

Abstract
Nutrient overload in combination with decreased energy dissipation promotes obesity and diabetes. Obesity results in a hormonal imbalance, which among others activates G protein‐coupled receptors utilizing diacylglycerol (DAG) as secondary messenger. Protein kinase D1 (PKD1) is a DAG effector, which integrates multiple nutritional and hormonal inputs, but its physiological role in adipocytes is unknown. Here, we show that PKD1 promotes lipogenesis and suppresses mitochondrial fragmentation, biogenesis, respiration, and energy dissipation in an AMP‐activated protein kinase (AMPK)‐dependent manner. Moreover, mice lacking PKD1 in adipocytes are resistant to diet‐induced obesity due to elevated energy expenditure. Beiging of adipocytes promotes energy expenditure and counteracts obesity. Consistently, deletion of PKD1 promotes expression of the β3‐adrenergic receptor (ADRB3) in a CCAAT/enhancer binding protein (C/EBP)‐α‐ and δ‐dependent manner, which leads to the elevated expression of beige markers in adipocytes and subcutaneous adipose tissue. Finally, deletion of PKD1 in adipocytes improves insulin sensitivity and ameliorates liver steatosis. Thus, depletion of PKD1 in adipocytes increases energy dissipation by several complementary mechanisms and might represent an attractive strategy to treat obesity and its related complications.
Keywords: AMP‐activated protein kinase, beige adipocytes, C/EBP, protein kinase D1, β3 adrenergic receptor
Subject Categories: Development & Differentiation, Metabolism
Introduction
Persistent imbalance between energy intake and dissipation results in obesity, insulin resistance and can lead to the development of type 2 diabetes (T2D) and liver steatosis (Spiegelman & Flier, 2001; Rosen & Spiegelman, 2006). Recent studies indicate that white adipocytes, which were previously seen only as a storage compartment of the organism, present a high degree of plasticity and can acquire a molecular machinery, which allows them to dissipate energy in form of heat. This process is often referred to as browning or beiging of white adipose tissue. Importantly, strategies to increase energy dissipation by adipocytes recently became central for the development of new anti‐obesity therapies (Muller et al, 2013; Kajimura & Saito, 2014; Kajimura et al, 2015; Kazak et al, 2015). On the molecular level, beige or brite adipocytes are characterized by the high mitochondrial content and expression of genes, which decouple the proton gradient from ATP production (Kajimura et al, 2015). Moreover, recent studies indicate that the induction of mitochondrial fragmentation also promotes decoupling activity in adipocytes, which protects mice from the development of diabetes and fatty liver disease (Wikstrom et al, 2014; Tol et al, 2016).
Obesity‐related metabolic overload leads to hormonal imbalance which promotes the activity of multiple cell surface receptors including G protein‐coupled receptors, which utilize diacylglycerol (DAG) as a secondary messenger (Samuel & Shulman, 2012). The impact of DAG‐evoked signaling on the adipocyte function remains poorly characterized. Protein kinase D (PKD) isoforms (PKD1, 2, and 3) are DAG and protein kinase C (PKC) effectors that integrate various nutritional and hormonal inputs and have been shown to be implicated in the regulation of multiple fundamental biological processes (Rozengurt, 2011). Previous studies implicated PKD1 in the regulation of muscle differentiation, pathophysiological heart remodeling, immune response, carcinogenesis, insulin secretion, actin remodeling, trans‐Golgi network dynamics, cell proliferation, and cell migration (Matthews et al, 2006; Fielitz et al, 2008; Kim et al, 2008; Sumara et al, 2009; Steiner et al, 2010; Rozengurt, 2011; Ferdaoussi et al, 2012; Gehart et al, 2012; Ittner et al, 2012; Goginashvili et al, 2015; Bergeron et al, 2018). However, the impact of PKD1 on energy dissipation and its physiological role in adipose tissue remains unknown.
Here, we show that the ablation of PKD1 in murine adipocytes results in a significant decrease of lipogenesis rate and accumulation of triglycerides (TG). At the same time, PKD1 deficiency results in an increased respiration and energy dissipation caused by mitochondrial fragmentation. On the other hand, adipocytes expressing a constitutive active form of PKD1 present a reversed phenotype. Importantly, mice lacking PKD1 specifically in adipocytes are resistant to diet‐induced obesity and T2D due to an elevated mitochondrial content and high expression of genes promoting energy dissipation in white adipocytes. Finally, our results indicate that PKD1 regulates adipocyte function primarily in an AMPK‐dependent manner. Taken together, our data demonstrate that PKD1 suppresses energy dissipation and drives lipogenesis and adiposity in mice. Therefore, ablation of PKD1 action in adipocytes might represent an attractive strategy to ameliorate obesity and T2D in the future.
Results
PKD1 promotes triglyceride accumulation in adipocytes
PKD1 is activated in response to the stimulation of multiple cell surface receptors, which utilize DAG as a secondary messenger (Rozengurt, 2011). Indeed, DAG can promote activation of PKD1 in undifferentiated 3T3L1 cells as well as in 3T3L1 cells, which were differentiated into adipocytes (Fig EV1A). Adiposity is associated with an increased rate of lipogenesis in all types of adipocytes. This raises the possibility that DAG accumulates in adipose tissue of obese mice. We measured steady‐state levels of DAG in white and brown adipose tissue of lean mice fed normal chow diet (ND) and obese mice fed high‐fat diet (HFD). However, diet regiment and degree of obesity did not change DAG levels in both tissues (Fig EV1B). Factors secreted by the peripheral nervous system (β‐adrenergic agonists) together with endocrine factors like insulin and gut‐derived serotonin as well as auto‐ and paracrine factors (adenosines: ADP and ATP) regulate adipocyte function (Sumara et al, 2012; Zechner et al, 2012; El‐Merahbi et al, 2015; Tozzi & Novak, 2017). We showed that among all of these factors only extracellular purines (ADP and ATP) robustly and specifically activate PKD1 in white adipocytes (Fig EV1C–F). In brown‐like adipocytes, extracellular purines also can activate PKD1. However, stimulation of these cells with insulin and serotonin also increases activity of PKD1 (Fig EV1G–I).
Figure EV1. Extracellular purines (ATP and ADP) promote PKD1 activity.

-
AWestern blot (WB) analysis of levels of indicated proteins in undifferentiated 3T3L1 cells and 3T3L1 cells differentiated into adipocytes stimulated with DAG (100 μM) for indicated time points.
-
BDAG content measured in subcutaneous white adipose tissue (sWAT) and brown adipose tissue (BAT) of wild‐type mice fed HFD or ND (n = 6/genotype).
-
C–EWB analysis using indicated antibodies in differentiated 3T3L1 cells stimulated with the indicated substances for the given time points.
-
FWB analysis using indicated antibodies in explants from sWAT of wild‐type mice treated with different concentrations of ATP for the given time points.
-
G–IWB analysis of levels of indicated proteins in differentiated T3TI cells which have been stimulated with the indicated substances for the given time points.
This result prompted us to investigate the impact of PKD1 on adipocyte function. Efficient shRNA‐mediated PKD1 knockdown in 3T3L1 cells (Appendix Fig S1A) led to a significant decrease in TG accumulation after 10 days of differentiation as compared to corresponding control cells (Fig 1A and B). Conversely, overexpression of constitutive active form of PKD1 (PKD1ca) in differentiated 3T3L1 cells (Appendix Fig S1B) resulted in an increased accumulation of TG compared to control cells (Fig 1C and D).
Figure 1. PKD1 promotes triglyceride (TG) accumulation and lipogenesis in adipocytes.

-
A, BQuantification of relative TG accumulation using AdipoRed reagent (A) and images of Oil Red O staining for neutral lipids (B) in 3T3L1 cells differentiated into adipocytes depleted from PKD1 using shRNA (shPKD1) or corresponding non‐targeting control (shNTC; n = 6; representative of three individual experiments).
-
C, DQuantification of relative TG accumulation using AdipoRed reagent (C) and images of Oil Red O staining for neutral lipids (D) in 3T3L1 cells which have been differentiated after overexpressing of wild‐type form of PKD1 (PKD1wt) as well as constitutive active form of PKD1 (PKD1ca) and corresponding controls (Myc) (n = 6; representative of three individual experiments).
-
E–GBasal and insulin‐induced lipogenesis rate (arbitrary units—AU) in differentiated 3T3L1 depleted of PKD1 by shRNA (E), PKD1‐deficient adipocytes derived from SVC (F), and 3T3L1 cells expressing PKD1ca (G) (n = 3; representative for three individual experiments).
To corroborate these findings, we generated mice lacking PKD1 specifically in adipocytes by using PKD1 floxed (PKD1f/f) mice (Fielitz et al, 2008) and a mouse strain expressing Cre recombinase under the control of the adiponectin promoter (Eguchi et al, 2011). We confirmed efficiency and specificity of PKD1 deletion in these mice on protein and mRNA levels (Appendix Fig S1C–F). None of the other two isoforms of PKD were affected by this deletion, neither in PKD1 depleted primary cells, nor in the different depots of adipose tissue (Appendix Fig S1G and H). Importantly, stroma vascular cells (SVC) isolated from subcutaneous white adipose tissue (sWAT) of PKD1adipoΔ/Δ mice and differentiated into adipocytes presented markedly lower TG content than corresponding control SVC isolated from PKD1f/f mice (Appendix Fig S1I and J). Interestingly, deletion of PKD1 in SVC did not affect the protein levels of adipocyte differentiation markers. Particularly, levels of a major transcription factor defining terminal differentiation of adipocytes, PPARγ, were not changed. Similarly, levels of enzymes and structure proteins specific for adipocytes (ATGL, HSL, and Perilipin) were unaffected (Appendix Fig S1K). Altogether, these data suggest that PKD1 promotes TG accumulation in adipocytes but does not regulate adipocyte differentiation markers.
PKD1 decreases mitochondrial respiration and promotes lipogenesis in adipocytes
Since PKD1 did not affect adipocyte differentiation, reduced TG accumulation in the absence of PKD1 can be caused by an increased rate of lipolysis, decreased lipogenesis, and/or enhanced rate of respiration in adipocytes. In differentiated SVC and 3T3L1 cells, the insulin‐stimulated lipogenesis rate was markedly reduced in the absence of PKD1 (Fig 1E and F). Conversely, cells expressing PKD1ca generated more TG under basal and insulin‐stimulated conditions (Fig 1G). However, the lipolysis rate was not altered in PKD1‐deficient adipocytes (Appendix Fig S2A and B). Next, we measured the cellular respiration rate using a Seahorse analyzer. Deficiency of PKD1 in differentiated 3T3L1 cells, as well as in SVC, resulted in increased oxygen consumption rate (OCR) during basal respiration, in response to inhibition of the mitochondrial ATP synthase by oligomycin as well as in response to FCCP, which stimulates the maximal respiration rate in cells. In response to antimycin A/rotenone (inhibitors of mitochondrial respiration), OCR was reduced in both PKD1‐deficient and control adipocytes as compared to the baseline (Fig 2A and Appendix Fig S2C). These results indicate that PKD1‐deficient adipocytes display an increased mitochondrial activity and dissipation of energy. Consistently, an increased mitochondrial content was observed in PKD1‐deficient adipocytes (Fig 2B). Conversely, expression of PKD1ca in 3T3L1‐derived adipocytes suppressed mitochondrial decoupling activity and OCR (Fig 2C). These experiments demonstrate that PKD1 promotes lipid storage and suppresses energy dissipation in white adipocytes.
Figure 2. Deletion of PKD1 increases energy dissipation and mitochondrial fragmentation in adipocytes.

-
AOxygen consumption rate (OCR) in response to indicated substances as well as OCR annotated to the indicated cellular processes in PKD1‐deficient SVC differentiated into adipocytes (n = 14; representative of three individual experiments).
-
BRatio of mitochondrial (mtDNA) and genomic DNA (gDNA) in SVC‐derived adipocytes with deletion of PKD1 (n = 5; representative of three individual experiments).
-
COCR upon indicated substances and OCR required for indicated processes in PKD1ca expressing 3T3L1 cells (n = 30; representative of three individual experiments).
-
D, ERepresentative microscopy images of the mitochondrial morphology and its respective quantification in PKD1‐deficient adipocytes derived from SVC (D) and differentiated 3T3L1 cells expressing PKD1ca (E).
To test whether PKD1 also affects brown adipocyte function, we isolated SVC from interscapular brown adipose tissue (BAT) and differentiated them into brown adipocytes (BSVC). Of note, deletion of PKD1 in brown adipocytes suppressed the lipogenesis rate but did not affect basal respiration or decoupling activity of these cells (Appendix Fig S2D and E). Taken together, our data show that PKD1 primarily affects white adipocyte function by decreasing mitochondrial respiration and promoting lipogenesis.
PKD1 suppresses mitochondrial fragmentation
Mitochondria are highly dynamic organelles that undergo constant fusion and fission in order to maintain optimal mitochondrial bioenergetics (Twig et al, 2008). Mitochondrial fragmentation was previously shown to support uncoupled respiration, and therefore, it increases energy expenditure in adipocytes (Wikstrom et al, 2014; Tol et al, 2016). Consistently, our results show that deletion of PKD1 in differentiated adipocytes increased mitochondrial fragmentation (Fig 2D and Appendix Fig S2F). On the other hand, there were more fused mitochondria in differentiated 3T3L1 cells expressing PKD1ca (Fig 2E). This indicates that PKD1 promotes mitochondrial fusion or suppresses mitochondrial fragmentation to suppress energy dissipation in adipocytes.
PKD1 regulates adipocyte function in an AMPK‐dependent manner
To unravel the underlying molecular mechanism of PKD1‐mediated regulation of adipocyte function, we tested the activation of AMPK, which is a multi‐subunit enzyme regulating a number of processes of energy homeostasis in adipocytes (Bijland et al, 2013). Importantly, previous studies showed that PKD1 phosphorylates AMPK alpha subunits and inactivates the kinase activity of the whole complex in muscle cells (Coughlan et al, 2016). Interestingly, AMPK alpha phosphorylation at putative PKD1 sites (S485/S491) was reduced in PKD1‐deficient 3T3L1 cells as well as SVC and enhanced in 3T3L1 cells expressing PKD1ca, while total AMPK alpha levels remained unchanged (Fig 3A, and Appendix Fig S3A and B). This result indicates that PKD1 suppresses AMPK activity in adipocytes. Consistently, phosphorylation of the classical AMPK targets, acetyl‐CoA carboxylase 1 and 2 (ACC1/ACC2) at S79/S221 (Bijland et al, 2013), was markedly enhanced in PKD1‐deficient adipocytes and reduced in cells expressing PKD1ca (Fig 3A and Appendix Fig S3A and B). Similarly, phosphorylation of AMPK alpha subunit on T172 was enhanced in cells depleted from PKD1 and reduced in cells expressing PKD1ca (Fig 3A and Appendix Fig S3A and B). Consistently, activity of AMPK complex was markedly reduced in cells expressing PKD1ca and enhanced in PKD1‐deficient adipocytes (Fig 3B and C). AMPK‐dependent phosphorylation of ACC inhibits lipogenesis (Bijland et al, 2013) and chemical activation of AMPK signaling by AICAR in cells expressing PKD1ca (Appendix Fig S3C) reduced the lipogenesis rate to the level observed in control cells (Fig 3D). In contrast, silencing of AMPK alpha subunits in differentiated 3T3L1 cells depleted of PKD1 (Appendix Fig S3D) increased the lipogenesis rate to the same level as seen in control cells (Fig 3E). Consistently, inhibition of AMPK activity with Compound C (CC) normalized reduced lipogenesis rate in PKD1‐deficient adipose tissue explants (Fig 3F). In line with these findings, silencing of AMPK alpha subunits in adipocytes depleted from PKD1 normalized TG content (Appendix Fig S3E and F), while AICAR treatment reduced TG accumulation in cells expressing PKD1ca to the level observed in control cells (Fig 3G and H). Silencing of AMPK alpha subunits (Appendix Fig S3G) also normalized enhanced respiration of PKD1‐deficient primary cells (Fig 3I), while AICAR treatment enhanced respiration of PKD1ca expressing cells to the level observed in control adipocytes (Fig 3J). Finally, activation of AMPK in PKD1ca expressing cells reversed mitochondrial fusion (Fig EV2). Taken together, these results show that PKD1 regulates adipocyte function primarily by suppressing AMPK.
Figure 3. PKD1 promotes lipogenesis, TG accumulation, and suppresses OCR in an AMPK‐dependent manner.

-
AWestern blot (WB) analysis using antibodies against indicated proteins in protein lysates of differentiated 3T3‐L1 cells lacking PKD1 and densitometric quantification of 3–4 replicates for each protein.
-
B, CAMPK activity in differentiated 3T3L1 cells expressing PKD1ca (B) and 3T3L1 cells depleted PKD1 (C). Compound C (CC)‐stimulated cells served as a negative control (n > 5; average of two individual experiments).
-
D, EBasal and insulin‐induced lipogenesis rate (arbitrary units—AU) in differentiated 3T3L1 cells expressing PKD1ca treated with AICAR (2 mM) for 2 h (D) and cells lacking PKD1 which have been transfected with control siRNA and siRNA against AMPKα1/α2 subunits (E) (n = 3; representative of three individual experiments).
-
FBasal and insulin‐induced lipogenesis rate (arbitrary units—AU) in explants derived from subcutaneous white adipose tissue (sWAT) of control mice and mice lacking PKD1 treated with Compound C (CC) (5 μM) for 4 h (n = 4; representative of two individual experiments).
-
G, HQuantification of relative TG accumulation using AdipoRed reagent (G) and images of Oil Red O staining for neutral lipids (H) in differentiated 3T3L1 cells expressing PKD1ca and corresponding controls treated with AICAR (0.5 mM) for 3 days (n = 3; representative of three individual experiments).
-
I, JOCR required for indicated processes in PKD1‐deficient 3T3l1 cells which have been transfected with control siRNA and siRNA against AMPKα1/α2 subunits (I) and PKD1ca expressing 3T3L1 cells treated with AICAR (0.5 mM) for 3 days (J) (n = 22; representative of two individual experiments).
Figure EV2. PKD1 promotes mitochondrial aggregation in an AMPK‐dependent manner.
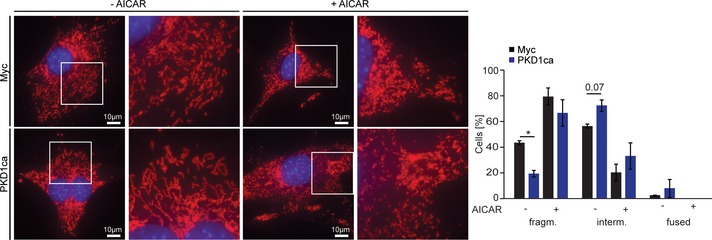
Representative microscopy images of the mitochondrial morphology and its respective quantification in differentiated 3T3L1 cells expressing PKD1ca, which have been treated with AICAR (2 mM) for 2 h. Data are presented as mean ± SEM. Data represent three independent experiments with 200 cells counted for each replicate. *P < 0.05 (unpaired, two‐tailed Student's t‐test).
PKD1 deletion in adipocytes protects against diet‐induced obesity
To understand the physiological consequences of PKD1‐mediated regulation of energy dissipation and lipogenesis, we investigated the impact of PKD1 action in adipocytes in vivo. Activation, abundance, and expression of PKD1 in adipocytes were induced upon feeding (Fig 4A and B). Moreover, in adipose tissue of mice lacking PKD1 in adipocytes (PKD1adipoΔ/Δ) fed ND we observed decreased AMPK alpha phosphorylation at S485/S491, increased phosphorylation of AMPK alpha subunit at T172, and enhanced phosphorylation of AMPK downstream target ACC at S79 (Fig EV3A), suggesting that PKD1 suppresses AMPK activity also in the in vivo settings. AMPK is a master activator of autophagy (Kim et al, 2011). Interestingly, we observed increased LC3 cleavage in adipose tissue of PKD1adipoΔ/Δ mice fed ND (Fig EV3A) suggesting enhanced autophagy.
Figure 4. Deletion of PKD1 in adipocytes protects against obesity by increasing energy dissipation.

-
AWB analysis of indicated proteins in sWAT from 24 h fasted and 24 h fasted‐refed mice including densitometric quantification of triplicates for each protein.
-
BqPCR analysis of PKD1 expression in sWAT from fasted and fasted‐refed mice (n = 3 mice per group).
-
CBody weight evolution of control and PKD1adipoΔ/Δ mice fed HFD and ND (n > 5/genotype).
-
DOrgan weight of control and PKD1adipoΔ/Δ mice fed HFD (n > 8/genotype).
-
E–GFood intake (E), voluntary movement (F), and energy expenditure (G) of control and PKD1adipoΔ/Δ mice fed HFD (n > 8/genotype).
-
H–LRepresentative H&E stainings of gonadal white adipose tissue (gWAT) (H), interscapular brown adipose tissue (BAT) (J), and sWAT (K) from PKD1adipoΔ/Δ and control mice fed HFD. Average adipocyte size in (I) gWAT and (L) sWAT of indicated mice fed HFD (n = 8/genotype).
Figure EV3. PKD1 deletion promotes AMPK signaling in adipose tissue of mice fed ND .

-
AWB analysis of indicated proteins in sWAT of control and PKD1‐deficient mice fed ND including densitometric quantification of 3–4 replicates each.
-
B–EFFA (B), glycerol (C), TG (D), and glucose (E) levels in blood of PKD1adipoΔ/Δ and control mice after 24 h fasting and 24 h fasting‐refeeding (n > 5/genotype).
Next, we subjected PKD1adipoΔ/Δ and control animals for fasting/refeeding experiment. The total TG, free fatty acids (FFA), glycerol, and glucose levels did not differ from PKD1adipoΔ/Δ compared to control animals at fasted and fasted‐refed stage (Fig EV3B–E). Also, the levels of phosphorylation of AMPK alpha subunits at S485/S491 and on T172 as well as phosphorylation of ACC at S79 were not altered by PKD1 deletion during fasting (Appendix Fig S4A). However, in adipose tissue of refed animals, deletion of PKD1 resulted in decreased phosphorylation of AMPK alpha subunit at S485/S491 and enhanced phosphorylation of AMPK alpha T172 as well as ACC at S79 (Appendix Fig S4B). Altogether, these findings suggest that PKD1 can promote adiposity in vivo. To test this hypothesis, we monitored mice lacking PKD1 in adipocytes (PKD1adipoΔ/Δ) for 24 weeks on ND and on HFD. PKD1adipoΔ/Δ mice showed significantly less weight gain when challenged with HFD, whereas no differences could be observed when PKD1adipoΔ/Δ mice were fed ND (Fig 4C). Body composition analysis revealed markedly smaller fat tissue depots as well as reduced liver weight, while weight of other organs was not altered (Fig 4D). Of note, mice with PKD1 deletion had less gonadal white adipose tissue (gWAT) weight even under ND feeding (Appendix Fig S4C). Consistently with our previous findings, we observed increased phosphorylation of ACC at S79 as well as reduced phosphorylation at S485/S491 of AMPK alpha in sWAT isolated from PKD1adipoΔ/Δ fed HFD (Fig EV4A). However, phosphorylation of AMPK alpha at T172 in sWAT of mice fed with HFD for 24 weeks was not altered (Fig EV4A). Interestingly, phosphorylation of AMPK alpha on S485/S49, T172, and ACC at S79 in BAT isolated from HFD and ND fed PKD1adipoΔ/Δ was not changed (Fig EV4B). Furthermore, in PKD1‐deficient SVC isolated from BAT no changes in phosphorylation of AMPK alpha on S485/S49, T172, and ACC at S79 were observed (Appendix Fig S5A). Metabolic analysis of PKD1adipoΔ/Δ mice showed no significant differences in food intake or activity compared to control animals when fed HFD (Fig 4E and F). Importantly, PKD1adipoΔ/Δ mice fed HFD showed an increase in energy expenditure compared to control animals (Fig 4G). Similar effects were observed in PKD1adipoΔ/Δ mice fed ND (Appendix Fig S5B), while other metabolic parameters were not changed (Appendix Fig S5C and D). Therefore, deletion of PKD1 specifically in adipocytes protects mice from diet‐induced obesity by increasing their energy expenditure.
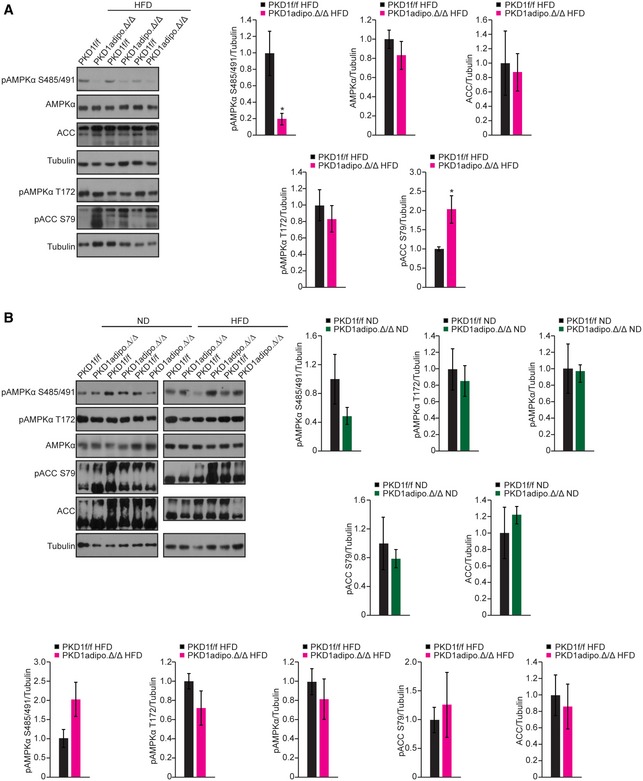
Figure EV4. PKD1 deletion in adipocytes does not affect AMPK signaling in BAT .
-
A, BWB analysis of indicated proteins in sWAT of control and PKD1‐deficient mice fed HFD (A) and BAT of the same mice fed ND and HFD (B) including densitometric quantification of four replicates each (A, B).
Consistent with these results, PKD1adipoΔ/Δ mice also showed profoundly changed adipose tissue morphology. The overall adipocyte size was smaller in PKD1adipoΔ/Δ mice as compared to control animals when fed HFD (Fig 4H–L). However, no changes were observed in different types of adipose tissue of mice fed ND (Appendix Fig S6A–E). Moreover, we did not observe ectopic myofiber formation within subcutaneous adipose tissue depots of PKD1adipoΔ/Δ mice (Appendix Fig S6F and G). Induction of energy expenditure by adipocytes requires a reprogramming of expression patterns of these cells. RNA sequencing revealed that sWAT from PKD1adipoΔ/Δ mice showed elevated expression of genes promoting the creatine‐driven substrate cycle, which was recently proposed to promote energy expenditure by adipocytes (Kazak et al, 2015; Fig 5A). Consistently, genes characteristic for beige adipocytes were also elevated (Fig 5B). Gene set enrichment analysis also revealed elevated expression of genes related to beiging of white adipocytes in sWAT lacking PKD1 (Fig 5A and B). qPCR analysis confirmed increased expression of genes promoting creatine cycle in the absence of PKD1 (Fig 5C). Moreover, PKD1‐deficient sWAT displayed elevated expression of genes promoting energy expenditure (Kajimura et al, 2015) including Adrb3, Ucp1, Ucp3, and Pgc‐1α, while expression of the master regulator of adipocyte differentiation Pparγ was not altered (Fig 5C). Similarly, expression of the major genes determining lipid metabolism in adipocytes (Fasn, Dgat, Lpl, Acc, Srebp1, Srebp2, Creb, and Chrebp) was not altered by PKD1 deletion (Appendix Fig S7A). Of note, mitochondrial content in sWAT was markedly elevated in the absence of PKD1 (Fig 5D). Although we did not observe a clear correlation between PKD1 deletion and appearance of multilocular adipocytes (Fig 4H and K), the protein level of the major beige adipocyte marker UCP1 was markedly elevated in PKD1‐deficient sWAT (Fig 5E). However, in BAT, expression of these genes was not enhanced upon deletion of PKD1 (Appendix Fig S7B). Acquisition of beige adipocytes is largely inhibited at the thermoneutral environment. Importantly, maintenance of PKD1adipoΔ/Δ mice at 30°C resulted in normalization of elevated energy expenditure and reduction of body weight in PKD1‐deficient mice (Appendix Fig S7C and D). Altogether, these data suggest that deficiency of PKD1 induces beiging in white adipocytes. This elevates whole‐body energy dissipation and protects PKD1adipoΔ/Δ mice from diet‐induced obesity.
Figure 5. PKD1 suppresses expression of genes characteristic for beiging.

- RNA‐sequencing based heat map of the expression patterns of browning‐related genes in sWAT from mice of the indicated genotypes (GSE analysis software; n = 4/genotype).
- MA‐plot showing RNA‐sequencing results. Differential expression and average abundance [indicated as log2 fold‐change (FC) and log counts‐per‐million (CPM)] of all genes was calculated with EdgeR. Beiging‐related genes are highlighted in red (n = 4/genotype).
- qPCR analysis of expression of indicated genes in sWAT from control and PKD1‐deficient mice (n = 6/genotype).
- Ratio of mtDNA and gDNA in sWAT of control and PKD1‐deficient mice (n = 6/genotype).
- WB analysis of UCP1 in sWAT from control and PKD1‐deficient mice fed HFD including densitometric quantification of four replicates each.
PKD1 suppresses ADRB3 expression in an AMPK–C/EBP‐α/C/EBP‐δ‐dependent manner
Next, we tested if the expression of beige/brown markers is altered in isolated adipocytes. The expression of the creatine kinase cycle‐related genes and selected muscle markers (Ckm, Ckmt2, Myh1, Myh2, Mck, and Myg) was enhanced in differentiated SVC deficient for PKD1 (Fig 6A). However, the expression of Ucp1 and other classical beige/brown markers (Ucp3, Cidea, Cideb, Prdm16, Bmp7, Slc27a1, Slc27a2, Pgc‐1α, Pparα, and Pparγ) was not altered by PKD1 deletion (Fig 6A). Interestingly, the expression of the gene encoding for β3‐adrenergic receptor (Adrb3) was markedly enhanced in cultured adipocytes lacking PKD1 (Fig 6A). Consistently, immunofluorescence staining revealed elevated levels of β3‐adrenergic ADRB3 on the surface of PKD1‐deficient adipocytes (Fig 6B). β‐adrenergic stimulation results in induction of UCP1 expression in adipocytes (Kajimura et al, 2015). In fact, stimulation of PKD1‐deficient adipocytes and control cells with a β‐agonist resulted in enhanced expression of Ucp1 in the absence of PKD1 (Fig 6C). Altogether, these findings suggest that UCP1 expression in adipose tissue is regulated by PKD1 in an ADRB3‐dependent manner. However, the transcription factor mediating PKD1‐dependent ADRB3 expression remains unknown. A number of transcription factors mediate beige adipocyte function (Inagaki et al, 2016). We measured protein abundance and/or phosphorylation of CtBP1/CtBP2, PGC‐1α, C/EBP‐α, C/EBP‐δ, Rb, FoxO1, C/EBP‐β, ERRα, ATF2, CREB, and PRDM16 in 3T3L1 adipocytes expressing PKD1ca (Fig EV5A). Interestingly, expression of PKD1ca resulted in decreased abundance of C/EBP‐α and C/EBP‐δ (Fig EV5A). Conversely, depletion of adipocytes from PKD1 resulted in elevation of C/EBP‐α and C/EBP‐δ levels (Fig 6D and E), whereas mRNA levels of both transcription factors remained unchanged (Fig EV5B and C). Of note, stimulation of PKD1ca expressing adipocytes with AICAR resulted in increased C/EBP‐α and C/EBP‐δ levels in both, control and PKD1ca cells (Fig 6F). Previous studies showed that C/EBP‐α and possibly C/EBP‐δ are required for ADRB3 expression in adipocytes (Dixon et al, 2001). Moreover, deletion of C/EBP‐α and C/EBP‐δ in mice results in reduction of UCP1 expression in adipose tissue (Tanaka et al, 1997). To test whether PKD1 regulates ADRB3 expression in C/EBP‐α‐ and/or C/EBP‐δ‐dependent manner, we silenced these transcription factors in SVC deficient for PKD1. Expression analysis revealed that silencing of both C/ebp‐α and C/ebp‐δ (Fig EV5B and C) is sufficient to normalize elevated expression of Adrb3 in cells lacking PKD1 (Fig 6G). Altogether, our data suggest that PKD1 suppresses C/EBP‐α and C/EBP‐δ abundance in an AMPK‐dependent manner and both of these transcription factors promote ADRB3 expression which is required for full induction of beige/brown gene expression.
Figure 6. PKD1 suppresses ADRB3 expression in an AMPK–C/EBP‐α/C/EBP‐δ‐dependent manner.

-
AqPCR analysis of expression of indicated genes in differentiated SVC derived from sWAT from control and PKD1‐deficient mice (n > 7).
-
BRepresentative immunofluorescent images of ADRB3 staining and quantification of fluorescent intensity in PKD1‐deficient and control adipocytes derived from SVC (n > 43; representative of two individual experiments).
-
CqPCR analysis of Ucp1 in differentiated SVC of indicated genotypes after 24 h stimulation with Isoproterenol (Iso) at indicated concentrations (n = 5, representative of two individual experiments).
-
D, EWB analysis of indicated proteins in differentiated 3T3L1 cells lacking PKD1 (D) and SVC from control and PKD1‐deficient mice (E) including densitometric quantification of triplicates each.
-
FWB analysis of indicated proteins in differentiated 3T3L1 expressing PKD1ca and treated with AICAR (2 mM) for 2 h.
-
HqPCR analysis of Adrb3 in SVC derived from sWAT of control and PKD1‐deficient mice transfected with control siRNA, siRNA against C/EBP‐α and/or C/EBP‐δ (n > 4).
Figure EV5. PKD1 attenuates C/EBP‐α and C/EBP‐δ abundance.

-
AWB analysis of indicated proteins in differentiated 3T3L1 cells expressing PKD1ca including densitometric quantification of four replicates each.
-
B, CqPCR analysis of C/ebp‐α (B) and C/ebp‐δ (C) in SVC derived from sWAT of control and PKD1‐deficient mice transfected with control siRNA, siRNA against C/EBP‐α and/or C/EBP‐δ (n > 4).
PKD1 deletion in adipocytes protects against diet‐induced fatty liver disease and type 2 diabetes
AMPK protects against fatty liver disease and insulin resistance and its activation can control adipokine levels in the blood (Mottillo et al, 2016; Zhu et al, 2016). Therefore, we tested these conditions in mice with specific PKD1 deletion in adipocytes.
Levels of adiponectin and resistin in the blood of PKD1adipoΔ/Δ mice were not altered as compared to control animals when fed ND or HFD (Appendix Fig S8A and B). However, leptin levels were reduced in PKD1adipoΔ/Δ mice when fed ND, whereas on HDF reduction of leptin levels did not reach significance (P = 0.18), most probably due to the high variations induced upon HFD feeding (Appendix Fig S8C). Consistently with the fact that PKD1adipoΔ/Δ mice presented decreased adipose tissue content, we also observed that circulating FFA levels were markedly lower in HFD fed mice lacking PKD1 in adipocytes compared to control animals. PKD1adipoΔ/Δ mice fed ND showed moderately reduced FFA levels in the circulation (P = 0.13; Fig 7A). Importantly, levels of another lipolysis product, glycerol, and TG levels were not altered in PKD1adipoΔ/Δ mice (Appendix Fig S8D and E). Next, we tested whether PKD1adipoΔ/Δ mice respond better to insulin as predicted by lower FFA levels. Indeed, HFD fed PKD1adipoΔ/Δ mice challenged with insulin injection displayed lower levels of glucose as compared to control animals at all time points tested. PKD1adipoΔ/Δ mice fed ND also showed a moderate but significant improvement of insulin action (Fig 7B). In line with these results, PKD1adipoΔ/Δ mice fed with HFD for 24 weeks displayed improved glucose tolerance, whereas no impact of PKD1 deletion was observed in mice fed ND (Fig 7C). Interestingly, no impact of PKD1 deletion on glucose tolerance has been observed in mice fed HFD for a short period (8 weeks; Appendix Fig S8F), indicating that improved glucose tolerance in mice fed HFD for 24 weeks is secondary to the decreased adiposity. Importantly, improved glucose tolerance in PKD1adipoΔ/Δ mice was specifically caused by a better peripheral insulin sensitivity, since insulin levels were not altered in the absence of PKD1 (Appendix Fig S8G). Consistently, activation of Akt kinase, a key signaling molecule in the insulin pathway, was markedly enhanced in PKD1‐deficient adipocytes and reduced in adipocytes with elevated PKD1‐dependent signaling (Fig 7D and E).
Figure 7. Deletion of PKD1 in adipocytes protects against peripheral insulin resistance and hepatic steatosis.

-
A–CFFAs levels (A), glucose (B), and insulin (C) tolerance test in PKD1adipoΔ/Δ and control mice fed ND or HFD (n > 5/genotype).
-
D, EWB analysis of indicated proteins in SVC‐derived adipocytes lacking PKD1 (D) and 3T3L1‐derived adipocytes expressing PKD1ca (E).
-
F, GOil Red O staining of liver sections (F) and liver TG content (G) from PKD1adipoΔ/Δ and control mice fed HFD (n > 8/genotype).
-
HCorrelation between PKD1 expression and HOMA‐IR levels in human sWAT.
-
IBasal and insulin‐induced lipogenesis rate (arbitrary units—AU) in differentiated human subcutaneous preadipocytes treated with CRT 0066101 (3 μM) or vehicle for 3 days (n = 4; representative of two individual experiments).
-
JOxygen consumption rate (OCR) in response to indicated substances as well as OCR annotated to the indicated cellular processes in differentiated human subcutaneous preadipocytes treated with CRT 0066101 (3 μM) for 3 days (n = 19; representative of two individual experiments).
Obesity is often associated with fatty liver disease. Indeed, we observed a reduced liver weight in PKD1adipoΔ/Δ mice fed HFD (Fig 4D), which could be explained by a reduced triglyceride content in this organ (Fig 7F and G).
To test whether PKD1 also regulates insulin sensitivity in humans, we measured expression of PKD1 in sWAT of insulin resistant and control patients. PKD1 levels correlated positively with HOMA‐IR (the major parameter for insulin resistance), indicating that PKD1 might regulate insulin sensitivity in human tissues (Fig 7H). Moreover, human adipocytes treated with a PKD‐inhibitor presented reduced lipogenesis rate (Fig 7I). Consistently with our previous findings, inhibition of PKD in human adipocytes also resulted in higher oxygen consumption at the basal conditions and after oligomycin stimulation (oxygen consumption caused by the decoupling activity; Fig 7J). Altogether, deletion of PKD1 specifically in adipocytes protects from diet‐induced T2D and lipid accumulation in liver. Moreover, the proposed cellular mechanisms of PKD1 action also seem to be valid in human adipocytes.
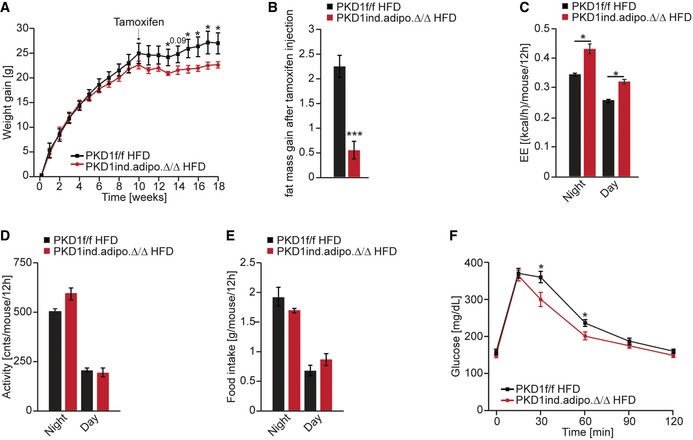
Next, we tested if abrogation of PKD1‐dependent signaling in adipose tissue of mice with established obesity can attenuate the course of this disease. For that purpose, we generated mice in which adipocyte‐specific deletion of PKD1 was induced by tamoxifen administration after 10 weeks of HFD feeding (Appendix Fig S8H). Of note, upon deletion of PKD1, mice did not further gain body weight compared to corresponding control animals (Fig 8A). Similarly, the gain of fat mass was almost completely abrogated upon PKD1 deletion while control animals kept accumulating fat (Fig 8B). Consistently with our previous data, PKD1 deletion resulted in a higher energy expenditure without affecting activity and food intake of these mice (Fig 8C–E). Finally, deletion of PKD1 in adipocytes of obese mice resulted in improved glucose tolerance (Fig 8F).
Figure 8. Inactivation of PKD1 in obese mice prevents further body weight gain.
-
ABody weight evolution of control and tamoxifen‐inducible PKD1 deletion (PKD1ind.adipoΔ/Δ) in mice fed HFD for 22 weeks (n > 6/genotype).
-
BFat mass gain after tamoxifen injection in control and PKD1ind.adipoΔ/Δ mice fed HFD (n > 6/genotype).
-
C–EEnergy expenditure (C), voluntary movement (D), and food intake (E) of control and PKD1ind.adipoΔ/Δ mice fed HFD (n > 6/genotype).
-
FGlucose tolerance test in PKD1ind.adipoΔ/Δ and control mice after 17 weeks of HFD (n > 6/genotype).
In conclusion, our results demonstrate that deletion of PKD1 promotes beiging in white adipocytes in an AMPK‐dependent manner. This results in an elevated whole‐body energy expenditure and is protective against diet‐induced obesity, development of T2D, and liver steatosis. Therefore, PKD1 could be a new therapeutic target to treat obesity.
Discussion
PKD1‐mediated signaling emerges as a master regulator of nutrient and energy homeostasis in health and disease (Sumara et al, 2009; Goginashvili et al, 2015; Bergeron et al, 2018). PKD1 is activated by DAG as well as PKCs (Rozengurt, 2011), and our data suggest that the levels and activity of PKD1 are induced in adipose tissue upon feeding. Moreover, we showed that in adipocytes PKD1 is activated specifically by extracellular purines. However, specific purinergic receptors responsible for PKD1 activation need to be identified. Adipocytes play a central role in the regulation of metabolic homeostasis. Excessive lipid accumulation in white adipocytes is a hallmark of obesity and promotes obesity‐related disorders. Importantly, formation of metabolically active beige adipocytes within the white adipose tissue protects against obesity (Kajimura et al, 2015; Kazak et al, 2015). Up to now, the physiological role of PKD1 in adipose tissue and its role in the relevant human diseases remained unknown. Here, we showed that PKD1 expressed in adipocytes plays a crucial role in the whole‐body energy homeostasis. PKD1adipoΔ/Δ mice are resistant to diet‐induced obesity. They display higher energy expenditure, lower content of adipose tissue, and smaller adipocytes. Moreover, sWAT depots contained higher numbers of mitochondria and showed increased expression of genes responsible for energy dissipation. Interestingly, we observed elevated expression of classical genes involved in adaptive thermogenesis (e.g., Adrb3, Ucp1, Pgc‐1α, and Ucp3) and genes promoting creatine‐driven substrate cycle (Ckm, Ckmt2), suggesting that PKD1 can suppress energy dissipation by adipocytes using several complementary mechanisms. Although we did not observe a clear correlation between the absence of PKD1 and appearance of multilocular cells within subcutaneous depot of adipose tissue, the protein abundance of UCP1 was clearly elevated in the absence of PKD1. Of note, in several other mouse models, increased UCP1 abundance was observed in unilocular adipocytes (Rossmeisl et al, 2002; Schneider et al, 2016; Zheng et al, 2017). However, in isolated PKD1‐deficient adipocytes we observed elevated gene expression of Adrb3, but Ucp1 levels were not altered at basal conditions. In fact, our data suggest that induction of UCP1 expression by PKD1 deletion requires presence of the ligand for β3‐adrenergic receptor, as addition of β‐agonists to the media of cultured adipocytes resulted in increased expression of UCP1 in the absence of PKD1. Furthermore, our data suggest that PKD1 regulates ADRB3 expression by targeting C/EBP‐α and C/EBP‐δ in an AMPK‐dependent manner. Of note, we did not observe enhanced expression of thermogenic genes in BAT of PKD1adipoΔ/Δ mice. Moreover, AMPK‐dependent signaling does not seem to be affected by the deletion of PKD1 in BAT. This result is in agreement with the fact that the deletion of PKD1 in isolated brown adipocytes did not affect basal respiration and energy dissipation. Taken together, our data indicate that PKD1 deletion promotes beiging in WAT, but does not affect brown adipocytes.
In addition, our data suggest an alternative way to support uncoupling respiration in adipocytes by regulating mitochondrial dynamics. A previous study revealed that β‐adrenergic stimulation increases mitochondrial fission in brown adipocytes leading to thermogenic activation and increased oxygen consumption (Wikstrom et al, 2014). Moreover, ablation of PPARγ target Bnip3 promotes mitochondrial fusion in white adipocytes, which results in suppression of energy dissipation (Tol et al, 2016). Our results indicate that the deletion of PKD1 increases mitochondrial fragmentation, which positively correlates with higher energy dissipation in adipocytes. Therefore, PKD1 represents the first identified signaling module (kinase) suppressing mitochondrial fragmentation to decrease energy dissipation by adipocytes.
Importantly, our data link PKD1 signaling to the key regulator of adipocyte function, AMPK. A recent study indicates that PKD1 suppresses activity of AMPK by phosphorylation of its alpha subunit in muscle cells in vitro (Coughlan et al, 2016). Our results show that PKD1 suppresses AMPK activity also in adipocytes. Of note, we showed that deletion of PKD1 in mice results in an enhanced AMPK signaling in subcutaneous depots of adipose tissue derived from mice fed ND or HFD. However, unlike other markers of AMPK activation, phosphorylation of T172 of AMPK alpha subunit in adipose tissue of mice fed HFD for 24 weeks was not clearly enhanced by PKD1 deletion. This indicates that prolonged HFD feeding might induce additional signaling cascades which partially mask the effect of PKD1 deletion. AMPK promotes mitochondrial biogenesis as well as the expression of genes involved in energy dissipation by adipocytes and suppresses lipogenesis (Bijland et al, 2013; Mottillo et al, 2016; Zhu et al, 2016). Consistently, PKD1 drives lipogenesis, reduces mitochondrial content, and lowers energy dissipation by adipose tissue. AMPK suppresses lipogenesis by phosphorylating and inhibiting the action of ACC (Bijland et al, 2013). Consistently, the ACC phosphorylation of S79 was elevated in the absence of PKD1, but the expression of genes implicated in lipid synthesis was not altered. AMPK activation in multiple cell lines leads to the induction of autophagy in an mTORC1‐dependent and mTORC1‐independent manner (Roach, 2011). In line with these data, we observed that PKD1 deletion leads to elevation of autophagy marker—LC3 cleavage. Induction of autophagy promotes beige to white adipocyte transition (Altshuler‐Keylin et al, 2016). Moreover, ATG7 deletion, which is required for induction of autophagy, results in a beiging‐like phenotype (Singh et al, 2009; Zhang et al, 2009). However, the activation of AMPK in adipocytes promotes energy expenditure and protects against obesity (Gaidhu et al, 2011; Zhang et al, 2014; Zhu et al, 2016). Therefore, our data support the notion that AMPK promotes energy dissipation by adipocytes by utilizing autophagy‐independent mechanisms. Additionally, in other cell types AMPK promotes mitochondrial fragmentation (Toyama et al, 2016), while our results indicate that PKD1 suppresses this process. Moreover, the effect of overexpression of PKD1ca on mitochondrial aggregation is fully reversible by manipulation of AMPK activity. A number of studies aimed to define the AMPK impact on the regulation of lipolysis. Both anti‐ and pro‐lipolytic effects of AMPK have been reported (Djouder et al, 2010; Bijland et al, 2013). A recent genetic study revealed that ablation of AMPK activity in adipocytes does not affect β‐adrenergic‐induced lipolysis in mice (Mottillo et al, 2016). However, during cancer‐associated cachexia, loss of AMPK activity promotes both lipolysis and lipogenesis, which results in energy dissipation by adipocytes (Rohm et al, 2016). Our results indicate that PKD1 does not affect basal and β‐adrenergic‐induced lipolysis in isolated adipocytes. Also, the levels of FFAs and glycerol of fasted young‐adult (8 weeks old) PKD1‐deficient mice are not altered. However, we showed that FFA levels are reduced in mice lacking PKD1, which is probably caused by the decreased adipose tissue content. Similarly to the deletion of PKD1, elevation of AMPK signaling in adipocytes by genetic manipulations or pharmacological interventions ameliorates diet‐induced obesity, insulin resistance, and hepatic steatosis by enhancing energy expenditure of animals (Gaidhu et al, 2011; Zhang et al, 2014; Zhu et al, 2016).
Expression of PKD1 in adipocytes promotes insulin resistance and mice lacking PKD1 present improved insulin sensitivity, glucose tolerance, and protection against liver steatosis. However, PKD1 in cardiomyocytes rather promotes insulin sensitivity (Dirkx et al, 2014), indicating that the impact of PKD1 on insulin signaling might depend on the cellular context. Alternatively, improved insulin sensitivity in the absence of PKD1 in adipocytes might be secondary to the reduced lipid accumulation. Strikingly, however, we demonstrated that PKD1 expression in human adipose tissue correlates with insulin sensitivity. Moreover, in line with our data obtained in rodents, we showed that inhibition of PKD in human adipocytes promotes energy dissipation and suppresses lipogenesis, which indicates the presented mechanisms are conserved in humans.
Taken together, our data provide evidence for the crucial role of PKD1 signaling in fat tissue and energy homeostasis in vivo. We predict that inhibition of PKD1 specifically in fat tissue represents an attractive strategy for the development of precision medicine to treat obesity and diabetes in the future.
Materials and Methods
Preadipocyte culture and differentiation
3T3L1 preadipocytes (ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM), 10% fetal calf serum (FCS), and 40 μg/ml gentamycin and differentiated into adipocytes as previously described (Cai et al, 2017). T37I cells were cultured in DMEM‐F12, 10% FCS, and 1% sodium pyruvate (SP). Differentiation was induced 2 days post‐confluence by adding 2 nM T3‐hormone and 1.5 μg/ml insulin for 7 days to achieve full differentiation as described before (Zennaro et al, 1998). Stromal vascular cells (SVC) from subcutaneous white adipose tissue (sWAT) and brown adipose tissue (BAT) of 5‐ to 9‐week‐old mice were isolated and differentiated into mature adipocytes. Concisely, sWAT or BAT was collected from mice, minced, and treated with 2 mg/ml collagenase D (11088882001, Roche) in PBS containing 5 mM CaCl2 and 1% BSA for 40 min. After hemolysis and filtration of adipose tissue through a 40‐μm mesh, the sample was washed in PBS by centrifugation and cultured in DMEM/F‐12 containing 10% FBS, 1% SP, 1% non‐essential amino acids (NEAA), and 1% Penicillin‐Streptomycin (P/S). Two days post‐confluence, SVC differentiation was induced by adding a cocktail including 0.2 μM indomethacin, 1 μM dexamethasone, 0.5 mM IBMX, and 1.5 μg/ml insulin for the first 4 days, followed by insulin treatment for additional 4 days to achieve differentiation into adipocytes. In SVC derived from BAT, additionally 100 nM T3 2 nM was added to the medium throughout differentiation. Before treatment, cells were serum starved for 2 h in DMEM supplemented with 0.5% BSA and afterward stimulated with 100 nM insulin for 10 min. In addition, in certain experiments 2 mM AICAR (Sigma) was added to the medium for 2 h. In case of PKD1 stimulation assessment, cells were serum starved for 2 h followed by 100 μM 1,2‐dioctanoyl‐sn‐glycerol (DAG; Enzo Life Science) stimulation for 2 or 4 h or stimulation with insulin (Sigma; 10/100 nM), carbachol (Tocris; 1/10 μM), isoproterenol (Sigma; 1/10 μM), serotonin (Sigma; 50/250 μM), ATP (Sigma; 10/300 mM), and ADP (Sigma; 100/300 mM). Afterward, cell lysates were used for Western blot analysis. In case of lipogenesis assay, cells were serum starved for 3 h. SVC isolated from sWAT were serum starved for 2 h followed by 24 h stimulation with Isoproterenol (0.1, 1, and 10 μM) for further qPCR analysis. Human subcutaneous preadipocytes (ATCC) were cultured and differentiated into adipocytes according to the manufacturer's protocol. Differentiated human preadipocytes were pretreated with 3 μM CRT 0066101 (Tocris) for 3 days and used to measure lipogenesis rate or mitochondrial fragmentation.
Explants derived from sWAT
Explants from freshly harvested mouse subcutaneous adipose tissue (sWAT) were removed from the mouse. After removal of fibrotic tissue and obvious vasculature, the tissue was cut into roughly 15 mg of tissue pieces, which were embedded in the individual tubes filled with DMEM and 0.5% BSA for 2 h at 37°C to equilibrate. Explants were either used for WB analysis afterward or lipogenesis assay was performed in these explants with additional Compound C (CC; Tocris; 5 μM) treatment for 4 h.
PKD1 flox/flox adiponectin Cre mice (PKD1adipo ∆/∆) and tamoxifen‐inducible strain (PKD1ind.adipo ∆/∆)
All animal studies were approved by the local institutional animal care (Regierung von Unterfranken, Germany) and conducted according to the guidelines and state regulations. Animal protocol number AK 55.2‐2531.01‐124/13 approved on 28.01.2014. PKD1flox/flox mice (Fielitz et al, 2008) were cross‐bred with adiponectin promoter‐driven Cre mice (Eguchi et al, 2011; Jackson Laboratory) or adiponectin promoter‐driven Cre‐ERT mice (Sassmann et al, 2010; Jackson Laboratory), which resulted in mice with targeted deletion of PKD1 in adipocytes of white and brown adipose tissue from the beginning or after induction with 100 mg/kg tamoxifen injection for five consecutive days. Genotyping protocols for the given mouse lines are available from Jackson Laboratory. Mice were kept in cages from Tecniplast (#GM500PFS) in a green Line IVC‐rack system and provided with sterile water and irradiated rodent normal diet (sniff Spezialdiäten) at all times, which was only removed or exchanged if necessary for an experiment. High‐fat diet (HFD; ResearchDiets) was given to mice when indicated. In case of thermoneutral conditions, mice were kept at 30°C during the experiment.
Human samples
The human studies were approved by the Ethics Committee of the University Hospital of Salamanca, and all subjects provided written informed consent to undergo subcutaneous fat biopsy under direct vision during surgery. The study population included adults who underwent elective bariatric surgery at the University Hospital of Salamanca. Patients were excluded if they had a history of alcohol abuse or excessive alcohol consumption (> 30 g/day in men and > 20 g/day in women), or chronic hepatitis C or B or diabetes. Data were collected on demographic information (age, sex, and ethnicity), anthropomorphic measurements (BMI), smoking and alcohol history, coexisting medical conditions, and medication use. Fasting venous blood samples were collected, and sWAT was obtained during surgery and frozen for posterior analysis.
AMP activity assay
To measure AMPK activity in cell lysates the immunoassay CycLex AMPK Kinase Assay Kit (MBL International Corporation) was used. For CycLex AMPK Kinase Assay, differentiated 3T3L1 cells were serum starved for 2 h followed by 100 nM Insulin stimulation for 10 min. Cell lysates were collected, and AMPK activity was measured following the manufacturer's instructions. Absorbance was read at 450/540 nm with (Zennaro et al, 1998) Spark 10 M microplate reader (Tecan). Results were normalized to protein content, after background substraction.
Quantification of triglyceride accumulation and lipolysis rate
The amount of triglycerides normalized to DNA content of the cells was assessed by double staining of AdipoRed reagent (Lonza) and Hoechst 33342 (Thermo Fischer Scientific) according to manufacturers’ protocols. Oil Red O staining was used to visualize the multiple intracellular lipid droplets in cells. Briefly, once fully differentiated, cells were fixed with 10% formaldehyde for 30 min and Oil Red O solution was added for 1 h. For certain experiments, cells were either transfected with siRNA against AMPKα1/α2 subunits for 48 h or pretreated with 0.5 mM AICAR for 3 days. To assess lipolysis rate, cells were serum starved for 2 h in phenol red‐free DMEM supplemented with 0.5% BSA and afterward stimulated with 10 μM isoproterenol (Iso; Sigma‐Aldrich) for 2 h in the fresh medium. FFAs in the medium were measured using NEFA reagents (Wako), and glycerol was quantified by free glycerol reagent (Sigma‐Aldrich) according to the manufacturers’ instructions.
Lipogenesis assay
Cells were incubated in 1 μCi/ml D‐[3‐3H]‐glucose (Perkin Elmer) for 3 h in the presence or absence of 100 nM insulin. Total intracellular lipids were extracted with a mixture of chloroform and methanol (2:1, v/v). After centrifugation, chloroform phase was transferred into a scintillation vial and 4 ml of scintillation liquid was added before counting. De novo lipogenesis rates were normalized to protein levels using Quick Start Bradford Protein Assay (Bio‐Rad). For certain experiments, cells were either transfected with siRNA against AMPKα1/α2 subunits for 48 h or pretreated with 2 mM AICAR for 2 h.
Mitochondrial respiration
Mitochondrial respiration was determined by measuring oxygen consumption rate (OCR) using the Seahorse XF Cell Mito Stress Test (103015‐100; Agilent Technologies) in a Seahorse XFe96 Analyzer according to the manufacturer's protocol. Briefly, cells were incubated for 1 h with 175 μl Seahorse assay medium containing 1 mM sodium pyruvate, 2 mM glutamine, and 5 mM glucose. Meanwhile, the Seahorse sensor cartridge ports were loaded with 25 μl of inhibitors to have a final concentration of 2 μM oligomycin (port A), 1 μM FCCP (port B), and 0.75 μM rotenone/antimycin A (port C). The experimental design was set up using the WAVE software program, and measurement was performed in the Seahorse XFe96 Analyzer. Normalization to the DNA content was conducted by fixing the cells in 75% ethanol, followed by staining with Crystal Violet (Sigma‐Aldrich) for 30 min, washed with water and air‐dried. Afterward, Crystal Violet was dissolved in 10% acidic acid and the absorption was measured at 590 nm in a Spark 10 M microplate reader (Tecan). For certain experiments, cells were either transfected with siRNA against AMPKα1/α2 subunits for 48 h or pretreated with 0.5 mM AICAR for 3 days.
Western blot
Western blot analysis was performed according to standard procedures, by running the proteins on an 8–10% SDS–PAGE, transferring it onto polyvinylidene fluoride (PVDF) membranes (Millipore), and then probing the membrane with primary antibodies against: AMPKα (D5A2) Rabbit mAb (Cell Signaling Technology #5831), Phospho‐AMPKα1 (Ser485)/AMPKα2 (Ser491) Rabbit Ab (Cell Signaling Technology #4185), Phospho‐AMPKα (Thr172; 40H9) Rabbit mAb (Cell Signaling Technology #2535), Acetyl‐CoA Carboxylase (C83B10) Rabbit mAb (Cell Signaling Technology #3676), Phospho‐Acetyl‐CoA Carboxylase (Ser79; D7D11) Rabbit mAb (Cell Signaling Technology #11818), PKD/PKCμ Rabbit Ab (Cell Signaling Technology #2052), Phospho‐PKD/PKCμ (Ser916) Rabbit Ab (Cell Signaling Technology #2051), Phospho‐PKD/PKCμ (Ser744/748) Rabbit Ab (Cell Signaling Technology #2054), β‐Actin Mouse mAb (Sigma‐Aldrich #A5441), GAPDH Rabbit Ab (Sigma‐Aldrich #G9545), α‐Tubulin Rabbit Ab (Cell Signaling Technology #2144), Phospho‐Akt (Ser473) Rabbit Ab (Cell Signaling Technology #4058), Myc‐Tag (9B11) Mouse mAb (Cell Signaling Technology #2276), PPARγ (81B8) Rabbit mAb (Cell Signaling Technology #2443), HSL Rabbit Ab (Cell Signaling Technology #4107), ATGL (30A4) Rabbit mAb (Cell Signaling Technology #2439), or Perilipin (D1D8) Rabbit mAb (Cell Signaling Technology #9349), Anti‐UCP1 antibody (Abcam #ab10983), Phospho‐CtBP1/CtBP2 (Ser158, Ser164) Polyclonal Antibody (Thermo Fischer Scientific #PA5‐64665), PGC‐1α (3G6) Rabbit mAb (Cell Signaling Technology #2178), C/EBPα (D56F10) Rabbit mAb (Cell Signaling Technology #8178), C/EBPδ Rabbit Ab (Cell Signaling Technology #2318), Phospho‐Rb (Ser807/811; D20B12) Rabbit mAb (Cell Signaling Technology #8516), Phospho‐FoxO1 (Ser256) Ab (Cell Signaling Technology #9461), C/EBPβ (LAP) Ab (Cell Signaling Technology #3087), ERRα (E1G1J) Rabbit mAb (Cell Signaling Technology #13826), Phospho‐ATF‐2 (Thr71; 11G2) Rabbit mAb (Cell Signaling Technology #5112), Phospho‐CREB (Ser133; 87G3) Rabbit mAb (Cell Signaling Technology #9198), Phospho‐C/EBPβ (Thr235) Ab (Cell Signaling Technology #3084), Anti‐PRDM16 antibody (Abcam #ab106410). After incubation with corresponding horseradish peroxidase (HRP)‐conjugated secondary antibodies, proteins were visualized by ECL (Bio‐Rad) using X‐ray films.
Real time PCR analysis
Total RNA was extracted from cells and tissue pieces according to the QIAzol Handbook using the RNeasy Plus Universal Tissue Mini Kit (Qiagen). RNA concentration was spectrophotometrically (Nanodrop, Thermo Fischer Scientific) determined and first‐strand cDNA was synthesized by First Strand cDNA Synthesis Kit (Thermo Fischer Scientific) according to the manufacturer's protocol. Real‐time quantitative polymerase chain reaction (RT–qPCR) was performed using SYBR green Universal PCR master mix (Thermo Fischer Scientific) on a QuantStudio 5 Real Time PCR System (Thermo Fischer Scientific). Expression of all genes was normalized to the Rpl13a housekeeping gene (Appendix Table S1). Mitochondrial DNA (mtDNA) copy number was analyzed from total DNA isolated from differentiated SVC as well as snap‐frozen sWAT samples, according to the manufacturer's instructions using the DNeasy Blood & Tissue Kit (Qiagen). mtDNA was normalized to genomic DNA (gDNA; Appendix Table S1) by RT–qPCR. For analysis of PKD1 expression in humans, sWAT was obtained during surgery and frozen to posterior analysis. From this tissue, 1 mg of RNA was extracted with RNeasy Plus Mini kit (Qiagen) following manufacturer's instructions and further transcribed to cDNA. RT–PCR was performed using Fast TaqMan probe (Thermo Fischer Scientific) and the appropriate TaqMan Assay (Thermo Fischer Scientific) in the 7900 Fast Real Time thermocycler. Relative mRNA expression was normalized to 18s mRNA measured in each sample.
Mitochondrial staining
For analysis of mitochondrial morphology, cells were plated on a cover slider overnight. Mitochondria were stained by adding 150 nM MitoTracker Red (Thermo Fischer Scientific) in the suitable growth medium for 30 min at 37°C. The cells were fixed with 4% PFA for 20 min. After washing with PBS 1×, cells were permeabilized by adding ice‐cold methanol for 5 min. Cover slides were mounted using ProLong Gold Antifade Mountant with DAPI (Thermo Fischer Scientific). In case of AICAR treatment, 2 mM AICAR was added 2 h prior to the fixation. Cells were imaged using a fluorescent microscope (Leica). To quantify mitochondrial morphology in stained differentiated adipocytes, grouping into the different categories was done blindly to genotype and treatment. Cells were defined as fragmented (fragm.) when mitochondria were spherical with no clear length or width. When mitochondria were highly interconnected and elongated, the cells were clustered as fused. All the cells in between were counted as intermediate (interm.).
Immunofluorescent staining of Adrb3
Cells were fixed in 4% PFA and stained according to the standard procedure. Primary antibody against ADRB3 (Anti‐beta 3‐adrenergic Receptor antibody; Abcam #ab59685) and secondary antibody (Alexa Fluor® 488 AffiniPure Donkey Anti‐Chicken IgY; Jackson Immuno Research #703‐545‐155) were used. ProLong Gold Antifade Mountant with DAPI (Thermo Fischer Scientific) was used for mounting. Cells were imaged using a fluorescent microscope (Leica). Fluorescent intensity of the single cells (about 120 cells per genotype) was quantified using ImageJ software.
RNA sequencing
RNeasy mini columns (Qiagen) with on‐column DNase I digestion were used to extract total RNA from tissue samples. PolyA+ RNA was extracted from total RNA using the NEBNext® Poly(A) mRNA Magnetic Isolation Module (NEB). Preparation of sequencing libraries was performed using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina (NEB) including the use of Agencourt AMPure XP Beads (Beckman Coulter) for size selection and purification of the libraries. The Experion Automated Electrophoresis System (Bio‐Rad) was used for library quantification and quality control. All samples were sequenced on a NextSeq500 System (Illumina), and base calling was done with Illumina's FASTQ Generation software v1.0.0. The FastQC script (Babraham Bioinformatics) was used to determine overall sequencing quality. Fastq files were mapped to mm9 using Bowtie2 with default settings, and the countOverlaps function from the R package GenomicRanges was used to count reads per gene. Following removal of weakly expressed genes (mean CPM over all samples < 1), the differential expression of the remaining genes was called using EdgeR. GSE analyses (Subramanian et al, 2005) were done using signal2noise metric, 1,000 permutations, and the C2 gene set collection (v6.0) of MSigDB. In some cases, the C2 gene set collection was spiked with a gene signature of published markers for browning.
Histological analysis
For histological analysis, adipose tissues were fixed in 10% formaldehyde and embedded in paraffin. The sections (4–5 μm) were stained with hematoxylin–eosin (H&E) according to the standard procedure. Livers were embedded in OCT‐Tissue‐Tek (SAKURA FINETEK USA INC), and frozen sections were stained according to the standard procedure. Briefly, frozen liver sections (5 μm) were stained in a 0.375% Oil Red O solution (3:2 isopropanol:ddH2O, v/v) for 10 min. Subsequently, the sections were rinsed under running tap water for 30 min and embedded in a water‐soluble mounting medium. For immunohistochemistry, primary antibody to heavy chain myosin (Abcam #124205) was used after rehydration and epitope retrieval and later detected with SignalStain® (Cell Signaling) according to the standard procedure. Quadriceps sections were used as positive controls.
Cell size quantification
Images from H&E‐stained sections of gonadal white adipose tissue (gWAT) and sWAT from mice under normal or high‐fat diet were taken with 20× magnification under the microscope. Four representative pictures were analyzed from each sample by manually measuring the area of each cell in a total area of 0.6 mm2. ImageJ was utilized for this measurement by using the tool, which allows manually encircling of each cell, and automatic measurement of the area.
Liver triglyceride extraction
Frozen liver pieces were homogenized in PBS, and lipids were extracted with a mixture of 2:1 chloroform:methanol (v/v). The lipid‐containing chloroform phase was dried under nitrogen flow, and lipids were re‐solubilized in 15% Triton X‐100. Liver triglycerides were quantified using the commercial glycerol and triglyceride reagents according to manufacturer's instructions and normalized to total protein amount.
Glucose and insulin tolerance tests
Glucose tolerance test (GTT) was performed in 4 h fasted mice at week 8 and 18 of high‐fat diet (HFD) and week 18 of normal diet (ND), and insulin tolerance test (ITT) after 20 weeks of ND or HFD feeding. A defined dose of glucose (Roth; 1 g/kg BW) or insulin (Sanofi‐Aventis; 0.25 U/kg BW) was injected and glucose removal from the blood was measured prior to the test, 15, 30, 60, 90, and 120 min after intraperitoneal injection. Blood glucose levels were assessed from tail vein blood by an automated glucometer (Accu‐Chek, Roche).
Indirect calorimetry analysis
Body weight gain of mice was monitored every other week, and food intake, energy expenditure, and voluntary movements were measured at the end of respective diets using the Phenomaster system (TSE) in mice housed at 23 or 30°C as described before (Trujillo Viera et al, 2016).
Analysis of blood samples in mice and humans
FFAs, glycerol, and triglycerides were determined from serum of mice using NEFA (Lonza), free glycerol, and triglyceride reagents (both Sigma‐Aldrich) according to the manufacturers’ instructions. Analysis of adiponectin, leptin, resistin, and insulin was performed by a magnetic‐bead based multiplex ELISA assay (Bio‐Rad) on a MagPix Systems (Bio‐Rad) according to the manufacturer's guidelines. We collected blood samples for measuring complete cell blood count, as well as glucose using a standard glucometer. Blood samples were also centrifuged, and plasma was frozen to detect insulin by Luminex assay (Appendix Table S2). Individually matched glucose and insulin levels were used to assess HOMA‐IR as previously described using the following calculations: HOMA‐IR = [glucose (mg/dl) × insulin (mU/l)/405].
Statistical analysis
All data, unless otherwise indicated, are presented as mean values ± standard error of the mean (SEM). Data set comparisons of more than two groups were analyzed using one‐way analysis of variance (ANOVA) followed by the post hoc Tukey test. Determination of significances between two independent groups was determined with unpaired, two‐tailed Student's t‐test. P‐values at the level of 0.05 were considered statistically significant. Each figure gives exact statistical indications.
Data availability
The RNA‐sequencing raw‐ and processed datasets as well as information on data processing have been deposited in NCBI Gene Expression Omnibus under the accession number: GSE104797.
Author contributions
MCL performed most of the experiments and largely contributed to writing as well as the experimental design. GS wrote the manuscript and was responsible for the experimental design. AEM, JTV, RE‐M, ALV, TK, WS, AS, ME, SJ‐R, NM, JLT, and MM performed part of the experiments. CPA performed high‐throughput sequencing and analysis of RNA‐sequencing data. GS, AS, and ME contributed to the experimental design.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Dr. Olga Sumara, Prof. Dr. Bernhard Nieswandt, and Dr. Izabela Sumara for critical comments to our manuscript. We thank Dr. Marc Lombes for kindly providing us the T37I cells. This project was supported by the Starting Grant (SicMetabol) from the European Research Council (ERC) and internal funds of the Rudolf Virchow Center. Additionally, J. Trujillo, R. El‐Merabih, and G. Sumara are supported by the Emmy Noether grant from the German Research Foundation (Number Su 820/1‐1). G. Sabio received funding from the European Union's Seventh Framework Program (FP7/2007‐2013) under grant agreement no ERC 260464, EFSD/Lilly, MINECO‐FEDER SAF2016‐79126‐R, and Comunidad de Madrid S2010/BMD‐2326; to M.M.: ISCIII and FEDER, PI10/01692, and I3SNS‐INT12/049.
The EMBO Journal (2018) 37: e99182
References
- Altshuler‐Keylin S, Shinoda K, Hasegawa Y, Ikeda K, Hong H, Kang Q, Yang Y, Perera RM, Debnath J, Kajimura S (2016) Beige adipocyte maintenance is regulated by autophagy‐induced mitochondrial clearance. Cell Metab 24: 402–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron V, Ghislain J, Vivot K, Tamarina N, Philipson LH, Fielitz J, Poitout V (2018) Deletion of protein kinase D1 in pancreatic beta‐cells impairs insulin secretion in high‐fat diet‐fed mice. Diabetes 67: 71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijland S, Mancini SJ, Salt IP (2013) Role of AMP‐activated protein kinase in adipose tissue metabolism and inflammation. Clin Sci (Lond) 124: 491–507 [DOI] [PubMed] [Google Scholar]
- Cai K, El‐Merahbi R, Loeffler M, Mayer AE, Sumara G (2017) Ndrg1 promotes adipocyte differentiation and sustains their function. Sci Rep 7: 7191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlan KA, Valentine RJ, Sudit BS, Allen K, Dagon Y, Kahn BB, Ruderman NB, Saha AK (2016) PKD1 inhibits AMPKalpha2 through phosphorylation of serine 491 and impairs insulin signaling in skeletal muscle cells. J Biol Chem 291: 5664–5675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirkx E, van Eys GJ, Schwenk RW, Steinbusch LK, Hoebers N, Coumans WA, Peters T, Janssen BJ, Brans B, Vogg AT, Neumann D, Glatz JF, Luiken JJ (2014) Protein kinase‐D1 overexpression prevents lipid‐induced cardiac insulin resistance. J Mol Cell Cardiol 76: 208–217 [DOI] [PubMed] [Google Scholar]
- Dixon TM, Daniel KW, Farmer SR, Collins S (2001) CCAAT/enhancer‐binding protein α is required for transcription of the β3‐adrenergic receptor gene during adipogenesis. J Biol Chem 276: 722–728 [DOI] [PubMed] [Google Scholar]
- Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, Viollet B, Makela TP, Wallimann T, Neumann D, Krek W (2010) PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J 29: 469–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J, Estall JL, Klein U, Maratos‐Flier E, Rosen ED (2011) Transcriptional control of adipose lipid handling by IRF4. Cell Metab 13: 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Merahbi R, Loffler M, Mayer A, Sumara G (2015) The roles of peripheral serotonin in metabolic homeostasis. FEBS Lett 589: 1728–1734 [DOI] [PubMed] [Google Scholar]
- Ferdaoussi M, Bergeron V, Zarrouki B, Kolic J, Cantley J, Fielitz J, Olson EN, Prentki M, Biden T, MacDonald PE, Poitout V (2012) G protein‐coupled receptor (GPR)40‐dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1. Diabetologia 55: 2682–2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielitz J, Kim MS, Shelton JM, Qi X, Hill JA, Richardson JA, Bassel‐Duby R, Olson EN (2008) Requirement of protein kinase D1 for pathological cardiac remodeling. Proc Natl Acad Sci USA 105: 3059–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidhu MP, Frontini A, Hung S, Pistor K, Cinti S, Ceddia RB (2011) Chronic AMP‐kinase activation with AICAR reduces adiposity by remodeling adipocyte metabolism and increasing leptin sensitivity. J Lipid Res 52: 1702–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehart H, Goginashvili A, Beck R, Morvan J, Erbs E, Formentini I, De Matteis MA, Schwab Y, Wieland FT, Ricci R (2012) The BAR domain protein Arfaptin‐1 controls secretory granule biogenesis at the trans‐Golgi network. Dev Cell 23: 756–768 [DOI] [PubMed] [Google Scholar]
- Goginashvili A, Zhang Z, Erbs E, Spiegelhalter C, Kessler P, Mihlan M, Pasquier A, Krupina K, Schieber N, Cinque L, Morvan J, Sumara I, Schwab Y, Settembre C, Ricci R (2015) Insulin granules. Insulin secretory granules control autophagy in pancreatic beta cells. Science 347: 878–882 [DOI] [PubMed] [Google Scholar]
- Inagaki T, Sakai J, Kajimura S (2016) Transcriptional and epigenetic control of brown and beige adipose cell fate and function. Nat Rev Mol Cell Biol 17: 480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner A, Block H, Reichel CA, Varjosalo M, Gehart H, Sumara G, Gstaiger M, Krombach F, Zarbock A, Ricci R (2012) Regulation of PTEN activity by p38delta‐PKD1 signaling in neutrophils confers inflammatory responses in the lung. J Exp Med 209: 2229–2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Saito M (2014) A new era in brown adipose tissue biology: molecular control of brown fat development and energy homeostasis. Annu Rev Physiol 76: 225–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Spiegelman BM, Seale P (2015) Brown and beige fat: physiological roles beyond heat generation. Cell Metab 22: 546–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazak L, Chouchani ET, Jedrychowski MP, Erickson BK, Shinoda K, Cohen P, Vetrivelan R, Lu GZ, Laznik‐Bogoslavski D, Hasenfuss SC, Kajimura S, Gygi SP, Spiegelman BM (2015) A creatine‐driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell 163: 643–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Fielitz J, McAnally J, Shelton JM, Lemon DD, McKinsey TA, Richardson JA, Bassel‐Duby R, Olson EN (2008) Protein kinase D1 stimulates MEF2 activity in skeletal muscle and enhances muscle performance. Mol Cell Biol 28: 3600–3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan K‐L (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews SA, Liu P, Spitaler M, Olson EN, McKinsey TA, Cantrell DA, Scharenberg AM (2006) Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes. Mol Cell Biol 26: 1569–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottillo EP, Desjardins EM, Crane JD, Smith BK, Green AE, Ducommun S, Henriksen TI, Rebalka IA, Razi A, Sakamoto K, Scheele C, Kemp BE, Hawke TJ, Ortega J, Granneman JG, Steinberg GR (2016) Lack of adipocyte AMPK exacerbates insulin resistance and hepatic steatosis through brown and beige adipose tissue function. Cell Metab 24: 118–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller TD, Lee SJ, Jastroch M, Kabra D, Stemmer K, Aichler M, Abplanalp B, Ananthakrishnan G, Bhardwaj N, Collins S, Divanovic S, Endele M, Finan B, Gao Y, Habegger KM, Hembree J, Heppner KM, Hofmann S, Holland J, Kuchler D et al (2013) p62 links beta‐adrenergic input to mitochondrial function and thermogenesis. J Clin Invest 123: 469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach PJ (2011) AMPK→ uLK1→ autophagy. Mol Cell Biol 31: 3082–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohm M, Schafer M, Laurent V, Ustunel BE, Niopek K, Algire C, Hautzinger O, Sijmonsma TP, Zota A, Medrikova D, Pellegata NS, Ryden M, Kulyte A, Dahlman I, Arner P, Petrovic N, Cannon B, Amri EZ, Kemp BE, Steinberg GR et al (2016) An AMP‐activated protein kinase‐stabilizing peptide ameliorates adipose tissue wasting in cancer cachexia in mice. Nat Med 22: 1120–1130 [DOI] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM (2006) Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444: 847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmeisl M, Barbatelli G, Flachs P, Brauner P, Zingaretti MC, Marelli M, Janovská P, Horáková M, Syrový I, Cinti S (2002) Expression of the uncoupling protein 1 from the aP2 gene promoter stimulates mitochondrial biogenesis in unilocular adipocytes in vivo . Eur J Biochem 269: 19–28 [DOI] [PubMed] [Google Scholar]
- Rozengurt E (2011) Protein kinase D signaling: multiple biological functions in health and disease. Physiology (Bethesda) 26: 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Shulman GI (2012) Mechanisms for insulin resistance: common threads and missing links. Cell 148: 852–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassmann A, Offermanns S, Wettschureck N (2010) Tamoxifen‐inducible Cre‐mediated recombination in adipocytes. Genesis 48: 618–625 [DOI] [PubMed] [Google Scholar]
- Schneider K, Valdez J, Nguyen J, Vawter M, Galke B, Kurtz TW, Chan JY (2016) Increased energy expenditure, Ucp1 expression, and resistance to diet‐induced obesity in mice lacking nuclear factor‐erythroid‐2‐related transcription factor‐2 (Nrf2). J Biol Chem 291: 7754–7766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ (2009) Autophagy regulates adipose mass and differentiation in mice. J Clin Invest 119: 3329–3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman BM, Flier JS (2001) Obesity and the regulation of energy balance. Cell 104: 531–543 [DOI] [PubMed] [Google Scholar]
- Steiner TS, Ivison SM, Yao Y, Kifayet A (2010) Protein kinase D1 and D2 are involved in chemokine release induced by toll‐like receptors 2, 4, and 5. Cell Immunol 264: 135–142 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumara G, Formentini I, Collins S, Sumara I, Windak R, Bodenmiller B, Ramracheya R, Caille D, Jiang H, Platt KA, Meda P, Aebersold R, Rorsman P, Ricci R (2009) Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis. Cell 136: 235–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumara G, Sumara O, Kim JK, Karsenty G (2012) Gut‐derived serotonin is a multifunctional determinant to fasting adaptation. Cell Metab 16: 588–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Yoshida N, Kishimoto T, Akira S (1997) Defective adipocyte differentiation in mice lacking the C/EBPβ and/or C/EBPδ gene. EMBO J 16: 7432–7443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tol MJ, Ottenhoff R, van Eijk M, Zelcer N, Aten J, Houten SM, Geerts D, van Roomen C, Bierlaagh MC, Scheij S, Hoeksema MA, Aerts JM, Bogan JS, Dorn GW II, Argmann CA, Verhoeven AJ (2016) A PPARgamma‐Bnip3 axis couples adipose mitochondrial fusion‐fission balance to systemic insulin sensitivity. Diabetes 65: 2591–2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Loson OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, Shaw RJ (2016) Metabolism. AMP‐activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351: 275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozzi M, Novak I (2017) Purinergic receptors in adipose tissue as potential targets in metabolic disorders. Front Pharmacol 8: 878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo Viera J, El‐Merahbi R, Nieswandt B, Stegner D, Sumara G (2016) Phospholipases D1 and D2 suppress appetite and protect against overweight. PLoS One 11: e0157607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Hyde B, Shirihai OS (2008) Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta 1777: 1092–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrom JD, Mahdaviani K, Liesa M, Sereda SB, Si Y, Las G, Twig G, Petrovic N, Zingaretti C, Graham A, Cinti S, Corkey BE, Cannon B, Nedergaard J, Shirihai OS (2014) Hormone‐induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J 33: 418–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, Madeo F (2012) FAT SIGNALS–lipases and lipolysis in lipid metabolism and signaling. Cell Metab 15: 279–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zennaro M‐C, Le Menuet D, Viengchareun S, Walker F, Ricquier D, Lombès M (1998) Hibernoma development in transgenic mice identifies brown adipose tissue as a novel target of aldosterone action. J Clin Invest 101: 1254–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S (2009) Adipose‐specific deletion of autophagy‐related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci USA 106: 19860–19865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang H, Li B, Meng X, Wang J, Zhang Y, Yao S, Ma Q, Jin L, Yang J, Wang W, Ning G (2014) Berberine activates thermogenesis in white and brown adipose tissue. Nat Commun 5: 5493 [DOI] [PubMed] [Google Scholar]
- Zheng Q, Lin J, Huang J, Zhang H, Zhang R, Zhang X, Cao C, Hambly C, Qin G, Yao J (2017) Reconstitution of UCP1 using CRISPR/Cas9 in the white adipose tissue of pigs decreases fat deposition and improves thermogenic capacity. Proc Natl Acad Sci USA 114: E9474–E9482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Ghoshal S, Rodrigues A, Gao S, Asterian A, Kamenecka TM, Barrow JC, Chakraborty A (2016) Adipocyte‐specific deletion of Ip6k1 reduces diet‐induced obesity by enhancing AMPK‐mediated thermogenesis. J Clin Invest 126: 4273–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The RNA‐sequencing raw‐ and processed datasets as well as information on data processing have been deposited in NCBI Gene Expression Omnibus under the accession number: GSE104797.