

Abstract
Smoking is the leading cause of preventable death worldwide. Though cigarette smoke is an established cause of head and neck cancer (including oral cancer), molecular alterations associated with chronic cigarette smoke exposure are poorly studied. To understand the signaling alterations induced by chronic exposure to cigarette smoke, we developed a cell line model by exposing normal oral keratinocytes to cigarette smoke for a period of 12 months. Chronic exposure to cigarette smoke resulted in increased cellular proliferation and invasive ability of oral keratinocytes. Proteomic and phosphoproteomic analyses showed dysregulation of several proteins involved in cellular movement and cytoskeletal reorganization in smoke exposed cells. We observed overexpression and hyperphosphorylation of protein kinase N2 (PKN2) in smoke exposed cells as well as in a panel of head and neck cancer cell lines established from smokers. Silencing of PKN2 resulted in decreased colony formation, invasion and migration in both smoke exposed cells and head and neck cancer cell lines. Our results indicate that PKN2 plays an important role in oncogenic transformation of oral keratinocytes in response to cigarette smoke. The current study provides evidence that PKN2 can act as a potential therapeutic target in head and neck squamous cell carcinoma, especially in patients with a history of smoking.
Electronic supplementary material
The online version of this article (10.1007/s12079-017-0442-2) contains supplementary material, which is available to authorized users.
Keywords: Orbitrap fusion, High-throughput, Carcinogenesis, Smoking, Cell adhesion
Introduction
Long term cigarette smoking is epidemiologically linked to development of various cancers (Ezzati et al. 2005). Cigarette smoke exposure has been associated with mutations in tumor suppressor genes, activation of oncogenic drivers and methylation changes in various cancers including oral cancer (Pfeifer et al. 2002; Chen et al. 2011). In addition, carcinogenic constituents of mainstream cigarette smoke such as polyaromatic hydrocarbons (PAHs) including Benzo[a]pyrene (BaP), 7,12-Dimethylbenz(a)anthracene (DMBA) and nitrosamines are associated with activation of oncogenic and survival pathways in different cell types such as breast, liver, lung and head and neck cells (Hecht 1998; Currier et al. 2005; Hardonniere et al. 2016; Chen et al. 2005).
The adverse effects of cigarette smoke are observed upon chronic rather than acute exposure in oral cells. However, only a few in vitro studies have focused on chronic exposure of cigarette smoke or its constituents in oral cells. Chronic exposure with cigarette smoke extract for up to 7 months in oral keratinocytes was seen to select for survival cues such as apoptotic dysfunction and activation of aerobic glycolysis with HIF1α accumulation (Chang et al. 2010; Sun et al. 2011). Another study documents the malignant transformation of an established dysplastic oral leukoplakia cell line upon intermittent treatment with a mixture of B(a)P and DMBA. Results from proteomics analysis in this study showed enrichment of pathways such as signaling by Rho family of GTPases in transformed cells compared to untreated cells (Dong et al. 2015).
Rho family of GTPases have been linked to increased cellular migration in various cancers (Parri and Chiarugi 2010). In addition, limited studies indicate a link between cigarette smoke exposure and signaling downstream of Rho/Rac proteins (Olivera et al. 2007). Cigarette smoke exposure in non-small cell lung cancer cells was shown to activate PAK6 expression which was directly associated with cellular motility, survival and cellular transformation (Raja et al. 2016). Signaling downstream of Rho family of GTPases has also been linked to cigarette smoke–induced tumorigenesis in human bronchial epithelial cells (Zhang et al. 2013).
Global proteomics and phosphoproteomics can aid in identifying dysregulated signaling modules in both cellular models and primary tissue samples. To the best of our knowledge there are no high throughput studies documenting the adverse effects of chronic cigarette smoke exposure on normal oral keratinocytes. We employed a high-throughput mass spectrometry-based approach to study the proteomic and phosphoproteomic alterations in normal oral keratinocytes upon chronic exposure to cigarette smoke. Our data indicate dysregulation of vital cellular processes including cell adhesion and cytoskeletal reorganization in oral cells upon exposure to cigarette smoke. In conjunction with previous studies, we observed an activation of signaling downstream of Rho GTPases through aberrant activation of protein kinase N2 (PKN2) in smoke exposed cells. We studied the role of PKN2 and its interactors in oncogenic transformation of oral cells chronically exposed to cigarette smoke.
Materials and methods
Cell culture and adaptation of normal oral keratinocytes to cigarette smoke condensate
Normal human oral keratinocytes, OKF6/TERT1, were generously gifted by Dr. James Rheinwald (Brigham and Women’s Hospital, Boston, MA). OKF6/TERT1 were grown and maintained in keratinocyte serum free medium (KSFM) (supplemented with bovine pituitary extract (25 μg/ml), epidermal growth factor (EGF) (0.2 ng/ml) (ThermoFisher Scientific, MA)), 1% penicillin/streptomycin and calcium chloride (0.4 mM). The cells were cultured at 37 °C in a humidified air incubator with 5% CO2.
FaDu and CAL 27 were procured from ATCC. JHU-O11, JHU-O22, JHU-O29 and FaDu were cultured in RPM1–1640 media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. CAL 27 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. All cell lines were grown in a humidified incubator with 5% CO2 at 37 °C. The cell lines used for the study were authenticated by short tandem repeat analysis at the Genetic Resources Core Facility of Johns Hopkins University School of Medicine.
Cigarette smoke condensate (CSC) was purchased from Murty Pharmaceuticals, Inc., KY. OKF6/TERT1 cells were cultured in the presence of CSC in a dedicated smoke incubator as described previously (Chang et al. 2010). Briefly, cells were treated with 0.1% CSC chronically for a period of 12 months. In the study, OKF6/TERT1 cells exposed to CSC have been referred to as OKF6/TERT1-Smoke cells. OKF6/TERT1 cells cultured in a regular incubator without any exposure to CSC have been referred to as OKF6/TERT1-Parental cells.
Sample preparation for mass spectrometric analysis
OKF6/TERT1-Parental and OKF6/TERT1-Smoke cells were grown to 80% confluence, serum starved for 8 h, washed with 1X PBS thrice and harvested in lysis buffer (2% SDS, 5 mM sodium fluoride, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate in 50 mM Triethyl ammonium bicarbonate (TEABC)). The cell lysates were sonicated, centrifuged and protein concentration was determined by BCA(Walker 1994).
In-solution digestion and TMT labeling
Equal amounts of cell lysate from all conditions were reduced using 5 mM dithiothreitol (DTT) and incubated at 60 °C for 20 mins. The reduced protein lysate was alkylated using iodoacetamide (IAA) (20 mM) and incubated for 10 mins in the dark at room temperature. We employed filter aided sample preparation (FASP) protocol as described earlier to remove SDS (Kim et al. 2014). Proteins were then subjected to trypsin digestion using sequencing grade trypsin (Promega, Madison, WI) at an enzyme to substrate ratio of 1:20. Trypsin digestion was carried out at 37 °C for 16 h. The digested peptides were lyophilized and labeled with TMT reagents as per manufacturers’ instructions. Briefly, peptide samples were dissolved in 50 mM TEABC (pH 8.0) and added to TMT reagents dissolved in anhydrous acetonitrile. Peptides from OKF6/TERT1-Parental were labeled with TMT tags 126 and 127 and OKF6/TERT1-Smoke cells were labeled with TMT tags 128 and 130. After incubation at room temperature for 1 h, the reaction was quenched with 5% hydroxylamine.
Basic reversed-phase liquid chromatography (bRPLC) and phosphopeptides enrichment using titanium dioxide (TiO2)
The labeled peptides were subjected to bRPLC fractionation generating 96 fractions, as described previously (Selvan et al. 2014). The fractions were concatenated into 12 fractions. From these pooled fractions, one-tenth volume equivalent peptides were taken for total proteome analysis. All fractions were lyophilized and subjected to TiO2-based enrichment (Radhakrishnan et al. 2016). The enriched and desalted peptide samples were further subjected to mass spectrometry analysis. LC-MS2 analysis was performed on Thermo Scientific™ Orbitrap Fusion™ Tribrid™ Mass Spectrometer.
LC-MS/MS analysis
A total of 12 bRPLC fractions were reconstituted in 0.1% formic acid and analyzed on Orbitrap Fusion™ Tribrid™ mass spectrometer (Thermo Electron, Bremen, Germany) interfaced with Easy-nLC 1000 nanoflow liquid chromatography system (Thermo Scientific, Odense, Denmark). Reconstituted peptides were initially loaded onto trap column packed (75 μm × 2 cm) with Magic C18 AQ (Michrom Bioresources, Inc., Auburn, CA) at a flow rate of 3 μL/min. Peptides were analyzed on mass spectrometer by resolving them on an analytical column (75 μm × 20 cm) at a flow rate of 300 nL/min using a gradient of 10% - 35% solvent B (0.1% formic acid in 95% acetonitrile) over 100 min. The total run time was set to 120 min. Survey scan MS (from m/z 400–1600) was acquired in time-dependent acquisition mode in the Orbitrap with resolution of 120,000 at 200 m/z. The AGC target was set to 200,000 with ion injection time of 100 ms. Precursor ions with charge 2–5 were considered for MS2 analysis and dynamic exclusion of fragmented precursor ion was enabled for 30s. Precursor ionswere isolated using Quadrupole mass filter (isolation width = 2 m/z) and subjected to HCD fragmentation mode with 32%NCE. Fragment ions were analyzed in Orbitrap using 100–2000 m/z scan range at 30 K resolution. AGC target of 50,000 and 200 ms maximun injection time was used for the MS2 scans. Internal calibration was done using lock-mass from ambient air (m/z 445.1200025). Phosphopeptide enriched fractions were analyzed using the same method described above.
Data analysis
Mass spectrometry data obtained from LC-MS analysis was searched against Human RefSeq70 database using Sequest and Mascot (version 2.4.1) search algorithms through Proteome Discoverer 2.0 (Thermo Scientific, Bremen, Germany). Enzyme specificity was set as trypsin with maximum one missed cleavage allowed. The minimum peptide length was specified to be 6 amino acids. Carbamidomethylation of cysteine and TMT modification at N-terminus of the peptide and at lysine (K) were specified as fixed modifications and oxidation of methionine was included as variable modification. For phosphoproteomic data, phosphorylation of serine, threonine, and tyrosine was considered as additional variable modifications. The mass error of parent ions was set to 10 ppm and 0.05 Da for fragment ions. The data was also searched against a decoy database and MS/MS identifications of <1% false discovery rate (FDR) score threshold was considered for further analysis.
Bioinformatics analysis
Information on the subcellular localization, molecular function, molecular class and biological processes of quantified proteins was obtained from Human Protein Reference Database(Goel et al. 2011) (HPRD; http://www.hprd.org). Ingenuity Pathway Analysis (version 31,813,283) (Qiagen, USA) was employed to study the interaction networks of proteins dysregulated and/or differentially phosphorylated (≥2 fold) in smoke exposed cells compared to control cells. Interaction networks were generated for Homo sapiens.
siRNA transfection
ON-TARGETplus SMARTpool control siRNA and PKN2 siRNA were obtained from Dharmacon (Lafayette, CO). OKF6/TERT1-Smoke, JHU-O11, JHU-O22, JHU-O29, FaDu and CAL 27 cells were transfected using RNAiMAX (Invitrogen, Grand Island, NY) following manufacturer’s instructions. Transfection was carried out as previously described (Chang et al. 2011). Cells were subjected to invasion assays and colony formation assays 48 h post-transfection.
Cellular proliferation assays
OKF6/TERT1-Smoke and OKF6/TERT1-Parental cells were seeded at a density of 25 × 103 cells per well in triplicate in 6-well plates. Cellular proliferation was monitored for 8 days where the cells were counted using trypan blue exclusion method.
OKF6/TERT1-Smoke, JHU-O11, JHU-O22, JHU-O29, FaDu and CAL 27 cells were seeded at a density of 4 × 103 cells per well in quadruplicate in 96-well plates. Cells were transfected with control or PKN2 siRNA. Cellular proliferation was monitored for 4 days as described previously (Chatterjee et al. 2006).
Western blot analysis
All cells were grown to 70% confluence and washed thrice with ice-cold 1X phosphate buffer saline (PBS). Following this, cells were harvested in RIPA lysis buffer (10 mM Tris pH 7.4, 150 mM NaCl, 5 mM Ethylenediaminetetraacetic acid (EDTA), 1% Triton-X-100, 0.1% Sodium Dodecyl Sulfate (SDS) containing protease and phosphatase inhibitor cocktails) and protein concentration was determined using Bicinchoninic acid (BCA) assays. Western blot analysis was carried out as described previously (Nanjappa et al. 2015). Briefly, 30 μg of the cell lysate was resolved by SDS-PAGE and transferred onto nitrocellulose membrane. The membrane was probed with primary antibodies and incubated overnight as per manufacturer’s instructions. Membranes were then probed with HRP-conjugated anti-mouse or anti-rabbit secondary antibodies (Santa Cruz Biotechnology, Dallas, TX). Proteins on the membrane were visualized using enhanced chemiluminescence detection kit as per manufacturer’s instructions. PRK2 (PKN2) and DSC3 antibodies were obtained from Santa Cruz (Santa Cruz Biotechnology, Dallas, TX). Phospho-antibodies for CD44 (S706) and PRK3 + PRK2 + PRK1 (T718 + T774 + T816) were procured from Abcam (Abcam, USA). Antibody for CD44, was purchased from Cell Signaling (Cell Signaling Technology, Danvers, MA). β-actin antibody was procured from Sigma (Sigma Aldrich, USA) and was used as loading control for all Western blots.
Cellular invasion assays
The invasive ability of OKF6/TERT1-Smoke cells and HNSCC cells was investigated using a transwell system (BD Biosciences, San Jose, CA) with Matrigel (BD Biosciences, San Jose, CA) coated filters. Cellular invasion was evaluated after 48 h. Briefly, invasiveness of the cells was assayed in the membrane invasion culture system using polyethylene terephthalate (PET) membrane (8-μm pore size) (BD Biosciences, San Jose, CA) coated with Matrigel. The cells were seeded at a density of 2.0 × 104 in 500 μl of serum free media on the Matrigel-coated PET membrane in the upper compartment and placed in a compartment filled with complete growth media. All plates were incubated at 37 °C for 48 h. Post incubation, upper surface of the membrane was wiped with a cotton-tip applicator to remove non-migratory cells. Cells that migrated to the lower side of the membrane were fixed and stained using 4% methylene blue. All experiments were performed in triplicate. Representative images were photographed at 10× magnification.
Colony formation assays
Colony formation assays were carried out as described previously (Radhakrishnan et al. 2016). Briefly, OKF6/TERT1-Smoke or HNSCC cells were seeded in triplicate at a density of 3 × 103, transfected with control or PKN2 siRNA. The resulting colonies were fixed with methanol, stained with 4% methylene blue and counted. The number of colonies formed was counted for ten randomly selected viewing fields and representative images were photographed at 3× magnification. All experiments were performed in triplicate.
Scratch wound assays
The wound migration assays were performed as described previously (Cormier et al. 2015). Briefly, the cells were seeded in 6-well dishes, allowed to grow till 90% confluence and multiple, uniform size scratch wounds were introduced using sterile 200 μl pipette tip. OKF6/TERT1-Smoke and HNSCC cells were transfected with control or PKN2 siRNA. The wound photomicrographs at 0 and 10 h were taken at 10× magnification. Average change in migration area was calculated for control and siRNA transfected cells and plotted. All experiments were performed in triplicate unless otherwise indicated.
Data availability
Mass spectrometric data generated in this study was submitted to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Vizcaino et al. 2013) with the dataset identifier PXD007637.
Results
We developed a cellular model of chronic smoke exposure with normal oral keratinocytes, OKF6/TERT1 where cells were exposed to cigarette smoke for duration of 12 months. Cells were studied for cellular and molecular changes induced upon chronic exposure to cigarette smoke.
Chronic exposure to cigarette smoke induces phenotypic changes in oral keratinocytes
Chronic exposure to cigarette smoke resulted in changes to cellular morphology of OKF6/TERT1 cells (Fig. 1a). OKF6/TERT1-Smoke cells were more elongated compared to parental cells. We also observed a number of phenotypic changes associated with cancer hallmarks in smoke exposed cells compared to parental cells. Cigarette smoke treatment resulted in an increase in rate of cellular proliferation in OKF6/TERT1-Smoke cells (Fig. 1b). Oncogenic transformation results in sustained proliferative growth of cells. We performed colony formation assays to evaluate these changes in smoke exposed compared to parental cells. Invasion assays are often used to investigate metastatic potential in transformed cells. Scratch wound assays reflect migratory pattern of cells in a monolayer which cannot be studied in invasion assays. Studies have previously discussed the development of collective migration phenotype in squamous carcinomas of epithelial origin (Rorth 2009). We observe the same in smoke exposed oral cells. As seen in Fig. 1c, d and e, smoke exposed cells displayed increased invasiveness and cell scattering compared to OKF6/TERT1-Parental cells. In addition, we also observed a distinct change in the migratory pattern displayed by OKF6/TERT1 cells upon chronic exposure to cigarette smoke (Fig. 1f).
Fig. 1. Chronic exposure to cigarette smoke induces phenotypic changes in oral keratinocytes.

a Cellular morphology of OKF6/TERT1-Parental cells and OKF6/TERT1-Smoke cells (magnification 20X) (b) Growth curve depicting cellular proliferation rates of OKF6/TERT1-Parental and OKF6/TERT1-Smoke cells. c Transwell-based invasion assays were performed using Matrigel-coated chambers where number of cells that invade into the lower chamber were visualized (10× magnification) with methylene blue staining in OKF6/TERT1-Parental and OKF6/TERT1-Smoke cells. d Invaded cells were counted and relative changes in invasive ability of OKF6/TERT1-Parental and OKF6/TERT1-Smoke cells were calculated and represented graphically (***p < 0.0001). e Colony formation assay of smoke exposed and parental cells visualized (3× magnification) after staining with methylene blue. f Wound migration assays were carried out using OKF6/TERT1-Smoke and parental cells between 0 and 10 h. Cells were imaged at 10× magnification
These phenotypic changes indicate a change in cellular signaling upon smoke exposure. To understand the altered signaling induced by cigarette smoke we studied the proteomic and phosphoproteomic changes in OKF6/TERT1-Smoke cells compared to OKF6/TERT1-Parental cells.
Cigarette smoke exposure results in global proteomic and phosphoproteomic changes in oral keratinocytes
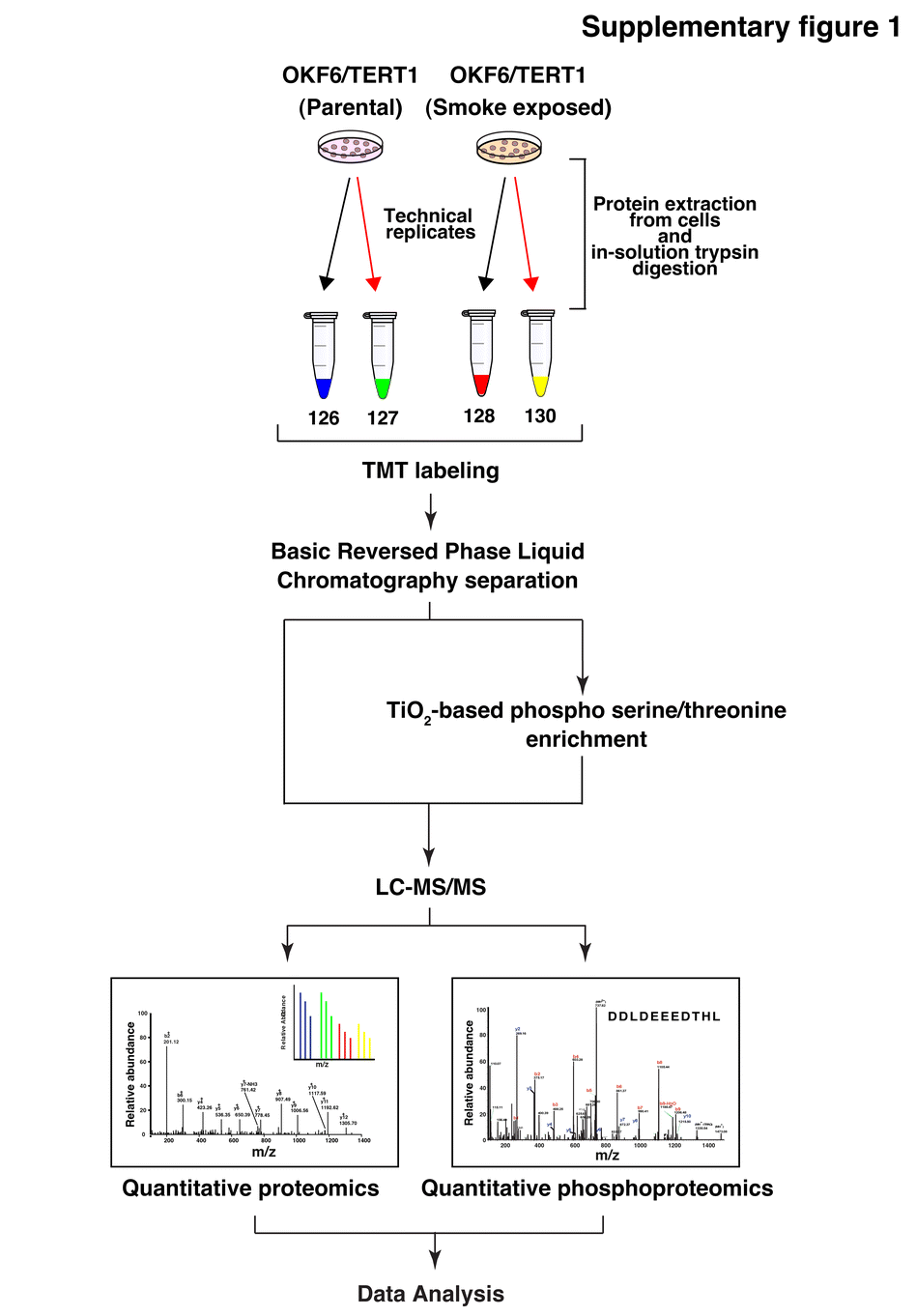
We employed quantitative Tandem Mass Tag (TMT)-based proteomic and phosphoproteomic approaches to better understand the molecular alterations induced by cigarette smoke in oral keratinocytes. The experimental strategy is depicted in Supplementary Fig. 1. Proteomic analysis resulted in the quantification of 5066 proteins in at least one replicate of which 40 proteins were overexpressed and 55 proteins were downregulated by 2-fold in both replicates. A complete list of quantified proteins is provided in Supplementary Table 1. Quantitative phosphoproteomic analysis resulted in the quantification of 3662 phosphosites corresponding to 1801 proteins (Supplementary Table 2). Employing a 2-fold cut-off in both replicates, we identified 127 hyperphosphorylated and 144 hypophosphorylated phosphopeptides corresponding to 95 and 86 proteins, respectively. Amongst the hyperphosphorylated proteins, 8 kinases were identified to be hyperphosphorylated (≥2 fold) in both replicates. These included proteins such as rho-associated protein kinase 2 (ROCK2), serine/threonine-protein kinase N2 (PKN2), STE20-like serine/threonine-protein kinase isoform 1 (SLK) and phosphorylase b kinase regulatory subunit beta isoform a (PHKB). A partial list of proteins dysregulated and/or differentially phosphorylated (≥2 fold) is provided in Table 1. The distribution of phosphorylation and protein quantitation in smoke exposed cells compared to parental cells in depicted in Fig. 2a.
Table 1.
Partial list of molecules differentially expressed and/or differentially phosphorylated in OKF6/TERT1-Smoke cells compared to OKF6/TERT1-Parental cells
| Gene symbol | Protein Name | Protein fold change (≥2 fold) | Phosphopeptide sequence | Phosphosite | Phosphopeptide fold change (≥2 fold) |
|---|---|---|---|---|---|
| ICAM1 | intercellular adhesion molecule 1 | 2.6 | GTPMKPNTQAtPP | T530 | 3.3 |
| PLCG1 | 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma-1 isoform a | 1.3 | AREGsFESR | S1248 | 4.2 |
| EVPL | envoplakin | 0.9 | SAsPTVPR | S2025 | 0.5 |
| TJP1 | tight junction protein ZO-1 isoform d | 0.9 | SEPSDHSRHsPQQPSNGSLR | S333 | 0.2 |
| LMNA | lamin isoform A | 0.7 | LRLsPsPTSQR | S390; S392 | 0.4 |
| DSG3 | desmoglein-3 | 0.5 | GsHTMLCTEDPCSR | S985 | 0.4 |
| COL17A1 | collagen alpha-1(XVII) chain | 0.5 | HAYEGSSSGNssPEYPRK | S117; S118 | 0.3 |
| PKP1 | plakophilin-1 isoform 1b | 0.3 | SSQSSTLSHsNR | S65 | 0.4 |
| LAD1 | ladinin-1 | 0.4 | IPSKEEEADMSsPTQR | S356 | 0.4 |
| KRT14 | keratin, type I cytoskeletal 14 | 0.4 | APSTYGGGLsVSSSR | S51 | 0.1 |
| KRT5 | keratin, type II cytoskeletal 5 | 0.3 | sLYNLGGSK | S64 | 0.1 |
| KRT16 | keratin, type I cytoskeletal 16 | 0.3 | EVFTSSsSSSSR | S448 | 0.2 |
| KRT6B | keratin, type II cytoskeletal 6B | 0.4 | SGFSSIsVSR | S37 | 0.2 |
| KRT6A | keratin, type II cytoskeletal 6A | 0.4 | AIGGGLSSVGGGsSTIK | S546 | 0.4 |
Fig. 2.

a Quadrant plot depicting log fold change of proteins at proteome and phosphoproteome level in OKF6/TERT1-Smoke cells compared to OKF6/TERT1-Parental cells. b Ingenuity pathway analysis of dysregulated proteins (≥2 fold) and differentially phosphorylated proteins (≥2 fold) in smoke exposed cells compared to parental cells. Top ten significant (p ≤ 0.05) molecular and cellular functions as observed in OKF6/TERT1-Smoke cells. c Top twelve significant (p ≤ 0.05) functions annotated under ‘Cellular Movement’
Chronic cigarette smoke exposure alters signaling processes associated with cytoskeletal reorganization and invasion in oral keratinocytes
We performed bioinformatics analysis of proteins that were dysregulated and/ or differentially phosphorylated by ≥2 fold in both replicates in our proteomic and phosphoproteomic data. Ingenuity Pathway Analysis revealed a strong concordance with results of cellular assays. The top molecular and cellular functions that were seen to be significantly (p < 0.01) affected were those involved in cellular movement, cellular assembly and organization (Fig. 2b). Under cellular movement, the specific cellular processes found to be activated (activation z-score > 2, p < 0.0001) included invasion of cells, cytoskeletal organization and cell migration (Fig. 2c).
We observed hyperphosphorylation of cortactin (CTTN) and 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma-1 isoform a (PLCG1) at Ser368 and Ser1248, respectively. Phosphorylation of CTTN at Ser368 and PLCG1 at Ser1248 has been linked to enhanced cellular motility (Wang et al. 2006; Kelley et al. 2010). Phosphorylation of CTTN at Ser368 has also been associated with activation of focal adhesion kinase (FAK) with a concurrent increase in cell scattering in human gastric adenocarcinoma cells (Tegtmeyer et al. 2011). In conjunction with this, OKF6/TERT1-Smoke cells displayed increased cell scattering and we also observed a 1.85 fold increase in FAK or PTK2 phosphorylation at Ser932 in these cells compared to parental cells. Hyperphosphorylation of FAK at Ser932 has been linked to cytoskeletal reorganization (Villa-Moruzzi 2007). In addition, proteins with established roles in cytoskeletal reorganization and structural integrity in keratinocytes such as keratins 5 and 14 (KRT5, KRT14), envoplakin (EVPL), plakophilin 1 (PKP1) and desmoglein 3 (DSG3) (Johnson et al. 2014) were downregulated and hypophosphorylated (≥2 fold) in OKF6/TERT1-Smoke cells compared to parental cells. Another protein known to be over expressed in cancers - intercellular adhesion molecule 1 (ICAM1) - was overexpressed and hyperphosphorylated (≥fold) in smoke cells (Rosette et al. 2005).
Serine/threonine protein kinases including ROCK2 and PKN2 are known downstream effectors of Rho and Rac GTPases in cytoskeletal rearrangements (Amano et al. 2010; Vincent and Settleman 1997). Both proteins were hyperphosphorylated by ≥2 fold in our phosphophoproteomics data. Active RhoA is involved in ROCK2 phosphorylation at Ser1374 resulting in its activation (Julian and Olson 2014). ROCK2 expression has been associated with invasion in non-small cell lung cancers (Vigil et al. 2012). PKN2 expression has been found to be essential for cellular migration and invasion in bladder cancer cells (Lachmann et al. 2011).
In concordance with mass spectrometry data, we observed an increase in endogenous expression of PKN2 in OKF6/TERT1-Smoke cells as well as a panel of HNSCC cell lines compared to OKF6/TERT1-Parental cells (Fig. 3a).
Fig. 3. PKN2 mediated signaling in smoke exposed cells.

a Western blot analysis shows endogenous expression of PKN2 in OKF6/TERT1-Smoke cells and in a panel of HNSCC cell lines compared to OKF6/TERT1-Parental cells (b) OKF6/TERT1 Smoke and HNSCC cell lines JHU-O11, FaDu, CAL 27, JHU-O22 and JHU-O29 were transfected with PKN2 siRNA. Immunoblot analysis of total PKN2, p-PKN2 (Thr816), CD44, p-CD44 (Ser706) and DSC3 was performed. β-actin was used as loading control
Silencing of PKN2 in OKF6/TERT1-Smoke cells and a panel of HNSCC cells was confirmed by Western blot analysis (Fig. 3b). PKN2 silencing resulted in a decrease in its phosphorylation at Thr816. In addition, we observed a decrease in CD44 expression levels with a concurrent downregulation of CD44 phosphorylation at Ser706 upon PKN2 silencing in OKF6/TERT1-Smoke cells as well as HNSCC cells. Signaling downstream of Rho GTPases has previously been linked to desmosomal protein expression and function (Johnson et al. 2014). We observed an increase in DSC3 expression levels in both smoke exposed and HNSCC cells upon siRNA mediated silencing of PKN2 (Fig. 3b). These observations indicate activation of PKN2 signaling cascade in response to cigarette smoke in HNSCC.
Silencing of PKN2 decreases cellular proliferation
siRNA-mediated silencing of PKN2 resulted in significant decrease in cellular proliferation of both smoke exposed oral cells (Fig. 4a) as well as in a panel of HNSCC cells (Fig. 4b–f) compared to control cells. PKN2 silencing also led to significant reduction in colony forming ability of smoke exposed cells as well as HNSCC cells compared to parental cells (Fig. 5a and b). Our data indicates that PKN2 plays a significant role in the proliferative ability of OKF6/TERT1-Smoke cells. In addition, decrease in proliferation of HNSCC cells upon PKN2 silencing suggests a crucial role of PKN2 in oncogenic transformation.
Fig. 4. Silencing of PKN2 decreases cellular proliferation.

Proliferation curve of (a) OKF6/TERT1-Smoke (b) JHU-O11 (c) JHU-O22 (d) JHU-O29 (e) FaDu (f) CAL 27 cells upon siRNA-mediated silencing of PKN2 (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001)
Fig. 5. Inhibition of PKN2 decreases the invasive and migratory ability of smoke exposed cells and HNSCC cells exposed to cigarette smoke.

a–b Colony formation assays were carried out using OKF6/TERT1-Smoke, JHU-O11, FaDu, CAL 27, JHU-O22 and JHU-O29 cells following siRNA-mediated silencing of PKN2 or control siRNA (scrambled siRNA), A graphical representation of the colony forming ability of the indicated cells upon PKN2 silencing (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001). Colonies were visualized at 3× magnification. c–d Invasion assays were carried out using OKF6/TERT1-Smoke, JHU-O11, FaDu and CAL 27 cells. Cells were transfected with either control (Scrambled) or PKN2 siRNA and invaded cells were photographed at 10× magnification. A graphical representation of the invasive ability of the cells upon PKN2 silencing (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001) (e) Wound migration assays were carried out using OKF6/TERT1-Smoke and HNSCC cells with or without PKN2 SiRNA. Distance between migrated cells were calculated between 0 and 10 h and represented as bar graph. (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001)
Inhibition of PKN2 decreases invasive and migratory ability of cells exposed to cigarette smoke
Colony forming ability is often associated with invasive capability of cancer cells (Nanjappa et al. 2015; Radhakrishnan et al. 2016). Since invasive potential reflects metastatic potential of tumor cells, we investigated the role of PKN2 in invasion and migration ability of OKF6/TERT1-Smoke and HNSCC cells. As depicted in Fig. 5c and d, PKN2 silencing led to significant decrease in the invasive ability of not only OKF6/TERT1-Smoke cells but also in a panel of HNSCC cells. In addition, we also observed a significant decrease in the migration ability of OKF6/TERT1-Smoke and HNSCC cells due to PKN2 silencing (Fig. 5e).
Taken together, our results from proteomic and phosphoproteomic data analysis as well as cellular assays indicate that PKN2 plays an important role in oncogenesis in oral cells especially due to chronic smoke exposure.
Discussion
Several in vitro studies have previously documented the adverse molecular effects of short term (acute) cigarette smoke exposure. Cigarette smoke and its constituents are known to affect signaling pathways linked to increased cellular proliferation, survival and migration (Dasgupta et al. 2009; Kim et al. 2010; Gumus et al. 2008; Yang et al. 2015). However, it is chronic rather than acute exposure that is associated with and leads to oncogenic transformation. A recent study by Vaz et al. highlights that chronic smoke exposure induces epigenetic alterations over time that sensitize human bronchial epithelial cells to oncogenic transformation by a single oncogene (Vaz et al. 2017). Using a cellular model, we too, demonstrate molecular changes that are prerequisites for progression to malignancy. This is evidenced by increased cellular proliferation, invasive and changed migratory ability of the smoke exposed cells.
High-throughput studies such as genome sequencing and transcriptome studies have characterized irreversible and reversible changes to key cellular processes such as DNA repair pathways, oxidative stress, xenobiotic metabolism and tumor suppression in smokers compared to non-smokers (Spira et al. 2004; Govindan et al. 2012). However, documenting molecular alterations in signaling molecules may better reflect the functional consequences of cigarette smoke exposure at a cellular level. Using quantitative proteomic and phosphoproteomic approaches, we elucidated the molecular changes in oral keratinocytes upon chronic smoke exposure. Bioinformatics analysis of proteomic data revealed dysregulation and differential phosphorylation of several proteins downstream of Rho/Rac signaling including PKN2, ROCK2, CTTN, PLCG1, members of the keratin family of cytoskeletal proteins and members of the plakin family of proteins. Proteins such as envoplakin, keratins 5 and 14, plakophilin and tight junction protein ZO-1, are involved in maintenance of epithelial integrity and cellular adhesion and were seen to be downregulated and/or hypophosphorylated (≥2 fold). Rho GTPases-associated signaling is known to control expression of proteins involved in desmosome formation (Johnson et al. 2014). In addition, loss of desmosomes has been reported as an early event in carcinogenesis (Dusek and Attardi 2011). Our data is in concordance with this phenomenon. PKN2 is a protein kinase downstream of Rac1 signaling that is associated with increased migration in bladder cancer cells (Lachmann et al. 2011). Silencing of PKN2 resulted in downregulation of proteins such as CD44 with a concurrent increase in levels of desmosomal protein DSC3. CD44 is a known marker of cancer progression in multiple cancers including gastric and oral cancer (Yuan et al. 2016; Judd et al. 2012). Restoration of desmosomal proteins has been shown to suppress tumorigenic potential in human chondrosarcoma cells (Galoian et al. 2015). Silencing of PKN2 significantly reduced cellular proliferation, invasion and migration in OKF6/TERT1 cells transformed by smoke exposure as well as in a panel of HNSCC cell lines. Our data indicates that chronic exposure to cigarette smoke results in distinct dysregulation of pathways associated with cellular adhesion and cytoskeletal reorganization. Further, silencing of PKN2, a key downstream effector of Rho GTPases, results in suppression of carcinogenesis induced by smoke exposure in oral cells. Our data suggests that PKN2 plays a major role in oncogenic transformation in HNSCC and can serve as a potential therapeutic target in head and neck cancer.
In conclusion, we report PKN2 activation in oral cells chronically exposed to cigarette smoke. Activation of the PKN2 signaling axis affects desmosomal integrity and cellular adhesion-related processes which aids in the oncogenic transformation of oral cells. Further studies are required to evaluate the prevalence of PKN2 activation in HNSCC among smokers and to determine the therapeutic potential of PKN2 in a clinical setting.
Electronic supplementary material
(PDF 6532 kb)
(PDF 6473 kb)
Schematic workflow employed to study the proteomic and phosphoproteomic alterations in normal oral keratinocytes chronically exposed to cigarette smoke (TIFF 1988 kb)
{kind=link}
Acknowledgements
We thank the Department of Biotechnology (DBT), Government of India for research support to the Institute of Bioinformatics (IOB), Bangalore. Pavithra Rajagopalan is a recipient of Senior Research Fellowship from the Council of Scientific and Industrial Research (CSIR), New Delhi, India. Kiran K. Mangalaparthi is a recipient of Senior Research Fellowship from University Grants Commission (UGC), New Delhi, India. Harsha Gowda is a Wellcome Trust/DBT India Alliance Intermediate Career Fellow. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- AGC
Automatic Gain Control
- BCA
Bicinchoninic acid
- EDTA
Ethylenediaminetetraacetic acid
- EMT
Epithelial–mesenchymal transition
- HCD
Higher energy collisional dissociation
- HPLC
High Pressure Liquid Chromatography
- SDS
Sodium Dodecyl Sulfate
- TEABC
Triethyl ammonium bicarbonate
- TMT
Tandem Mass Tag
Author’s contribution
AC and HG participated in study conception and study design. PR and VN were involved in cell culture and performed all assays and experiments. APJ and KKM carried out fractionation and mass spectrometric analysis of samples. PR and KP prepared the manuscript and manuscript figures. PR, KP and AHP were involved in data analyses and interpretation. AC, HG, DS, JAC, PPM, TSKP and BN edited, critically read and revised the manuscript. All the authors have read and approved the final manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that no competing financial interests exist.
Contributor Information
Pavithra Rajagopalan, Phone: 91-80-28416140, Email: pavithra@ibioinformatics.org.
Vishalakshi Nanjappa, Phone: 91-80-28416140, Email: vishalakshi@ibioinformatics.org.
Krishna Patel, Phone: 91-80-28416140, Email: krishnapatel@ibioinformatics.org.
Ankit P. Jain, Phone: 91-80-28416140, Email: ankit@ibioinformatics.org
Kiran K. Mangalaparthi, Phone: 91-80-28416140, Email: kiran@ibioinformatics.org
Arun H. Patil, Phone: 91-80-28416140, Email: arun@ibioinformatics.org
Bipin Nair, Phone: 91-476 2803000, Email: bipin@amrita.edu.
Premendu P. Mathur, Phone: 91-80-67618624, Email: ppmathur@yahoo.com
T. S. Keshava Prasad, Phone: 91-80-28416140, Email: keshav@ibioinformatics.org
Joseph A. Califano, Phone: (858) 822-6197, Email: jcalifano@ucsd.edu
David Sidransky, Phone: 1 (410) 502-5144, Email: dsidrans@jhmi.edu.
Harsha Gowda, Phone: +91 80 28416140, Email: harsha@ibioinformatics.org.
Aditi Chatterjee, Phone: +91 80 28416140, Email: aditi@ibioinformatics.org.
References
- Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 2010;67:545–554. doi: 10.1002/cm.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SS, Jiang WW, Smith I, Glazer C, Sun WY, Mithani S, Califano JA. Chronic cigarette smoke extract treatment selects for apoptotic dysfunction and mitochondrial mutations in minimally transformed oral keratinocytes. Int J Cancer. 2010;126:19–27. doi: 10.1002/ijc.24777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang X, Ravi R, Pham V, Bedi A, Chatterjee A, Sidransky D. Adenylate kinase 3 sensitizes cells to cigarette smoke condensate vapor induced cisplatin resistance. PLoS One. 2011;6:e20806. doi: 10.1371/journal.pone.0020806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Mambo E, Zhang Y, Deweese T, Sidransky D. Targeting of mutant hogg1 in mammalian mitochondria and nucleus: effect on cellular survival upon oxidative stress. BMC Cancer. 2006;6:235. doi: 10.1186/1471-2407-6-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Yan Y, Li J, Ma Q, Stoner GD, Ye J, Huang C. Differential requirement of signal pathways for benzo[a]pyrene (B[a]P)-induced nitric oxide synthase (iNOS) in rat esophageal epithelial cells. Carcinogenesis. 2005;26:1035–1043. doi: 10.1093/carcin/bgi052. [DOI] [PubMed] [Google Scholar]
- Chen RJ, Chang LW, Lin P, Wang YJ. Epigenetic effects and molecular mechanisms of tumorigenesis induced by cigarette smoke: an overview. J Oncol. 2011;2011:654931. doi: 10.1155/2011/654931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier N, Yeo A, Fiorentino E, Paxson J (2015) Optimization of the wound scratch assay to detect changes in murine mesenchymal stromal cell migration after damage by soluble cigarette smoke extract. J Vis Exp e53414 [DOI] [PMC free article] [PubMed]
- Currier N, Solomon SE, Demicco EG, Chang DL, Farago M, Ying H, Dominguez I, Sonenshein GE, Cardiff RD, Xiao ZX, Sherr DH, Seldin DC. Oncogenic signaling pathways activated in DMBA-induced mouse mammary tumors. Toxicol Pathol. 2005;33:726–737. doi: 10.1080/01926230500352226. [DOI] [PubMed] [Google Scholar]
- Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, Haura E, Chellappan S. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer. 2009;124:36–45. doi: 10.1002/ijc.23894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Zhao Q, Ma X, Ma G, Liu C, Chen Z, Yu L, Liu X, Zhang Y, Shao S, Xiao J, Li J, Zhang W, Fu M, Dong L, Yang X, Guo X, Xue L, Fang F, Zhan Q, Zhang L. Establishment of a new OSCC cell line derived from OLK and identification of malignant transformation-related proteins by differential proteomics approach. Sci Rep. 2015;5:12668. doi: 10.1038/srep12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusek RL, Attardi LD. Desmosomes: new perpetrators in tumour suppression. Nat Rev Cancer. 2011;11:317–323. doi: 10.1038/nrc3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzati M, Henley SJ, Lopez AD, Thun MJ. Role of smoking in global and regional cancer epidemiology: current patterns and data needs. Int J Cancer. 2005;116:963–971. doi: 10.1002/ijc.21100. [DOI] [PubMed] [Google Scholar]
- Galoian K, Qureshi A, Wideroff G, Temple HT. Restoration of desmosomal junction protein expression and inhibition of H3K9-specific histone demethylase activity by cytostatic proline-rich polypeptide-1 leads to suppression of tumorigenic potential in human chondrosarcoma cells. Mol Clin Oncol. 2015;3:171–178. doi: 10.3892/mco.2014.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel R, Muthusamy B, Pandey A, Prasad TS. Human protein reference database and human proteinpedia as discovery resources for molecular biotechnology. Mol Biotechnol. 2011;48:87–95. doi: 10.1007/s12033-010-9336-8. [DOI] [PubMed] [Google Scholar]
- Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, Maher CA, Fulton R, Fulton L, Wallis J, Chen K, Walker J, McDonald S, Bose R, Ornitz D, Xiong D, You M, Dooling DJ, Watson M, Mardis ER, Wilson RK. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumus ZH, Du B, Kacker A, Boyle JO, Bocker JM, Mukherjee P, Subbaramaiah K, Dannenberg AJ, Weinstein H. Effects of tobacco smoke on gene expression and cellular pathways in a cellular model of oral leukoplakia. Cancer Prev Res (Phila) 2008;1:100–111. doi: 10.1158/1940-6207.CAPR-08-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardonniere K, Saunier E, Lemarie A, Fernier M, Gallais I, Helies-Toussaint C, Mograbi B, Antonio S, Benit P, Rustin P, Janin M, Habarou F, Ottolenghi C, Lavault MT, Benelli C, Sergent O, Huc L, Bortoli S, Lagadic-Gossmann D. The environmental carcinogen benzo[a]pyrene induces a Warburg-like metabolic reprogramming dependent on NHE1 and associated with cell survival. Sci Rep. 2016;6:30776. doi: 10.1038/srep30776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Najor NA, Green KJ. Desmosomes: regulators of cellular signaling and adhesion in epidermal health and disease. Cold Spring Harb Perspect Med. 2014;4:a015297. doi: 10.1101/cshperspect.a015297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judd NP, Winkler AE, Murillo-Sauca O, Brotman JJ, Law JH, Lewis JS, Jr, Dunn GP, Bui JD, Sunwoo JB, Uppaluri R. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res. 2012;72:365–374. doi: 10.1158/0008-5472.CAN-11-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julian L, Olson MF. Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases. 2014;5:e29846. doi: 10.4161/sgtp.29846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LC, Hayes KE, Ammer AG, Martin KH, Weed SA. Cortactin phosphorylated by ERK1/2 localizes to sites of dynamic actin regulation and is required for carcinoma lamellipodia persistence. PLoS One. 2010;5:e13847. doi: 10.1371/journal.pone.0013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Huang Y, Lee J, Zhong X, Jiang WW, Ratovitski EA, Sidransky D. Cellular transformation by cigarette smoke extract involves alteration of glycolysis and mitochondrial function in esophageal epithelial cells. Int J Cancer. 2010;127:269–281. doi: 10.1002/ijc.25057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, Thomas JK, Muthusamy B, Leal-Rojas P, Kumar P, Sahasrabuddhe NA, Balakrishnan L, Advani J, George B, Renuse S, Selvan LD, Patil AH, Nanjappa V, Radhakrishnan A, Prasad S, Subbannayya T, Raju R, Kumar M, Sreenivasamurthy SK, Marimuthu A, Sathe GJ, Chavan S, Datta KK, Subbannayya Y, Sahu A, Yelamanchi SD, Jayaram S, Rajagopalan P, Sharma J, Murthy KR, Syed N, Goel R, Khan AA, Ahmad S, Dey G, Mudgal K, Chatterjee A, Huang TC, Zhong J, Wu X, Shaw PG, Freed D, Zahari MS, Mukherjee KK, Shankar S, Mahadevan A, Lam H, Mitchell CJ, Shankar SK, Satishchandra P, Schroeder JT, Sirdeshmukh R, Maitra A, Leach SD, Drake CG, Halushka MK, Prasad TS, Hruban RH, Kerr CL, Bader GD, Iacobuzio-Donahue CA, Gowda H, Pandey A. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann S, Jevons A, De Rycker M, Casamassima A, Radtke S, Collazos A, Parker PJ. Regulatory domain selectivity in the cell-type specific PKN-dependence of cell migration. PLoS One. 2011;6:e21732. doi: 10.1371/journal.pone.0021732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanjappa V, Renuse S, Sathe GJ, Raja R, Syed N, Radhakrishnan A, Subbannayya T, Patil A, Marimuthu A, Sahasrabuddhe NA, Guerrero-Preston R, Somani BL, Nair B, Kundu GC, Prasad TK, Califano JA, Gowda H, Sidransky D, Pandey A, Chatterjee A. Chronic exposure to chewing tobacco selects for overexpression of stearoyl-CoA desaturase in normal oral keratinocytes. Cancer Biol Ther. 2015;16:1593–1603. doi: 10.1080/15384047.2015.1078022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera DS, Boggs SE, Beenhouwer C, Aden J, Knall C. Cellular mechanisms of mainstream cigarette smoke-induced lung epithelial tight junction permeability changes in vitro. Inhal Toxicol. 2007;19:13–22. doi: 10.1080/08958370600985768. [DOI] [PubMed] [Google Scholar]
- Parri M, Chiarugi P. Rac and rho GTPases in cancer cell motility control. Cell Commun Signal. 2010;8:23. doi: 10.1186/1478-811X-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21:7435–7451. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A, Nanjappa V, Raja R, Sathe G, Chavan S, Nirujogi RS, Patil AH, Solanki H, Renuse S, Sahasrabuddhe NA, Mathur PP, Prasad TS, Kumar P, Califano JA, Sidransky D, Pandey A, Gowda H, Chatterjee A. Dysregulation of splicing proteins in head and neck squamous cell carcinoma. Cancer Biol Ther. 2016;17:219–229. doi: 10.1080/15384047.2016.1139234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja R, Sahasrabuddhe NA, Radhakrishnan A, Syed N, Solanki HS, Puttamallesh VN, Balaji SA, Nanjappa V, Datta KK, Babu N, Renuse S, Patil AH, Izumchenko E, Prasad TS, Chang X, Rangarajan A, Sidransky D, Pandey A, Gowda H, Chatterjee A. Chronic exposure to cigarette smoke leads to activation of p21 (RAC1)-activated kinase 6 (PAK6) in non-small cell lung cancer cells. Oncotarget. 2016;7:61229–61245. doi: 10.18632/oncotarget.11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorth P. Collective cell migration. Annu Rev Cell Dev Biol. 2009;25:407–429. doi: 10.1146/annurev.cellbio.042308.113231. [DOI] [PubMed] [Google Scholar]
- Rosette C, Roth RB, Oeth P, Braun A, Kammerer S, Ekblom J, Denissenko MF. Role of ICAM1 in invasion of human breast cancer cells. Carcinogenesis. 2005;26:943–950. doi: 10.1093/carcin/bgi070. [DOI] [PubMed] [Google Scholar]
- Selvan LD, Renuse S, Kaviyil JE, Sharma J, Pinto SM, Yelamanchi SD, Puttamallesh VN, Ravikumar R, Pandey A, Prasad TS, Harsha HC. Phosphoproteome of Cryptococcus neoformans. J Proteome. 2014;97:287–295. doi: 10.1016/j.jprot.2013.06.029. [DOI] [PubMed] [Google Scholar]
- Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X, Palma J, Brody JS. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci U S A. 2004;101:10143–10148. doi: 10.1073/pnas.0401422101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Chang SS, Fu Y, Liu Y, Califano JA. Chronic CSE treatment induces the growth of normal oral keratinocytes via PDK2 upregulation, increased glycolysis and HIF1alpha stabilization. PLoS One. 2011;6:e16207. doi: 10.1371/journal.pone.0016207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegtmeyer N, Wittelsberger R, Hartig R, Wessler S, Martinez-Quiles N, Backert S. Serine phosphorylation of cortactin controls focal adhesion kinase activity and cell scattering induced by helicobacter pylori. Cell Host Microbe. 2011;9:520–531. doi: 10.1016/j.chom.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Vaz M, Hwang SY, Kagiampakis I, Phallen J, Patil A, O'Hagan HM, Murphy L, Zahnow CA, Gabrielson E, Velculescu VE, Easwaran HP, Baylin SB. Chronic cigarette smoke-induced epigenomic changes precede sensitization of bronchial epithelial cells to single-step transformation by KRAS mutations. Cancer Cell. 2017;32(360–376):e366. doi: 10.1016/j.ccell.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil D, Kim TY, Plachco A, Garton AJ, Castaldo L, Pachter JA, Dong H, Chen X, Tokar B, Campbell SL, Der CJ. ROCK1 and ROCK2 are required for non-small cell lung cancer anchorage-independent growth and invasion. Cancer Res. 2012;72:5338–5347. doi: 10.1158/0008-5472.CAN-11-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa-Moruzzi E. Targeting of FAK Ser910 by ERK5 and PP1delta in non-stimulated and phorbol ester-stimulated cells. Biochem J. 2007;408:7–18. doi: 10.1042/BJ20070058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent S, Settleman J. The PRK2 kinase is a potential effector target of both rho and Rac GTPases and regulates actin cytoskeletal organization. Mol Cell Biol. 1997;17:2247–2256. doi: 10.1128/MCB.17.4.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, O'Kelly G, Schoenegger A, Ovelleiro D, Perez-Riverol Y, Reisinger F, Rios D, Wang R, Hermjakob H. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–D1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JM. The bicinchoninic acid (BCA) assay for protein quantitation. Methods Mol Biol. 1994;32:5–8. doi: 10.1385/0-89603-268-X:5. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wu J, Wang Z. Akt binds to and phosphorylates phospholipase C-gamma1 in response to epidermal growth factor. Mol Biol Cell. 2006;17:2267–2277. doi: 10.1091/mbc.e05-10-0918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Long M, Tachado SD, Seng S. Cigarette smoke modulates PC3 prostate cancer cell migration by altering adhesion molecules and the extracellular matrix. Mol Med Rep. 2015;12:6990–6996. doi: 10.3892/mmr.2015.4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Zhang X, Zhang W, Liang W, Zhang P, Shi H, Zhang B, Shao M, Yan Y, Qian H, Xu W. SALL4 promotes gastric cancer progression through activating CD44 expression. Oncogene. 2016;5:e268. doi: 10.1038/oncsis.2016.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Gallup M, Zlock L, Finkbeiner WE, McNamara NA. Rac1 and Cdc42 differentially modulate cigarette smoke-induced airway cell migration through p120-catenin-dependent and -independent pathways. Am J Pathol. 2013;182:1986–1995. doi: 10.1016/j.ajpath.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 6532 kb)
(PDF 6473 kb)
Schematic workflow employed to study the proteomic and phosphoproteomic alterations in normal oral keratinocytes chronically exposed to cigarette smoke (TIFF 1988 kb)
Data Availability Statement
Mass spectrometric data generated in this study was submitted to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Vizcaino et al. 2013) with the dataset identifier PXD007637.