

Abstract
Key points
Nicotine (NIC) modulates cognition and memory function by targeting the nicotinic ACh receptor and releasing different transmitter systems postsynaptically.
With both NIC‐generated mechanisms, calcium influx and calcium permeability can be regulated, which is a key requirement for the induction of long‐term potentiation, comprising the physiological basis of learning and memory function.
We attempt to unmask the underlying mechanism of nicotinic effects on anodal transcranial direct current stimulation (tDCS)‐induced long‐term potentiation‐like plasticity based on the hypothesis of calcium‐dependency.
Abolished tDCS‐induced neuroplasticity as a result of NIC administration is reversed by calcium channel blockade with flunarizine in a dose‐dependent manner.
The results of the present study suggest that there is a dose determination of NIC/NIC agonists in therapeutical settings when treating cognitive dysfunction, which partially explains the heterogeneous results on cognition observed in subjects in different experimental settings.
Abstract
Nicotine (NIC) modulates neuroplasticity and improves cognitive performance in animals and humans mainly by increased calcium permeability and modulation of diverse transmitter systems. NIC administration impairs calcium‐dependent plasticity induced by non‐invasive brain stimulation with transcranial direct current stimulation (tDCS) in non‐smoking participants probably as a result of intracellular calcium overflow. To test this hypothesis, we analysed the effect of calcium channel blockade with flunarizine (FLU) on anodal tDCS‐induced cortical excitability changes in healthy non‐smokers under NIC. We applied anodal tDCS combined with NIC patch and FLU at three different doses (2.5, 5 and 10 mg) or with placebo medication. NIC abolished anodal tDCS‐induced neuroplasticity. Under medium dosage (but not under low and high dosage) of FLU combined with NIC, plasticity was re‐established. For FLU alone, the lowest dosage weakened long‐term potentiation (LTP)‐like plasticity, whereas the highest dosage again abolished tDCS‐induced plasticity. The medium dosage turned LTP‐like plasticity in long‐term depression‐like plasticity. The results of the present study suggest a key role of calcium influx and calcium levels in nicotinic effects on LTP‐like plasticity in humans. This knowledge might be relevant for the development of new therapeutic strategies in cognitive dysfunction.
Keywords: nicotine, synaptic plasticity, Calcium channel, tdcs
Key points
Nicotine (NIC) modulates cognition and memory function by targeting the nicotinic ACh receptor and releasing different transmitter systems postsynaptically.
With both NIC‐generated mechanisms, calcium influx and calcium permeability can be regulated, which is a key requirement for the induction of long‐term potentiation, comprising the physiological basis of learning and memory function.
We attempt to unmask the underlying mechanism of nicotinic effects on anodal transcranial direct current stimulation (tDCS)‐induced long‐term potentiation‐like plasticity based on the hypothesis of calcium‐dependency.
Abolished tDCS‐induced neuroplasticity as a result of NIC administration is reversed by calcium channel blockade with flunarizine in a dose‐dependent manner.
The results of the present study suggest that there is a dose determination of NIC/NIC agonists in therapeutical settings when treating cognitive dysfunction, which partially explains the heterogeneous results on cognition observed in subjects in different experimental settings.
Introduction
Nicotine (NIC), the main component of tobacco, is not only an addictive substance, but also has relevant effects on cognitive functions. It improves working memory performance, episodic memory performance and motor functions (Hahn & Stolerman, 2002, Heishman et al. 2010; Kumari et. al. 2003; Grundey et al. 2015) in both animals and humans. Conversely, NIC withdrawal in tobacco smokers is often associated with the deterioration of working and verbal memory and neuroplasticity, whereas NIC re‐administration can restitute those withdrawal‐dependent deficits (Jacobsen et al. 2005; Cole et al. 2010; Grundey et al. 2012a; 2012b). The physiological foundation of these cognitive effects has begun to be revealed in recent years. The main targets of NIC are nicotinic ACh receptors (nAChRs) of which the α4β2 and the α7 receptors are predominantly located in the central nervous system (Wallace & Porter, 2011). Both receptors serve as ligand‐gated ion channels with an increase of calcium permeability and modulation of a wide diversity of transmitter pathways, including serotonin, dopamine, NMDA and GABA, by post‐ and presynaptic mechanisms (Burnashev, 1998; Daja‐Bailador & Wonnacott, 2004). They are involved in the induction and modulation of neuroplasticity (Levin & Simon, 1998; Lisman, 2001; Gotti & Clementi, 2004) by facilitating long‐term potentiation (LTP) and reversal of GABAergic inhibition (Fujii et al. 2000; Matsuyama et al. 2000). Because LTP is a relevant physiological basis for learning and memory formation (Rioult‐Pedotti et al. 1998; 2000; Nakauchi et al. 2007), the nicotinic activation of nAChR is considered to play an important role in the cognition‐enhancing properties of NIC, presumably mainly via regulation of calcium permeability and neurotransmitter release (Huang et al. 2010).
In humans, non‐invasive brain stimulation protocols offer the opportunity to generate LTP‐like and long‐term depression (LTD)‐like plasticity (Ziemann et al. 2008). Transcranial direct current stimulation (tDCS) and paired associative stimulation (PAS) allow the generation of long‐lasting cortical excitability modifications that are both Ca2+ and NMDA receptor‐dependent (Nitsche & Paulus, 2000; 2001; Nitsche et al. 2003, Stefan et al. 2000 and 2002; Liebetanz et al. 2002; Nitsche et al. 2003b, Nitsche et al. 2003b, 2004a). Because tDCS affects neuronal populations non‐selectively by subthreshold resting membrane potential modulation, it is assumed to induce non‐focal plasticity (Nitsche et al. 2008). PAS, on the other hand, mainly affects synapses between motor and somatosensory neurons (Carson & Kennedy, 2013) and is thus assumed to induce focal, synapse‐specific plasticity.
Recent studies by our group have focused on the nicotinic and cholinergic impact on cortical neuroplasticity in healthy humans. Global cholinergic activation with rivastigmine (a cholinesterase inhibitor) produces a focusing effect with increased focal LTP‐like plasticity induced by PAS and abolished non‐focal tDCS‐induced LTP‐like plasticity (Kuo et al. 2007), whereas LTD‐like plasticity was prolonged and preserved by both focal and non‐focal LTD‐like plasticity induction procedures. This focusing effect on LTP‐like plasticity might explain its beneficial impact on cognition. NIC, as an agonist of nicotinic ACh receptors, leads to similar results for LTP‐like plasticity in non‐smokers (Thirugnanasambandam et al. 2011; Grundey et al. 2012b) by abolishing tDCS‐like plasticity. We assumed therefore that the nicotinic effects on neuroplasticity are controlled by nAChRs, specifically nAChRs with calcium channel properties. Accordingly, varenicline, a high‐affinity partial agonist to α4β2 and full agonist to α7 receptors, has similar effects on tDCS and PAS‐induced plasticity (Batsikadze et al. 2015).
The present study aimed to further evaluate the mechanism of the focusing effect of NIC on neuroplasticity in humans, especially the contribution of calcium‐dependent mechanisms. We speculate that the combination of NIC and excitatory anodal tDCS with a long and diffuse stimulation mechanism leads to a calcium overflow preventing the induction of non‐focal, LTP‐like plasticity by tDCS. Studies with dextrometorphan, an NMDA receptor antagonist, provide further evidence that calcium‐dependency plays a major role in nicotinic effects on neuroplasticity (Lugon et al. 2017). We therefore aimed to clarify the role of calcium ions in tDCS‐induced excitatory changes by blocking specific calcium channels. For this reason, we combined anodal, non‐focal facilitatory tDCS, with NIC and flunarizine (FLU), a non‐selective calcium channel blocker. NIC alone has been shown to abolish tDCS‐induced LTP‐like plasticity in non‐smokers. If these changes are the result of calcium overflow, the reduction of calcium concentration by administration of FLU should reverse these effects and re‐establish plasticity under specific dosages. We hypothesized that blocking calcium receptors to different degrees would have non‐linear effects on anodal tDCS‐induced plasticity. Medium dosages of calcium‐blockade would re‐establish facilitatory plasticity because of a gradual diminution of calcium influx, whereas higher dosages might again abolish facilitatory plasticity because of a stronger diminution of calcium influx.
Methods
The experiments were approved by the Ethics Committee of the University of Göttingen and conformed with the standards set by the latest revision of the Declaration of Helsinki, except for registration in a database (nr. 24/6/15).
Participants
We conducted a power analysis (G*‐Power 3.1; http://www.gpower.hhu.de) based on our former studies with nicotinic effects (effect sizes/Cohen's d between 1 and 1.05) and calculated a sample size of 12; thus, 12 healthy volunteers participated in this experiment (six males/six females). The mean ± SD age was 25.33 ± 3.3 years. Participants were right‐handed according to the Edinburgh handedness inventory (Oldfield, 1971) and were recruited from a student population. Exclusion criteria were aged <18 years or >50 years, a current history of neurological or psychiatric disease, current or previous drug abuse, alcohol abuse, bronchial asthma or allergies to components in the NIC patch or FLU tablets, present pregnancy or metallic head implants. All subjects were non‐smokers; none of them had smoked tobacco or even occasionally consumed NIC products for at least 5 years prior to the beginning of the study. All participants provided their written informed consent before participation.
tDCS
Direct current stimulation was administered by a battery‐driven constant current stimulator (neuroConn GmbH, Ilmenau, Germany). The current was delivered through a pair of rubber electrodes covered with saline‐soaked sponges (35 cm2). The target electrode was placed over the primary motor cortex representing the right abductor digiti minimi muscle (ADM) and the return electrode over the contralateral supraorbital region. All subjects received 1 mA of anodal stimulation for 13 min combined with NIC patch plus placebo (PLC), NIC patch plus FLU (2.5 , 5 and 10 mg), FLU (2.5, 5 and 10 mg) plus PLC patch, or PLC (tablets and patch) alone (Fig. 1). The stimulation intensity of 1 mA and duration (13 min) generates enhanced cortical excitability for ∼60 min post‐stimulation (Nitsche & Paulus, 2001; Nitsche et al. 2013a). The experimental sessions were separated by an interval of at least 1 week.
Figure 1. Experimental procedures.

The course of the experiments. First, TMS intensity was adjusted to elicit MEP amplitudes of 1 mV (bl1). NIC patch or a PLC patch was administered. Four hours after patch administration, FLU (2.5, 5 or 10 mg)/PLC was ingested by the participants. Again, 2 h later, 25 MEPs were recorded at the adjusted baseline stimulus intensity (bl2/3). Then, anodal tDCS was administered followed by the immediate recording of 25 MEPs at 0, 5, 10, 15, 20, 25, 30, 60, 90, 120 and 240 min, as well as the next morning (nm) and the next evening (ne), after tDCS.
Assessing motor cortex excitability
Single TMS pulses were delivered by a Magstim 200 stimulator (Magstim Company, Whitland, Dyfed, UK) to measure excitability changes in the representional motor cortical area of the right ADM via the amplitude of the motor‐evoked potential (MEP). The TMS pulses were given at a frequency of 0.25 Hz with a figure of eight‐shaped coil (diameter of one winding 70 mm; peak magnetic field 2.2 T). The coil was held tangentially to the scalp at an angle of 45° to the sagittal plane with the coil handle pointing laterally and posteriorly. This induced a posterior–anterior current flow that optimally activates the corticospinal system monosynaptically (Di Lazarro et al. 1998). The optimal position was defined as the site where the stimulation resulted consistently in the largest MEPs. Surface EMG was recorded from the right ADM in a belly–tendon montage. The signals were filtered and amplified with a low‐pass filter of 2.5 kHz and a time constant of 10 ms. Digitization was carried out at an analogue‐to‐digital rate of 5 kHz, and then signals were further relayed into a laboratory computer using the Signal software and CED 1401 hardware (Cambridge Electronic Design, Cambridge, UK). The intensity was adjusted to elicit, on average, baseline MEPs of 1 mV peak‐to‐peak amplitude. This TMS‐intensity was kept constant throughout the experiment. Changes in mean MEP amplitude over time reflected alterations of cortical excitability in the primary motor cortex.
Pharmacological intervention
Each subject participated in eight sessions in a randomized order. NIC transdermal patches (Nicorette Depotpflaster; Pfizer, New York City, NY, USA; releasing 15 mg over 16 h) or PLC patches were applied to all subjects in combination with FLU (2.5, 5 or 10 mg) or PLC capsules under anodal tDCS. A NIC patch dosage of 15 mg has been shown to affect cognition, attention and working memory (Min et al. 2001; Wignall & de Wit, 2011; Grundey et al. 2015) and tDCS‐ and PAS‐induced neuroplasticity in humans (Thirugnanasambandam et al. 2011; Grundey et al. 2012a; 2012b). The patch was applied 6 h before the start of tDCS. This is the approximate time needed for the plasma level to reach its maximum following patch application (Nørregard et al. 1992). To avoid nicotinic side‐effects (dizziness, vomiting, diarrhoea, etc.), subjects also received 20 mg of domperidone, a peripheral‐acting dopamine D2 receptor antagonist, if needed. It is assumed that the substance does not relevantly cross the blood–brain barrier (Barone, 1999) and it has shown no effects on motor cortical excitability at a dosage of 20 mg (Grundey et al. 2013).
FLU (Acis Arzneimittel GmbH; Grünwald, Germany) was administered in dosages of 2.5, 5 and 10 mg in different sessions of the experiments 2 h before tDCS. FLU is an L‐type calcium channel antagonist that diminishes intracellular calcium levels, which is relevant for the induction of anodal tDCS‐induced after‐effects in humans (Nitsche et al. 2003). A dosage of 10 mg of FLU has been shown to abolish tDCS‐induced excitatory after‐effects; thus, it has relevant effects on neuroplasticity. Maximum plasma levels are reached 2 h after oral intake (Holmes et al. 1984).
Course of the experiment
First, we determined the TMS‐evoked baseline MEPs before any pharmacological intervention. The subjects were seated in a reclined position in a comfortable chair with a head‐ and arm‐rest (dentist chair). They were asked to relax completely. EMG electrodes were placed over the right ADM as described above and the exact position was marked with a pen. TMS was then applied over the left representational area of the right ADM to identify the spot with the consistently highest MEP amplitudes in the resting ADM (‘hotspot’). The spot was also marked with a waterproof pen. TMS intensity was adjusted to elicit MEPs with a peak‐to‐peak amplitude averaging 1 mV. Twenty‐five MEPs were then recorded at this stimulus intensity and the mean MEP amplitude was calculated as the baseline (baseline 1; bl1). A NIC patch/PLC patch was administered on the skin of the upper arm and remained there until the last measurement was accomplished the next evening. Four hours after application of the NIC patch, either FLU, at the dosages mentioned above, or PLC medication were administered. Two hours later, the baseline MEP amplitude was controlled (baseline 2; bl2) and, if needed, TMS intensity was adjusted to 1 mV (baseline 3; bl3). Subsequently, the anodal tDCS protocol was administered, followed by the recording of at least 25 MEPs at 0, 5, 10, 15, 20, 25, 30, 60, 90, 120 and 240 min. Further MEP recordings were made the next morning (nm) and next evening (Fig. 1). The experiments were performed in randomized order. The subjects were blinded for both stimulation and medication conditions, whereas the experimenter was blinded for the medication condition only.
Statistical analysis
The individual means of 25 MEP amplitudes recorded at each time point were calculated for all subjects and all conditions separately. The post‐intervention mean MEP amplitudes were then normalized to the respective individual mean baseline MEP amplitude (quotient of post‐intervention vs. pre‐intervention MEP amplitudes (bl2 or bl3, respectively)). A separate repeated measurement ANOVA was performed on the normalized data for the MEP values of NIC plus different FLU dosages (2.5, 5 or 10 mg) and for PLC plus different dosages of FLU. Normalized MEP amplitudes were the dependent variable, including all time points up to the next evening. Medication (NIC/PLC), FLU (2.5, 5 and 10 mg of FLU/PLC) and time points were included as within‐subject factors. Mauchly's sphericity test was performed and Greenhouse–Geisser correction applied when necessary. Fisher's post hoc tests (P < 0.05) were calculated to compare the the time course of MEP amplitudes for different conditions (NIC/PLC) and FLU dosages and also to compare the effect of medications at single time points in the case of significant results in the ANOVA. All results are given as the mean ± SEM and the 95% confidence interval.
To compare the main effects of NIC and different dosages of FLU on plasticity, averaged MEPs were calculated for the first 30 min after stimulation for each subject per experimental session and normalized to bl2 or bl3. A repeated measurement ANOVA was then performed. Averaged MEPs values were the dependent variable. Medication (NIC/PLC) and FLU (PLC; 2.5, 5 and 10 mg) were included as within‐subject factors. Based on significant results, post hoc Fisher's least significant difference (LSD) (P < 0.05) was carried out to compare the average MEP values for each dosage condition against one another. A similar statistical calculation was conducted for the pooled MEP values at later time points (from 60 min to the next evening). On the basis of the means, SDs and correlations, the effect size (Cohen's d) was further calculated for the NIC and FLU condition (Table 1).
Table 1.
Cohen's d
| Condition | Timepoint (min) | Correlation | Cohen's d |
|---|---|---|---|
| NIC vs. PLC | 0 | 0.29 | 0.6 |
| 5 | 0.41 | 0.5 | |
| 10 | 0.68 | 0.9 | |
| 15 | 0.55 | 0.5 | |
| 20 | 0.28 | 1.2 | |
| 25 | 0.18 | 0.5 | |
| 30 | 0.18 | 0.5 | |
| 60 | 0.0 | 0.4 | |
| 90 | 0.32 | 0.4 | |
| 120 | 0.31 | 0.5 | |
| 240 | 0.03 | 0.0 | |
| nm | 0.34 | 0.2 | |
| ne | 0.28 | 0.4 | |
| NIC vs. NIC plus FLU 2.5 mg | 0 | 0.24 | 0.1 |
| 5 | 0.18 | 0.2 | |
| 10 | 0.31 | 0.1 | |
| 15 | 0.28 | 0.2 | |
| 20 | 0.32 | 0.1 | |
| 25 | 0.22 | 0.1 | |
| 30 | 0.33 | 0.1 | |
| 60 | 0.38 | 0.1 | |
| 90 | 0.43 | 0.2 | |
| 120 | 0.23 | 0.0 | |
| 240 | 0.15 | 0.0 | |
| nm | 0.02 | 0.1 | |
| ne | 0.29 | 0.2 | |
| NIC vs. NIC plus FLU 5 mg | 0 | 0.58 | 0.4 |
| 5 | 0.02 | 0.4 | |
| 10 | 0.05 | 0.4 | |
| 15 | 0.25 | 0.7 | |
| 20 | 0.11 | 0.9 | |
| 25 | 0.30 | 0.8 | |
| 30 | 0.37 | 0.9 | |
| 60 | 0.61 | 1.0 | |
| 90 | 0.33 | 0.2 | |
| 120 | 0.25 | 0.5 | |
| 240 | 0.26 | 0.1 | |
| nm | 0.52 | 0.1 | |
| ne | 0.58 | 0,3 | |
| NIC vs. NIC plus FLU 10 mg | 0 | 0.54 | 0.4 |
| 5 | 0.54 | 0.1 | |
| 10 | 0.65 | 0.1 | |
| 15 | 0.29 | 0.2 | |
| 20 | 0.41 | 0.2 | |
| 25 | 0.03 | 0.2 | |
| 30 | 0.56 | 0.1 | |
| 60 | 0.29 | 0.2 | |
| 90 | 0.50 | 0.1 | |
| 120 | 0.43 | 0.1 | |
| 240 | 0.12 | 0.0 | |
| nm | 0.48 | 0.2 | |
| ne | 0.29 | 0.2 | |
| PLC vs. PLC plus FLU 2.5 mg | 0 | 0.18 | 0.3 |
| 5 | 0.03 | 0.1 | |
| 10 | 0.70 | 0.6 | |
| 15 | 0.26 | 0.1 | |
| 20 | 0.35 | 0.2 | |
| 25 | 0.36 | 0.1 | |
| 30 | 0.51 | 0.1 | |
| 60 | 0.14 | 0.1 | |
| 90 | 0.10 | 0.3 | |
| 120 | 0.04 | 0.1 | |
| 240 | 0.37 | 0.2 | |
| nm | 0.17 | 0.2 | |
| ne | 0.33 | 0.2 | |
| PLC vs. PLC plus FLU 5 mg | 0 | 0.01 | 0.8 |
| 5 | 0.05 | 0.7 | |
| 10 | 0.02 | 0.4 | |
| 15 | 0.66 | 0.9 | |
| 20 | 0.26 | 1.4 | |
| 25 | 0.37 | 1.1 | |
| 30 | 0.56 | 1.3 | |
| 60 | 0.46 | 1.2 | |
| 90 | 0.17 | 0.9 | |
| 120 | 0.32 | 0.4 | |
| 240 | 0.33 | 0.7 | |
| nm | 0.27 | 0.3 | |
| ne | 0.25 | 0.7 | |
| PLC vs. PLC plus FLU 10 mg | 0 | 0.16 | 0.5 |
| 5 | 0.57 | 0.7 | |
| 10 | 0.02 | 0.3 | |
| 15 | 0.55 | 0.7 | |
| 20 | 0.22 | 0.8 | |
| 25 | 0.3 | 0.5 | |
| 30 | 0.21 | 0.3 | |
| 60 | 0.32 | 0.2 | |
| 90 | 0.34 | 0.9 | |
| 120 | 0.11 | 0.0 | |
| 240 | 0.34 | 0.0 | |
| nm | 0.11 | 0.0 | |
| ne | 0.22 | 0.3 |
The effect size (Cohen's d) is calculated on the basis of mean, SD and correlation for the different conditions and time points. Nm, next morning; ne, next evening.
To exclude differences between baseline values of different conditions, and also between first and second baseline values, an equivalence testing (TOST procedure; Walker & Nowacki, 2011) was carried out separately for the the respective MEP values and the percentage of maximal stimulator output (MSO). The defined equivalence interval for the MSO was set to 20%; for the MEP value to 10% (error probability 0.05).
Results
All participants completed the entire study. Six subjects complained about mild nausea and dizziness after NIC patch administration and additional intake of FLU. One participant experienced vomiting in the NIC‐only condition. All side effects were well controlled by domperidone and subsided before the tDCS intervention. Two subjects had itching and a slight redness underneath the patch that did not interfere with the experiments. All other subjects tolerated the drugs well.
The equivalence test confirmed that the average baseline MEP values did not differ between different medication conditions and were not affected by medication (Table 2)
Table 2.
MEP amplitudes and stimulation intensity before and after drug administration
| TMS parameter | Medication condition | bl1 | bl2 | bl3 |
|---|---|---|---|---|
| MEP | PLC/PLC | 1.06 ± 0.09 | 1.15 ± 0.13 | 1.05 ± 0.07 |
| MSO in % | 48 ± 10.8 | 48,5 ± 11.3 | ||
| MEP | NP/PLC | 1.06 ± 0.06 | 1.04 ± 0.16 | 1.06 ± 0.06 |
| MSO in % | 48.3 ± 10.9 | 49.6 ± 11.9 | ||
| MEP | PLC + FLU 2.5 | 1.08 ± 0.06 | 1.14 ± 0.18 | 1.10 ± 0.05 |
| MSO in % | 48.25 ± 10.1 | 48.25 ± 9.2 | ||
| MEP | PLC + FLU 5 | 1.06 ± 0.09 | 1.11 ± 0.18 | 1.07 ± 0.08 |
| MSO in % | 50.1 ± 13 | 49.3 ± 11.7 | ||
| MEP | PLC + FLU 10 | 1.04 ± 0.09 | 1.12 ± 0.19 | 1.03 ± 0.08 |
| MSO in % | 50.8 ± 10.9 | 50.8 ± 10.7 | ||
| MEP | NP + FLU 2.5 | 1.05 ± 0.08 | 1.15 ± 0.28 | 1.03 ± 0.08 |
| MSO in % | 50.8 ± 10.1 | 49.9 ± 10.6 | ||
| MEP | NP + FLU 5 | 1.05 ± 0.06 | 1.19 ± 0.21 | 1.01 ± 0.05 |
| MSO in % | 48.4 ± 10.7 | 47.3 ± 11.0 | ||
| MEP | NP + FLU 10 | 1.06 ± 0.09 | 1.13 ± 0.22 | 1.03 ± 0.08 |
| MSO in % | 50.1 ± 11.8 | 49.1 ± 12.6 |
The principal ANOVA conducted for the main experiment revealed a significant main effect for the factor time point (F 13,143 = 3.715; P = 0.0005) and significant interactions across medication and FLU (F 3,33 = 9.448; P = 0.0005) and medication × FLU time points (F 39,429 = 2.619; P < 0.0001) (Table 3).
Table 3.
Results of the repeated measurement ANOVA
| Factor | (d.f.N/d.f.D) | F value | P value |
|---|---|---|---|
| Medication (NIC/PLC) | (1/11) | 0.654 | 0.436 |
| Flunarizine (2.5, 5 and 10 mg/PLC) | (3/33) | 0.121 | 0.947 |
| Time point | (13/143) | 3.715 | 0.000 a |
| Medication × Flunarizine | (3/33) | 9.448 | 0.000 a |
| Medication × Time point | (13/143) | 0.728 | 0.733 |
| Flunarizine × Time point | (39/429) | 0.658 | 0.945 |
| Medication × Flunarizine × Time point | (39/429) | 2.619 | 0.000 a |
Significant results at P < 0.05.
d.f.N = numerator degrees of freedom; d.f.D = denominator of freedom.
Effects of calcium channel blockade on tDCS‐induced excitatory plasticity
According to post hoc Fisher's LSD tests, anodal tDCS enhanced MEP values for 30 min after stimulation in the PLC medication condition (PLC patch plus PLC medication). Low dosage FLU (2.5 mg) plus PLC patch reduced the effect of anodal tDCS. A significant MEP‐enhancement from baseline MEP values did not initiate before 20 min and lasted only a further 10 min. The MEP values, however, did not differ significantly from those obtained under PLC medication at any time point. Post hoc Fisher's LSD tests further revealed that the medium dosage (5 mg) of FLU (plus PLC patch) differed significantly from other FLU dosages and the PLC medication condition. Although anodal tDCS in the PLC medication condition enhanced the MEP amplitudes compared to the baseline value, a medium dosage of FLU (5 mg) reduced the MEP values significantly at 20, 60, 90 and 240 minutes, as well as at the next morning and next evening, compared to baseline values and thus the well‐known effect of anodal tDCS was reversed. The effect of 5 mg of FLU on MEP values was highly significant at 0, 5, 10, 15, 20, 25, 30, 60 and 90 min compared to PLC/PLC medication. For the highest dosage of FLU (10 mg), Fisher's LSD revealed that MEP amplitudes did not differ from baseline values after anodal tDCS stimulation. All results are shown in Fig. 2.
Figure 2. Effect of different dosages of FLU on anodal tDCS‐induced excitability with and without nicotinic effects.
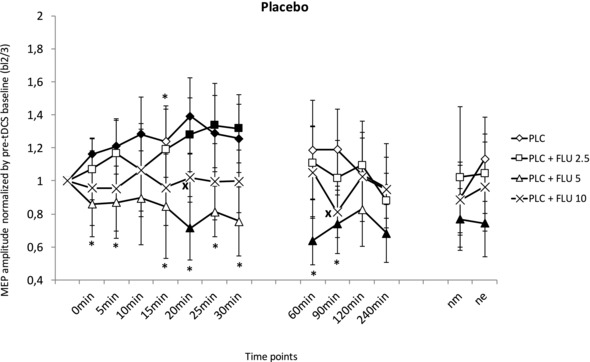
Standardized (to bl2/3) MEP amplitudes on the y‐axis plotted against the time points after atDCS under PLC or FLU medication (2.5, 5 or 10 mg). In the PLC condition, anodal tDCS leads to enhanced MEP amplitudes for 30 min post‐stimulation. The administration of 5 and 10 mg of FLU abolished the tDCS‐induced MEP enhancements and partially turned excitability enhancement into inhibition. Intake of 2.5 mg of FLU weakened the excitability changes after anodal tDCS. Excitability elevations lasted from 20 min to 30 min. Filled symbols indicate statistically significant deviations of post‐stimulation MEPs from the respective baseline values. *Significant differences between the PLC and 5 mg of FLU conditions. ×Significant differences between the PLC and 10 mg of FLU conditions (Fisher's least significant difference, P < 0.05). Vertical bars indicate the 95% confidence interval. nm, next morning; ne, next evening.
Nicotinic impact on tDCS‐induced plasticity under calcium‐channel blockade
As revealed by Fisher's LSD, applying a NIC patch alone completely abolished the tDCS‐induced excitability enhancement. MEP amplitudes after tDCS did not differ from baseline MEP amplitudes. However, combining a NIC patch with 5 mg of FLU partially re‐established the tDCS‐induced excitatory after‐effects. The MEPs were significantly enhanced from after 15 min up to 30 min compared to the respective baseline values. MEPs in this intervention condition differed significantly from NIC without FLU at 15, 20 and 30 min after tDCS (P < 0.05). Low‐dose (2.5 mg) and high‐dose FLU (10 mg) did not re‐establish tDCS‐induced excitability enhancements. For both dosages, MEP amplitudes did not differ from the MEP amplitudes obtained under NIC alone (Fig. 3).
Figure 3. Nicotinergic impact on anodal tDCS‐induced LTP‐like plasticity and its interaction with calcium channel blockade.
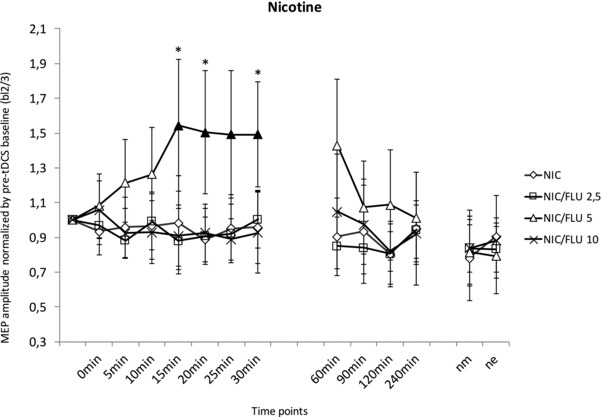
Standardized (to bl2/3) MEP amplitudes on the y‐axis plotted against the time points following anodal tDCS under NIC patch administration and different FLU dosages (2.5, 5 or 10 mg) and PLC, respectively. Under administration of NIC/PLC, tDCS‐induced excitability changes were completely abolished. NIC/FLU 5 restores facilitatory plasticity with significantly enhanced MEP amplitudes from 15–30 min after anodal tDCS compared to the respective baseline values. The MEPs of 2.5 and 10 mg of NIC/FLU were not different from the baseline value and the NIC/PLC condition. Filled symbols indicate statistically significant deviations of post‐stimulation MEPs from the respective baseline values. *Significant differences between the NIC/PLC and 5 mg of NIC/FLU conditions (Fisher's least significant difference, P < 0.05). Vertical bars indicate the 95% confidence interval. nm, next morning; ne, next evening.
Pooled time bins after anodal tDCS for NIC and different dosages of FLU (up to 30 min and from 60 min to next evening) after anodal tDCS
For the early pooled time bin (from 0 min to 30 min), the repeated measurement ANOVA revealed a significant interaction between medication (NIC/FLU) and dosage (F 3,9 = 11.083; P < 0.001). Fisher's LSD displayed significant effects of FLU at a medium dosage (5 mg) compared to the lowest applied FLU dosage (P = 0.001) and the PLC condition (P = 0.011). A dosage of 5 mg of FLU significantly converted excitability enhancements to excitability diminutions compared to the PLC and FLU 2.5 condition. For a dosage of 10 mg of FLU, a trendwise abolishing effect on tDCS‐induced neuroplasticity could be detected (P = 0.105). With NIC patch administration, the effects of FLU were different from the PLC patch condition. Fishers's LSD indicated that medium‐dose FLU (5 mg) re‐established tDCS‐induced neuroplasticity (P < 0.05), which was abolished by NIC alone (P = 0.032). Low (2.5 mg) and high (10 mg) dosages of FLU did not affect nicotinic changes in tDCS‐induced plasticity (Fig. 4).
Figure 4. Impact of NIC and different dosages of FLU on anodal tDCS‐induced facilitatory plasticity for MEP values pooled for 30 min post‐stimulation.
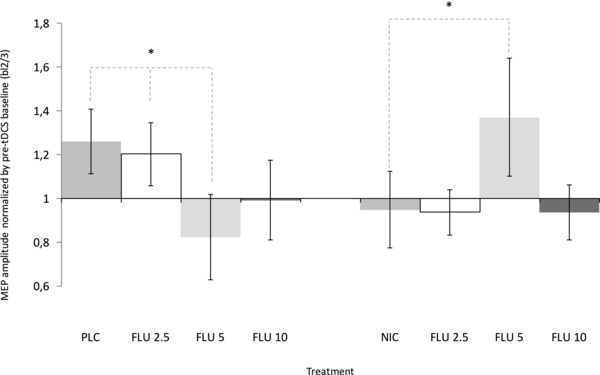
Mean of the standardized MEP values pooled for 30 min on the y‐axis plotted against the different FLU conditions with NIC and PLC, respectively. In the PLC condition, 5 and 10 mg of FLU abolished tDCS‐induced excitability enhancements, whereas 2.5 mg of FLU had no effect on plasticity. Under NIC administration, facilitatory plasticity is re‐established by 5 mg of FLU, whereas NIC alone abolished tDCS‐induced neuroplasticity. Low and high dosages of FLU did not affect abolished facilitatory plasticity. Each column represents the mean of baseline‐normalized MEP ± 95% confidence interval until 30 min after stimulation. *Significant differences between the PLC/FLU and PLC/PLC or NIC/FLU and NIC/PLC condition (Fisher's least significant difference, P < 0.05).
For the later time points (from 60 min until evening), the repeated measurement ANOVA revealed no significant effects and interactions.
Discussion
The results of the present study show that NIC affects anodal tDCS‐induced excitability changes and that these alterations are primarily controlled by calcium influx. Consistent with previous studies (Batsikadze, 2017), NIC alone prevented the induction of LTP‐like plasticity in healthy non‐smoking participants. This effect appears to be caused at least partially by calcium overflow because the combination of NIC with a medium dosage of FLU (5 mg), a calcium channel blocker, re‐established tDCS‐induced LTP‐like plasticity. The present study emphasizes how the impact of NIC on LTP‐like plasticity depends on the calcium level. Because LTP is a physiological correlate of memory formation and learning processes, this might explain the partially heterogeneous effects of NIC in cognitive studies.
Proposed mechanisms of action
NIC is an agonist of nAChRs. NAChRs increase the intracellular calcium level directly by serving as a ligand‐gated ion channel and indirectly by modulation of transmitter release, including dopamine, glutamate, GABA and noradrenaline (Alkandon et al. 1997; Summers et al. 1997; Albuquerque et al. 2009; Huang et al. 2010). On the basis of these wide‐reaching mechanisms, nicotinic effects on LTP and LTD are plausible. In animal experiments, NIC enhances LTP by activation of α7 nicotinic receptors in the dentate gyrus (Welsby et al. 2006) and facilitates NMDA‐dependent LTP induction (Yamazaki et al. 2005; Prestori et al. 2013). Because nAChRs gate the transport of calcium ions, changes in calcium concentrations driven by nAChRs are a possible explanation for these effects (Fayuk & Yakel, 2005; Zhong et al. 2013). In humans, the results are only partially equivalent. NIC was found to prolong focal PAS‐LTP‐like plasticity but abolish tDCS‐induced non‐focal plasticity (Thirugnanasambandam et al. 2011). The mechanisms of these focusing effects of NIC on LTP‐like plasticity in humans remain to be explored. tDCS‐induced LTP‐like plasticity depends on NMDA receptors and calcium influx because blockade of either NMDA receptors and/or calcium channels abolishes tDCS‐induced excitatory after‐effects (Liebetanz et al. 2002; Nitsche et al. 2003b). However, with an increasing calcium concentration intracellularly, non‐linear dose‐dependent opposing effects of calcium concentration on LTP and LTD can occur. Enhancement of intracellular calcium concentration can either induce LTD, no plasticity at all or LTP, depending on the amount of calcium enhancement. Low calcium enhancement results in LTD, whereas high calcium enhancement results in LTP. Further enhancement of calcium concentration might again abolish LTP or turn it into inhibition as a result of the counter‐regulatory activation of potassium channels (Lisman et al. 2001, Misonou et al. 2004). Accordingly, it can be speculated that nicotinic receptor activation in the case of anodal tDCS results in a calcium enhancement too large for the induction of LTP, and thus the focusing effect of NIC on LTP would be caused by NIC‐driven calcium overflow. This concept is supported by the main results of the present study, which show a dose‐dependent re‐establishing effect of calcium channel blockade on anodal tDCS‐induced LTP‐like plasticity under NIC. Specifically, adding FLU, a calcium channel blocker at a medium dosage (5 mg) to NIC exposition re‐establishes facilitatory plasticity, presumably as a result of the calcium‐decreasing effects of FLU that then allow LTP‐induction. Lower dosages of FLU (2.5 mg) did not restore LTP‐like plasticity, presumably because of an insufficient reduction of calcium influx (see explanation above). Likewise, the highest dosage of FLU (10 mg) applied in the present study did not re‐establish compromised LTP‐like plasticity under NIC (Figs 3 and 4). Previous studies have already shown that 10 mg of FLU blocks tDCS‐generated plasticity independent of NIC administration (Nitsche et al. 2003b); thus, this dosage most probably resulted in a calcium influx reduction that was too large to be compensated for by the calcium influx from nicotinic receptor activation. Re‐establishment of tDCS‐induced facilitatory plasticity was also accomplished with dextrometorphan, an NMDA‐receptor blocker, in an earlier study (Lugon et al. 2017). Here, similar mechanisms of action were discussed. The results of the present study suggest that the calcium level governing the focusing effect of NIC on LTP‐like plasticity in humans is not solely transmitted via NMDA receptors.
Apart from this main finding of the present study, the results provide additional evidence for the calcium‐concentration dependence hypothesis of plasticity. Anodal tDCS combined with different dosages of FLU alone, and thus different amounts of calcium channel blockade, resulted in non‐linear neuroplastic effects. Low‐dose FLU did not have a relevant impact on tDCS‐induced plasticity, whereas medium dosages converted LTP‐like plasticity into LTD‐like plasticity. Although the low dose of FLU was probably insufficient to induce a relevant modulation of intracellular calcium, the results obtained with medium concentrations suggest that it reduces the calcium concentration from the high LTP‐ to the lower LTD‐inducing range. The reduction of calcium concentration accomplished by high‐dosage FLU might have been too large to induce any plasticity (Fig. 5). Although consistent with results of animal model studies, the respective conclusions are indirect at present. Taken together, the results of the present study strongly support the concept that the effects of NIC on LTP‐like plasticity are calcium‐dependent. The mediating impact of nicotinic receptor activation on neuronal calcium influx furthermore explains the focussing and non‐linear effects of NIC on plasticity. On a functional level, it can be speculated that lacking/reduced LTP‐like plasticity is linked to the deterioration of cognitive functions. A previous study by our group has reported attenuation of motor learning performance after NIC administration in non‐smokers (Grundey et al. 2017).
Figure 5. Suggested effects of PLC, FLU, NIC and the combination of NIC and FLU on intracellular calcium concentration and tDCS‐induced LTP‐like plasticity.
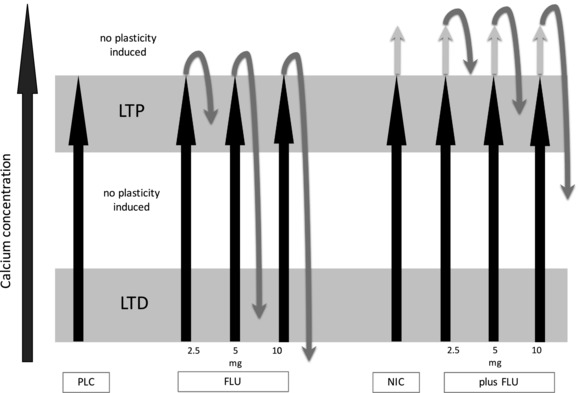
Assumed association between the changes of intracellular calcium concentration (x‐axis) and induction of tDCS‐induced excitability changes. Gradual enhancement of calcium concentration can either induce LTD, LTP or no plasticity. The combination of tDCS and PLC leads to LTP (black arrow alone). Additional administration of FLU in increasing doses gradually lowers intracellular calcium levels to either weaker LTP‐, LTD‐ or no plasticity induction (black and grey arrows). The combination of tDCS and NIC leads to a calcium overflow that prevents LTP induction (black and grey arrow). Gradually decreasing calcium concentration with additional administration of FLU re‐establishes tDCS‐induced LTP‐like plasticity in medium dosages (black and grey arrows).
Limitations
Some limitations of the present study should be taken into account. Because we did not monitor calcium influx directly, the explanation of calcium‐dependent nicotinic effects is hypothetical at present and remains to be explored in the future. Similar considerations apply with respect to the links to cognitive effects. Here, a systematic work‐up is needed to clearly understand how NIC affects cognitive processes based on our understanding of physiological effects, including non‐linearities, state‐ and task‐dependency. Furthermore, we monitored nicotinic effects on neuroplasticity after a single dose of NIC, although we did not explore possible dose‐dependent effects or the impact of chronic administration. Other neuromodulators such as dopamine reveal a non‐linear, dose‐dependent impact on neuroplasticity, which might also be expected for NIC (Monte‐Silva et al. 2010; Fresnoza et al. 2014). This dose‐dependency may play a role in NIC dependency and warrants more detailed investigation in the future.
Conclusions
The present study demonstrates that the abolishment of tDCS‐induced LTP‐like plasticity by administration of NIC is reversed by calcium channel blockade in a dose‐dependent manner. These results suggest a key role of calcium influx and calcium levels in the nicotinic effects on LTP. The present study provides a possible explanation for the partially heterogeneous nicotinic effects (with both improvement and deterioration of performance) on various cognitive processes (Levin, 2002, Swan & Lessov‐Schlagar, 2009, Grundey et al. 2015, 2017). These differences might depend on the actual calcium concentration. With regard to potential clinical application, agonists of nAChRs, especially the α7 and α4β2 receptors, have already been tested as cognitive enhancing drugs (e.g. in Alzheimer animal models and schizophrenic patients) (Bitner et al. 2010; Gee et al. 2017). The present study suggests that substance administration and dose determination need to be carefully addressed because too high or too low calcium concentrations might compromise functions.
Future perspectives
The application of nicotinic agents in patients is still at an early stage, although initial promising steps have been taken. However, before any final conclusions can be made about the effectiveness of this therapeutic option, a thorough work‐up is needed in terms of possible clinical application areas, long‐term side effects, pharmacology (possible genetic polymorphisms that are related to age‐dependent effects of calcium blockade) (Xu et al. 2017) and probable age‐dependent effects in healthy humans and patients.
Additonal information
Competing interests
MN is a member of the Advisory Board of Neuroelectrics.
Funding
MN receives support from the EC Horizon 2020 Program, FET Grant, 686764 as grants from the German Ministry of Research and Education (GCBS grant 01EE1403C, TRAINSTIM grant 01GQ1424E). JG was further supported by the Heidenreich von Siebold program of the University Medical Centre Goettingen. This work was supported by DFG grant NI 683/4‐2.
Author contributions
JG and MN designed and conceived the study. JB and JG acquired and analysed the data. MN, WP, MFK, GB and JG wrote the paper and revised the original work. All authors approved the final version of the manuscript submitted for publication.
Biography
Jessica Grundey is a neurologist at Göttingen Medical Center (Department of Clinical Neurophysiology). She has been working with non‐invasive brain stimulation methods, particularly transcranial direct current stimulation and paired associative stimulation, for >15 years (working group of Professor Michael Nitsche). Recently, she has been investigating nicotinic effects on neuroplasticity in healthy non‐smoking and smoking humans and the correlation with working memory and impicit motor learning performance. The latest studies have focussed on the underlying pharmacalogical mechanisms of nicotinic effects on neuroplasticity and calcium‐dependency.

Edited by: Ole Paulsen & Richard Carson
This is an Editor's Choice article from the 15 November 2018 issue.
References
- Albuquerque EX, Pereira EF, Alkondon M, Rogers SW (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89, 73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Barbosa CT, Albuquerque EX (1997). Neuronal nicotinic acetylcholine receptor activation modulates gamma‐aminobutyric acid release from CA1 neurons of rat hippocampal slices. J Pharmacol Exp Ther 283, 1396–1411. [PubMed] [Google Scholar]
- Barone JA (1999). Domperidone: a peripherally acting dopamine2‐receptor antagonist. Ann Pharmacother 33, 429–440. [DOI] [PubMed] [Google Scholar]
- Batsikadze G, Paulus W, Grundey J, Kuo MF, Nitsche MA (2015). Effect of the nicotinic α4β2‐receptor partial agonist varenicline on non‐invasive brain stimulation‐induced neuroplasticity in the human motor cortex. Cereb Cortex 25, 3249–3259. [DOI] [PubMed] [Google Scholar]
- Batsikadze G, Paulus W, Hasan A, Grundey J, Kuo MF, Nitsche MA (2017). Compromised neuroplasticity in cigarette smokers under nicotine withdrawal is restituted by the nicotinic α4β2‐receptor partial agonist varenicline. Sci Rep 7, 1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitner RS, Bunnelle WH, Decker MW, Drescher KU, Kohlhaas KL, Markosyan S, Marsh KC, Nikkel AL, Browman K, Radek R, Anderson DJ, Buccafusco J, Gopalakrishnan M (2010). In vivo pharmacological characterization of a novel selective alpha7 neuronal nicotinic acetylcholine receptor agonist ABT‐107: preclinical considerations in Alzheimer's disease. J Pharmacol Exp Ther 334, 875–886. [DOI] [PubMed] [Google Scholar]
- Burnashev N (1998). Calcium permeability of ligand‐gated channels. Cell Calcium 24, 325–332. [DOI] [PubMed] [Google Scholar]
- Carson RG, Kennedy NC (2013). Modulation of human corticospinal excitability by paired associative stimulation. Front Hum Neurosci 7, 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DM, Beckmann CF, Long CJ, Matthews PM, Durcan MJ, Beaver JD (2010). Nicotine replacement in abstinent smokers improves cognitive withdrawal symptoms with modulation of resting brain network dynamics. Neuroimage 52, 590–599. [DOI] [PubMed] [Google Scholar]
- Dajas‐Bailador F, Wonnacott S (2004). Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci 25, 317–324. Review. [DOI] [PubMed] [Google Scholar]
- Di Lazzaro V, Oliviero A, Profice P, Saturno E, Pilato F, Insola A, Mazzone P, Tonali P, Rothwell JC (1998). Comparison of descending volleys evoked by transcranial magnetic and electric stimulation in conscious humans. Electroencephalogr Clin Neurophysiol 109, 397–401. [DOI] [PubMed] [Google Scholar]
- Fayuk D, Yakel JL (2005). Ca2+ permeability of nicotinic acetylcholine receptors in rat hippocampal CA1 interneurones. J Physiol 566, 759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresnoza S, Paulus W, Nitsche MA, Kuo MF (2014). Nonlinear dose‐dependent impact of D1 receptor activation on motor cortex plasticity in humans. J Neurosci 34, 2744–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Jia Y, Yang A, Sumikawa K (2000). Nicotine reverses GABAergic inhibition of long‐term potentiation induction in the hippocampal CA1 region. Brain Res 863, 259–265. [DOI] [PubMed] [Google Scholar]
- Gee KW, Olincy A, Kanner R, Johnson L, Hogenkamp D, Harris J, Tran M, Edmonds SA, Sauer W, Yoshimura R, Johnstone T, Freedman R (2017). First in human trial of a type I positive allosteric modulator of alpha7‐nicotinic acetylcholine receptors: pharmacokinetics, safety, and evidence for neurocognitive effect of AVL‐3288. J Psychopharmacol 31, 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Clementi F (2004). Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol 74, 363–396. [DOI] [PubMed] [Google Scholar]
- Grundey J, Thirugnanasambandam N, Kaminsky K, Drees A, Skwirba AC, Lang N, Paulus W, Nitsche MA (2012). Neuroplasticity in cigarette smokers is altered under withdrawal and partially restituted by nicotine exposition. J Neurosci 32, 4156–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundey J, Thirugnanasambandam N, Kaminsky K, Drees A, Skwirba AC, Lang N, Paulus W, Nitsche MA (2012). Rapid effect of nicotine intake on neuroplasticity in non‐smoking humans. Front Pharmacol 3, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundey J, Freznosa S, Klinker F, Lang N, Paulus W, Nitsche MA (2013). Cortical excitability in smoking and not smoking individuals with and without nicotine. Psychopharmacology (Berl) 229, 653–664. [DOI] [PubMed] [Google Scholar]
- Grundey J, Amu R, Ambrus GG, Batsikadze G, Paulus W, Nitsche MA (2015). Double dissociation of working memory and attentional processes in smokers and non‐smokers with and without nicotine. Psychopharmacology (Berl) 232, 2491–2501. [DOI] [PubMed] [Google Scholar]
- Grundey J, Amu R, Batsikadze G, Paulus W, Nitsche MA (2017). Diverging effects of nicotine on motor learning performance: improvement in deprived smokers and attenuation in non‐smokers. Addict Behav 74, 90–97. [DOI] [PubMed] [Google Scholar]
- Hahn B, Shoaib M, Stolerman IP (2002). Nicotine‐induced enhancement of attention in the five‐choice serial reaction time task: the influence of task demands. Psychopharmacology (Berl) 162, 129–137. [DOI] [PubMed] [Google Scholar]
- Heishman SJ, Kleykamp BA, Singleton EG (2010). Meta‐analysis of the acute effects of nicotine and smoking on human performance. Psychopharmacology (Berl) 210, 453–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes B, Brogden RN, Heel RC, Speight TM, Avery GS (1984).Flunarizine. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use. Drugs 27, 6–44. [DOI] [PubMed] [Google Scholar]
- Huang LT, Sherwood JL, Sun YJ, Lodge D, Wang Y (2010). Activation of presynaptic alpha7 nicotinic receptors evokes an excitatory response in hippocampal CA3 neurones in anaesthetized rats: an in vivo iontophoretic study. Br J Pharmacol 159, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen LK, Krystal JH, Mencl WE, Westerveld M, Frost SJ, Pugh KR (2005). Effects of smoking and smoking abstinence on cognition in adolescent tobacco smokers. Biol Psychiatry 57, 56–66. [DOI] [PubMed] [Google Scholar]
- Kumari V, Gray JA, Ffytche DH, Mitterschiffthaler MT, Das M, Zachariah E, Vythelingum GN, Williams SC, Simmons A, Sharma T (2003). Cognitive effects of nicotine in humans: an fMRI study. Neuroimage 19, 1002–1013 [DOI] [PubMed] [Google Scholar]
- Kuo MF, Grosch J, Fregni F, Paulus W, Nitsche MA (2007). Focusing effect of acetylcholine on neuroplasticity in the human motor cortex. J Neurosci 27, 14442–14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Simon BB (1998). Nicotinic acetylcholine involvement in cognitive function in animals. Psychopharmacology (Berl) 138, 217–230. [DOI] [PubMed] [Google Scholar]
- Levin ED (2002). Nicotinic receptor subtypes and cognitive function. J Neurobiol 53, 633–640. [DOI] [PubMed] [Google Scholar]
- Liebetanz D, Nitsche MA, Tergau F, Paulus W (2002). Pharmacological approach to the mechanisms of transcranial DC‐stimulation‐induced after‐effects of human motor cortex excitability. Brain 125, 2238–2247. [DOI] [PubMed] [Google Scholar]
- Lisman JE (2001). Three Ca2+ levels affect plasticity differently: the LTP zone, the LTD zone and no man's land. J Physiol 532, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugon MD, Batsikadze G, Fresnoza S, Grundey J, Kuo MF, Paulus W, Nakamura‐Palacios EM, Nitsche MA (2017). Mechanisms of Nicotinic Modulation of Glutamatergic Neuroplasticity in Humans. Cereb Cortex 27, 544–553. [DOI] [PubMed] [Google Scholar]
- Matsuyama S, Matsumoto A, Enomoto T, Nishizaki T (2000). Activation of nicotinic acetylcholine receptors induces long‐term potentiation in vivo in the intact mouse dentate gyrus. Eur J Neurosci 12, 3741–3747. [DOI] [PubMed] [Google Scholar]
- Min SK, Moon IW, Ko RW, Shin HS (2001). Effects of transdermal nicotine on attention and memory in healthy elderly non‐smokers. Psychopharmacology (Berl) 159, 83–88. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS (2004). Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci 7, 711–718. [DOI] [PubMed] [Google Scholar]
- Monte‐Silva K, Liebetanz D, Grundey J, Paulus W, Nitsche MA (2010). Dosage‐dependent non‐linear effect of L‐dopa on human motor cortex plasticity. J Physiol 588, 3415–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakauchi S, Brennan RJ, Boulter J, Sumikawa K (2007). Nicotine gates long‐term potentiation in the hippocampal CA1 region via the activation of alpha2* nicotinic ACh receptors. Eur J Neurosci 25, 2666–2681. [DOI] [PubMed] [Google Scholar]
- Nitsche MA, Paulus W (2000). Excitability changes induced in the human motor cortex by weak transcranial direct current stimulation. J Physiol 527, 633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsche MA, Paulus W (2001). Sustained excitability elevations induced by transcranial DC motor cortex stimulation in humans. Neurology 57, 1899–1901. [DOI] [PubMed] [Google Scholar]
- Nitsche MA, Liebetanz D, Lang N, Antal A, Tergau F, Paulus W (2003). Safety criteria for transcranial direct current stimulation (tDCS) in humans. Clin Neurophysiol 114, 2220–2222. [DOI] [PubMed] [Google Scholar]
- Nitsche MA, Fricke K, Henschke U, Schlitterlau A, Liebetanz D, Lang N, Henning S, Tergau F, Paulus W (2003b). Pharmacological modulation of cortical excitability shifts induced by transcranial direct current stimulation in humans. J Physiol 553, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsche MA, Jaussi W, Liebetanz D, Lang N, Tergau F, Paulus W (2004). Consolidation of human motor cortical neuroplasticity by D‐cycloserine. Neuropsychopharmacology 29, 1573–1578. [DOI] [PubMed] [Google Scholar]
- Nitsche MA, Cohen LG, Wassermann EM, Priori A, Lang N, Antal A, Paulus W, Hummel F, Boggio PS, Fregni F, Pascual‐Leone A (2008). Transcranial direct current stimulation: state of the art 2008. Brain Stimul 1, 206–223. [DOI] [PubMed] [Google Scholar]
- Nørregaard J, Tønnesen P, Simonsen K, Säwe U (1992). Long‐term nicotine substitution after application of a 16‐hour nicotine patch in smoking cessation. Eur J Clin Pharmacol 43, 57–60. [DOI] [PubMed] [Google Scholar]
- Oldfield RC. (1971). The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia 9, 97–113. [DOI] [PubMed] [Google Scholar]
- Prestori F, Bonardi C, Mapelli L, Lombardo P, Goselink R, De Stefano ME, Gandolfi D, Mapelli J, Bertrand D, Schonewille M, De Zeeuw C, D'Angelo E (2013). Gating of long‐term potentiation by nicotinic acetylcholine receptors at the cerebellum input stage. PLoS ONE 8, e64828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioult‐Pedotti MS, Friedman D, Hess G, Donoghue JP. (1998). Strengthening of horizontal cortical connections following skill learning. Nat Neurosci 1, 230–234. [DOI] [PubMed] [Google Scholar]
- Rioult‐Pedotti MS, Friedman D, Donoghue JP (2000). Learning‐induced LTP in neocortex. Science 290, 533–536. [DOI] [PubMed] [Google Scholar]
- Stefan K, Kunesch E, Cohen LG, Benecke R, Classen J (2000). Induction of plasticity in the human motor cortex by paired associative stimulation. Brain 123, 572–584. [DOI] [PubMed] [Google Scholar]
- Stefan K, Kunesch E, Benecke R, Cohen LG, Classen J (2002). Mechanisms of enhancement of human motor cortex excitability induced by interventional paired associative stimulation. J Physiol 543, 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers KL, Kem WR, Giacobini E (1997). Nicotinic agonist modulation of neurotransmitter levels in the rat frontoparietal cortex. Jpn J Pharmacol 74, 139–146. [DOI] [PubMed] [Google Scholar]
- Swan GE, Lessov‐Schlaggar CN (2009). Tobacco addiction and pharmacogenetics of nicotine metabolism. J Neurogenet 23, 262–271. [DOI] [PubMed] [Google Scholar]
- Thirugnanasambandam N, Grundey J, Adam K, Drees A, Skwirba AC, Lang N, Paulus W, Nitsche MA (2011). Nicotinergic impact on focal and non‐focal neuroplasticity induced by non‐invasive brain stimulation in non‐smoking humans. Neuropsychopharmacology 36, 879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace TL, Porter RH (2011). Targeting the nicotinic alpha7 acetylcholine receptor to enhance cognition in disease. Biochem Pharmacol 82, 891–903. [DOI] [PubMed] [Google Scholar]
- Walker E & Nowacki AS (2011). Understanding equivalence and noninferiority testing. J Gen Intern Med 26, 192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsby P, Rowan M, Anwyl R (2006). Nicotinic receptor‐mediated enhancement of long‐term potentiation involves activation of metabotropic glutamate receptors and ryanodine‐sensitive calcium stores in the dentate gyrus. Eur J Neurosci 24, 3109–3118. [DOI] [PubMed] [Google Scholar]
- Wignall ND, de Wit H (2011). Effects of nicotine on attention and inhibitory control in healthy nonsmokers. Exp Clin Psychopharmacol 19, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Boström AE, Saeed M, Dubey RK, Waeber G, Vollenweider P, Marques‐Vidal P, Mwinyi J, Schiöth HB (2017). A genetic variant in the catechol‐O‐methyl transferase (COMT) gene is related to age‐dependent differences in the therapeutic effect of calcium‐channel blockers. Medicine (Baltimore) 96, e7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Jia Y, Hamaue N, Sumikawa K (2005). Nicotine‐induced switch in the nicotinic cholinergic mechanisms of facilitation of long‐term potentiation induction. Eur J Neurosci 22, 845–860. [DOI] [PubMed] [Google Scholar]
- Ziemann U, Paulus W, Nitsche MA, Pascual‐Leone A, Byblow WD, Berardelli A, Siebner HR, Classen J, Cohen LG, Rothwell JC (2008). Consensus: motor cortex plasticity protocols. Brain Stimul 1, 164–182. [DOI] [PubMed] [Google Scholar]
- Zhong C, Talmage DA, Role LW (2013). Nicotine elicits prolonged calcium signaling along ventral hippocampal axons. PLoS ONE 8, e82719. [DOI] [PMC free article] [PubMed] [Google Scholar]