

Abstract
Key points
Chronic hypercapnia per se has distinct effects on the mechanisms regulating steady‐state ventilation and the CO2/H+ chemoreflex.
Chronic hypercapnia leads to sustained hyperpnoea that exceeds predicted ventilation based upon the CO2/H+ chemoreflex.
There is an integrative ventilatory, cardiovascular and metabolic physiological response to chronic hypercapnia.
Chronic hypercapnia leads to deterioration of cognitive function.
Abstract
Respiratory diseases such as chronic obstructive pulmonary disease (COPD) often lead to chronic hypercapnia which may exacerbate progression of the disease, increase risk of mortality and contribute to comorbidities such as cognitive dysfunction. Determining the contribution of hypercapnia per se to adaptations in ventilation and cognitive dysfunction within this patient population is complicated by the presence of multiple comorbidities. Herein, we sought to determine the role of chronic hypercapnia per se on the temporal pattern of ventilation and the ventilatory CO2/H+ chemoreflex by exposing healthy goats to either room air or an elevated inspired CO2 (InCO2) of 6% for 30 days. A second objective was to determine whether chronic hypercapnia per se contributes to cognitive dysfunction. During 30 days of exposure to 6% InCO2, steady‐state (SS) ventilation ( I) initially increased to 335% of control, and then within 1–5 days decreased and stabilized at ∼230% of control. There was an initial respiratory acidosis that was partially mitigated over time due to increased arterial [HCO3 −]. There was a transient decrease in the ventilatory CO2/H+ chemoreflex, followed by return to pre‐exposure levels. The SS I during chronic hypercapnia was greater than predicted from the acute CO2/H+ chemoreflex, suggesting separate mechanisms regulating SS I and the chemoreflex. Finally, as assessed by a shape discrimination test, we found a sustained decrease in cognitive function during chronic hypercapnia. We conclude that chronic hypercapnia per se results in: (1) a disconnect between SS I and the CO2/H+ chemoreflex, and (2) deterioration of cognitive function.
Keywords: Hypercapnia, Chemoreflex, Cognition
Key points
Chronic hypercapnia per se has distinct effects on the mechanisms regulating steady‐state ventilation and the CO2/H+ chemoreflex.
Chronic hypercapnia leads to sustained hyperpnoea that exceeds predicted ventilation based upon the CO2/H+ chemoreflex.
There is an integrative ventilatory, cardiovascular and metabolic physiological response to chronic hypercapnia.
Chronic hypercapnia leads to deterioration of cognitive function.
Introduction
Respiratory disease patients who hypoventilate and develop chronic hypercapnia often show poorer prognoses, decreased quality of life, poor cognitive function and higher mortality rates compared to similar disease patients who maintain pulmonary ventilation and arterial CO2 (Incalzi et al. 1993; Costello et al. 1997; Schou et al. 2012; Slenter et al. 2012). A characteristic common to a subset of these patients is a decline in the CO2/H+ chemoreflex, as assessed by the ventilatory response to acutely elevated inspired CO2 (InCO2) (Kepron & Cherniack, 1973; Montes de Oca & Celli, 1998). Contributing factors to the decline in the chemoreflex of these patients may be mechanical limitations, such as elevated airway resistance, or the presence of adaptations in the central or peripheral chemoreceptor reactivity following chronically elevated levels of CO2. However, determining the individual consequence of sustained hypercapnia on the temporal pattern of steady‐state ventilation, the CO2/[H+] chemoreflex, and the multiple morbidities within these patients is complicated by the presence of confounding factors, such as airway/pulmonary mechanical limitations and the variable presence of concurrent hypoxaemia.
Studies of chronic experimental hypercapnia have not provided a consensus regarding the temporal pattern of steady‐state ventilation during chronically increased InCO2. Within minutes upon exposure to elevated InCO2 there is a uniform, initial hyperpnoea (Schaefer, 1949, 1958; Schaefer et al. 1963a; Clark et al. 1971; Lai et al. 1973; Jennings & Chen, 1976; Pingree, 1977; Guillerm & Radziszewski, 1979; Jennings & Davidson, 1984; Kondo et al. 2000) that is followed by either a maintenance of the hyperpnoea (Kondo et al. 2000), an attenuation in ventilation from initial exposure levels that is maintained for the duration of hypercapnic exposure (Schaefer, 1949; Schaefer et al. 1963a; Clark et al. 1971; Pingree, 1977; Guillerm & Radziszewski, 1979; Jennings & Davidson, 1984), or a triphasic response consisting of an attenuation followed by a secondary elevation of ventilation toward initial exposure levels (Jennings & Chen, 1976). Additionally, these changes in ventilation during chronic hypercapnia correlate poorly with measurable changes in either arterial or cerebrospinal fluid CO2/[H+] (Jennings & Chen, 1976; Jennings & Davidson, 1984). This dissociation between ventilation, and [H⁺] led Jennings & Davidson (1984) to conclude, ‘The mechanism(s) by which the increase in PCO2 during chronic respiratory acidosis results in a sustained elevation of ventilation remains to be resolved.’ Likewise, Dempsey & Forster (1982) concluded, ‘It seems unlikely that measurable chemical stimuli acting at the carotid and [CNS] chemoreceptors can account for the hyperpnea’ during chronically elevated InCO2.
Studies of the acute chemoreflex during chronic experimental hypercapnia also lack consensus, and have reported both a decrease (Schaefer, 1949; Chapin, 1955; Schaefer et al. 1963a; Guillerm & Radziszewski, 1979), as well as minimal/no change in the slope of the ventilatory response to acute increases in InCO2 above steady‐state levels (Falchuk et al. 1966; Clark et al. 1971; Jennings & Chen, 1976; Jennings, 1979) during chronic hypercapnia. Importantly, the pattern of the acute chemoreflex during chronic hypercapnia does not always coincide with the pattern of steady‐state ventilation, which suggests a disconnect between the effect of chronic hypercapnia on steady‐state ventilation and the acute ventilatory CO2 chemoreflex. In other words, the acute chemoreflex (∆ I /∆ or ∆ I /∆[H+]) during chronic experimental hypercapnia may not predict steady‐state changes in ventilation. This apparent disconnect is further exemplified by studies of chronic hypercapnia induced by carotid body denervation (CBD). For example, during the 30 days following CBD in goats, there is sustained hypoventilation that leads to chronic hypercapnia. However, despite the sustained hypoventilation during the steady state, the acute CO2 chemoreflex shows a triphasic response, with a recovery to control by day 15 before significantly declining again by day 30 following CBD (Miller et al. 2013, 2014). Additionally, CBD in rats showed a sustained hypoventilation at rest without any measurable change in the acute ventilatory CO2 chemoreflex (Mouradian et al. 2012). The dissociation between steady‐state ventilation and the acute hypercapnic ventilatory response has also been observed in humans following CBD resulting from resection surgery, where a sustained steady‐state hypoventilation was observed for up to 32 months after CBD, despite a near‐complete recovery of the hypercapnic ventilatory response by 18 months after CBD (Dahan et al. 2007).
The apparent disconnect between steady‐state ventilation and the acute ventilatory CO2 chemoreflex during chronic experimental hypercapnia and CBD‐induced hypercapnia suggests separate mechanisms determining steady‐state ventilation and the acute ventilatory chemoreflex during chronic hypercapnia. Accordingly, one objective herein was to test the hypothesis that chronic hypercapnia per se will indeed cause a disconnect between steady‐state ventilation and the acute chemoreflex.
A second objective was to test the hypothesis that chronic hypercapnia per se will decrease cognitive function. This hypothesis was based on findings of reduced cognitive function in COPD patients, which parallels the progression of the disease (Incalzi et al. 1993; Antonelli‐Incalzi et al. 2006; Hung et al. 2009; Villeneuve et al. 2012), and studies of CO2 stresses, such as studies of environmental medicine showing an association between elevated InCO2 and cognitive decline (Fothergill et al. 1991; Satish et al. 2012; Allen et al. 2016). Patients with respiratory disease who also show cognitive decline have significantly higher mortality rates than patients with similar disease severity but normal cognitive function (Antonelli‐Incalzi et al. 2006; Chang et al. 2012). The specific aspects of cognition affected by respiratory diseases such as COPD vary widely, but include cognitive tasks such as verbal memory, attention, visuospatial memory, and executive functioning (Incalzi et al. 1997; Antonelli‐Incalzi et al. 2008; Klein et al. 2010; Torres‐Sanchez et al. 2015). The cause(s) of cognitive decline in these patients is largely unknown, but has been proposed to correlate with multiple comorbid factors in COPD patients, such as hypoxaemia, hypertension, hypercapnia, inflammation and many others (Dodd et al. 2010; Andrianopoulos et al. 2017). Determining the contributions of these associated factors on cognitive impairment within this patient population has been a major challenge given the presence of multiple comorbidities occurring simultaneously during COPD progression. However, the independent effect of chronic hypercapnia on cognitive function is unknown, and thus there is a need to determine whether hypercapnia per se leads to decreased cognitive function.
Methods
Ethical approval
All study protocols were reviewed and approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee which complies with the Public Health Services Policy on Humane Care and Use of Laboratory Animals (PHS Policy) and by extension all applicable provisions of the Animal Welfare Act and other Federal statutes and regulations relating to animals. The Medical College of Wisconsin has remained continuously accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, International (AAALACi) since 1968 (AALAC #000129). The investigators understand the ethical principles under which the journal operates, and the work herein complies with the journal animal ethics checklist.
Study population and conditions
Data were obtained from 18 female goats weighing 40–50 kg. All goats were reared and transported under conditions specified by the USDA. The goats were chronically housed and studied individually in two specially constructed plexiglass environmental chambers (3.5 × 4 × 6 ft); one where the goat was maintained under normocapnic conditions and another in which the CO2 levels could be increased. The temperature and relative humidity in the chambers were controlled and maintained within normal limits, and the photoperiods were fixed between 06.00 and 18.00 h daily. The goats were given access to feed and water ad libitum except during periods of study and 24 h fasting prior to surgery.
Surgical procedure
The goats underwent surgery to elevate the carotid arteries to subcutaneous levels for serial blood sampling, implant EMG wires into the diaphragm muscle and implant a data logger (StarOddi MilliHRT, Gardabaer, Iceland) subcutaneously near the axilla for continuous body temperature measurements. Briefly, the goats were anaesthetized with ketamine (i.v.), intubated, and mechanically ventilated with 2% isoflurane in 100% O2. For carotid elevation, carotid arteries were isolated from the vagi, elevated superficially from the muscle and sutured in place underneath the skin. For EMG implantation, the diaphragm was visualized between the 9th and 10th ribs, EMG electrodes were woven into the diaphragm, and externalized through the overlaying skin. Data loggers were implanted subcutaneously near the axilla. Following surgery, goats were administered flunixin meglumine (2 mg/kg, i.m.) once daily for 48 h for analgesia, and ceftiofur sodium (4 mg/kg, i.m.) daily to minimize infection. Following 2 weeks of recovery from surgery, carotid arteries were chronically catheterized through an indwelling catheter, which were flushed daily with both heparinized saline (0.1% heparin in saline) and heparin as a lock solution.
Measurements
For all physiological experiments, a mask was taped to the goat's snout to which a breathing valve was attached with the inspiratory port connected to a pneumotach, and the expiratory port connected to a Tissot gasometer. Inspiratory flow and diaphragmatic EMG activity were continually measured, and the data recorded and analysed digitally breath‐by‐breath through the software LabChart (ADInstruments, Colorado Springs, CO, USA) or Windaq (Dataq, Akron, OH, USA). Expired air was collected in a Tissot gasometer, and mixed expired gas composition was determined through a gas analyser (OxiGraph, Sunnyvale, CA, USA). Diaphragmatic EMG activity was amplified, collected, digitally filtered with a 100 Hz high‐pass filter, averaged, rectified, integrated and assessed breath by breath through LabChart. A chronically placed catheter into the carotid artery was used to sample blood and assess heart rate and blood pressure. Arterial blood gas and pH measurements were made through a Siemens blood gas analyser (Rapid Lab 248, Bayer Health Care, Leverkusen, Germany) and the data corrected for body temperature and ambient pressure at the time of blood draw. Arterial blood electrolytes were analysed in whole blood through an ABL800 FLEX (Radiometer, Copenhagen, Denmark).
Experimental procedure
Following surgery, the goats were allowed 2 weeks of recovery during which they were acclimatized to the environmental chambers while breathing room air. Thereafter, control studies were completed while breathing room air to establish baseline physiological variables. Inspiratory minute ventilation ( I), breathing frequency (f), tidal volume (V T), heart rate and blood pressure were assessed over the course of 1–3 h between 08.00 and 12.00 h. Arterial blood from the carotid artery was sampled every 20–30 min, placed on ice, and assessed for blood gasses and electrolytes upon completion of studies. During arterial blood sampling, mixed expired air was collected every 5 min and assessed for the fractional concentration of expired CO2 () and the fractional concentration of expired O2 (). Oxygen consumption (), CO2 excretion (), mixed expired oxygen (), and mixed expired CO2 () were subsequently calculated from the expired air.
The acute CO2/[H+] chemoreflex was assessed at the end of each 1–3 h study by elevating the InCO2 to 3%, 5% and 7% InCO2 for 5 min each, and arterial blood was sampled during minutes 3–5 of each exposure. CO2/[H+] sensitivity was calculated as the change in ventilation, integrated diaphragmatic EMG and ventilatory drive (V T/T I) responses as a function of the change in and arterial [H+] during acute elevations in InCO2.
Cognitive function was assessed utilizing a daily visual discrimination test, whereby goats were presented 10 times with two shapes (X and O) on the outside wall of the environmental chamber. One shape was randomly assigned as the correct shape, and selection of the correct shape by the goat's snout resulted in a food reward. Shape discrimination was chosen to assess cognitive function because goats have been shown to excel at this particular cognitive task (Baldwin, 1979; Blakeman & Friend, 1986; Langbein et al. 2004, 2006, 2007, 2008). Additionally, shape discrimination assesses aspects of cognitions such as executive function (Mar et al. 2013), which has been shown to be affected in COPD patients (Dodd et al. 2010; Villeneuve et al. 2012).
Following completion of room air control studies, InCO2 in the hypercapnic goat's chamber was elevated to 6% InCO2, where it remained for 30 days with the exception of brief (∼15 min) periods of room air exposure daily that were required for feeding, medication and flushing of the arterial catheter. We chose 6% InCO2 for chronic exposure, as this level of InCO2 elicits elevations in found in patients with moderate COPD severity, as well as previous studies of CBD‐induced hypercapnia in the goat (Miller et al. 2013, 2014). Steady‐state physiological studies were repeated as described above every 3–4 days during the hypercapnic exposure. For CO2/[H+] chemosensitivity assessment (during the hypercapnia period), InCO2 was elevated to 7% and 8% InCO2 for 5 min each at the end of the studies. The rationale for using 7% and 8% InCO2 was to allow for elevation above steady‐state InCO2. InCO2 of 8% was not used for chemosensitivity assessment during the room air control period because the goats would not behaviourally tolerate this level of InCO2 until chronically exposed to 6% InCO2 for at least 24 h. Lastly, predicted minute ventilation for each day of chronic hypercapnia for subsequent comparison to recorded minute ventilation was calculated as the difference between steady‐state arterial [H+] at room air and 6% InCO2 multiplied by the slope of the relationship between minute ventilation and arterial [H+] during acute chemosensitivity assessment. This value represented the predicted elevation in minute ventilation above control room air values and was added to the previously recorded room air control ventilation to derive the predicted I.
Euthanasia
Upon completion of exposure to 30 days of 6% InCO2 or room air, goats were sedated with a ketamine/xylazine mixture (24:1 v/v/) and killed by pentobarbital sodium and phenytoin sodium (B‐euthanasia).
Data and statistical analysis
Steady‐state ventilatory data
Steady‐state ventilatory data were acquired breath‐by‐breath, averaged into 5 min bins, and averaged over 1–3 h throughout each study. Arterial blood values, , , respiratory exchange ratio (RER), heart rate, blood pressure, mixed expired CO2, predicted ventilatory and electrolyte values measured over the course of each 1–3 h study were averaged to derive a mean value for each parameter during each day of study. The days in which the goats were studied during hypercapnia was maintained as consistent as possible, but small deviations were necessary due on occasion to non‐functional arterial catheters. As such, the days of study during hypercapnia were binned as follows: days 1, 2, 4–6, 7–9, 10–12, 14–16, 17–19, 20–22, 23–26 and 27–30. Steady‐state ventilation, arterial blood gasses, acid–base, metabolic, heart rate, blood pressure, mixed expired CO2, predicted ventilation and electrolyte data were then averaged across goats for each binned time range. Steady‐state ventilation, arterial blood gasses, acid–base, metabolic, heart rate, blood pressure, mixed expired CO2, and electrolyte data were subjected to a one‐way repeated measures ANOVA, with a Holm–Sidak post hoc test, and predicted ventilation to a two‐way ANOVA with time and condition (predicted or actual ventilation) as factors. Body temperature was recorded every 5 min throughout the protocol, and averaged for each hour of the day. Differences across days were subject to a two‐way repeated measures ANOVA, with time of day and duration of hypercapnia as factors.
Acute CO2/H+ chemoreflexes
The effect of chronic hypercapnia on the acute CO2/H+ chemoreflex during the first week of hypercapnia varied temporally between goats, thus individual nadir values for each index was used to represent a nadir point during the first week of hypercapnia. Subsequent days following the first week were binned as described for other physiological variables listed above. Differences across days were subject to a one‐way repeated measures ANOVA with a Holm‐Sidak post hoc test.
I/[H+] relationship
Differences in the I/[H+] relationship during chronic hypercapnia were assessed using an analysis of covariance (ANCOVA).
Cognitive function
Cognitive function scores during the control period were averaged and compared to scores during chronic hypercapnia using a one‐way repeated measures ANOVA with a Holm‐Sidak post hoc test.
Results
Steady‐state ventilatory adaptations during chronic hypercapnia
I (Fig. 1 A) initially increased to 335% of control on Day 1 of hypercapnia, but declined to 249% of control by Day 2 of hypercapnia (P < 0.001). Ventilation decreased to a nadir near Day 8 (∼230% of control) with minimal changes thereafter. The initial hyperpnoea upon exposure to 6% InCO2 was mediated through both an increase in breathing frequency (f) and tidal volume (V T) (P < 0.001) (Fig. 1 B,C). However, f showed a progressive decline during the first week of hypercapnia, whereas V T initially declined between the first 2 days of hypercapnia but stayed near initial CO2 exposure levels. This temporal pattern of ventilatory adaptation during chronic hypercapnia was paralleled by a reduction in expiratory time (T E) that was sustained throughout the hypercapnic exposure (P < 0.001), and an initial small decline (P < 0.001) and return toward normal in inspiratory time (T I), leading to a sustained elevation in ventilatory drive (V T/T I; P < 0.001; Fig. 2 A–C). Minimal changes occurred in any ventilatory variable during 30 days exposure to room air in the control goats (Table 1).
Figure 1.
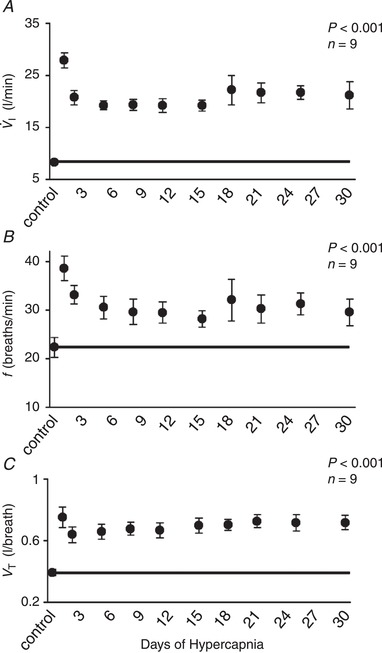
Temporal pattern of minute ventilation ( I), breathing frequency (f) and tidal volume (V T) during 30 days of exposure to 6% inspired CO2
Upon exposure to 6% InCO2 there is an initial increase in I to 335% of room air control values (A), and thereafter V T progressively declined to a nadir of ∼230% of control near day 8 and changed minimally thereafter (A). The initial hyperpnoea was mediated through both increased f and V T, but f returned toward control levels while V T remained elevated throughout the hypercapnic exposure (B, C). The solid lines provide a reference to control values obtained at room air prior to 6% InCO2.
Figure 2.
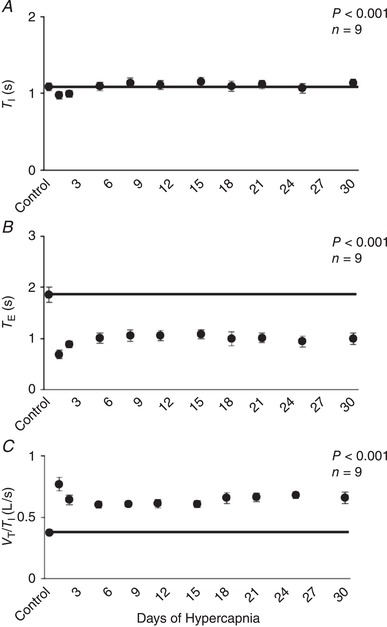
Temporal pattern of inspiratory time (T I), expiratory time (T E) and ventilatory drive (V T/T I) during 30 days of exposure to 6% inspired CO2
There was a small but significant reduction in T I during initial exposure to 6% InCO2, followed by a recovery to control during the first week of hypercapnia, with minimal changes thereafter (A). In contrast, T E remained significantly below control during 30 days of chronic hypercapnia (B), and there was a sustained elevation in ventilatory drive (C). The solid lines provide a reference to control values obtained at room air prior to 6% InCO2 exposure. P values shown were derived from a one‐way repeated measures ANOVA (time as factor).
Table 1.
Physiological parameters during 30 days of exposure to room air
| Control | Day 1 | Day 2 | Days 4–6 | Days 7–9 | Days 10–12 | Days 14–16 | Days 17–19 | Days 20–22 | Days 23–26 | Days 27–30 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| I (L/min) | 7.37 ± 0.63 | 7.41 ± 0.51 | 7.45 ± 0.68 | 6.79 ± 0.61 | 7.36 ± 0.67 | 7.42 ± 0.8 | 7.21 ± 0.75 | 7.18 ± 0.83 | 7.68 ± 0.94 | 7.39 ± 0.76 | 6.92 ± 0.83 |
| f (breaths/min) | 21.76 ± 1.34 | 21.33 ± 1.64 | 20.73 ± 1.92 | 19.26 ± 1.21 | 21.11 ± 2.04 | 20.27 ± 1.97 | 19.9 ± 2.14 | 20.83 ± 1.84 | 20.59 ± 2.00 | 22.28 ± 1.94 | 20.66 ± 0.94 |
| V T (L) | 0.34 ± 0.02 | 0.36 ± 0.02 | 0.37 ± 0.02 | 0.36 ± 0.02 | 0.36 ± 0.01 | 0.37 ± 0.01 | 0.37 ± 0.01 | 0.35 ± 0.02 | 0.38 ± 0.02 | 0.34 ± 0.02 | 0.34 ± 0.03 |
| T I (s) | 1.12 ± 0.06 | 1.13 ± 0.06 | 1.17 ± 0.06 | 1.21 ± 0.05 | 1.17 ± 0.07 | 1.21 ± 0.06 | 1.20 ± 0.06 | 1.17 ± 0.07 | 1.14 ± 0.09 | 1.06 ± 0.10 | 1.15 ± 0.08 |
| T E (s) | 1.86 ± 0.13 | 1.81 ± 0.14 | 1.89 ± 0.18 | 2.02 ± 0.16 | 1.85 ± 0.22 | 1.99 ± 0.23 | 2.07 ± 0.21 | 1.91 ± 0.21 | 1.81 ± 0.15 | 1.83 ± 0.28 | 1.82 ± 0.12 |
| V T/T I (L/s) | 0.32 ± 0.02 | 0.33 ± 0.02 | 0.32 ± 0.02 | 0.30 ± 0.02 | 0.31 ± 0.02 | 0.32 ± 0.02 | 0.32 ± 0.02 | 0.32 ± 0.02 | 0.32 ± 0.03 | 0.33 ± 0.03 | 0.30 ± 0.03 |
| (mmHg) | 40.08 ± 1.21 | 39.97 ± 0.62 | 39.38± 1.42 | 40.43 ± 1.27 | 41.78± 1.95 | 38.17 ± 1.35 | 39.17 ± 0.89 | 41.38± 1.82 | 40.62 ± 0.95 | 40.15 ± 1.07 | 39.8 ± 1.22 |
| (mmHg) | 93.77 ± 2.84 | 93.24 ± 2.98 | 92.23 ± 2.38 | 87.3 ± 3.20 | 94.14 ± 3.39 | 97.45 ± 1.24 | 93.38 ± 3.61 | 93.03 ± 2.98 | 96.28 ± 5.18 | 89.99 ± 3.82 | 85.86 ± 3.91 |
| pH | 7.45 ± 0.01 | 7.45 ± 0.01 | 7.45 ± 0.01 | 7.45 ± 0.01 | 7.45 ± 0.01 | 7.45 ± 0.01 | 7.44 ± 0.01 | 7.45 ± 0.01 | 7.45 ± 0.01 | 7.45 ± 0.01 | 7.45 ± 0.01 |
| [H+] (nmol/L) | 35.88 ± 0.47 | 35.62 ± 0.54 | 35.66 ± 0.71 | 35.62 ± 0.50 | 35.84 ± 0.78 | 35.42 ± 0.76 | 36.02 ± 0.98 | 35.24 ± 0.60 | 35.41 ± 0.96 | 35.64 ± 0.86 | 35.23 ± 0.53 |
| [HCO3 +] (mEq/L) | 26.72 ± 0.98 | 26.80 ± 0.70 | 26.37 ± 1.07 | 27.05 ± 0.75 | 27.75 ± 0.86 | 25.71 ± 1.14 | 26.00 ± 0.96 | 28.01 ± 1.34 | 27.45 ± 1.03 | 26.89 ± 0.89 | 26.98 ± 1.15 |
| [H+]/ (nmol.L/mmHg) | 0.90 ± 0.03 | 0.89 ± 0.02 | 0.91 ± 0.04 | 0.89 ± 0.02 | 0.86 ± 0.03 | 0.93 ± 0.04 | 0.92 ± 0.03 | 0.86 ± 0.04 | 0.87 ± 0.03 | 0.89 ± 0.03 | 0.89 ± 0.04 |
| [Cl–] (mmol/L) | 107.6 ± 0.83 | 109.3 ± 1.45 | 107.8 ± 1.07 | 108.3 ± 1.50 | 107.5 ± 1.5 | 108.0 ± 0.58 | 108.8 ± 1.25 | 107.0 ± 1.35 | 106.8 ± 0.58 | 107.5 ± 1.32 | 104.8 ± 1.78 |
| [Na+] (mmol/L) | 143.3 ± 0.66 | 143.6 ± 0.67 | 143.2 ± 0.73 | 144.8 ± 0.48 | 145.0 ± 1.0 | 143.3 ± 0.89 | 143.8 ± 0.48 | 145 ± 0.41 | 143.6 ± 0.24 | 143.5 ± 0.87 | 143.8 ± 0.49 |
| [Haemoglobin] (g/dL) | 8.19 ± 0.35 | 7.80 ± 0.76 | 7.84 ± 0.53 | 7.70 ± 0.64 | 7.75 ± 0.95 | 6.90 ± 0.62 | 7.30 ± 0.44 | 7.13 ± 0.55 | 7.86 ± 0.60 | 7.43 ± 0.58 | 7.12 ± 0.39 |
| [K+] (mmol/L) | 4.11 ± 0.07 | 4.10 ± 0.10 | 4.18 ± 0.07 | 4.15 ± 0.12 | 4.00 ± 0.10 | 4.13 ± 0.12 | 4.13 ± 0.05 | 3.85 ± 0.20 | 4.06 ± 0.09 | 4.00 ± 0.07 | 3.86 ± 0.10 |
| [Ca2+] (mmol/L) | 1.07 ± 0.01 | 1.12 ± 0.01 | 1.06 ± 0.02 | 1.06 ± 0.01 | 1.06 ± 0.02 | 1.03 ± 0.02 | 1.05 ± 0.01 | 1.00 ± 0.03 | 1.05 ± 0.01 | 1.04 ± 0.01 | 1.02 ± 0.02 |
| Blood glucose (mg/dL) | 66.0 ± 1.65 | 70.67 ± 13.20 | 70.20 ± 7.39 | 64.00 ± 4.24 | 58.50 ± 3.50 | 58.00 ± 1.15 | 58.50 ± 1.04 | 60.75 ± 2.25 | 60.6 ± 2.73 | 62.75 ± 2.17 | 66.00 ± 7.78 |
| ∆ I/∆ (L.min/mmHg) | 1.72 ± 0.20 | – | 1.93 ± 0.22 | 1.80 ± 0.24 | 2.30 ± 0.40 | 2.06 ± 0.51 | 2.10 ± 0.36 | 1.47 ± 0.26 | 1.81 ± 0.26 | 1.58 ± 0.28 | 1.57 ± 0.16 |
| ∆ I/∆[H+] (L.min/nmol.L) | 2.44 ± 0.20 | – | 2.48 ± 0.31 | 2.26 ± 0.34 | 2.38 ± 0.39 | 2.40 ± 0.48 | 2.78 ± 0.56 | 1.96 ± 0.10 | 2.47 ± 0.14 | 2.09 ± 0.25 | 2.18 ± 0.35 |
| ΔVT/T I/Δ (L.s/mmHg) | 0.04 ± 0.00 | – | 0.05 ± 0.00 | 0.04 ± 0.00 | 0.05 ± 0.01 | 0.05 ± 0.01 | 0.05 ± 0.01 | 0.04 ± 0.01 | 0.04 ± 0.01 | 0.05 ± 0.00 | 0.04 ± 0.00 |
| ΔV T/T I/Δ[H+] (L.s/nmol.L) | 0.06 ± 0.01 | – | 0.06 ± 0.00 | 0.05 ± 0.01 | 0.05 ± 0.01 | 0.06 ± 0.01 | 0.06 ± 0.01 | 0.05 ± 0.00 | 0.06 ± 0.00 | 0.06 ± 0.00 | 0.05 ± 0.01 |
| Δ∫Diaphragm EMG/Δ (mV/nmol.L) | 0.01 ± 0.02 | – | 0.01 ± 0.01 | 0.01 ± 0.01 | 0.07 ± 0.02 | 0.01 ± 0.02 | 0.04 ± 0.04 | 0.01 ± 0.02 | 0.03 ± 0.02 | 0.06 ± 0.02 | 0.05 ± 0.01 |
| Δ∫Diaphragm EMG /Δ[H+] (mV/nmol.L) | 0.01 ± 0.03 | – | 0.01 ± 0.01 | 0.01 ± 0.01 | 0.01 ± 0.02 | 0.01 ± 0.02 | 0.01 ± 0.06 | 0.01 ± 0.02 | 0.04 ± 0.03 | 0.07 ± 0.02 | 0.06 ± 0.02 |
During 30 days of exposure to room air, there were minimal changes in any of the physiological parameters studied. The exception was a significant increase in [Ca2+] during day 1 of room air exposure, which returned to control thereafter.
Arterial blood adaptations during chronic hypercapnia
increased 10 torr upon initial exposure to 6% InCO2 (Fig. 3 A). Thereafter there was a further 5 torr increase in during the first week of hypercapnia, so that remained ∼15 torr above control for the duration of hypercapnic exposure (P < 0.001). The level of inspired O2 was not controlled within the environmental chamber, and thus remained near an of 19.5% during hypercapnic exposure. However, due to the increase in ventilation, remained above control levels throughout the chronic hypercapnia (P < 0.001) (Fig. 3 D). Minimal changes occurred in (P = 0.112) and (P = 0.302) during 30 days of exposure to room air in the control goats (Table 1).
Figure 3. Arterial blood adaptations during 30 days of exposure to 6% InCO2 .
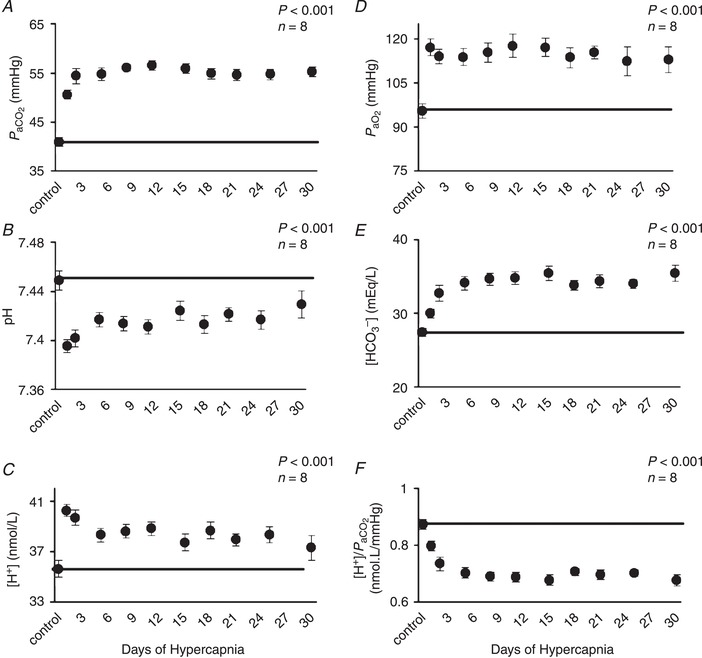
A, increased by 10 mmHg during the first day of increased InCO2, followed by an additional 5 mmHg increase between days 1 and 2 of hypercapnia so that was ∼15 mmHg above control for the remainder of hypercapnic exposure. Arterial pH (B) initially decreased 0.05 units on the first day of hypercapnia, followed by a return towards control due to a nearly 8 mEq/L increase in arterial [HCO3 −] (D), leading to a ∼0.03 unit acidosis throughout hypercapnic exposure. C, changes in arterial [H+] mirrored changes in pH. F, the increase in HCO3 − was reflected by the increase in steady‐state buffering capacity. D, InO2 was not controlled during exposure to 6% InCO2, and thus fell to ∼19.5% InO2, although remained ∼20 mmHg above control throughout hypercapnic exposure. The solid lines provide a reference to control values obtained at room air prior to 6% InCO2 exposure. P values shown were derived from a one‐way repeated measures ANOVA (time as factor).
Arterial pH initially decreased (−0.05 pH units) relative to control during the first hour of hypercapnic exposure (P < 0.001) (Fig. 3 B), but thereafter there was a time‐dependent return towards control due to a ∼8 mEq/L increase in [HCO3 −] during the first week of hypercapnic exposure (Fig. 3 E). Changes in the chronic steady‐state arterial [H+] (Fig. 3 C) presumably reflect changes in the ventilatory stimulus level at the carotid and intracranial chemoreceptors, based upon the assumption that changes in CSF and arterial [H+] during the chronic state are similar during experimental (elevated InCO2) hypercapnia (Siesjo, 1972). Based on this assumption, the stimulus to breathe is initially elevated above control during the initial exposure to 6% InCO2 (P < 0.001) but then is partially buffered during the first week of hypercapnia due to the large increase in arterial [HCO3 −] (P < 0.001) or the increase in arterial buffering capacity, represented by a sustained reduction in [H+]/ during chronic hypercapnia (P < 0.001; Fig. 3 F). Importantly, the buffering of the arterial blood, while greatly elevated during chronic hypercapnia, does not completely restore acid–base balance resulting in a sustained slight acidosis (−0.03 pH units or ∼2.5 nmol/L in [H+]; Fig. 3 B,C). In contrast, we found minimal or no changes in pH (P = 0.962), [HCO3 −] (P = 0.066), [H+] (P = 0.959) or buffering capacity ([H+]/) (P = 0.055) throughout the 30 days of exposure to room air in the control goats (Table 1).
Arterial blood electrolytes during chronic hypercapnia
The largest change in blood electrolytes was a decrease in arterial [Cl−] during the first week of chronic hypercapnia (P < 0.001; Fig. 4 A), which was concurrent with the observed increases in arterial [HCO3 −]. Other electrolyte adaptations during chronic hypercapnia included a small, but significant increase in arterial [K+] (P = 0.006) (Fig. 4 D), and a decrease in arterial haemoglobin (P < 0.001) (Fig. 4 C) without effects in arterial [Na+] (P = 0.748), [Ca2+] (P = 0.335) or blood glucose (P = 0.727) (Fig. 4 B,E,F). During 30 days of room air exposure, there were no changes in measured electrolytes throughout the 30‐day protocol with the exception of a small but significant increase in arterial [Ca2+] during the first day (P = 0.005; Table 1).
Figure 4. Arterial blood electrolyte adaptations during 30 days of chronic exposure to 6% InCO2 .
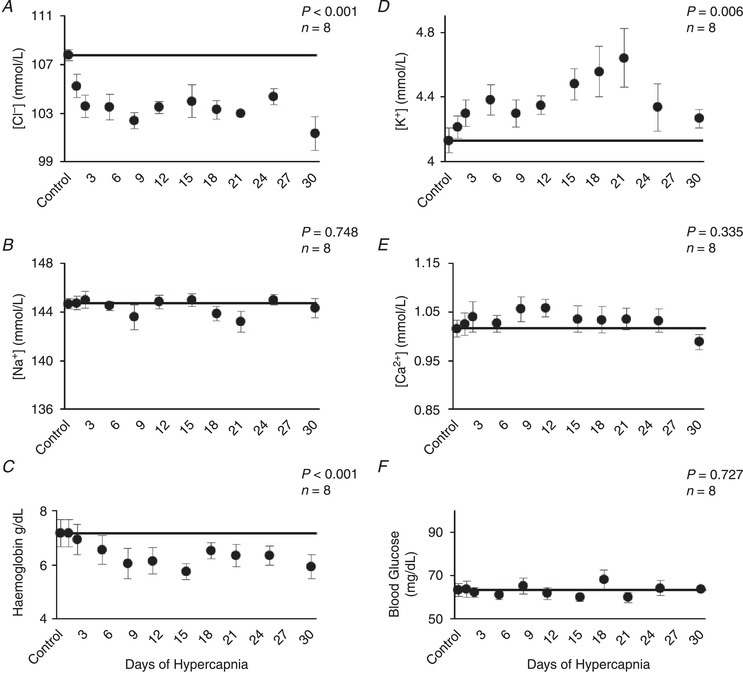
Arterial chloride decreased ∼5 mmol/L during the first week of chronic hypercapnia (A), coincident with increases in arterial [HCO3 −] (Figure 3). Thereafter, arterial chloride remained below control for the duration of chronic hypercapnia. Arterial potassium showed a small but significant increase during the first week of hypercapnia (D), that was maintained throughout hypercapnic exposure. Haemoglobin decreased ∼1 g/dL during the first week of hypercapnia and remained below control (C). Minimal changes were noted in arterial sodium (B), calcium (E) and blood glucose (F), during chronic hypercapnia. The solid lines provide a reference to control values obtained at room air prior to 6% InCO2 exposure. P values shown were derived from a one‐way repeated measures ANOVA (time as factor).
Metabolic rate, heart rate, blood pressure and body temperature adaptations during chronic hypercapnia
followed a similar pattern to ventilation during chronic hypercapnia, which increased during initial exposure to the elevated InCO2 (P < 0.001) followed by a sustained attenuation for the remainder of the 30 days (Fig. 5 A). Despite the large increase in during chronic hypercapnia, remained at control levels for the duration of the protocol (P = 0.161), resulting in a significant reduction in the RER (P = 0.011) (Fig. 5 B,C). Heart rate and blood pressure increased ∼10 beats/min (P < 0.001) and 10 mmHg (P < 0.001), respectively, above control at Day 30 of hypercapnia (Fig. 6 A,B), whereas there were no changes in heart rate (P = 0.994) or blood pressure (P = 0.755) in control goats during 30 days of room air exposure (Fig. 6 C,D). There were minimal changes in body temperature from baseline to Day 3 of chronic hypercapnia, although body temperature was reduced ∼0.5°C by Day 30 of chronic hypercapnia (P = 0.041; Fig. 7 A). In contrast, we found minimal changes in body temperature in control room air‐exposed goats (P = 0.867; Fig. 7 B).
Figure 5. Oxygen consumption (), CO2 excretion () and respiratory exchange ratio (RER) during 30 days of chronic exposure to 6% InCO2 .
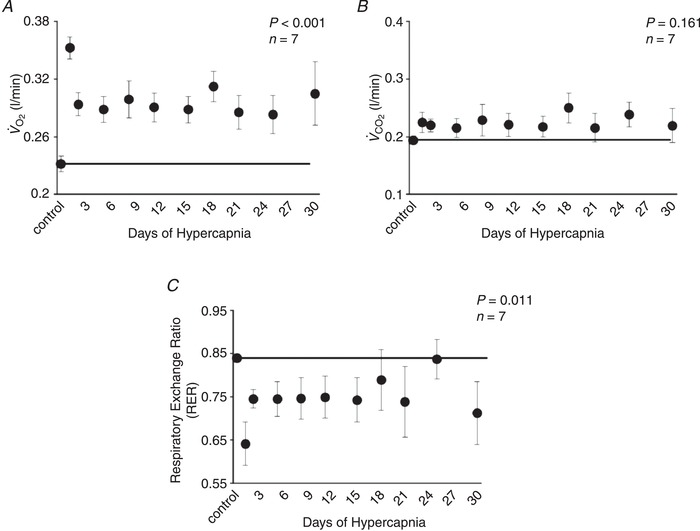
increased 0.12 L/min from control during initial exposure to 6% InCO2 (A), followed by an attenuation between days 1 and 2 of chronic hypercapnia. Thereafter, remained ∼0.06 L/min above control for the remainder of hypercapnic exposure. In contrast, (B) remained near control levels during 30 days of chronic hypercapnia, resulting in a sustained reduction in RER (C), which is consistent with increased tissue storage of CO2. The solid lines provide a reference to control values obtained at room air prior to 6% InCO2 exposure. P values shown were derived from a one‐way repeated measures ANOVA (time as factor).
Figure 6. Heart rate and mean arterial blood pressure during 30 days of exposure to 6% InCO2 or room air.
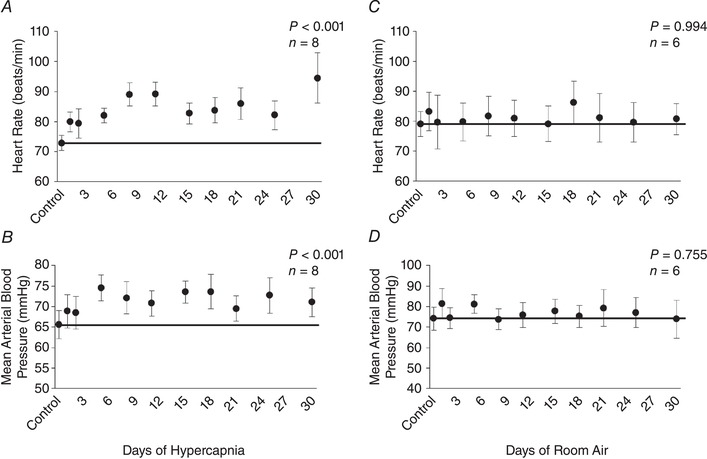
Heart rate increased during initial exposure to 6% InCO2 (A) and remained ∼10 beats/min above control throughout 30 days of chronic hypercapnia (A). Similarly, mean arterial blood pressure increased by ∼10 mmHg during the first week of hypercapnia, and remained elevated thereafter (B). Minimal changes were noted in either heart rate or mean arterial blood pressure during 30 days of room air exposure (C, D). The solid lines provide a reference to control values obtained at room air prior to 30 days of 6% InCO2 or room air exposure. P values shown were derived from a one‐way repeated measures ANOVA (time as factor).
Figure 7. Body temperature (°C) during 30 days of chronic exposure to 6% InCO2 or room air.
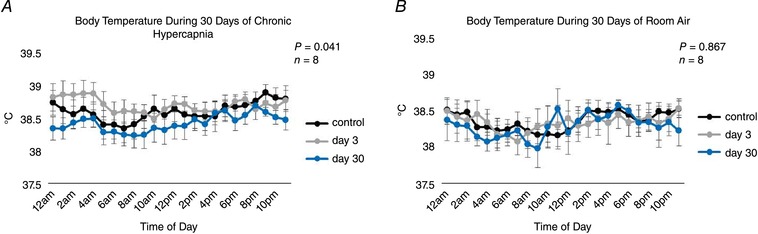
There were minimal changes in body temperature during the first 3 days of hypercapnic exposure, but body temperature declined throughout the remainder of chronic hypercapnia and was ∼0.5°C below control by day 30 of chronic hypercapnia (A). No changes in body temperature were noted during 30 days of exposure to room air (B). P values shown represent the interaction term between time of day and duration of hypercapnia with a two‐way repeated measures ANOVA.
Arterial–mixed expired difference during chronic hypercapnia
The arterial–mixed expired difference declined from a control value of 19.0 mmHg during room air exposure to near 0 mmHg during the steady state upon initial exposure to hypercapnia (P < 0.001) and remained near this level for the duration of the 30 days of hypercapnic exposure (Fig. 8 A). Similarly, the arterial–mixed expired difference observed during the acute chemoreflex challenges was either near or less than 0 mmHg during acute exposure to 7% and 8% InCO2, respectively (Fig. 8 C,D).
Figure 8. Arterial–mixed expired CO2 difference during chronic exposure to 6% InCO2 and acute exposure to 7% and 8% InCO2 .
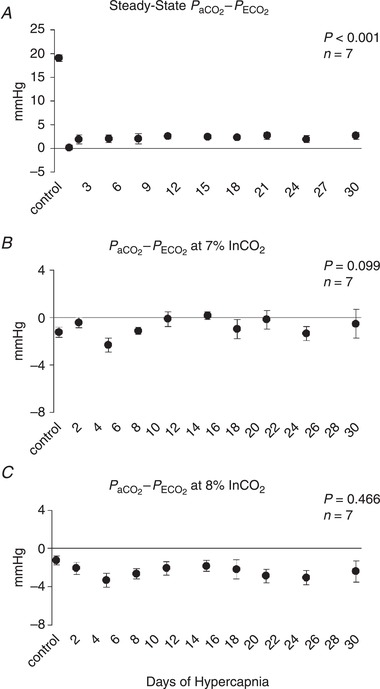
During 30 days of exposure to 6% InCO2, the arterial–mixed expired CO2 difference fell to near 0 mmHg (A), suggesting an increased contribution of gastric CO2 to the mixed expired CO2. Acute exposure of 5 min each to 7% (B) and 8% InCO2 (C) resulted in a near (7%) or below (8%) 0 mmHg arterial–mixed expired CO2 difference, with minimal changes in this response during 30 days of chronic hypercapnia. P values shown were derived from a one‐way repeated measures ANOVA (time as factor).
Acute CO2/[H+] chemosensitivities
Periodically throughout the experiments, InCO2 was elevated acutely above steady‐state levels (3, 5 and 7% InCO2 during control periods; 7% and 8% InCO2 during chronic hypercapnia) to determine the acute ventilatory CO2/[H+] chemosensitivity, represented as the slope of the Δ I/Δ and Δ I/Δ[H+] relationship. The acute CO2 chemoreflex response of each goat decreased during the first week of hypercapnia, but this reduction occurred at variable times during the first week in each goat. When the responses are plotted as the acute chemosensitivities during the control period, the nadir during the first week, and across the remainder of chronic hypercapnia, there is a significant decline in both the Δ I/Δ and Δ I/Δ[H+] relationship during the first week of hypercapnia (P < 0.05), followed by a return to control levels (Fig. 9 A,B). Similar responses were noted in the slopes of ventilatory drive to H+ (ΔV T/T I/Δ[H+]) and (ΔV T/T I/Δ) responses during acute CO2 chemoreflex challenges (P < 0.05) (Fig. 9 C,D). In contrast, the slopes of diaphragm activity relative to H+ and (Δ∫Dia EMG/Δ[H+] and Δ∫Dia EMG/Δ, respectively) showed a sustained reduction throughout chronic hypercapnia (P < 0.01), suggesting further recruitment of other respiratory muscles to maintain the acute ventilatory responses during hypercapnia (Fig. 9 E,F). There were no changes in any index of acute CO2 chemosensitivity during 30 days of exposure to room air (P > 0.05; Table 1).
Figure 9.
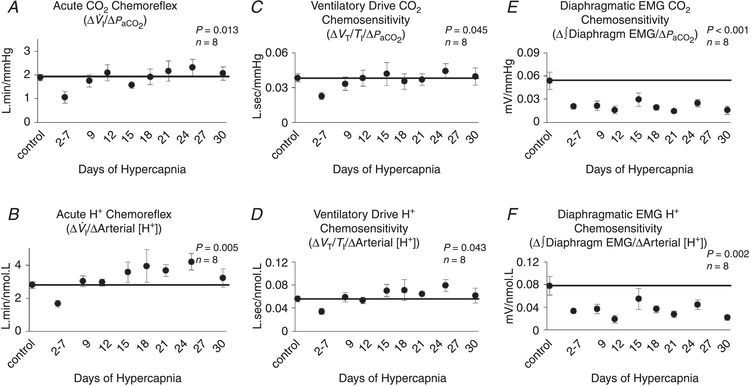
Temporal pattern of the acute ventilatory CO2/H+ chemoreflex during 30 days of chronic exposure to 6% InCO2 or room air
The acute CO2 (∆ I/∆) and H+ (∆ I/∆[H+]) chemoreflexes were significantly decreased during the first week of chronic hypercapnia (A, B). The time at which this nadir occurred within the first week varied temporally between goats, and thus the nadir for each goat is represented between days 2 and 7 of hypercapnia. The initial decline was followed by a recovery to at or near control levels by the end of the first week for both indices of chemosensitivity (A and B). Ventilatory drive (V T/T I) CO2 and H+ chemosensitivity followed a similar pattern with a decline during the first week of hypercapnia, followed by recover to at or near control levels (C, D). Lastly, diaphragm muscle EMG activity during chemoreflex challenges remained below control at nearly all time points (E, F), indicating further respiratory muscle recruitment to maintain ventilation during the chemoreflex challenges. The solid lines provide a reference to control values obtained prior to chronic hypercapnic exposure. P values shown were derived from a one‐way repeated measures ANOVA (time as factor).
Predicted vs. actual ventilation during chronic hypercapnia
Based upon the levels of arterial [H+] and the acute ventilatory [H+] chemosensitivity slopes (Δ I/Δ[H+]) measured during chronic hypercapnia, we calculated the predicted I during chronic hypercapnia (Fig. 10). Due to the incomplete arterial blood buffering and sustained slight elevation in arterial [H+] during chronic hypercapnia, steady‐state ventilation would be predicted to be ∼7 L/min above control throughout chronic hypercapnia. However, the measured ventilation significantly exceeded the predicted I for each day of chronic hypercapnia (P < 0.001), suggesting a disconnect between steady‐state ventilation and the acute CO2/[H+] chemoreflex responses during chronic hypercapnia.
Figure 10. Actual vs. predicted minute ventilation during 30 days of chronic exposure to 6% InCO2 .
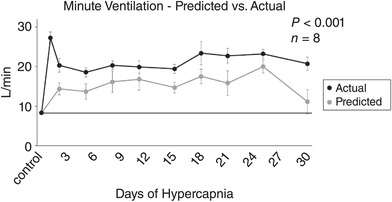
Minute ventilation of the goats during 30 days of chronic hypercapnia remained significantly above the level of ventilation that would be predicted from the steady‐state level of arterial [H+] and the results from the acute CO2/[H+] chemoreflex for each day of chronic hypercapnia. The sustained elevation of measured ventilation above the predicted values represents a disconnect between steady‐state ventilation and the acute chemoreflex during chronic hypercapnia, and a shift in the ventilatory set‐point to an elevated level. The solid line provides a reference to control values obtained prior to 6% InCO2 exposure. The P value shown was derived from a two‐way ANOVA (predicted vs. actual ventilation and time as factors).
Shift in ventilatory set‐point during chronic hypercapnia
The ventilatory set‐point represents the relationship between ventilation and ventilatory stimuli ([H+]) during a steady state. Shown in Fig. 11 is the relationship between ventilation and arterial [H+] during steady state, and during the acute CO2/[H+] chemoreflex challenges. This relationship is plotted both during the control period prior to hypercapnic exposure, and following 30 days of chronic hypercapnia. The first point in each line represents the steady‐state ventilatory set‐point, and the slope of the relationship during increasing [H+] represents the gain of the system to challenges of H+ that exceed this set‐point. In other words, the set‐point value represents homeostasis during the steady state, and the slope represents the function of the error sensing mechanism to deviations in blood gases/acid–base status. During chronic hypercapnia, this relationship was leftward‐shifted so that ventilation for any given arterial [H+] is at an elevated level (P < 0.001). However, the slope of the ventilatory response to acute increases in InCO2 remains unaltered, consistent with a shift in set‐point to an elevated level with no change in gain of the system (Fig. 11).
Figure 11. Relationship between ventilation and arterial [H+] during 30 days of exposure to 6% InCO2 .
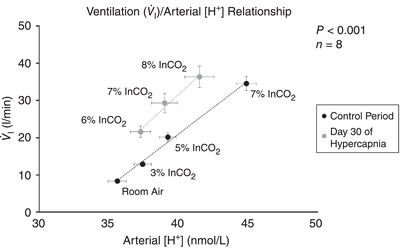
Following 30 days of chronic exposure to 6% InCO2 there is a leftward shift in the relationship between ventilation and arterial [H+], suggesting a shift in the ventilatory set‐point to an elevated level, such that ventilation for a given concentration of H+ in the arterial blood is greater during chronic hypercapnia than during control. However, the gain of the error‐sensing mechanism to acute changes in arterial H+ from homeostasis, represented by the slope of the I/H+ relationship during acute increases in arterial H+, remains unchanged following 30 days of chronic hypercapnia. The P value represents the significance term from an analysis of covariance (ANCOVA).
Cognitive function declines during chronic hypercapnia
Cognitive function assessments based upon a shape discrimination test declined by nearly 50% during the first week of hypercapnia, and remained below control throughout the 30 days of chronic hypercapnia (P < 0.001; Fig. 12 A). Minimal changes in cognitive function performance were noted in goats exposed to 30 days of room air (P = 0.380; Fig. 12 B).
Figure 12. Number of correct shape selections during 30 days of exposure to 6% InCO2 or room air.
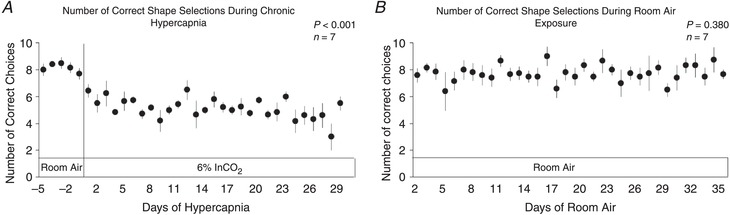
Daily, goats were presented 10 times with two shapes (X and O) and positively rewarded with food immediately upon a correct choice. There was a significant decline in shape selection performance during 30 days of chronic exposure to 6% InCO2 (A), with minimal changes in performance during 30 days of exposure to room air (B). P values shown were derived from a one‐way repeated measures ANOVA (time as factor).
Discussion
Consistent with some of the previous studies using chronic increased InCO2 (Schaefer, 1949; Schaefer et al. 1963a; Clark et al. 1971; Pingree, 1977; Guillerm & Radziszewski, 1979; Lai et al. 1981; Jennings & Davidson, 1984), we demonstrated here in adult goats a biphasic hyperpnoea during 30 days of chronic hypercapnia, where steady‐state ventilation increased and then partially returned toward baseline where it remained elevated thereafter. The initial respiratory acidosis (during the uncompensated phase of hypercapnia exposure) was also partially mitigated over time due to an apparent renal compensation providing a large increase in arterial [HCO3 −]. During chronic hypercapnia, there was a transient decrease in the ventilatory CO2/H+ chemoreflex, but this response returned toward normal despite the sustained increase in steady‐state ventilation, resulting in a disconnect between the acute CO2/H+ chemoreflex and steady‐state ventilation. Finally, we found a substantial and sustained decrease in cognitive function during chronic hypercapnia.
Dissociation of steady‐state breathing and acute CO2 chemoreflexes during chronic hypercapnia
Of note are two important assumptions that were made regarding the chemical control of ventilation and our interpretation of the relationship between the acid–base status and the temporal pattern of ventilation: (1) the primary stimulus for ventilation is [H+](Winterstein, 1956; Nattie, 2011), and (2) changes in CSF [H+] may be inferred by changes in arterial [H+] during the steady state in experimental hypercapnia (Siesjo, 1972), i.e. arterial [H+] provided a close approximation of the major ventilatory stimulus within the CNS. Accordingly, the greatest hyperpnoea occurred within minutes to hours of exposure to 6% InCO2, presumably due to the ensuing acidosis from the increase in and minimal initial change in [HCO3 −]. continued to rise over the next 2–5 days of chronic hypercapnia, and remained near that level for the remainder of hypercapnic exposure. Despite this secondary increase in , the initial hyperpnoea was attenuated, probably due to a decrease in steady‐state [H+] and thus stimuli for ventilation as a result of renal compensation to increase [HCO3 −], causing an increase in buffering capacity (shown by a smaller [H+]/). Thereafter, ventilation and arterial [H+] were essentially ‘clamped’ at this new steady‐state level, leading to a sustained hyperpnoea that remained for the duration of hypercapnic exposure.
In contrast, the temporal pattern of the acute ventilatory CO2/H+ chemoreflex during chronic hypercapnia showed a different pattern than the steady‐state ventilation, and/or arterial [H+]. Initially, there was a decline in ventilatory responses to acute increases in CO2 and [H+], but after ∼1 week both indices of acute ventilatory chemosensitivity returned to baseline values during the remainder of the study. These results suggest that the acute CO2/H+ chemoreflex does not predict steady‐state ventilation during chronic hypercapnia, consistent with a disconnect between the acute CO2/H+ chemoreflex and steady‐state ventilation during chronic hypercapnia. The disconnect becomes particularly apparent by the significant difference between the predicted ventilation compared to measured ventilation throughout chronic hypercapnia, where measured ventilation exceeded predicted ventilation (Fig. 10). Furthermore, there was a leftward shift in the I/[H+] relationship, without a change in the slope of the I/[H+] relationship following ∼1 week of chronic hypercapnia (Fig. 11). These data suggest that the relationship between ventilation and any given amount of ventilatory stimulus ([H+]) during the steady state was elevated, consistent with the conclusion of a shift in the ventilatory set‐point to an elevated level.
What mechanism(s) can account for a shift in the ventilatory set‐point to an elevated level during chronic hypercapnia without changing the gain/slope of the acute CO2 chemoreflex? First, our data show that the ventilatory CO2 chemoreflex is unaffected or transiently reduced (in the early phase of the response), suggesting that a generalized increase in cellular CO2/pH sensitivity does not constitute a mechanism for an increased ventilatory set‐point during chronic hypercapnia. CNS cell populations with intrinsic cellular CO2/pH sensitivity, such as glutamatergic retrotrapezoid nucleus (RTN), serotonergic raphe and catecholaminergic locus coeruleus (among other) neurons, are thought to be major contributors to the acute ventilatory CO2 chemoreflex through changes in firing rates that ‘encode’ CO2/pH. However, increases in cellular sensitivity to respiratory acidosis would probably affect resting ventilation and the slope of the CO2 response through generalized increases in tonic activity during chronic acidosis and/or excitatory neuromodulation.
It has long been questioned as to why there are presumably multiple ‘sites’ of central respiratory chemoreceptors within the CNS. One postulate is that there are multiple sites in order to match homostatic demands under changing conditions, providing a dynamic and responsive system that attempts to maintain pH homeostasis. One possible explanation for the shift in set‐point could be that during chronic changes in the environment, or in the context of disease, various cell populations take on new roles in the control of breathing and pH regulation. For example, the observed shift in the ventilatory set‐point during chronic hypercapnia may involve a presynaptic mechanism involving both slowly adapting tonic, and rapidly adapting phasic chemosensitive populations of neurons that ‘drive’ the ventilatory control network, acting to distinctly modulate steady‐state ventilation and the acute chemoreflex (Fig. 13). Separate tonic and phasic chemosensitive neuronal populations have been identified within the brainstem (Fukuda et al. 1980; Arita et al. 1988; Nattie et al. 1993), and thus it is possible that some of the known CO2/pH chemoreceptor neurons may alter their contributions during chronic hypercapnia whereby the tonically active chemosensitive neurons would remain highly active during chronic hypercapnia, while phasic chemosensitive neurons may ‘reset’ to this newly established set‐point.
Figure 13. Proposed mechanism(s) accounting for the shift in ventilatory set‐point during 30 days of exposure to 6% InCO2 .
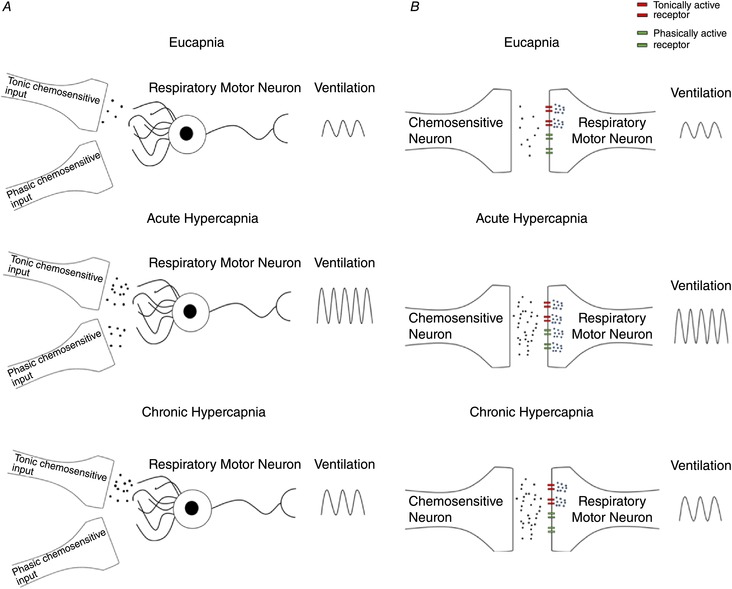
The shift in ventilatory set‐point may involve either presynaptic and/or postsynaptic mechanisms. A, the proposed presynaptic mechanism involving both tonic and phasic chemosensitive input into a respiratory motor neuron. Under eucapnic conditions, the primary input onto the respiratory motor neuron is from tonic presynaptic chemosensitive input. During acute hypercapnia, input from the tonic presynaptic input is elevated, alongside increased input from the phasically chemosensitive presynaptic neuron. During chronic hypercapnia, the phasically active chemosensitive input is silenced, whereas elevated input from the tonic chemosensitive presynaptic neuron is maintained. B, the alternative postsynaptic mechanism accounting for a shift in the ventilatory set‐point during chronic hypercapnia. In this schematic, there is the presence of both tonically active and phasically active receptors upon the postsynaptic membrane of a respiratory motor neuron. Under eucapnic conditions, activity from the presynaptic chemosensitive neuron activates the tonically active receptors upon the postsynaptic membrane of the respiratory motor neuron. However, during acute hypercapnia, there is both elevated activity of the tonically active receptors, and contribution from the phasically active receptors. During chronic hypercapnia, the phasically active receptors are silenced, whereas activity from the tonically active receptors is maintained at an elevated level.
An alternative explanation for the shift in set‐point could be the presence of a postsynaptic mechanism with both tonic and phasically active receptors of respiratory motor/pre‐motor neurons receiving input from chemosensitive cells within the respiratory network (Fig. 13). For example, the presence of tonically active receptors receiving input from chemosensitive nuclei may be constitutively active during the steady state by responding to a tonic homeostatic input from chemosensitive nuclei. In contrast, phasically active receptors within the same neurons may provide a mechanism to translate information about rapid changes in signal intensity, as would be the case during acute CO2/H+ challenge. However, phasically active receptors would reset during sustained input to the neuron(s), as would be the case during chronic hypercapnia, in order to give responsiveness on top of a tonically elevated input signal.
We note that our data suggest both hypothesized mechanisms, but provide no direct evidence of any proposed molecular mechanism. Further work investigating the network properties of neuronal and/or glial responses to chronic hypercapnia is needed to determine the phenomena accounting for the shift in the ventilatory set‐point observed during chronic hypercapnia in our model. Results from this work may uncover properties governing separate mechanisms regulating steady‐state encoding of chemical ventilatory stimuli to maintain homeostasis and defence/error‐sensing mechanisms that provide responsiveness at any given ventilatory homeostatic set‐point.
Integrated physiological responses to chronic hypercapnia
There were several changes in measured variables that indicate a systemic, integrated response to chronic hypercapnia. First, with the additional reclamation of renal HCO3 − to add buffering capacity, there was a predictable reduction in arterial [Cl−]. This change is a well‐known phenomenon which aims to maintain electrical neutrality (balance between cations such as Na+ with anions) in the face of an increase in anions, which clinically is referred to as the Anion Gap. While this decrease in Cl− anions was predicted, there was a greater increase in HCO3 − than a reduction in Cl− in our model of chronic hypercapnic acidosis. Thus, it follows that further reductions in other unmeasured anions or increases in unmeasured cations must account for the maintenance of electroneutrality of the blood. While our data show a decrease in haemoglobin during hypercapnia, which may partially contribute to the maintenance of electroneutrality, it remains unclear how ionic flux may ultimately be affected.
Second, consistent with some (Schaefer et al. 1963a; Lai et al. 1981), but not other (Schaefer et al. 1975; Jennings & Davidson, 1984) studies of chronic hypercapnia, we observed an increase in during chronic hypercapnia. Contributing to this increase was probably the increased work of breathing from the biphasic, sustained hyperpnoea observed, although the exact contribution remains to be determined. In contrast to , there was no change in CO2 excretion () during exposure to chronic hypercapnia. The separation between and during chronic hypercapnia may seem paradoxical. However, if CO2 storage within body tissues (muscle, bone, fat, etc.) was increased during hypercapnia, which has been observed in other studies (Schaefer et al. 1963b; Reichart et al. 1976; Schaefer, 1982), this may result in an additional CO2 buffering mechanism and account for the minimal changes in and subsequent decrease in RER observed in our model.
Third, another potential CO2 buffering mechanism to remove CO2 from the body during chronic hypercapnia is gastric CO2 excretion. As explained by Dean (2011) in the theory of gastric CO2 ventilation, during respiratory acidosis CO2 may be consumed during gastric acid production, reconstituted to CO2 within the stomach, and subsequently removed by bulk flow through the oesophagus, contributing significantly to the mixed expired CO2 concentration measured at the mouth. Under normal conditions, exceeds due to contributions of deadspace ventilation to . However, as the contribution of gastric CO2 increases, may equal or even exceed values. Important to note is that the measurement of values equal to or exceeding occur only when ventilation is measured oronasally, in which the expired air consists of contributions from both the trachea and the oesophagus. Measurement of from an endotracheal tube bypasses the contributions of expired air from the oesophagus. Findings of values equal to or exceeding have been noted by others during hypercapnia (Jennings & Chen, 1975; Forster et al. 1986). During chronic hypercapnia we observed values that were near values during the steady state, and values that exceeded values during acute chemosensitivity challenges, suggesting involvement of gastric CO2 excretion as a further CO2 buffering mechanism during chronic hypercapnia.
Fourth, heart rate and blood pressure increased during the first week of hypercapnia and remained elevated throughout the remainder of chronic hypercapnia. This response has been noted in COPD patients who retain CO2 (Fontana et al. 2000), suggesting that hypercapnia per se may elicit pressor responses within the cardiovascular system in healthy but chronically hypercapnic mammals. Mechanisms for this pressor response have been hypothesized to result from increased sympatho‐adrenergic tone, as evident by positive correlations between hypercapnia and catecholamine secretion (Rose et al. 1983; Low et al. 1993).
Finally, body temperature declined during 30 days of chronic hypercapnia, despite the increase in heat production as indicated by the sustained increase in . The mechanism contributing to the decline in temperature within our model is unknown, but the fall in temperature was probably not from increased convective heat loss presumably accompanying the sustained hyperpnoea as there was no change in temperature by day 3 of chronic hypercapnia, despite the sustained hyperpnoea. The decline in body temperature observed during 30 days of exposure to 6% InCO2 was consistent with previous reports of a decline in body temperature during acute hypercapnic exposure in dogs (Jennings, 1979). However, this acute response in dogs was followed by a recovery of temperature during chronic hypercapnic exposure, contrasting with the results from this study on goats. Interestingly, a major factor in the control of breathing, the acute CO2 chemoreflex and body temperature control is the brainstem serotonin system. Deficiencies in serotonergic neurons in genetically modified mice indicate poor heat production and reductions in the ventilatory CO2 chemoreflex, without effects on eupnoeic ventilation or basal . In contrast, CBD‐induced hypoventilation was also associated with a major suppression of the rate‐limiting enzyme in serotonin synthesis, tryptophan hydroxylase, in addition to the serotonin reuptake transporter SERT. Thus, while it is unclear if chronic hypercapnia induces a downregulation of the brainstem serotonin system, it may represent an integrative centre that could be differentially affected in the chronic hypercapnic state.
A decline in cognitive function results from chronic hypercapnia in otherwise healthy adult goats
Respiratory diseases resulting in chronic hypercapnia lead to an increase in morbidities that substantially decrease the quality of life and increase the risk of mortality. Morbidities associated with CO2 retention in respiratory diseases such as COPD include hypertension, heart failure, osteoporosis, diabetes, muscle weakness, heightened inflammation and cognitive dysfunction (Sin et al. 2003; Donaldson et al. 2005; Holguin et al. 2005; Joppa et al. 2006; Mannino et al. 2008). Previous studies investigating the causal role of individual aspects of respiratory disease to the development of these morbidities have been limited by the presence of multiple potential causal factors such as hypercapnia, hypoxia and lung injury. Thus, there is a need to determine within an experimental model the individual role of these factors to the development of morbidities within respiratory disease. Herein we studied the role of chronic hypercapnia per se to the development of morbidities associated with respiratory diseases such as COPD. Consistent with clinical cases of respiratory disease patients, we observed a decline in cognitive function during 30 days of chronic hypercapnia. The mechanisms contributing to the association of chronic hypercapnia with cognitive dysfunction in our model are unknown. However, we hypothesize that the decline in cognition in our model of chronic hypercapnia suggests that hypercapnia per se may alter cellular function within higher brain centres, leading to maladaptations that potentially underlie alterations in cognition often observed in pathological cases of hypercapnia. Studies investigating the effects of chronic hypercapnia on specific brain regions important for cognition, such as the hippocampal formation, orbitofrontal cortex, medial pre‐frontal cortex and the insular cortex are needed to determine the underlying mechanism of the hypercapnia‐induced cognitive dysfunction. One potential mechanism of hypercapnia‐induced cognitive decline could be the effects on serotonin (5‐HT) in cortical regions. We previously reported that CBD‐induced hypercapnia in goats reduced the central enzyme (tryptophan hydroxylase) for 5‐HT synthesis by 50% (Miller et al. 2013). Others have shown that decreasing central 5‐HT by tryptophan depletion impairs stimulus reward learning (Rogers et al. 1999). Another potential mechanism may be from hypercapnia‐induced alterations in cerebral blood flow. Previous studies investigating cognitive function in mice have shown that alterations in cerebral blood flow lead to a form of cognitive dysfunction known as vascular cognitive impairment (Zuloaga et al. 2016). Additionally, chronic hypercapnia has been shown to alter sleep patterns (Fraigne et al. 2008), which may directly influence cognitive performance in our model if sleep patterns were disrupted from the chronic hypercapnia. Accordingly, our goat models of chronic hypercapnia will be useful in gaining an understanding of the role of 5‐HT depletion, cerebral blood flow, sleep disruption and other mechanisms to explain cognitive dysfunction. In addition, results from our study, and future studies investigating causal factors between chronic hypercapnia and cognitive dysfunction may have implications towards the treatment of respiratory disorders by suggesting the need to correct arterial CO2 levels within patients retaining CO2, in addition to the correction of hypoxaemia, which is traditionally the standard of care.
Summary, conclusions and importance
The results from this study provide a comprehensive dataset that provides insight into the multi‐system physiological responses that occur during chronic hypercapnic exposure. First, our data demonstrate that chronic hypercapnia uncovers an uncoupling between steady‐state ventilation and the acute CO2/H+ chemoreflex, suggesting that under these conditions, steady‐state ventilation may adapt independently of acute error‐sensing mechanisms in order to meet altered homeostatic demands, whilst retaining reactivity to acute challenges in the face of new homeostatic set‐points. Second, we have shown an integrated systemic response to chronic hypercapnia within the same animals over 30 days of chronic hypercapnia, including an apparent renal compensation to buffer the respiratory acidosis, along with a proposed increased tissue CO2 storage, and increase in gastric CO2 excretion. There was also a sustained tachycardia and modest hypertension, suggesting a pressor and potential sympatho‐adrenal response resulting from the hypercapnia. Lastly, we also show that chronic hypercapnia per se leads to a decline in cognitive function in an otherwise healthy animal, suggesting the cognitive decline in respiratory disease patients with COPD may result from hypercapnia alone. The significance/importance is that the findings herein provide an animal model/preparation for future studies to elucidate changes that occur in the brain that underlie physiological/cognitive changes due to hypercapnia per se. This is particularly important for better understanding the physiological/neurological consequences that occur during times of CO2 retention, including not only disease states such as COPD, but also the study of environmental CO2 exposure that occurs in fields such as aerospace, undersea and military research. Future studies investigating the causal mechanism for these findings may provide novel therapeutic targets to further our understanding of respiratory disease progression and allow for advancement of respiratory disease management and reversal of cognitive decline.
Additional information
Conflict of interest
The authors declare no significant conflicts of interest financial or otherwise.
Funding
Funding for this work was provided by the National Heart, Lung, and Blood Institute grant HL‐007852 and the Department of Veterans Affairs. Our funding sources were not involved in the preparation of the manuscript, experimental design or data collection, data analysis or interpretation, writing the report, or decision to submit the manuscript for publication.
Author contributions
N.J.B. performed surgeries and experiments, analysed data, created figures, and wrote the manuscript. S.E.N. performed experiments and manuscript editing. K.B performed experiments. T.M.L. performed surgeries. M.R.H. performed surgeries, contributed to intellectual discussions, and manuscript editing. L.P. performed surgeries and contributed to intellectual discussions. H.V.F. contributed to intellectual discussions and manuscript writing and editing. All experiments were performed at Medical College of Wisconsin, Milwaukee, Wisconsin, U.S.A.
Translational Perspective.
Patients with pulmonary or other diseases often present with respiratory insufficiency characterized by high arterial carbon dioxide (), reduced ventilatory sensitivity to CO2 and cognitive impairment. Patients with CO2 retention have poorer prognoses and life expectancy than patients without CO2 retention. However, the contributions of hypercapnia per se to reduced ventilatory CO2 sensitivity and impaired cognition are not known. Here, we measured the ventilatory and integrated physiological consequences of chronic hypercapnia (30 days) in a large animal species (goat) to test the hypothesis that hypercapnia per se alters ventilatory CO2 sensitivity and impairs cognitive function. Chronic increases in inspired CO2 ( = 6%) led to multiphasic ventilatory responses in both steady‐state ventilation and acute ventilatory CO2/H+ chemoreflex sensitivity, where steady‐state ventilation initially increased, followed by a sustained attenuation, and ventilatory CO2 sensitivity decreased transiently and returned to normal. This response differs greatly from the time‐dependent increase in both steady‐state ventilation and the ventilatory CO2/O2 chemoreflexes observed during chronic hypoxia, suggesting different mechanisms of plasticity underlying acclimatization to the different conditions. Future studies investigating the different mechanisms of plasticity may identify unique targets susceptible to differential therapeutic manipulation between the two conditions. Additional physiological adaptations to chronic hypercapnia included shifts in blood electrolytes, increases in tissue CO2 storage and gastric CO2 excretion, elevations in heart rate and blood pressure, and decreases in body temperature. Lastly, chronic hypercapnia was associated with a decline in cognitive performance. These findings in our large animal model suggest that hypercapnia per se does not directly alter ventilatory CO2 sensitivity, but may directly or indirectly drive multiple adaptive/maladaptive changes in integrated responses and impair cognitive function. Thus, treatments aiming to correct CO2 retention in patients with disease may prevent maladaptive physiological effects and cognitive decline to potentially improve quality of life in patients suffering from diseases associated with hypercapnia.
Biography
After completing his undergraduate degree in human biology from the University of Wisconsin‐Green Bay, Nicholas J. Burgraff began graduate school at the Medical College of Wisconsin. He is currently in his fourth year of graduate school working in the lab of Dr Hubert Forster, studying chronic adaptations to hypercapnia in goats. Following completion of graduate school, he plans to continue with a career in physiological research and academia, with an ultimate goal of becoming an independent investigator studying the interactions between the control of breathing and neuroplasticity.

Edited by: Scott Powers & Frank Powell
This is an Editor's Choice article from the 15 November 2018 issue.
References
- Allen JG, MacNaughton P, Satish U, Santanam S, Vallarino J & Spengler JD (2016). Associations of cognitive function scores with carbon dioxide, ventilation, and volatile organic compound exposures in office workers: a controlled exposure study of green and conventional office environments. Environ Health Perspect 124, 805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrianopoulos V, Gloeckl R, Vogiatzis I & Kenn K (2017). Cognitive impairment in COPD: should cognitive evaluation be part of respiratory assessment? Breathe (Sheff) 13, e1–e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli‐Incalzi R, Corsonello A, Pedone C, Trojano L, Acanfora D, Spada A, Izzo O & Rengo F (2006). Drawing impairment predicts mortality in severe COPD. Chest 130, 1687–1694. [DOI] [PubMed] [Google Scholar]
- Antonelli‐Incalzi R, Corsonello A, Trojano L, Acanfora D, Spada A, Izzo O & Rengo F (2008). Correlation between cognitive impairment and dependence in hypoxemic COPD. J Clin Exp Neuropsychol 30, 141–150. [DOI] [PubMed] [Google Scholar]
- Arita H, Kogo N & Ichikawa K (1988). Locations of medullary neurons with non‐phasic discharges excited by stimulation of central and/or peripheral chemoreceptors and by activation of nociceptors in cat. Brain Res 442, 1–10. [DOI] [PubMed] [Google Scholar]
- Baldwin B (1979). Operant studies on shape discrimination in goats. Physiol Behav 23, 455–459. [DOI] [PubMed] [Google Scholar]
- Blakeman N & Friend T (1986). Visual discrimination at varying distances in Spanish goats. Appl Anim Behav Sci 16, 279–283. [Google Scholar]
- Chang SS, Chen S, McAvay GJ & Tinetti ME (2012). Effect of coexisting chronic obstructive pulmonary disease and cognitive impairment on health outcomes in older adults. J Am Geriatr Soc 60, 1839–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapin JL, Otis AB & Rahn H (1955). Changes in the sensitivity of the respiratory center in man after prolonged exposure to 3% CO2 . Wright Air Development Center Technical Report 250.
- Clark JM, Sinclair RD & Welch BE (1971). Rate of acclimatization to chronic hypercapnia in man In Underwater Physiology, ed. Lambertsen CJ, pp. 399–408. Academic Press, New York. [Google Scholar]
- Costello R, Deegan P, Fitzpatrick M & McNicholas WT (1997). Reversible hypercapnia in chronic obstructive pulmonary disease: a distinct pattern of respiratory failure with a favorable prognosis. Am J Med 102, 239–244. [DOI] [PubMed] [Google Scholar]
- Dahan A, Nieuwenhuijs D & Teppema L (2007). Plasticity of central chemoreceptors: effect of bilateral carotid body resection on central CO2 sensitivity. PLoS Med 4, e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JB (2011). Theory of gastric CO2 ventilation and its control during respiratory acidosis: implications for central chemosensitivity, pH regulation, and diseases causing chronic CO2 retention. Respir Physiol Neurobiol 175, 189–209. [DOI] [PubMed] [Google Scholar]
- Dempsey JA & Forster HV (1982). Mediation of ventilatory adaptations. Physiol Rev 62, 262–346. [DOI] [PubMed] [Google Scholar]
- Dodd JW, Getov SV & Jones PW (2010). Cognitive function in COPD. Eur Resp J 35, 913–922. [DOI] [PubMed] [Google Scholar]
- Donaldson GC, Seemungal TA, Patel IS, Bhowmik A, Wilkinson TM, Hurst JR, Maccallum PK & Wedzicha JA (2005). Airway and systemic inflammation and decline in lung function in patients with COPD. Chest 128, 1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchuk KH, Lamb TW & Tenney SM (1966). Ventilatory response to hypoxia and CO2 following CO2 exposure and NaHCO3 ingestion. J Appl Physiol 21, 393–398. [DOI] [PubMed] [Google Scholar]
- Fontana F, Bernardi P, Tartuferi L, Boschi S, De Iasio R & Merlo Pich E (2000). Mechanisms of hypertension in patients with chronic obstructive pulmonary disease and acute respiratory failure. Am J Med 109, 621–627. [DOI] [PubMed] [Google Scholar]
- Forster HV, Pan LG, Bisgard GE, Flynn C & Hoffer RE (1986). Effect of reducing anatomic dead space on arterial PCO2 during CO2 inhalation. J Appl Physiol (1985) 61, 728–733. [DOI] [PubMed] [Google Scholar]
- Fothergill DM, Hedges D & Morrison JB (1991). Effects of CO2 and N2 partial pressures on cognitive and psychomotor performance. Undersea Biomed Res 18, 1–19. [PubMed] [Google Scholar]
- Fraigne JJ, Dunin‐Barkowski WL & Orem JM (2008). Effect of hypercapnia on sleep and breathing in unanesthetized cats. Sleep 31, 1025–1033. [PMC free article] [PubMed] [Google Scholar]
- Fukuda Y, See WR & Honda Y (1980). H+‐sensitivity and pattern of discharge of neurons in the chemosensitive areas of the ventral medulla oblongata of rats in vitro. Pflugers Arch 388, 53–61. [DOI] [PubMed] [Google Scholar]
- Guillerm R & Radziszewski E (1979). Effects on man of 30‐day exposure to a PICO2 of 14 torr (2 %): application to exposure limits. Undersea Biomed Res 6 (Suppl.), S91–114. [PubMed] [Google Scholar]
- Holguin F, Folch E, Redd SC & Mannino DM (2005). Comorbidity and mortality in COPD‐related hospitalizations in the United States, 1979 to 2001. Chest 128, 2005–2011. [DOI] [PubMed] [Google Scholar]
- Hung WW, Wisnivesky JP, Siu AL & Ross JS (2009). Cognitive decline among patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 180, 134–137. 29 [DOI] [PubMed] [Google Scholar]
- Incalzi RA, Gemma A, Marra C, Capparella O, Fuso L & Carbonin P (1997). Verbal memory impairment in COPD: its mechanisms and clinical relevance. Chest 112, 1506–1513. [DOI] [PubMed] [Google Scholar]
- Incalzi RA, Gemma A, Marra C, Muzzolon R, Capparella O & Carbonin P (1993). Chronic obstructive pulmonary disease: an original model of cognitive decline. Am Rev Respir Dis 148, 418–424. [DOI] [PubMed] [Google Scholar]
- Jennings DB (1979). Body temperature and ventilatory responses to CO2 during chronic respiratory acidosis. J Appl Physiol Respir Environ Exerc Physiol 46, 491–497. [DOI] [PubMed] [Google Scholar]
- Jennings DB & Chen CC (1975). Negative arterial‐mixed expired PCO2 gradient during acute and chronic hypercapnia. J Appl Physiol 38, 382–388. [DOI] [PubMed] [Google Scholar]
- Jennings DB & Chen CC (1976). Ventilation in conscious dogs during acute and chronic hypercapnia. J Appl Physiol 41, 839–847. [DOI] [PubMed] [Google Scholar]
- Jennings DB & Davidson JS (1984). Acid‐base and ventilatory adaptation in conscious dogs during chronic hypercapnia. Respir Physiol 58, 377–393. [DOI] [PubMed] [Google Scholar]
- Joppa P, Petrasova D, Stancak B & Tkacova R (2006). Systemic inflammation in patients with COPD and pulmonary hypertension. Chest 130, 326–333. [DOI] [PubMed] [Google Scholar]
- Kepron W & Cherniack RM (1973). The ventilatory response to hypercapnia and to hypoxemia in chronic obstructive lung disease. Am Rev Respir Dis 108, 843–850. [DOI] [PubMed] [Google Scholar]
- Klein M, Gauggel S, Sachs G & Pohl W (2010). Impact of chronic obstructive pulmonary disease (COPD) on attention functions. Respir Med 104, 52–60. [DOI] [PubMed] [Google Scholar]
- Kondo T, Kumagai M, Ohta Y & Bishop B (2000). Ventilatory responses to hypercapnia and hypoxia following chronic hypercapnia in the rat. Respir Physiol 122, 35–43. [DOI] [PubMed] [Google Scholar]
- Lai YL, Lamm JE & Hildebrandt J (1981). Ventilation during prolonged hypercapnia in the rat. J Appl Physiol Respir Environ Exerc Physiol 51, 78–83. [DOI] [PubMed] [Google Scholar]
- Lai YL, Martin ED, Attebery BA & Brown EB, Jr . (1973). Mechanisms of extracellular pH adjustments in hypercapnia. Respir Physiol 19, 107–114. [DOI] [PubMed] [Google Scholar]
- Langbein J, Nürnberg G & Manteuffel G (2004). Visual discrimination learning in dwarf goats and associated changes in heart rate and heart rate variability. Physiol Behav 82, 601–609. [DOI] [PubMed] [Google Scholar]
- Langbein J, Nurnberg G, Puppe B & Manteuffel G (2006). Self‐controlled visual discrimination learning of group‐housed dwarf goats (Capra hircus): behavioral strategies and effects of relocation on learning and memory. J Comp Psychol 120, 58–66. [DOI] [PubMed] [Google Scholar]
- Langbein J, Siebert K & Nuernberg G (2008). Concurrent recall of serially learned visual discrimination problems in dwarf goats (Capra hircus). Behav Processes 79, 156–164. [DOI] [PubMed] [Google Scholar]
- Langbein J, Siebert K Nurnberg G, & Manteuffel G (2007). Learning to learn during visual discrimination in group housed dwarf goats (Capra hircus). J Comp Psychol 121, 447–456. [DOI] [PubMed] [Google Scholar]
- Low JM, Gin T, Lee TW & Fung K (1993). Effect of respiratory acidosis and alkalosis on plasma catecholamine concentrations in anaesthetized man. Clin Sci (Lond) 84, 69–72. [DOI] [PubMed] [Google Scholar]
- Mannino DM, Thorn D, Swensen A, & Holguin F (2008). Prevalence and outcomes of diabetes, hypertension and cardiovascular disease in COPD. Eur Respir J 32, 962–969. [DOI] [PubMed] [Google Scholar]
- Mar AC, Horner AE, Nilsson SR, Alsio J, Kent BA, Kim CH, Holmes A, Saksida LM & Bussey TJ (2013). The touchscreen operant platform for assessing executive function in rats and mice. Nat Protoc 8, 1985–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JR, Neumueller S, Muere C, Olesiak S, Pan L, Bukowy JD, Daghistany AO, Hodges MR & Forster HV (2014). Changes in glutamate receptor subunits within the medulla in goats after section of the carotid sinus nerves. J Appl Physiol (1985) 116, 1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JR, Neumueller S, Muere C, Olesiak S, Pan L, Hodges MR & Forster HV (2013). Changes in neurochemicals within the ventrolateral medullary respiratory column in awake goats after carotid body denervation. J Appl Physiol (1985) 115, 1088–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes de Oca M, & Celli BR (1998). Mouth occlusion pressure, CO2 response and hypercapnia in severe chronic obstructive pulmonary disease. Eur Respir J 12, 666–671. [DOI] [PubMed] [Google Scholar]
- Mouradian GC, Forster HV & Hodges MR (2012). Acute and chronic effects of carotid body denervation on ventilation and chemoreflexes in three rat strains. J Physiol 590, 3335–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie E (2011). Julius H. Comroe, Jr., distinguished lecture: central chemoreception: then … and now. J Appl Physiol (1985) 110, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie EE, Fung ML, Li A & St John WM (1993). Responses of respiratory modulated and tonic units in the retrotrapezoid nucleus to CO2 . Respir Physiol 94, 35–50. [DOI] [PubMed] [Google Scholar]
- Pingree BJ (1977). Acid‐base and respiratory changes after prolonged exposure to 1% carbon dioxide. Clin Sci Mol Med 52, 67–74. [DOI] [PubMed] [Google Scholar]
- Reichart E, Claudon F & Sabliere S (1976). [CO2 storage in various organs during chronic experimental hypercapnia (author's transl)]. Bull Eur Physiopathol Respir 12, 19–32. [PubMed] [Google Scholar]
- Rogers RD, Blackshaw AJ, Middleton HC, Matthews K, Hawtin K, Crowley C, Hopwood A, Wallace C, Deakin JF, Sahakian BJ & Robbins TW (1999). Tryptophan depletion impairs stimulus‐reward learning while methylphenidate disrupts attentional control in healthy young adults: implications for the monoaminergic basis of impulsive behaviour. Psychopharmacology (Berl) 146, 482–491. [DOI] [PubMed] [Google Scholar]
- Rose CE, Jr , Althaus JA, Kaiser DL, Miller ED & Carey RM (1983). Acute hypoxemia and hypercapnia: increase in plasma catecholamines in conscious dogs. Am J Physiol Hear Circ Physiol 245, H924–929. [DOI] [PubMed] [Google Scholar]
- Satish U, Mendell MJ, Shekhar K, Hotchi T, Sullivan D, Streufert S & Fisk WJ (2012). Is CO2 an indoor pollutant? Direct effects of low‐to‐moderate CO2 concentrations on human decision‐making performance. Environ Health Perspect 120, 1671–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer KE (1949). Respiration and acid base balance during prolonged exposure to 3% CO2 . Pflugers Arch 251, 689–715. [Google Scholar]
- Schaefer KE (1958). Respiratory pattern and respiratory response to CO2 . J Appl Physiol 13, 1–14. [DOI] [PubMed] [Google Scholar]
- Schaefer KE (1982). Effects of increased ambient CO2 levels on human and animal health. Experientia 38, 1163–1168. [DOI] [PubMed] [Google Scholar]
- Schaefer KE, Hastings BJ, Carey CR & Nichols G, Jr (1963a). Respiratory acclimatization to carbon dioxide. J Appl Physiol 18, 1071–1078. [DOI] [PubMed] [Google Scholar]
- Schaefer KE, Messier AA, Morgan C & Baker GT, 3rd. (1975). Effect of chronic hypercapnia on body temperature regulation. J Appl Physiol 38, 900–906. [DOI] [PubMed] [Google Scholar]
- Schaefer KE, Nichols G, Jr. & Carey CR (1963b). Calcium phosphorus metabolism in man during acclimatization to carbon dioxide. J Appl Physiol 18, 1079–1084. [DOI] [PubMed] [Google Scholar]
- Schou L, Østergaard B, Rasmussen LS, Rydahl‐Hansen S & Phanareth K (2012) Cognitive dysfunction in patients with chronic obstructive pulmonary disease – A systematic review. Respir Med 106, 1071–1081. [DOI] [PubMed] [Google Scholar]
- Siesjo BK (1972). Symposium on acid–base homeostasis. The regulation of cerebrospinal fluid pH. Kidney Int 1, 360–374. [DOI] [PubMed] [Google Scholar]
- Sin DD, Man JP & Man SF (2003). The risk of osteoporosis in Caucasian men and women with obstructive airways disease. Am J Med 114, 10–14. [DOI] [PubMed] [Google Scholar]
- Slenter R, Sprooten R, Kotz D, Wesseling G, Wouters E & Rohde G (2012). Predictors of 1‐year mortality at hospital admission for acute exacerbations of chronic obstructive pulmonary disease. Respiration 85, 15–26. [DOI] [PubMed] [Google Scholar]
- Torres‐Sanchez I, Rodriguez‐Alzueta E, Cabrera‐Martos I, Lopez‐Torres I, Moreno‐Ramirez MP & Valenza MC (2015). Cognitive impairment in COPD: a systematic review. J Bras Pneumol 41, 182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve S, Pepin V, Rahayel S, Bertrand JA, de Lorimier M, Rizk A, Desjardins C, Parenteau S, Beaucage F, Joncas S, Monchi O & Gagnon JF (2012). Mild cognitive impairment in moderate to severe COPD: a preliminary study. Chest 142, 1516–1523. [DOI] [PubMed] [Google Scholar]
- Winterstein H (1956). Chemical control of pulmonary ventilation. III. The reaction theory of respiratory control. N Engl J Med 255, 331–337. [DOI] [PubMed] [Google Scholar]
- Zuloaga KL, Johnson LA, Roese NE, Marzulla T, Zhang W, Nie X, Alkayed FN, Hong C, Grafe MR, Pike MM, Raber J & Alkayed NJ (2016). High fat diet‐induced diabetes in mice exacerbates cognitive deficit due to chronic hypoperfusion. J Cereb Blood Flow Metab 36, 1257–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]