00424-002-0854-5. [DOI] [PubMed] [Google Scholar]
- Dulhunty AF, Haarmann CS, Green D, Laver DR, Board PG, Casarotto MG. Interactions between dihydropyridine receptors and ryanodine receptors in striated muscle. Prog Biophys Mol Biol. 2002;79:45–75. doi: 10.1016/s0079-6107(02)00013-5. [DOI] [PubMed] [Google Scholar]
- Dulhunty AF, Wei-La PL, Casarotto MG, Beard NA. Core skeletal muscle ryanodine receptor calcium release complex. Clin Exper Pharmacol Physiol. 2017;44:3–12. doi: 10.1111/1440-1681.12676. [DOI] [PubMed] [Google Scholar]
- Durham WJ, Aracena-Parks P, Long C, Rossi AE, Goonasekera SA, Boncompagni S, Galvan DL, Gilman CP, Baker MR, Shirokova N, Protasi F, Dirksen R, Hamilton SL. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1knockin mice. Cell. 2008;133:53–65. doi: 10.1016/j.cell.2008.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltit JM, Hidalgo J, Liberona JL, Jaimovich E. Slow calcium signals after tetanic electrical stimulation in skeletal myotubes. Biophys J. 2004;86:3042–3051. doi: 10.1016/S0006-3495(04)74353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M. Conditions required for calcium-induced release of calcium from the sarcoplasmic reticulum. Proc Jap Acad. 1975;51:467–472. [Google Scholar]
- Endo M (1981) Mechanism of calcium-induced calcium release in the SR membrane. Academic, pp 257–264
- Endo M. Calcium-induced calcium release in skeletal muscle. Physiol Rev. 2009;89:1153–1176. doi: 10.1152/physrev.00040.2008. [DOI] [PubMed] [Google Scholar]
- Endo M, Tanaka M, Ogawa Y. Calcium induced release of calcium from the sarcoplasmic reticulum of skinned skeletal muscle fibres. Nature. 1970;228:34–36. doi: 10.1038/228034a0. [DOI] [PubMed] [Google Scholar]
- Eu JP, Sun J, Xu L, Stamler JS, Meissner G. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102:499–509. doi: 10.1016/s0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Appraisal of the physiological relevance of two hypotheses for the mechanism of calcium release from the mammalian cardiac sarcoplasmic reticulum: calcium-induced release versus charge coupled release. Mol Cell Biochem. 1989;89:135–140. doi: 10.1007/BF00220765. [DOI] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J Physiol Lond. 1975;249:469–495. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell EF, Antaramian A, Rueda A, Gomez AM, Valdivia HH. Sorcin inhibits calcium release and modulates excitation-contraction coupling in the heart. J Biol Chem. 2003;278:34660–34666. doi: 10.1074/jbc.M305931200. [DOI] [PubMed] [Google Scholar]
- Fauconnier J, Thireau J, Reiken S, Cassan C, Richard S, Matecki S, Marks AR, Lacampagne A. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc Natl Acad Sci. 2010;107:1559–1564. doi: 10.1073/pnas.0908540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero TG, Zable AC, Bowman MB, Thompson A, Abramson JJ. Metabolic end products inhibit sarcoplasmic reticulum Ca2+ release and [3H] ryanodine binding. J Appl Physiol. 1995;78:1665–1672. doi: 10.1152/jappl.1995.78.5.1665. [DOI] [PubMed] [Google Scholar]
- Felix R, Gurnett CA, Waard DM, Campbell KP. Dissection of functional domains of the voltage-dependent Ca2+ channel α2δ subunit. J Neurosci. 1997;17:6884–6891. doi: 10.1523/JNEUROSCI.17-18-06884.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Liu G, Allen PD, Pessah IN. Transmembrane redox sensor of ryanodine receptor complex. J Biol Chem. 2000;275:35902–35907. doi: 10.1074/jbc.C000523200. [DOI] [PubMed] [Google Scholar]
- Ferreira R, Neuparth MJ, Vitorino R, Appell HJ, Amado F, Duarte JA. Evidences of apoptosis during the early phases of soleus muscle atrophy in hindlimb suspended mice. Physiol Res. 2008;57:601–611. doi: 10.33549/physiolres.931272. [DOI] [PubMed] [Google Scholar]
- Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- Flucher BE, Tuluc P. How and why are calcium currents curtailed in the skeletal muscle voltage-gated calcium channels? J Physiol. 2017;595:1451–1463. doi: 10.1113/JP273423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana J, Fritz N, Brismar H, Aperia A. Apoptosis caused by excessive mitochondrial albumin uptake in renal cells is initiated by increased mitochondrial calcium concentration. FASEB J. 2015;29:845–830. [Google Scholar]
- Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Protasi F, Ramesh V. Comparative ultrastructure of Ca2+ release units in skeletal and cardiac muscle. Ann N Y Acad Sci. 1998;16:20–30. doi: 10.1111/j.1749-6632.1998.tb08253.x. [DOI] [PubMed] [Google Scholar]
- Furuichi T, Furutama D, Hakamata Y, Nakai J, Takeshima H, Mikoshiba K. Multiple types of ryanodine receptor/Ca2+ release channels are differentially expressed in rabbit brain. The Journal of Neuroscience. 1994;14(8):4794–4805. doi: 10.1523/JNEUROSCI.14-08-04794.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlert S, Bloch W, Suhr F. Ca2+ dependent regulations and signaling in skeletal muscle: from electro-mechanical coupling. Int J Mol Sci. 2015;16:1066–1095. doi: 10.3390/ijms16011066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrig SM, van der Poel C, Sayer TA, Schertzer JD, Henstridge DC, Church JE, Lamon S, Russell AP, Davies KE, Febbraio MA, Lynch GS. Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature. 2012;484:394–398. doi: 10.1038/nature10980. [DOI] [PubMed] [Google Scholar]
- Giannini G, Conti A, Mammarella S, Scrobogna M, Sorrentino V. The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J Cell Biol. 1995;128:893–904. doi: 10.1083/jcb.128.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goonasekera SA, Lam CK, Millay DP, Sargent MA, Hajjar RJ, Kranias EG, Molkentin JD. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J Clin Invest. 2011;121:1044–1052. doi: 10.1172/JCI43844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakamata Y, Nakai J, Takeshima H, Imoto K. Primary structure and distribution of a novel ryanodine receptor/calcium release channel from rabbit brain. FEBS Lett. 1992;312:229–235. doi: 10.1016/0014-5793(92)80941-9. [DOI] [PubMed] [Google Scholar]
- Hidalgo C, Sanchez G, Barrientos G, Aracena-Parks P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S-glutathionylation. J Biol Chem. 2006;281:26473–26482. doi: 10.1074/jbc.M600451200. [DOI] [PubMed] [Google Scholar]
- Hoang-Trong TM, Ullah A, Jafri SM. Calcium sparks in the heart: dynamics and regulation. Res Rep Biol. 2015;6:203–214. doi: 10.2147/RRB.S61495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstick EJ, Linsley JW, Dowling JJ, Hauser MA, McDonald KK, Ashley-Koch A, Saint-Amant L, Satish A, Cui WW, Zhou W, Sprague SM, Stamm DS, Powell CM, Speer MC, Franzini-Armstrong C, Hirata H, Kuwada JY. Stac3 is a component of the excitation–contraction coupling machinery and mutated in native American myopathy. Nature Comm. 2013;4:1952. doi: 10.1038/ncomms2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MJ. Strategies for reducing oxidative damage in ageing skeletal muscle. Adv Drug Deliv Rev. 2009;61:1363–1368. doi: 10.1016/j.addr.2009.07.018. [DOI] [PubMed] [Google Scholar]
- Jaggar JH, Stevenson AS, Nelson MT. Voltage dependence of Ca2+ sparks in intact cerebral arteries. Am J Physiol Cell Physiol. 1998;274:C1755–C1761. doi: 10.1152/ajpcell.1998.274.6.C1755. [DOI] [PubMed] [Google Scholar]
- Jaggar JH, Valerie A, Porter W, Lederer J, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol Cell Physiol. 2000;278:C235–C256. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- Jain K, Suryakumar G, Prasad R, Singh SM, Ganju L. Differential activation of myocardial ER stress response: a possible role in hypoxic tolerance. Int J Cardiol. 2013;168:4667–4677. doi: 10.1016/j.ijcard.2013.07.180. [DOI] [PubMed] [Google Scholar]
- Jain K, Suryakumar G, Prasad R, Singh SM, Ganju L. Myocardial ER chaperone activation and protein degradation occurs due to synergistic, not individual, cold and hypoxic stress. Biochimie. 2013;95:1897–1908. doi: 10.1016/j.biochi.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Jayaraman T, Brillantes AM, Timerman AP, Fleischer S, Erdjument-Bromage H, Tempst P, Marks AR. FK506 binding protein associated with the calcium release channel (ryanodine receptor) J Biol Chem. 1992;267:9474–9477. [PubMed] [Google Scholar]
- Jeong HN, Park HJ, Lee JH, Shin HY, Kim SH, Kim SM, Choi YC. Clinical and pathologic findings of Korean patients with RYR1-related congenital myopathy. J Clin Neurol. 2018;14(1):58–65. doi: 10.3988/jcn.2018.14.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatous SB, Mammen PPA, Rosenberg PB, Martin CM, White MD, DiMaio JM, Huang G, Muallem S, Garry DJ. Hypoxia reprograms calcium signaling and regulates myoglobin expression. Am J Physiol-Cell Physiol. 2009;296(3):C393–C402. doi: 10.1152/ajpcell.00428.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Nakamori M, Lueck JD, Pouliquin P, Aoike F, Fujimura H, Dirksen RT, Takahashi MP, Dulhunty AF, Sakoda S. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum Mol Genet. 2005;14:2189–2200. doi: 10.1093/hmg/ddi223. [DOI] [PubMed] [Google Scholar]
- Kiviluoto S, Decuypere JP, De Smedt H, Missiaen L, Parys JB, Bultynck G. STIM1 as a key regulator for Ca2+ homeostasis in skeletal-muscle development and function. Skelet Muscle. 2011;1(1):16. doi: 10.1186/2044-5040-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein MG, Cheng H, Santana LF, Jiang YH, Lederer WJ, Schneider MF. Two mechanisms of quantized calcium release in skeletal muscle. Nature. 1996;379:455–458. doi: 10.1038/379455a0. [DOI] [PubMed] [Google Scholar]
- Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwajima G, Futatsugi A, Niinobe M, Nakanishi S, Mikoshiba K. Two types of ryanodine receptors in mouse brain: skeletal muscle type exclusively in Purkinje cells and cardiac muscle type in various neurons. Neuron. 1992;9:1133–1142. doi: 10.1016/0896-6273(92)90071-k. [DOI] [PubMed] [Google Scholar]
- Lai FA, Dent M, Wickenden C, Xu L, Kumari G, Misra M, Lee HB, Sar M, Meissner G. Expression of a cardiac Ca2+-release channel isoform in mammalian brain. Biochem J. 1992;288:553–564. doi: 10.1042/bj2880553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung FP, Yung LM, Yao X, Laher I, Huang Y. Store-operated calcium entry in vascular smooth muscle. Br J Pharmacol. 2008;153:846–857. doi: 10.1038/sj.bjp.0707455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Harlow L, Graham ZA, Bauman WA, Cardoz C. Spinal cord injury leads to Hyperoxidation and Nitrosylation of skeletal muscle ryanodine Receptor-1 associated with upregulation of NADH oxidase 4. J Neurotrauma. 2016;34(12):2069–2074. doi: 10.1089/neu.2016.4763. [DOI] [PubMed] [Google Scholar]
- Lunde PK, Dahlstedt AJ, Bruton JD, Lannergren J, Thoren P, Sejersted OM, Westerblad (2001) Contraction and intracellular Ca2+ handling in isolated skeletal muscle of rats with congestive heart failure. Circ Res 88:1299–1305 [DOI] [PubMed]
- MacLennan DH, Rice WJ, Green NM. The mechanism of Ca2þ transport by sarco (endo) plasmic reticulum Ca2+ ATPases. J Biol Chem. 1997;272:28815–28818. doi: 10.1074/jbc.272.46.28815. [DOI] [PubMed] [Google Scholar]
- MacLennan DH, Asahi M, Tupling AR. The regulation of SERCA-type pumps by phospholamban and sarcolipin. Ann N Y Acad Sci. 2003;986:472–480. doi: 10.1111/j.1749-6632.2003.tb07231.x. [DOI] [PubMed] [Google Scholar]
- Marks AR, Tempst P, Hwang KS, Taubman MB, Inui M, Chadwick C, Fleischer S, Nadal-Ginard B. Molecular cloning and characterization of the rynodine receptor/junctional channel complex cDNA from skeletal muscle sarcoplasmic reticulum. Proc Natl Acad Sci. 1989;86:8683–8687. doi: 10.1073/pnas.86.22.8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martonosi AN, Pikula S. The network of calcium regulation in muscle. Acta Biochim Pol. 2003;50:1–30. [PubMed] [Google Scholar]
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Melzer W, Herrmann-Frank A, Luttgau HC. The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim Biophys Acta. 1995;1241:59–116. doi: 10.1016/0304-4157(94)00014-5. [DOI] [PubMed] [Google Scholar]
- Michalak M, Robert Parker JM, Opas M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium. 2002;32:269–278. doi: 10.1016/s0143416002001884. [DOI] [PubMed] [Google Scholar]
- Milner RE, Famulski KS, Michalak M. Calcium binding proteins in the sarcoplasmic/endoplasmic reticulum of muscles and non muscles cells. Mol Cell Biochem. 1992;112:1–13. doi: 10.1007/BF00229637. [DOI] [PubMed] [Google Scholar]
- Missiaen L, De Smedt H, Droogmans G, Casteels R. Ca2+ release induced by inositol 1, 4, 5-trisphosphate is a steady-state phenomenon controlled by luminal Ca2+ in permeabilized cells. Nature. 1992;357:599–602. doi: 10.1038/357599a0. [DOI] [PubMed] [Google Scholar]
- Moore CP, Rodney G, Zhang JZ, Santacruz-Toloza L, Strasburg G, Hamilton SL. Apocalmodulin and Ca2+ calmodulin bind to the same region on the skeletal muscle Ca2+ release channel. Biochemistry. 1999;38:8532–8537. doi: 10.1021/bi9907431. [DOI] [PubMed] [Google Scholar]
- Mosca B, Eckhardt J, Bergamelli L, Treves S, Bongianino R, Negri MD, Priori SG, Protasi F, Zorzato F. Role of the JP45-Calsequestrin complex on calcium entry in slow twitch skeletal muscles. J Biol Chem. 2016;291(39):20824. doi: 10.1074/jbc.A115.709071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free Radic Biol Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Kuwajima G, Mikoshiba K. Immunohistochemical localization of ryanodine receptors in mouse central nervous system. Neurosci Res. 1992;15:130–142. doi: 10.1016/0168-0102(92)90026-9. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- Neylon CB, Richards SM, Larsen MA, Agrotis A, Bobik A. Multiple types of ryanodine receptor/Ca2+ release channels are expressed in vascular smooth muscle. Biochem Biophys Res Commun. 1995;215:814–821. doi: 10.1006/bbrc.1995.2536. [DOI] [PubMed] [Google Scholar]
- Nixon GF, Mignery GA, Somlyo AV. Immunogold localization of inositol 1,4,5-triphosphate receptors and characterization of ultrastructural features of the sarcoplasmic reticulum in phasic and tonic smooth muscle. J Muscle Res Cell Motil. 1994;15:682–700. doi: 10.1007/BF00121075. [DOI] [PubMed] [Google Scholar]
- Oba T, Murayama T, Ogawa Y. Redox states of type 1 ryanodine receptor alters Ca2+ release channel response to modulators. Am J Phys. 2002;282:C684–C692. doi: 10.1152/ajpcell.01273.2000. [DOI] [PubMed] [Google Scholar]
- Ohrtman J, Ritter B, Polster A, Beam KG, Papadopoulos S. Sequence differences in the IQ motifs of CaV1.1 and CaV1.2 strongly impact calmodulin binding and calcium-dependent inactivation. The J Biochem. 2008;283(43):29301–29311. doi: 10.1074/jbc.M805152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu K, Willard HF, Khanna VK, Zorzato F, Green NM, MacLennan DH. Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J Biol Chem. 1990;265:13472–13483. [PubMed] [Google Scholar]
- Ottini L, Marziali G, Conti A, Charlesworth A, Sorrentino V. Alpha and beta isoforms of ryanodine receptor from chicken skeletal muscle are the homologues of mammalian RyR1 and RyR3. Biochem J. 1996;315:207–216. doi: 10.1042/bj3150207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp S, Dziak E, Michalak M, Opas M. Is all of the endoplasmic reticulum created equal? The effects of the heterogeneous distribution of endoplasmic reticulum Ca2+ handling proteins. J Cell Biol. 2003;160:475–479. doi: 10.1083/jcb.200207136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB. Store-operated Ca2+ entry: dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane. J Physiol. 2003;547:333–348. doi: 10.1113/jphysiol.2002.034140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85(2):757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Periasamy M, Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve. 2007;35(4):430–442. doi: 10.1002/mus.20745. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Ravel-Chapuis A, Bélanger G, Côté J, Michel RN, Jasmin BJ. Misregulation of calcium-handling proteins promotes hyperactivation of calcineurin-NFAT signaling in skeletal muscle of DM1 mice. Hum Mol Genet. 2017;26(12):2192–2206. doi: 10.1093/hmg/ddx109. [DOI] [PubMed] [Google Scholar]
- Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C, Huang F, Gaburjakova M, Gaburjakova J, Rosemblit N, Warren MS, He KL, Yi GH, Wang J, Burkhoff D, Vassort G, Marks AR. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Cell Biol. 2003;160:919–928. doi: 10.1083/jcb.200211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuben JP, Brandt PW, Grundfest H. Regulation of myoplasmic calcium concentration in intact crayfish muscle fibers. J Mechano Chem Cell Motil. 1974;12:269–285. [PubMed] [Google Scholar]
- Rios E, Pizarro G. Voltage sensor of excitation-contraction coupling in skeletal muscle. Physiol Rev. 1991;71:849–908. doi: 10.1152/physrev.1991.71.3.849. [DOI] [PubMed] [Google Scholar]
- Roos J, PJ DG, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Veliçelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169(3):435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama G, Menshikova EV, Abramson JJ. Molecular interaction between nitric oxide and ryanodine receptors of skeletal and cardiac sarcoplasmic reticulum. Antioxid Redox Signal. 2000;2:5–16. doi: 10.1089/ars.2000.2.1-5. [DOI] [PubMed] [Google Scholar]
- Sammels E, Parys JB, Missiaen L, De Smedt H, Bultynck G. Intracellular Ca2+ storage in health and disease: a dynamic equilibrium. Cell Calcium. 2010;47:297–314. doi: 10.1016/j.ceca.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Samso M. 3D structure of the Dihydropyridine receptor of skeletal muscle. Eur J Transl Myol. 2015;25(1):4840. doi: 10.4081/ejtm.2015.4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schartner V, Romero NB, Donkervoort S, Treves S, Munot P, Pierson TM, Dabaj I, Malfatti E, Zaharieva IT, Zorzato F, Abath Neto O, Brochier G, Lornage X, Eymard B, Taratuto AL, Böhm J, Gonorazky H, Ramos-Platt L, Feng L, Phadke R, Bharucha-Goebel DX, Sumner CJ, Bui MT, Lacene E, Beuvin M, Labasse C, Dondaine N, Schneider R, Thompson J, Boland A, Deleuze JF, Matthews E, Pakleza AN, Sewry CA, Biancalana V, Quijano-Roy S, Muntoni F, Fardeau M, Bönnemann CG, Laporte J. Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. 2017;133(4):517–533. doi: 10.1007/s00401-016-1656-8. [DOI] [PubMed] [Google Scholar]
- Scherer NM, Deamer DW. Oxidative stress impairs the function of sarcoplasmic reticulum by oxidation of sulfhydryl groupsin the Ca2+- ATPase. Arch Biochem Biophys. 1986;246:589–601. doi: 10.1016/0003-9861(86)90314-0. [DOI] [PubMed] [Google Scholar]
- Schneider MF. Control of calcium release in functioning skeletal muscle fibers. Annu Rev Physiol. 1994;56:463–484. doi: 10.1146/annurev.ph.56.030194.002335. [DOI] [PubMed] [Google Scholar]
- Schreiber D, Donoghue P, Reilly C, Ohlendieck K. Role of Calsequestrin and related luminal Ca2+-binding proteins as mediators of excitation-contraction coupling. Basic Appl Myol. 2004;14:313–322. [Google Scholar]
- Sencer S, Papineni RV, Halling DB, Pate P, Krol J, Zhang JZ, Hamilton SLJ. Coupling of RYR1 and L-type calcium channels via calmodulin binding domains. J Biol Chem. 2001;276:38237–38241. doi: 10.1074/jbc.C100416200. [DOI] [PubMed] [Google Scholar]
- Seta KA, Yuan Y, Spicer Z, Lu G, Bedard J, Ferguson TK, Pathrose P, Cole-Strauss A, Kaufhold A, Millhorn DE. The role of calcium in hypoxia-induced signal transduction and gene expression. Cell Calcium. 2004;36:331–340. doi: 10.1016/j.ceca.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010;120:4375–4387. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, Lehnart SE, Lindegger N, Mongillo M, Mohler PJ, Marks AR. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest. 2010;120:4388–4398. doi: 10.1172/JCI32726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Coronado R, Meissner G. Single-channel measurement of the calcium release channel from skeletal muscle sarcoplasmic reticulum. J Gen Physiol. 1986;88:573–588. doi: 10.1085/jgp.88.5.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–237. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- Sun JH, Xu L, Eu JP, Stamler JS, Meissner G. Classes of thiols that influence the activity of the skeletal muscle calcium release channel. J Biol Chem. 2001;276:15625–15630. doi: 10.1074/jbc.M100083200. [DOI] [PubMed] [Google Scholar]
- Sun J, Xin C, Eu JP, Stamler JS, Meissner G. Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. PNAS. 2001;98:11158–11162. doi: 10.1073/pnas.201289098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima H. Primary structure and expression from cDNAs of the ryanodine receptor. Ann N Y Acad Sci. 1993;707:165–177. doi: 10.1111/j.1749-6632.1993.tb38051.x. [DOI] [PubMed] [Google Scholar]
- Treves S, Jungbluth H, Voermans N, Muntoni F, Zorzato F. Ca2+ handling abnormalities in early-onset muscle diseases: novel concepts and perspectives. Semin Cell Dev Biol. 2016;64:201–212. doi: 10.1016/j.semcdb.2016.07.017. [DOI] [PubMed] [Google Scholar]
- Tsugorka A, Rios E, Blatter LA. Imaging elementary events of calcium release in skeletal muscle cells. Science. 1995;269:1723–1726. doi: 10.1126/science.7569901. [DOI] [PubMed] [Google Scholar]
- Tupling AR, Bombardier E, Gupta SC, Hussain D, Vigna C, Bloemberg D, Quadrilatero J, Trivieri MG, Babu GJ, Backx PH, Periasamy M, MacLennan DH, Gramolini AO. Enhanced Ca2+ transport and muscle relaxation in skeletal muscle from sarcolipin-null mice. Am J Physiol Cell Physiol. 2011;301:C841–C849. doi: 10.1152/ajpcell.00409.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukcevic M, Broman M, Islander G, Bodelsson M, Ranklev-Twetman E, CR M¨l, Treves S. Functional properties of RYR1 mutations identified in Swedish patients with malignant hyperthermia and central Core disease. Anesthesia Analgesia. 2010;111:185–190. doi: 10.1213/ANE.0b013e3181cbd815. [DOI] [PubMed] [Google Scholar]
- Wei L, Dirksen RT. Ryanodinopathies: RyR-linked muscle diseases. Curr Top Membr. 2010;66:139–167. doi: 10.1016/S1063-5823(10)66007-3. [DOI] [PubMed] [Google Scholar]
- Wu X, Gao Y, Zhao X, Cui J. Effects of tetramethylpyrazine on nitric oxide synthase activity and calcium ion concentration of skeletal muscle in hindlimb unloading rats. Zhong hua yi xue za zhi. 2012;92:2075–2077. [PubMed] [Google Scholar]
- Wuytack F, Raeymaekers L, De Smedt H, Eggermont JA, Missiaen L, Van Den Bosch L, De Jaegere S, Verboomen H, Plessers L, Casteels R. Ca2+ transport ATPases and their regulation in muscle and brain. Ann N Y Acad Sci. 1992;671:82–91. doi: 10.1111/j.1749-6632.1992.tb43786.x. [DOI] [PubMed] [Google Scholar]
- Xia R, Stangler T, Abramson JJ. Skeletal muscle ryanodine receptor is a redox sensor with a well defined redox potential that is sensitive to channel modulators. J Biol Chem. 2000;275:36556–36561. doi: 10.1074/jbc.M007613200. [DOI] [PubMed] [Google Scholar]
- Xia R, Webb JA, Gnall LL, Cutler K, Abramson JJ. Skeletal muscle sarcoplasmic reticulum contains a NADH-dependent oxidase that generates superoxide. Am J Phys. 2003;285:C215–C221. doi: 10.1152/ajpcell.00034.2002. [DOI] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Fedotovskaya O, Cheng AJ, Cornachione AS, Minozzo FC, Aulin C, Fridén C, Turesson C, Andersson DC, Glenmark B, Lundberg IE, Rassier DE, Westerblad H, Lanner JT. Nitrosative modifications of the Ca2+ release complex and actin underlie arthritis-induced muscle weakness. Ann Rheum Dis. 2015;74(10):1907–1914. doi: 10.1136/annrheumdis-2013-205007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi N, Xin C, Meissner G. Identification of apocalmodulin and Ca2+-calmodulin regulatory domain in skeletal muscle Ca2+ release channel, ryanodine receptor. J Biol Chem. 2001;276(25):22579–22585. doi: 10.1074/jbc.M102729200. [DOI] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol. 2007;9(6):636–645. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalk R, Lehnart SE, Marks AR. Modulation of the rynodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367–385. doi: 10.1146/annurev.biochem.76.053105.094237. [DOI] [PubMed] [Google Scholar]
- Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF, Muallem S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell. 2008;32(3):439–448. doi: 10.1016/j.molcel.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]