

Abstract
Whereas the p38 MAP kinase has largely been associated with anti-proliferative functions, several observations have indicated that it may also have positive effects on proliferation. In hepatocytes, we have found that p38 has opposing effects on DNA synthesis when activated by EGF and HGF. Here we have studied the function of p38 in EGF- and HGF-induced DNA synthesis in the two pancreatic carcinoma cell lines AsPC-1 and Panc-1. In Panc-1 cells, the MEK inhibitor PD98059 reduced EGF- and HGF-induced DNA synthesis, while the p38 inhibitor SB203580 strongly increased the basal DNA synthesis and reduced expression of the cyclin-dependent kinase inhibitor (CDKI) p21. In contrast, in AsPC-1 cells, EGF- and HGF-induced DNA synthesis was not significantly reduced by PD98059 but was inhibited by SB203580. Treatment with SB203580 amplified the sustained ERK phosphorylation induced by these growth factors and caused a marked upregulation of the expression of p21, which could be blocked by PD98059. These results suggest that while DNA synthesis in Panc-1 cells is enhanced by ERK and strongly suppressed by p38, in AsPC-1 cells, p38 exerts a pro-mitogenic effect through MEK/ERK-dependent downregulation of p21. Thus, p38 may have suppressive or stimulatory effects on proliferation depending on the cell type, due to differential cross-talk between the p38 and MEK/ERK pathways.
Electronic supplementary material
The online version of this article (10.1007/s12079-017-0444-0) contains supplementary material, which is available to authorized users.
Keywords: Pancreatic carcinoma, EGF, HGF, p38, ERK, p21
Introduction
The p38 mitogen-activated protein kinases (MAPK), which include p38α, p38β, p38γ and p38δ, belong to a family of serine/threonine-directed kinases. Activation of the p38 MAPKs has been observed in response to various extracellular stimuli including cytokines and growth factors (Ono and Han 2000). Of the four members, p38α and p38β are the most widely expressed, found in nearly all cell types, and show high similarity in terms of substrate specificity and sensitivity to inhibitors (Nebreda and Porras 2000; Ono and Han 2000). Generally, p38α has been described as a tumor suppressor based on its role in negative regulation of cell cycle progression and in induction of apoptosis (Bulavin and Fornace 2004; Xu et al. 2014). p38α may modulate the expression or activity of cell cycle regulators including cyclin D1, the cyclin-dependent kinase inhibitors (CDKI) p16 and p21, and the tumor suppressor p53 in ways that inhibit tumor development (Bulavin and Fornace 2004). Nevertheless, several studies indicate that its role in tumorigenesis is much more complex. p38α may in some cancer cells positively regulate proliferation (Recio and Merlino 2002; Halawani et al. 2004; Ricote et al. 2006; Wagner and Nebreda 2009). In addition, high levels of phosphorylated p38α have been correlated with malignancy in several cancer types (Dolado and Nebreda 2007). Furthermore, there is evidence that p38α may increase the invasiveness of various cancers (Kim et al. 2003; Chen et al. 2004; Dreissigacker et al. 2006). These findings suggest that p38α may have suppressive effects on tumor initiation, whereas in cancer progression, it may have oncogenic functions.
p38α may cross-talk with several signaling pathways including the JNK, MEK/ERK and PI3K/Akt pathways (Cuadrado and Nebreda 2010). The negative effect of p38α on MEK/ERK signaling has been documented in several studies (Liu and Hofmann 2004; Junttila et al. 2008). In hepatocytes, we have found that pharmacological inhibition of p38 reduces ERK activation and DNA synthesis induced by EGF but, in contrast, inhibition of p38 enhanced these effects in response to HGF (Aasrum et al. 2016), showing that in the same cells, p38 may have inhibitory or stimulatory effects on mitogenic signalling depending on the growth factor. Differential cross-talk between p38 and MEK/ERK might be involved in this phenomenon.
In pancreatic cancer, the role of p38 in the regulation of proliferation by EGF and HGF has previously not been studied. It has been reported that oncogenic K-Ras enhances migration and invasiveness of pancreatic carcinoma cells through activation of p38 (Dreissigacker et al. 2006). Some pancreatic cancer cells also have endogenous activation of the p38 pathway, and p38 inhibition has been found to increase the growth of these cells (Zhong et al. 2014). Several mechanisms regulating cell proliferation converge at the CDKI proteins, including p16 and p21, which restrain the cell cycle activity by inhibiting CDKs (Pei and Xiong 2005). Mutated, inactivated, CDKN2A (the p16 gene) is one of the four major drivers in pancreatic oncogenesis, together with mutated KRAS, TP53, and SMAD4 (Dunne and Hezel 2015; Kamisawa et al. 2016). In contrast, p21 is rarely mutated but is regulated at the transcriptional and posttranscriptional level and exerts inhibition of the cell cycle by broad interference with CDKs (Abbas and Dutta 2009; Georgakilas et al. 2017). Upregulation of p21 has been found to mediate inhibition of pancreatic cell proliferation elicited by several physiological and pathophysiological mechanisms as well as by experimental and clinical pharmacological agents (Donadelli et al. 2006; Wiseman et al. 2007; Jia et al. 2008; Chen et al. 2010). In the present study we have examined the role of p38 in the regulation of proliferation in pancreatic cancer cells and some of the mechanisms conveying its effects, with focus on ERK and p21. The results suggest that in Panc-1 cells, p38 acts as a negative regulator of DNA synthesis, whereas in AsPC-1 cells, p38 enhances the mitogenic signalling.
Materials and methods
Materials
ATCC-modified Roswell Park medium (RPMI) was from Gibco (Grand Island, NY). Fetal bovine serum, glutamine and penicillin/streptomycin were from Lonza (Verviers, Belgium). Dulbecco’s modified Eagle’s medium and EGF (recombinant human) were from Sigma (St. Louis, MO). HGF (recombinant human) was from R&D (Minneapolis, MN). SB203580 and PD98059 were from Calbiochem (La Jolla, CA). The primary antibodies against phospho-p38 (Thr180/Tyr182), phospho-ERK1/2 (Thr202/Tyr204), GAPDH and p21were from Cell Signaling Technologies (Danvers, MA). The secondary HRP-conjugated antibody goat anti-rabbit IgG was from Bio-Rad Laboratories (Hercules, CA). [6-3H]thymidine was from Perkin Elmer (Waltham, MA).
Cell culture
The pancreatic cancer cell lines AsPC-1 and Panc-1 were obtained from ATCC (Manassas, VA). Both cell lines have activating KRAS mutations and inactivating p53 mutations; although the data on CDKN2A and SMAD4 are not fully consistent, most studies report that both these genes in AsPC-1, and CDKN2A in Panc-1, are inactivated by point mutations or homozygous deletions, while SMAD4 is wild type in Panc-1 (Deer et al. 2010). AsPC-1 cells were maintained in RPMI containing 10% FBS supplemented with penicillin (67 μg/ml) and streptomycin (100 μg/ml). Panc-1 cells were maintained in DMEM (4.5 g glucose/l) containing 10% FBS supplemented with penicillin (67 μg/ml) and streptomycin (100 μg/ml). The cultures were kept in a humidified 5% CO2 incubator. For western blotting and DNA synthesis experiments, cells were seeded in 12-well Costar plates (Corning Life Sciences, Acton, MA) at a density of 50,000 cells/cm2 in serum-containing medium. After 24 h, the medium was changed and cells cultured under serum-free conditions for 24 h prior to stimulation.
Western blot analysis
Total cell lysates were prepared in Laemmli buffer (4% SDS, 20% glycerol, 120 mM Tris-HCl, pH 6.8) and aliquots of 10 μg electrophoresed on 10% polyacrylamide TGX gels (Bio-Rad Laboratories, Hercules, CA). Proteins were transferred to nitrocellulose membranes. The blots were blocked in Tris-buffered saline containing 0.1% Tween 20 (TTBS) containing 5% non-fat dry milk and thereafter incubated with the primary antibodies as indicated in TTBS with 5% non-fat dry milk or 5% BSA overnight at 4 °C. The blots were washed twice in TTBS before incubation with the secondary antibody for 1 h at room temperature. LumiGLO (KPL, Gaithersburg, MA) was used for visualisation of the blots. Densitometric analyses were done using Labworks Software (UVP, Cambridge, UK). When quantifying western blots, the data were normalized using the signal strength of GAPDH or the respective non-phosphorylated protein.
DNA synthesis
Serum-starved cells were stimulated with EGF and HGF for 24 h. When using inhibitors, these were added 30 min before start of stimulation. [6-3H]thymidine was added 4 h before harvesting the cells. The amount of [6-3H]thymidine incorporated into the DNA was measured by scintillation counting as previously described (Refsnes et al. 1994).
Statistical analysis
The data shown are the mean ± SEM of at least 3 independent experiments. The statistical significance of differences was analysed by unpaired t-test using the GraphPad Software (La Jolla, CA).
Results
EGF and HGF induce phosphorylation of p38 in AsPC-1 and Panc-1 cells
Figure 1 shows that EGF and HGF induced phosphorylation of p38 in both AsPC-1 and Panc-1 cells. In both cell lines, EGF induced a maximal phosphorylation at 10 min, whereas HGF induced a maximal phosphorylation somewhat later. A higher concentration of EGF (10 nM) than HGF (1 nM) was used, since, on a molar basis, HGF is 10–20 times more potent than EGF (Gohda et al. 1990; Stolz and Michalopoulos 1994; Brusevold et al. 2012; Aasrum et al. 2013).
Fig. 1.

EGF- and HGF-induced phosphorylation of p38. Immunoblots show p38 phosphorylation induced by 10 nM EGF and 1 nM HGF in AsPC-1 (a) and Panc-1 (b) cells. The graphs show densitometric quantification of p38 phosphorylation based on three independent experiments ± SEM. The signal strength of p38 was used as a loading control and the amount of p-p38 was normalized to total p38
Effects of p38 inhibition on EGF- and HGF induced DNA synthesis
EGF has been found to induce DNA synthesis in both AsPC-1 and Panc-1 cells (Matsuda et al. 2002; Tveteraas et al. 2016). HGF-induced DNA synthesis has been demonstrated in AsPC-1 cells (Ohba et al. 1999; Pothula et al. 2016), while in Panc-1 cells, concentrations of HGF up to 0.5 nM had no effect (Ohba et al. 1999). Here we found that both EGF and HGF induced DNA synthesis in AsPC-1 as well as in Panc-1 cells (Fig. 2a, b).
Fig. 2.

Effect of p38 inhibition on EGF- and HGF-induced DNA synthesis in AsPC-1 (a) and Panc-1 (b) cells. Various doses of EGF or HGF was added to AsPC-1 (a) and Panc-1 (b) cells 30 min after adding SB203580 (10 μM) and DNA synthesis measured after 24 h of stimulation. The data represent the mean ± SEM of three independent experiments. * indicates p ≤ 0.05 and ** indicates p ≤ 0.01 (growth factor alone versus growth factor plus SB203580). # indicates p ≤ 0.05 and ## indicates p ≤ 0.01 (non-stimulated versus growth factor stimulated)
To investigate the possible role of the p38 pathway in the mitogenic signalling induced by EGF and HGF in the two pancreatic cancer cell lines, we inhibited p38 using 10 μM SB203580, which we previously found to completely block the activation of the p38 downstream target MAPKAP kinase-2 (Aasrum et al. 2016). Figure 2a shows that SB203580 reduced both EGF- and HGF-induced DNA synthesis in AsPC-1 cells. However, in marked contrast, SB203580 strongly enhanced the DNA synthesis in Panc-1 cells, and this effect consisted entirely of a large increase of the basal activity while the responses to EGF or HGF were not amplified (Fig. 2b). These results show that there are opposing effects of p38 on the regulation of DNA synthesis depending on the cell type.
Effect of MEK/ERK inhibition on DNA synthesis
We further examined the role of the MEK/ERK pathway in EGF- and HGF-induced DNA synthesis in AsPC-1 and Panc-1 cells, using the MEK inhibitor PD98059. As seen in Fig. 3a, the DNA synthesis induced by EGF or HGF in AsPC-1 cells was not affected significantly by PD98059. In contrast, in Panc-1 cells, both EGF- and HGF-induced DNA synthesis was reduced by PD98059 (Fig. 3b). Also the basal DNA synthesis was inhibited in the Panc-1 cells. These results suggest that the MEK/ERK pathway is involved in the upregulation of both basal and EGF- and HGF-induced DNA synthesis in Panc-1 cells, but not in AsPC-1.
Fig. 3.

Effect of MEK/ERK inhibition on EGF- and HGF-induced DNA synthesis. EGF (10 nM) or HGF (1 nM) was added to AsPC-1 (a) and Panc-1 (b) cells 30 min after adding PD98059 (20 μM) and DNA synthesis measured after 24 h of stimulation. The data represent the mean ± SEM of three independent experiments. * indicates p ≤ 0.05 and **indicates p ≤ 0.01 versus without PD98059
Effect of p38 inhibition on ERK phosphorylation
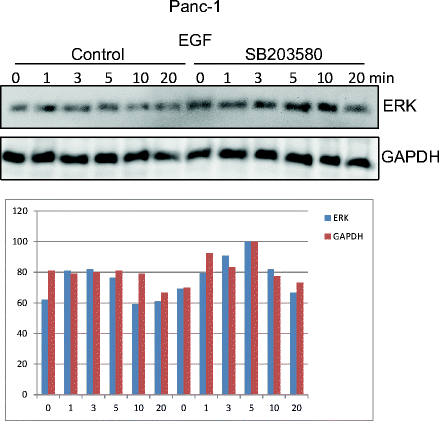
Findings in several cells indicate that a prolonged activation of ERK is important for cell cycle progression (Marshall 1995; Thoresen et al. 2003; Murphy and Blenis 2006; Aasrum et al. 2016).We previously found that in hepatocytes, p38 inhibition increased HGF-stimulated ERK phosphorylation, particularly the sustained response measured 24 h after exposure to HGF, and that this increase was involved in the enhancement of HGF-induced DNA synthesis (Aasrum et al. 2016). We here show that in AsPC-1 cells, p38 inhibition did not significantly affect the early phase (20 min) of ERK phosphorylation (Fig. 4a), whereas in Panc-1 cells, the responses to both EGF and HGF were moderately increased (Fig. 4b). This suggests that for the time course studied here, growth factor-induced p38 activation negatively affects the ERK pathway in Panc-1 cells but not in the AsPC-1 cells. However, to explore the role of a sustained ERK activity in the regulation of DNA synthesis by SB203580, we next examined the ERK phosphorylation 24 h after stimulation with EGF or HGF. As seen in Fig. 5a, no ERK phosphorylation was detected at 24 h in the Panc-1 cells, with or without SB203580. In contrast, in AsPC-1 cells, the sustained ERK phosphorylation in response to EGF and HGF was markedly increased by SB203580 (Fig. 5a, b). 3–4 times more protein was loaded for the Panc-1 cells, to clarify if there was an ERK signal. Compared to the loading of protein, the ERK signal is in Panc-1 can hardly be detected. These findings suggest that sustained ERK activity is not always induced by EGF and HGF and that this activity could play differential roles in different cell lines.
Fig. 4.

Effect of p38 inhibition on ERK phosphorylation. Immunoblots show ERK phosphorylation induced by 10 nM EGF and 1 nM HGF in AsPC-1 (a) and Panc-1 (b) with or without SB203580 (10 μM) pretreatment. The graphs show densitometric quantification of ERK phosphorylation based on three independent experiments ± SEM. pERK was normalized to the expression of GAPDH. * indicates p ≤ 0.05 versus growth factor without SB203580
Fig. 5.

Effect of p38 inhibition on the expression of p21. AsPC-1 and Panc-1 cells were stimulated by EGF (10 nM) or HGF (1 nM) for 24 h with or without SB203580 (10 μM) pretreatment for 30 min. (a) Immunoblots showing p21 expression and ERK phosphorylation. (b) Densitometric quantification of ERK phosphorylation in AsPC-1 cells based on three independent experiments. The level of pERK was normalized to total ERK. (c) Densitometric quantification of p21 expression in AsPC-1 and Panc-1 cells based on three independent experiments. p21 expression was normalized to the level of GAPDH. The data represent the mean ± SEM of three independent experiments. * indicates p ≤ 0.05, **indicates p ≤ 0.01 and *** indicates p ≤ 0.001 versus without SB203580
p38 inhibition upregulates the expression of p21 in AsPC-1 through an ERK-dependent mechanism
The cyclin-dependent kinase inhibitor p21 exerts inhibitory control of cell cycle progression (Abbas and Dutta 2009). In some cells, p21 expression has been shown to be positively regulated by the MEK/ERK pathway (Beier et al. 1999; Shirako et al. 2008). We found that in AsPC-1 cells, the increase of the sustained ERK phosphorylation in response to EGF or HGF in SB203580-treated cells was accompanied by upregulation of growth factor-induced p21 expression (Fig. 5a, c). In Panc-1 cells, there was a tendency for downregulation of p21 by SB203580.
This result could suggest that in AsPC-1 cells, p38 inhibition leads to an increase in the sustained ERK activity which positively regulates p21 expression. To determine to which extent the MEK/ERK pathway is involved in the upregulation of p21 expression in the presence of the p38 inhibitor in AsPC-1 cells, p21 expression was analysed in cells pretreated with both SB203580 and PD98059. As shown in Fig. 6a, b, PD98059 reduced the SB203580-mediated upregulation of p21 expression induced by EGF and HGF. This finding suggests that in the AsPC-1 cells, p21 expression is upregulated by a mechanism requiring sustained ERK activity.
Fig. 6.

Role of MEK/ERK in the upregulation of p21 expression in AsPC-1 cells. Cells were stimulated by EGF (10 nM) or HGF (1 nM) for 24 h with or without SB203580 (10 μM) and/or PD98059 (20 μM) pretreatment for 30 min. (a) Immunoblots showing p21 expression and ERK phosphorylation. (b) Densitometric quantification of p21 expression based on three independent experiments. p21 expression was normalized to the level of GAPDH. The data represent the mean ± SEM of three independent experiments. * indicates p ≤ 0.05
Discussion
The results of this report show that in two different pancreatic cancer cell lines, p38 acts differently in the regulation of mitogenic signalling. Thus, p38 affects EGF-and HGF-induced DNA synthesis negatively in Panc-1 but positively in AsPC-1 cells. We have previously reported that in hepatocytes, p38 may act both as an activator and an inhibitor of proliferation depending on the growth factor stimulus (Aasrum et al. 2016). Here we show that p38 can act as a negative or a positive regulator of growth factor- induced DNA synthesis, depending on the cell line.
In this study we have been using the MEK inhibitor PD98059 and the p38 inhibitor SB203580. Although these two kinase inhibitors generally are considered as highly specific, it is still worth to mention that caution must be taken when using such inhibitors, especially at high doses. SB203580 has been demonstrated to block a few other protein kinases including PI3K, GAC and CK1, but does not block other MAPKs (Bain et al. 2007). When it comes to PD98059, it has been demonstrated to inhibit MAPK kinase 5 (MKK5), but no unspecific effects on the p38 pathway has been shown (Dudley et al. 1995; Bain et al. 2007).
Generally, p38 has been considered as an inhibitor of cell proliferation, as documented in many cell types, including fibroblasts, cardiomyocytes and hepatocytes (Lavoie et al. 1996; Engel et al. 2005; Hui et al. 2007). The inhibitory effect of p38 on cell cycle progression at G1/S and G2/M transition involves several mechanisms, including upregulation of CDKIs and downregulation of cyclins (Ambrosino and Nebreda 2001; Bulavin and Fornace 2004; Thornton and Rincon 2009). However, several reports indicate that p38 may also act as a positive regulator of proliferation. In cytokine-stimulated hematopoietic cells and heregulin-stimulated breast cancer cells, p38 inhibition led to reduced cell cycle progression (Rausch and Marshall 1999; Neve et al. 2002). Also in HGF-stimulated melanoma cells, p38 positively affects proliferation. In these cells, p38 upregulates cyclin D1 expression through the activation of the transcription factor ATF-2 (Recio and Merlino 2002).
The role of p38 in the regulation of proliferation of pancreatic cancer cells has only briefly been studied previously. Especially, the role of p38 in growth factor-stimulated proliferation of these cells is poorly explored. One report has studied the role of p38 in EGF-stimulated proliferation of several pancreatic cancer cell lines, including AsPC-1 and Panc-1 (Matsuda et al. 2002). In contrast to our results, where we see an activating role of p38 in AsPC-1 and inhibition in Panc-1 cells, these authors found that p38 blockade using SB203580 inhibited EGF-induced cell growth of both AsPC-1 and Panc-1 cells. In that study, no effect of p38 inhibition was seen on basal DNA synthesis of Panc-1. We here show that SB203580 substantially upregulated the basal DNA synthesis in Panc-1. This result is in agreement with another report indicating that basal proliferation of Panc-1 cells is negatively regulated by p38 (Ding and Adrian 2001). Furthermore, in a variety of other pancreatic cancer cells, Zhong et al. found that cells with high basal levels of phosphorylated p38, had restrained growth (Zhong et al. 2014). Interestingly, they also showed that in patients with pancreatic cancer, a strong immunohistochemical labelling in the tumours significantly correlated with improved post-resection survival. Since the Panc-1 and AsPC-1 cells showed quite similar levels of phosphorylated p38 in the unstimulated state, this most likely could not explain the differential role of p38 seen in our experiments.
The p38 pathway may crosstalk with several other signalling pathways (Cuadrado and Nebreda 2010). With respect to the MEK/ERK pathway, p38 may inhibit ERK activation by various mechanisms (Liu and Hofmann 2004; Junttila et al. 2008). Inhibitory effects of p38 on ERK activity have been demonstrated in various cells, including cardiac myocytes, hepatoma cells, and both human and rat hepatocytes (Singh et al. 1999; Liu and Hofmann 2004; Henklova et al. 2008; Aasrum et al. 2016). In primary hepatocytes, we have previously shown that p38 inhibition leads to an increase in HGF-induced ERK phosphorylation, and this increase may partly account for the increase seen in HGF-induced DNA synthesis when p38 is inhibited (Aasrum et al. 2016). Here we see a quite similar phenomenon in both EGF- and HGF-stimulated Panc-1 cells. In the presence of SB203580, the early phase of the growth factor-induced ERK phosphorylation and the DNA synthesis was increased. We also demonstrate that the MEK/ERK pathway was involved in the regulation of EGF-and HGF-induced DNA synthesis in the Panc-1 cells. In contrast, in the AsPC-1 cells, SB203580 did not affect the early phase of ERK phosphorylation, in agreement with the finding that the MEK/ERK pathway did not play a role in EGF- and HGF-induced DNA synthesis.
Since we previously have shown that a sustained ERK signal is required for the induction of DNA synthesis in hepatocytes, and that p38 inhibition may increase the sustained ERK activity (Thoresen et al. 2003; Aasrum et al. 2016), we speculated if sustained ERK activity was involved in the strong increase in DNA synthesis seen in Panc-1 cells in the presence of SB203580. Surprisingly, no sustained ERK phosphorylation was seen in the Panc-1 cells at 24 h in the absence or presence of SB203580. In contrast, SB203580-treated AsPC-1 cells showed an increase in the sustained ERK phosphorylation, associated with an increase in the expression of p21. The CDK inhibitor p21 is a protein that exerts cell cycle arrest in response to a variety of stimuli (Abbas and Dutta 2009). p21 transcription may be regulated by several pathways, both p53-dependent and -independent (Jung et al. 2010). Since in AsPC-1 cells, MEK inhibition reduced the EGF- and HGF-induced expression of p21 seen in the presence of SB203580 in AsPC-1 cells, we suggest that p21 expression was upregulated due to a sustained ERK signal when p38 was inactivated. Several reports have previously suggested a role for the MEK/ERK pathway in the regulation of p21 expression. In for example chondrocytes, MEK inhibition reduces the level of p21 expression (Beier et al. 1999), whereas in HepG2 cells, both ERK-dependent and ERK independent pathways were found to be involved in the expression of p21 (Shirako et al. 2008). Especially, long-term ERK activation has been demonstrated to promote accumulation of p21 (Chambard et al. 2007). In addition, it is suggested that various cell types may need a different threshold of ERK activity to affect the balance between proliferation and growth arrest, and that intense ERK activation proves to be essential for p21 upregulation (Chambard et al. 2007). That p38 negatively affects the expression of p21 has previously not been shown. In contrast, p38 activity has been demonstrated to induce p21 expression in CHO-K1 cells (Chinese hamster ovary) (Alderton et al. 2001) and to increase the stability of p21 in HD3 cells (human colon carcinoma) (Kim et al. 2002).
In conclusion, we have demonstrated that p38 may affect EGF- and HGF-induced DNA synthesis positively or negatively, depending on the cell line. The cross-talk between the p38 and MEK/ERK pathways may at least partially explain this phenomenon. When p38 is inactivated, an increase in the early phase ERK activity may positively affect DNA synthesis in Panc-1 cells, whereas in AsPC-1 cells, an increase in the sustained ERK activity upregulates p21 expression, which in turn negatively may affect DNA synthesis.
Electronic supplementary material
{kind=link}
(GIF 31 kb)
References
- Aasrum M, Odegard J, Sandnes D, Christoffersen T. The involvement of the docking protein Gab1 in mitogenic signalling induced by EGF and HGF in rat hepatocytes. Biochim Biophys Acta. 2013;1833:3286–3294. doi: 10.1016/j.bbamcr.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Aasrum M, Brusevold IJ, Christoffersen T, Thoresen GH. HGF-induced DNA synthesis in hepatocytes is suppressed by p38. Growth Factors. 2016;3:217–223. doi: 10.1080/08977194.2017.1285765. [DOI] [PubMed] [Google Scholar]
- Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderton F, Humphrey PP, Sellers LA. High-intensity p38 kinase activity is critical for p21(cip1) induction and the antiproliferative function of G(i) protein-coupled receptors. Mol Pharmacol. 2001;59:1119–1112. doi: 10.1124/mol.59.5.1119. [DOI] [PubMed] [Google Scholar]
- Ambrosino C, Nebreda AR. Cell cycle regulation by p38 MAP kinases. Biol Cell. 2001;93:47–51. doi: 10.1016/S0248-4900(01)01124-8. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, Mclauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier F, Taylor AC, Luvalle P. The Raf-1/MEK/ERK pathway regulates the expression of the p21(Cip1/Waf1) gene in chondrocytes. J Biol Chem. 1999;274:30273–30279. doi: 10.1074/jbc.274.42.30273. [DOI] [PubMed] [Google Scholar]
- Brusevold IJ, Aasrum M, Bryne M, Christoffersen T. Migration induced by epidermal and hepatocyte growth factors in oral squamous carcinoma cells in vitro: role of MEK/ERK, p38 and PI-3 kinase/Akt. J Oral Pathol Med. 2012;41:547–558. doi: 10.1111/j.1600-0714.2012.01139.x. [DOI] [PubMed] [Google Scholar]
- Bulavin DV, Fornace AJ., Jr p38 MAP kinase's emerging role as a tumor suppressor. Adv Cancer Res. 2004;92:95–118. doi: 10.1016/S0065-230X(04)92005-2. [DOI] [PubMed] [Google Scholar]
- Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta. 2007;1773:1299–1310. doi: 10.1016/j.bbamcr.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Chen L, He HY, Li HM, Zheng J, Heng WJ, You JF, Fang WG. ERK1/2 and p38 pathways are required for P2Y receptor-mediated prostate cancer invasion. Cancer Lett. 2004;215:239–247. doi: 10.1016/j.canlet.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Chen J, Amos CI, Merriman KW, Wei Q, Sen S, Killary AM, Frazier ML. Genetic variants of p21 and p27 and pancreatic cancer risk in non-Hispanic Whites: a case-control study. Pancreas. 2010;39:1–4. doi: 10.1097/MPA.0b013e3181bd51c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- Deer EL, Gonzalez-Hernandez J, Coursen JD, Shea JE, Ngatia J, Scaife CL, Firpo MA, Mulvihill SJ. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425–435. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding XZ, Adrian TE. MEK/ERK-mediated proliferation is negatively regulated by P38 map kinase in the human pancreatic cancer cell line, PANC-1. Biochem Biophys Res Commun. 2001;282:447–453. doi: 10.1006/bbrc.2001.4595. [DOI] [PubMed] [Google Scholar]
- Dolado I, Nebreda AR. Regulation of tumorigenesis by p38α MAP kinase. In: Posas F, Nebreda AR, editors. Stress-activated protein kinases. Berlin: Springer-Verlag; 2007. pp. 99–128. [Google Scholar]
- Donadelli M, Dalla Pozza E, Costanzo C, Scupoli MT, Piacentini P, Scarpa A, Palmieri M. Increased stability of P21(WAF1/CIP1) mRNA is required for ROS/ERK-dependent pancreatic adenocarcinoma cell growth inhibition by pyrrolidine dithiocarbamate. Biochim Biophys Acta. 2006;1763:917–926. doi: 10.1016/j.bbamcr.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Dreissigacker U, Mueller MS, Unger M, Siegert P, Genze F, Gierschik P, Giehl K. Oncogenic K-Ras down-regulates Rac1 and RhoA activity and enhances migration and invasion of pancreatic carcinoma cells through activation of p38. Cell Signal. 2006;18:1156–1168. doi: 10.1016/j.cellsig.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunne RF, Hezel AF. Genetics and biology of pancreatic ductal adenocarcinoma. Hematol Oncol Clin North Am. 2015;29:595–608. doi: 10.1016/j.hoc.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–1187. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakilas AG, Martin OA, Bonner WM. p21: a two-faced genome guardian. Trends Mol Med. 2017;23:310–319. doi: 10.1016/j.molmed.2017.02.001. [DOI] [PubMed] [Google Scholar]
- Gohda E, Yamasaki T, Tsubouchi H, Kurobe M, Sakiyama O, Aoki H, Niidani N, Shin S, Hayashi K, Hashimoto S, et al. Biological and immunological properties of human hepatocyte growth factor from plasma of patients with fulminant hepatic failure. Biochim Biophys Acta. 1990;1053:21–26. doi: 10.1016/0167-4889(90)90020-E. [DOI] [PubMed] [Google Scholar]
- Halawani D, Mondeh R, Stanton LA, Beier F. p38 MAP kinase signaling is necessary for rat chondrosarcoma cell proliferation. Oncogene. 2004;23:3726–3731. doi: 10.1038/sj.onc.1207422. [DOI] [PubMed] [Google Scholar]
- Henklova P, Vrzal R, Papouskova B, Bednar P, Jancova P, Anzenbacherova E, Ulrichova J, Maurel P, Pavek P, Dvorak Z. SB203580, a pharmacological inhibitor of p38 MAP kinase transduction pathway activates ERK and JNK MAP kinases in primary cultures of human hepatocytes. Eur J Pharmacol. 2008;593:16–23. doi: 10.1016/j.ejphar.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Hui L, Bakiri L, Stepniak E, Wagner EF. p38alpha: a suppressor of cell proliferation and tumorigenesis. Cell Cycle. 2007;6:2429–2433. doi: 10.4161/cc.6.20.4774. [DOI] [PubMed] [Google Scholar]
- Jia D, Sun Y, Konieczny SF. Mist1 regulates pancreatic acinar cell proliferation through p21 CIP1/WAF1. Gastroenterology. 2008;135:1687–1697. doi: 10.1053/j.gastro.2008.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YS, Qian Y, Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal. 2010;22:1003–1012. doi: 10.1016/j.cellsig.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22:954–965. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388:73–85. doi: 10.1016/S0140-6736(16)00141-0. [DOI] [PubMed] [Google Scholar]
- Kim GY, Mercer SE, Ewton DZ, Yan Z, Jin K, Friedman E. The stress-activated protein kinases p38 alpha and JNK1 stabilize p21(Cip1) by phosphorylation. J Biol Chem. 2002;277:29792–29802. doi: 10.1074/jbc.M201299200. [DOI] [PubMed] [Google Scholar]
- Kim MS, Lee EJ, Kim HR, Moon A. p38 kinase is a key signaling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res. 2003;63:5454–5461. [PubMed] [Google Scholar]
- Lavoie JN, L'allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996;271:20608–20616. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- Liu Q, Hofmann PA. Protein phosphatase 2A-mediated cross-talk between p38 MAPK and ERK in apoptosis of cardiac myocytes. Am J Physiol Heart Circ Physiol. 2004;286:H2204–H2212. doi: 10.1152/ajpheart.01050.2003. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Matsuda K, Idezawa T, You XJ, Kothari NH, Fan H, Korc M. Multiple mitogenic pathways in pancreatic cancer cells are blocked by a truncated epidermal growth factor receptor. Cancer Res. 2002;62:5611–5617. [PubMed] [Google Scholar]
- Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006;31:268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Nebreda AR, Porras A. p38 MAP kinases: beyond the stress response. Trends Biochem Sci. 2000;25:257–260. doi: 10.1016/S0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- Neve RM, Holbro T, Hynes NE. Distinct roles for phosphoinositide 3-kinase, mitogen-activated protein kinase and p38 MAPK in mediating cell cycle progression of breast cancer cells. Oncogene. 2002;21:4567–4576. doi: 10.1038/sj.onc.1205555. [DOI] [PubMed] [Google Scholar]
- Ohba N, Funatomi H, Seki T, Makino R, Mitamura K. Hepatocyte growth factor stimulates cell growth and enhances the expression of transforming growth factor alpha mRNA in AsPC-1 human pancreatic cancer cells. J Gastroenterol. 1999;34:498–504. doi: 10.1007/s005350050303. [DOI] [PubMed] [Google Scholar]
- Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/S0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- Pei XH, Xiong Y. Biochemical and cellular mechanisms of mammalian CDK inhibitors: a few unresolved issues. Oncogene. 2005;24:2787–2795. doi: 10.1038/sj.onc.1208611. [DOI] [PubMed] [Google Scholar]
- Pothula SP, Xu Z, Goldstein D, Biankin AV, Pirola RC, Wilson JS, Apte MV. Hepatocyte growth factor inhibition: a novel therapeutic approach in pancreatic cancer. Br J Cancer. 2016;114:269–280. doi: 10.1038/bjc.2015.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch O, Marshall CJ. Cooperation of p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways during granulocyte colony-stimulating factor-induced hemopoietic cell proliferation. J Biol Chem. 1999;274:4096–4105. doi: 10.1074/jbc.274.7.4096. [DOI] [PubMed] [Google Scholar]
- Recio JA, Merlino G. Hepatocyte growth factor/scatter factor activates proliferation in melanoma cells through p38 MAPK, ATF-2 and cyclin D1. Oncogene. 2002;21:1000–1008. doi: 10.1038/sj.onc.1205150. [DOI] [PubMed] [Google Scholar]
- Refsnes M, Thoresen GH, Dajani OF, Christoffersen T. Stimulation of hepatocyte DNA synthesis by prostaglandin E2 and prostaglandin F2 alpha: additivity with the effect of norepinephrine, and synergism with epidermal growth factor. J Cell Physiol. 1994;159:35–40. doi: 10.1002/jcp.1041590106. [DOI] [PubMed] [Google Scholar]
- Ricote M, Garcia-Tunon I, Bethencourt F, Fraile B, Onsurbe P, Paniagua R, Royuela M. The p38 transduction pathway in prostatic neoplasia. J Pathol. 2006;208:401–407. doi: 10.1002/path.1910. [DOI] [PubMed] [Google Scholar]
- Shirako E, Hirayama N, Tsukada Y, Tanaka T, Kitamura N. Up-regulation of p21CIP1 expression mediated by ERK-dependent and -independent pathways contributes to hepatocyte growth factor-induced inhibition of HepG2 hepatoma cell proliferation. J Cell Biochem. 2008;104:176–188. doi: 10.1002/jcb.21614. [DOI] [PubMed] [Google Scholar]
- Singh RP, Dhawan P, Golden C, Kapoor GS, Mehta KD. One-way cross-talk between p38(MAPK) and p42/44(MAPK). Inhibition of p38(MAPK) induces low density lipoprotein receptor expression through activation of the p42/44(MAPK) cascade. J Biol Chem. 1999;274:19593–19600. doi: 10.1074/jbc.274.28.19593. [DOI] [PubMed] [Google Scholar]
- Stolz DB, Michalopoulos GK. Comparative effects of hepatocyte growth factor and epidermal growth factor on motility, morphology, mitogenesis, and signal transduction of primary rat hepatocytes. J Cell Biochem. 1994;55:445–464. doi: 10.1002/jcb.240550405. [DOI] [PubMed] [Google Scholar]
- Thoresen GH, Guren TK, Christoffersen T. Role of ERK, p38 and PI3-kinase in EGF receptor-mediated mitogenic signalling in cultured rat hepatocytes: requirement for sustained ERK activation. Cell Physiol Biochem. 2003;13:229–238. doi: 10.1159/000072426. [DOI] [PubMed] [Google Scholar]
- Thornton TM, Rincon M. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int J Biol Sci. 2009;5:44–51. doi: 10.7150/ijbs.5.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tveteraas IH, Aasrum M, Brusevold IJ, Odegard J, Christoffersen T, Sandnes D. Lysophosphatidic acid induces both EGFR-dependent and EGFR-independent effects on DNA synthesis and migration in pancreatic and colorectal carcinoma cells. Tumour Biol. 2016;37:2519–2526. doi: 10.1007/s13277-015-4010-1. [DOI] [PubMed] [Google Scholar]
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- Wiseman DA, Werner SR, Crowell PL. Cell cycle arrest by the isoprenoids perillyl alcohol, geraniol, and farnesol is mediated by p21(Cip1) and p27(Kip1) in human pancreatic adenocarcinoma cells. J Pharmacol Exp Ther. 2007;320:1163–1170. doi: 10.1124/jpet.106.111666. [DOI] [PubMed] [Google Scholar]
- Xu Y, Li N, Xiang R, Sun P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends Biochem Sci. 2014;39:268–276. doi: 10.1016/j.tibs.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Naito Y, Cope L, Naranjo-Suarez S, Saunders T, Hong SM, Goggins MG, Herman JM, Wolfgang CL, Iacobuzio-Donahue CA. Functional p38 MAPK identified by biomarker profiling of pancreatic cancer restrains growth through JNK inhibition and correlates with improved survival. Clin Cancer Res. 2014;20:6200–6211. doi: 10.1158/1078-0432.CCR-13-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(GIF 31 kb)