

Abstract
The endoplasmic reticulum‐associated protein degradation (ERAD) is responsible for recognizing and retro‐translocating protein substrates, misfolded or not, from the ER for cytosolic proteasomal degradation. HMG‐CoA Reductase (HMGCR) Degradation protein—HRD1—was initially identified as an E3 ligase critical for ERAD. However, its physiological functions remain largely undefined. Herein, we discovered that hepatic HRD1 expression is induced in the postprandial condition upon mouse refeeding. Mice with liver‐specific HRD1 deletion failed to repress FGF21 production in serum and liver even in the refeeding condition and phenocopy the FGF21 gain‐of‐function mice showing growth retardation, female infertility, and diurnal circadian behavior disruption. HRD1‐ERAD facilitates the degradation of the liver‐specific ER‐tethered transcription factor CREBH to downregulate FGF21 expression. HRD1‐ERAD catalyzes polyubiquitin conjugation onto CREBH at lysine 294 for its proteasomal degradation, bridging a multi‐organ crosstalk in regulating growth, circadian behavior, and female fertility through regulating the CREBH‐FGF21 regulatory axis.
Keywords: CREBH, ER‐associated degradation, FGF21, HRD1, multi‐organ crosstalk
Subject Categories: Membrane & Intracellular Transport; Metabolism; Post-translational Modifications, Proteolysis & Proteomics
Introduction
The liver undergoes a postprandial metabolic reprogramming to assimilate the glucose and triggers liver–brain–periphery axis to turn off the fat utilization (Izumida et al, 2013). Hepatokine fibroblast growth factor 21 (FGF21) is a metabolic hormone induced by various metabolic stresses, including fasting, high‐carbohydrate diets and exercise that regulates energy homeostasis(Gaich et al, 2013; Galman et al, 2008; Kim et al, 2013a,b; Lan et al, 2017; Solon‐Biet et al, 2016). The transgenic overexpression FGF21 mice showed female infertility and circadian behavior disruption (Bookout et al, 2013; Owen et al, 2013), and FGF21 treatment has also been shown to modulate the growth hormone, insulin‐like growth factor 1 (IGF1) axis, and stimulate bone loss (Talukdar et al, 2016; Wang et al, 2015; Wei et al, 2012b; Zhang et al, 2012). Therefore, FGF21 expression should be tightly controlled. It is well established that stress‐inducible hepatic transcriptional factor CREBH interacts with the nuclear receptor PPARα to synergistically regulate FGF21 expression during fasting (Galman et al, 2008; Kim et al, 2014). But it is not clear how the FGF21 expression is negatively regulated in controlling the physiological functions.
It has been suggested that the unfold protein response (UPR) is rapidly and temporarily activated upon refeeding to switch from glucose production to glucose assimilation through directly regulating the gluconeogenic and lipogenic gene expression (Malhi & Kaufman, 2011). The UPR does not only modulate the hepatic metabolic reprogramming but also increases ER‐associated protein degradation (ERAD; Christianson et al, 2011). However, the metabolic function of the ERAD system is largely uncharacterized. HRD1, the HMG‐CoA Reductase (HMGCR) Degradation protein, was initially identified as an E3 ligase to control cholesterol production through regulation of rate‐limiting enzyme HMGCR turnover in yeast (Hampton & Rine, 1994). As the key member of the ERAD system, HRD1 was found to catalyze ubiquitin conjugation of IRE1α, an ER stress sensor, to inhibit cell apoptosis and control intestinal stem cell homeostasis (Sun et al, 2015). The IRE1α downstream transcription factor Xbp‐1 has been shown to regulate HRD1 mRNA transcription, providing an interesting feedback loop for HRD1‐IRE1α during ER stress response (Gao et al, 2008). We recently found that HRD1 is involved in immune regulation in dendritic cell antigen presentation, as well as in the activation of both T and B lymphocytes (Kong et al, 2016; Xu et al, 2016; Yang et al, 2014). However, it is not clear whether HRD1 is regulated upon metabolic stresses. The metabolic roles of HRD1 remain largely uncharacterized.
In this study, we demonstrate that hepatic HRD1 plays a critical role in negatively regulating FGF21 expression and links a multi‐organ crosstalk in the regulation of growth, diurnal circadian behavior, and female fertility. Using the RNA‐seq and quantitative proteomic methods, we identified 737 proteins were increased at protein level without altering their mRNA expression in HRD1‐null liver, including transcriptional factor CREBH, which are potential HRD1 substrates. Further data showed that E3 ligase HRD1 decreases CREBH stability through mediating its ubiquitination. We also found that hepatic FGF21 deletion and high‐fat diet rescued the FGF21 gain‐of‐function‐like phenotypes.
Results
Deletion of HRD1 specifically in liver leads to growth retardation
To explore novel mechanism underlying hepatic reprogramming after refeeding, we used a fasting–refeeding model and analyzed the hepatic mRNA expression profile. Upon refeeding, 360 genes were decreased by more than 50% and 303 genes were increased for at least twofold in mouse liver. Gene Ontology characterization showed, consistent with previous studies (Malhi & Kaufman, 2011; Mihaylova & Shaw, 2011), that the upregulated genes were involved in facilitating the cholesterol and lipid biosynthetic process, but the genes for the long‐chain fatty acid and triglyceride (TG) metabolic process were downregulated upon refeeding (Appendix Fig S1A and B). Interestingly, the expression of genes involved in ERAD, including HRD1, was largely altered by fasting and refeeding (Fig 1A and Appendix Fig S1A–C), implying a critical role of liver HRD1‐ERAD in response to fasting and refeeding. To investigate the potential role of HRD1 in hepatic metabolic reprogramming, we generated a strain of liver‐specific HRD1 knockout (LKO, Hrd1 f/fAlbumin‐Cre+) mice (Appendix Fig S1D and E). Despite their body weights were comparable before the age of 4 weeks, both male and female HRD1 LKO mice were markedly smaller than their wild‐type littermate controls from 6 weeks with significant reductions in body weight (Fig 1B and C), body height, and tibia length (Fig 1D). Importantly, the STAT5 phosphorylation, which is the main transcription factor of the growth hormone signaling, was dramatically changed in liver by HRD1 deletion (Fig 1E). Consequently, STAT5 target genes, including Igf1, Hsd3b5, Cyp2d9, Cyp7b1, Cyp4a12, Mup1, and Mup3 (Inagaki et al, 2008), were decreased in the livers from HRD1 LKO mice (Fig 1F and G). Therefore, our results suggest that hepatic HRD1 appears to achieve its biological functions in regulating systemic growth.
Figure 1. Deletion of HRD1 specifically in liver leads to growth retardation.
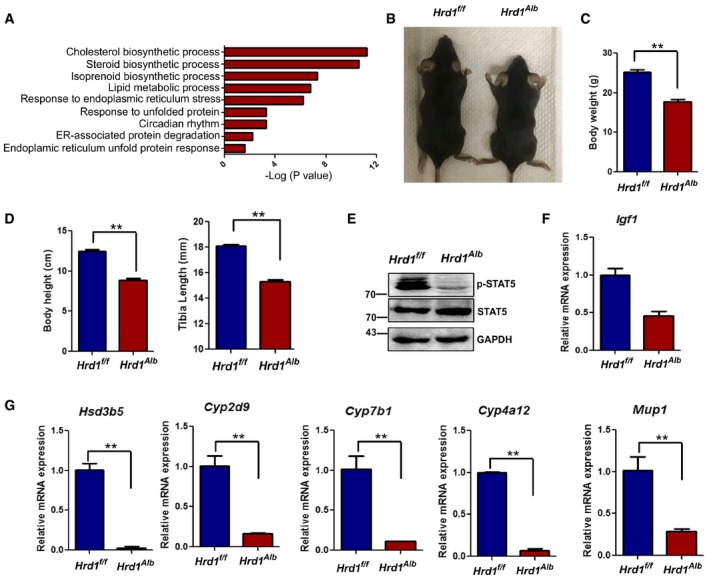
-
AGO functional analysis of the differential genes in the fasted and refed condition.
-
B, CBody weight of the WT and L‐HRD KO mice at the age of 12 weeks (n = 5 for each group).
-
DBody heights and tibia lengths of the WT and L‐HRD KO mice (n = 5 for each group).
-
EPhosphorylation of STAT5 in WT and HRD1 LKO liver.
-
F, GHepatic growth hormone signaling target gene mRNA levels in the WT and L‐HRD1 KO mice (n = 5 for each group).
Genetic ablation of hepatic HRD1 elevated hepatic mRNA and circulating FGF21
To investigate molecular mechanisms of hepatic HRD1 in regulating the growth, we analyzed the differential mRNA expression profile between WT and HRD1 KO livers using RNA‐seq. Notably, the hepatic FGF21 mRNA was dramatically upregulated by HRD1 deletion both in the fasting and in the refeeding conditions (Fig 2A), which was further confirmed by qPCR (Fig 2B). Consistent with previous studies (Bookout et al, 2013; Owen et al, 2013), our data show that FGF21 transcription is tightly controlled by fasting–refeeding in WT mice. However, refeeding failed to suppress Fgf21 expression in HRD1 LKO mice (Fig 2C), indicating that liver HRD1 gene deletion results in growth retardation due to FGF21 overexpression, which is similar to the FGF21 transgenic mice (Sakaguchi et al, 2017). Indeed, a strong positive correlation in the liver gene expression between HRD1 LKO and FGF21 transgenic overexpression mice was confirmed (Fig 1D and Appendix Fig S1F and G).
Figure 2. Genetic ablation of hepatic HRD1‐elevated hepatic mRNA and circulating Fgf21.
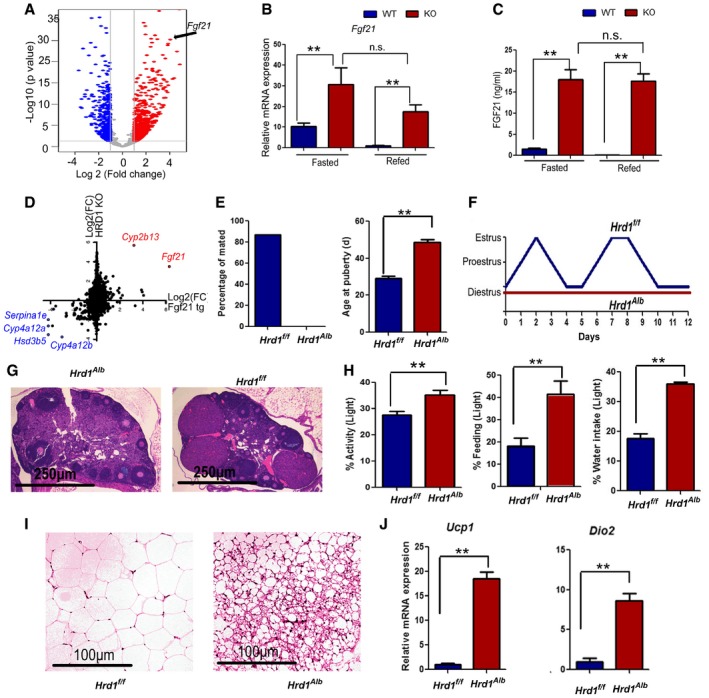
- Volcano plot of differential genes between WT and HRD1 LKO livers.
- Hepatic Fgf21 mRNA in the WT and L‐HRD1 KO mice (n = 5 for each group).
- Serum FGF21 protein levels in the WT and L‐HRD1 KO mice (n = 5 for each group).
- Correlation analysis of the gene expression (red: upregulated genes and blue: downregulated genes) between HRD1 LKO mice and FGF21 transgenic mice.
- Percentage of mated with proven stud males and age at onset of puberty in the female WT and L‐HRD KO mice (n = 10 for each group).
- Examples of ovarian histology from WT and HRD1 LKO mice.
- Representative examples of estrous cycles in WT and L‐HRD1 KO mice as determined by vaginal cytology.
- Percentage of feeding, water intake, and activity in the light time (n = 8 for each group).
- H&E stain of white adipose tissue WT and HRD1 LKO mice 14 weeks after HFD feeding.
- Ucp1 and Dio2 mRNA levels of adipose tissue from (I) (n = 5 for each group).
Genetic ablation of hepatic HRD1 phenocopy of Fgf21 transgenic gain‐of‐function mice
Interestingly, similar to mice with transgenic FGF21 expression (Bookout et al, 2013; Owen et al, 2013), we observed that female HRD1 LKO mice are infertile. Characterization of the cause of infertility revealed a delay in the onset of puberty and a failure to mate with proven stud males (Fig 2E). H&E staining showed that the follicles in chow‐fed HRD1 LKO mice rarely became postovulation corpora lutea (Fig 2G). Vaginal cytology methods showed abnormalities consistent with anovulatory hypogonadism (Fig 2F and Appendix Fig S2A). HRD1 LKO females rarely entered the ovulatory estrous phase of the cycle and displayed a prolonged diestrus (Fig 2F and Appendix Fig S2A). In addition, HRD1 LKO mice displayed severely fragmented activity (Fig 2H and Appendix Fig S2C), which were also found in the FGF21 transgenic overexpression mice under the entrainment of 12:12‐h LD condition (Bunger et al, 2000). In parallel with the changes in activity, HRD1 LKO mice had a dampened diurnal rhythm of feeding and drinking activity compared to WT mice (Fig 2H and Appendix Fig S2B–D). WT mice exhibited a robust diurnal rhythm with their activity and feeding largely (~ 80%) occurring during the dark period, whereas HRD1 LKO mice exhibited a phenotype of attenuation in rhythmicity, including consuming a higher percentage of daily food intake, water intake, and activity to 40% during the resting (light) period (Fig 2H). Collectively, our data indicate that HRD1 bridges a crosstalk of liver with its distal organs through controlling FGF21 production.
FGF21 has emerged as an important regulator of metabolic process and a new therapeutic target for the treatment of obesity and diabetes (Gaich et al, 2013). Indeed, glucose and TG levels were dramatically decreased in the HRD1 LKO mice (Appendix Fig S2E). To further investigate whether mice with liver HRD1 deletion display other key beneficial effects of FGF21 overexpression, WT and HRD1 LKO mice were fed with high‐fat diet for 14 weeks. As expected, the HRD1 LKO mice gained a significantly less body weight than that of the control mice (Appendix Fig S2F). Further, the accumulation of subcutaneous white adipose tissue and lipid levels were dramatically lower in HRD1 LKO mice compared to WT mice (Appendix Fig S2G and H). Histological analysis detected a significant increase in browning of the white adipose tissue in HRD1 LKO mice (Fig 2I). Moreover, mRNA of mitochondrial uncoupling protein 1 (Ucp1) and iodothyronine deiodinase 2 (Dio2) were also elevated in the adipose tissue of HRD1 LKO mice (Fig 2J). These results clearly indicate that liver‐specific HRD1 deletion protects mice from HFD‐induced obesity and hepatic steatosis.
Identification of potential HRD1‐ERAD substrates by protein‐level enrichment
As a ubiquitin ligase, HRD1 may regulate FGF21 expression through targeting protein degradation. We then aimed to identify proteins whose expressions are altered at post‐transcriptional levels in HRD1 LKO mouse liver (Fig 3A and B). We established a highly reproducible label‐free quantitative proteomic approach with an average Pearson correlation coefficient of 0.93 and detected a total of 5,985 proteins in liver from both WT and HRD1 LKO mice, out of which 838 proteins were dramatically increased and 218 were dramatically decreased (Fig 3C and Appendix Fig S3A and B). While the data were analyzed with a missing value of protein intensity replaced with the minimum value, a consistent result was obtained when the missing value of protein intensity was replaced with 1 (Appendix Fig S3C), further validating our proteomic studies. Meanwhile, the RNA‐seq analysis identified 15,503 genes with 959 increased and 484 decreased in the LKO liver (Appendix Fig S4A). GO functional analysis of the differential proteins showed that the upregulated proteins in the KO livers are critical for ER stress signaling and protein folding, which were reported before (Fig 3D and Appendix Fig S4B). Of note, 737 proteins were increased at protein level without altering their mRNA expression in HRD1‐null liver, which are potential HRD1 substrates (Fig 3E). Interestingly, 96 of those 737 proteins were involved in mRNA transcription including transcription factor and cofactor, and epigenetic regulator such as CREB/ATF family (CREB3, ATF6, ATF7, and CREBH) and RXR family (RXRα and RXRβ; Appendix Fig S4C and D). Moreover, the protein levels of IRE1α, NRF1, RXRβ, and NOTCH1, which have been reported as HRD1 substrates, were confirmed to be dramatically increased by hepatic HRD1 deletion (Fig 3F), while their mRNA levels were unaltered (Appendix Fig S4E).
Figure 3. 737 candidate HRD1 substrates are identified by protein‐level enrichment.
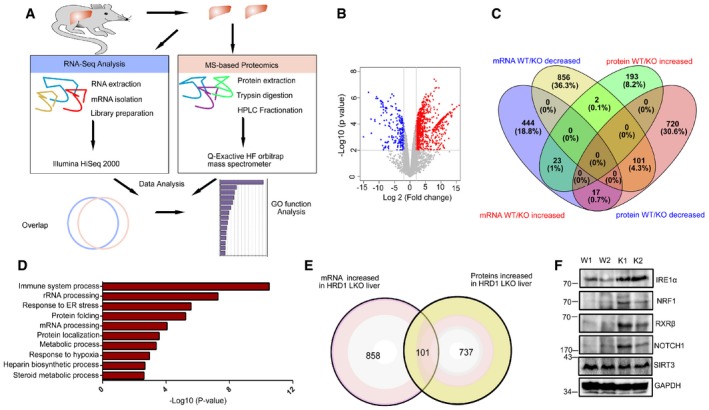
- Flowchart for the RNA‐seq and proteomic identification of differential mRNA and proteins.
- Volcano plot of differential proteins between WT and HRD1 LKO livers.
- Venn diagram of the overlaps of the differential genes in mRNA and protein levels.
- GO functional analysis of the differential proteins from proteomic screening.
- Overlap of the increased genes identified between RNA‐seq and proteomics.
- Protein levels of IRE1α, NRF1, RXRβ, NOTCH1, SIRT3, and GAPDH in the liver of WT and HRD1 LKO mice under the refed condition (n = 6 for each group).
Source data are available online for this figure.
HRD1‐ERAD inhibits FGF21 production through CREBH
It was reported that FGF21 expression is regulated by the ER stress‐associated transcription factors including ATF4 and CREBH (Kim et al, 2014, 2013a,b). Our mRNA profile indicated that the ATF4 signaling pathway was dramatically increased after HRD1 deletion (Appendix Fig S5A). The levels of Chop, Atf3, Asns, and Psat1 mRNA, as well as the protein expression of PERK, ATF4, and its target gene CHOP, were increased after HRD1 deletion (Appendix Fig S5B and C). In contrast, the mRNA of PERK between WT and HRD1 LKO mice was comparable (Appendix Fig S5D and E). These results imply a possibility that HRD1 inhibits FGF21 expression through ATF4 suppression. However, pharmacological suppression of PERK, the upstream kinase for ATF4 transcriptional activation, while inhibited Chop, Asns, Psat1 transcription and ATF4 protein expression as expected, failed to suppress FGF21 expression (Appendix Fig S5F and G), largely excluding the possibility that HRD1 regulates FGF21 transcription through targeting ATF4. FGF21 is also regulated by the IRE1‐Xbp1 branch of the unfolded protein response. We found that Xbp1s levels were comparable between WT and HRD1 LKO mice (Appendix Fig S5H).
Liver‐enriched transcription factor CREBH is one of the main FGF21 expression regulator10. Interestingly, our compared proteomic and RNA‐seq analysis showed that CREBH protein but not its mRNA expression was also increased in the HRD LKO livers (Appendix Fig S4C). Western blotting further confirmed that both the full‐length and the transcriptionally activated forms of CREBH were significantly elevated in the HRD1‐null hepatocytes (Fig 4A and B). CREBH interacted with peroxisome proliferator‐activated receptor α to regulate FGF21 expression. As expected, our chromatin immunoprecipitation (ChIP) analysis detected a significant increase in CREBH binding to the FGF21 gene promoters in the livers of HRD1 LKO mice (Appendix Fig S6A). However, the binding of PPARα to FGF21 gene promoters in the livers of HRD1 LKO mice was unaltered (Appendix Fig S6A). Interestingly, both the hepatic mRNA and protein levels of PPARα were decreased in the HRD1 LKO mice (Appendix Fig S6B and C). Therefore, we concluded that CREBH but not PPARα is the main transcription factor, which mediated FGF21 overexpression in the HRD1 LKO mice.
Figure 4. HRD1‐ERAD decreases the stability of CREBH through mediating its ubiquitination.
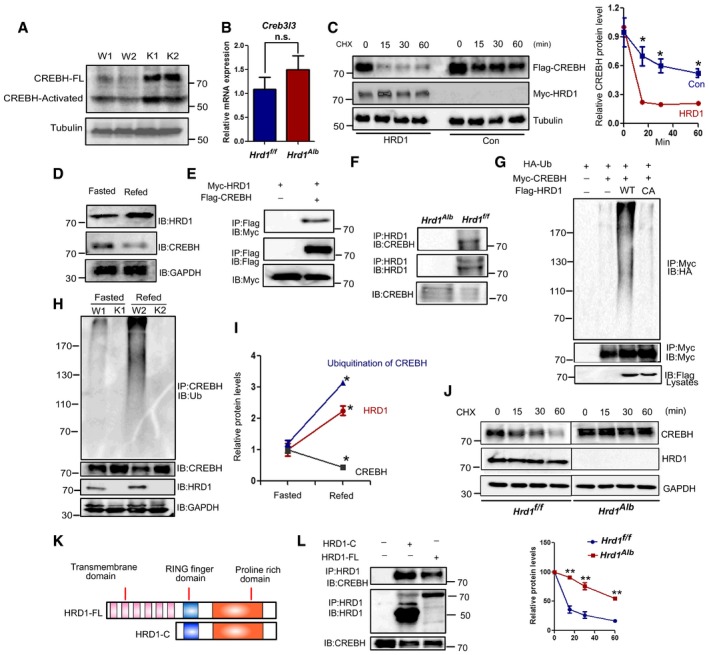
-
A, BHepatic CREBH protein (A) and mRNA (B) levels in the WT and L‐HRD1 KO mice (n = 6 for each group).
-
CWestern blot analysis of CREBH protein stability after HRD1 overexpression.
-
DWestern blot analysis of hepatic CREBH and HRD1 protein levels in WT and HRD1 LKO mice. The mice were fasted 16 h and refed for another 4 h.
-
EWestern blot analysis of interaction of CREBH and HRD1 after immunoprecipitates of Flag‐agarose in transfected HEK293T.
-
FEndogenous interaction between CREBH and HRD1 in liver.
-
GWestern blot analysis of ubiquitination of CREBH after immunoprecipitates of Flag‐agarose in transfected HEK293T.
-
HHepatic CREBH ubiquitination level in the WT and L‐HRD1 KO mice from (D).
-
IRelative protein levels of CREBH, HRD1, and ubiquitination of CREBH.
-
JPrimary hepatocytes were isolated from WT and HRD1 LKO mice and Western blot analysis CREBH protein stability.
-
KFull‐length HRD1 and C‐terminal HRD1 were generated.
-
LInteractions of full‐length HRD1 and C‐terminal HRD1 with CREBH were measured by Co‐IP.
HRD1 interacts with CREBH and promotes its ubiquitination‐mediated degradation in hepatocytes
In contrast to protein elevation, CREBH mRNA expression was not altered by HRD1 deletion (Fig 4A and B). These results suggest that HRD1 functions as a possible E3 ubiquitin ligase to promote CREBH degradation. Supporting this notion, the half‐life of the CREBH protein was dramatically decreased in cells co‐transfected with HRD1 (Fig 4C). Physiologically, protein levels of CREBH are inversely correlated with HRD1 in liver during fasting–refeeding (Fig 4D and I). Further, Co‐IP and Western blotting analysis detected that HRD1 interacted with CREBH and its overexpression dramatically enhanced CREBH ubiquitination, and mutation of the critical cysteine in the RING finger of HRD1 completely abolished CREBH ubiquitination (Fig 4E and G). In addition, HRD1 could endogenously interact with CREBH in mouse liver (Fig 4F). Consistent to the results that HRD1 expression was dramatically increased in the refeeding condition, we found that CREBH ubiquitination was also increased in refeeding condition and loss of HRD1 expression largely diminished the ubiquitination of CREBH in the HRD1 LKO hepatocytes (Fig 4H and I). To further determine whether HRD1 regulates CREBH turnover, primary hepatocytes were isolated from WT and HRD1 LKO mice.Twenty four hours after isolation, the primary hepatocytes were treated with CHX for indicated time. As shown in Fig 4J, the half‐life of CREBH protein dramatically increased in KO primary hepatocytes.
HRD1 protein contains six transmembrane domains, and its cytoplasmic tail carries an E3 ligase catalytic RING finger and a long proline‐rich C terminus (Fig 4K). However, deletion of the transmembrane domains in HRD1 did not affect its interaction with CREBH (Fig 4L), and HRD1‐C expression sufficiently promoted CREBH ubiquitination (Fig 4D), indicating that the RING finger‐containing cytoplasmic domain of HRD1 (HRD1‐C) is sufficient to regulate CREBH stability (Appendix Fig S7C). Consistent with this, HRD1‐C largely localizes in the cytoplasm and partially co‐localizes with the full‐length CREBH (Appendix Fig S7A). As a positive control, a partial colocalization of the full‐length HRD1 and CREBH was confirmed (Appendix Fig S7A). In contrast, as reported before, the cleaved‐CREBH are exclusively localized in the nucleus of the cell (Appendix Fig S7B) and could not interact with HRD1‐FL as shown in Fig 5B. Accordingly, HRD1‐C overexpression failed to promote the degradation of cleaved‐CREBH (Appendix Fig S7D). Collectively, these results indicate that HRD1 is a ubiquitin ligase for CREBH protein destruction in hepatocytes.
Figure 5. HRD1‐ERAD increases CREBH ubiquitination at the lysine residue 294 through C‐terminal RING finger and proline‐rich domain.
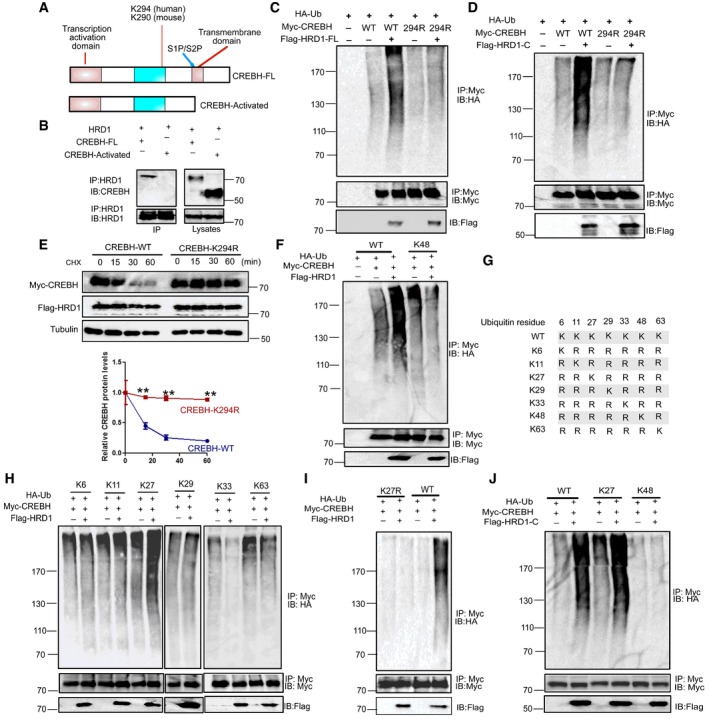
-
AFull‐length and activated forms of CREBH were generated.
-
BInteractions of full‐length and activated forms of CREBH with HRD1 were measured by Co‐IP.
-
C, DUbiquitination of CREBH and CREBH K294R was measured after co‐transfected C‐terminal of HRD1.
-
EWestern blot analysis of CREBH or CREBH‐K294R protein stability after HRD1 protein overexpression.
-
FWestern blot analysis of the CREBH WT‐ or K48‐only ubiquitination level after HRD1 C‐terminal co‐expression.
-
GSchematic presentation of wild‐type Ub and its mutants.
-
HWestern blot analysis of the CREBH K6‐, K11‐, K27‐, K29‐, and K33‐only ubiquitination level after HRD1 co‐expression.
-
IWestern blot analysis of the CREBH WT, K27R ubiquitination level after HRD1 full‐length co‐expression.
-
JWestern blot analysis of the CREBH WT‐, K27‐, and K48‐only ubiquitination level after HRD1 C‐terminal co‐expression.
HRD1‐ERAD catalyzes CREBH polyubiquitination
CREBH protein contains a transmembrane domain to localize at the ER membrane, which is spliced to produce a transcriptionally activated form by S1P (Zhang et al, 2006; Fig 5A). The fact that both the full‐length and activated forms of CREBH are increased in HRD1‐null hepatocytes implies a possibility that HRD1 may target both forms for ubiquitination‐mediated degradation. Surprisingly, only the full length but not its activated form of CREBH could interact with HRD1 (Fig 5B), indicating that transmembrane domain and ER localization of CREBH are essential for the HRD1‐CREBH interaction. Cleaved‐CREBH transferred to the nucleus in the overexpression and refeeding condition (Appendix Fig S7B, E, and F). However, HRD1‐C mainly localized in the cytoplasm (Appendix Fig S7A). Accordingly, HRD1‐C could not promote the degradation of cleaved‐CREBH (Appendix Fig S7D).
In addition, mutation of the lysine residue 294, a critical lysine acetylated by the acetyltransferase PCAF for CREBH transcriptional activation and FGF21 expression (Kim et al, 2015), totally abolished full‐length HRD1 and C‐terminal‐mediated ubiquitination (Fig 5C and D). The protein stability of CREBH/K294R was not affected by HRD1 expression (Fig 5E). Therefore, HRD1‐ERAD catalyzes CREBH ubiquitination at the lysine 294 for degradation.
It has been shown that the AAA+ ATPase p97 specifically recognize the K48‐ or K11‐linked polyubiquitinated substrates for ERAD (Locke et al, 2014), implying that ERAD ubiquitin ligases catalyze protein ubiquitination in a K48‐ or K11‐linked manner. However, HRD1 appears not to catalyze CREBH ubiquitination via the canonical K48 linkage (Fig 5F). We then defined the polyubiquitin chain topology using the serial mutations of ubiquitin plasmids as reported (Livingston et al, 2009; Fig 5G). Unexpectedly, HRD1‐mediated polyubiquitination of CREBH is largely in a K27‐linked manner (Fig 5H and J). Moreover, CREBH polyubiquitination mediated by HRD1 was not detected when using ubiquitin K27R (Fig 5I and Appendix Fig S8). Therefore, our studies suggested a previously unappreciated polyubiquitin topology by HRD1‐ERAD in regulating liver FGF21 expression through K27‐linked ubiquitination of CREBH.
Hepatic ablation of CREBH and FGF21 rescues the phenotypes induced by hepatic HRD1 deletion
To further determine whether HRD1 regulates FGF21 expression through CREBH, adenovirus expressing shRNA specific to mouse CREBH was delivered into wild‐type or HRD1 LKO mice through i.v. infusion (Appendix Fig S9A). We confirmed that CREBH mRNA was dramatically decreased 5 days after adenovirus injection (Appendix Fig S9B). Indeed, hepatic FGF21 expression induced by HRD1 deletion was reversed by CREBH knockdown (Appendix Fig S9C and D), confirming that HRD1 regulates FGF21 expression in a CREBH‐dependent manner. As the adenovirus‐mediated gene expression can only maintain for a short time period and it triggers robust inflammation, Adeno‐associated viral shCrebh was also generated and administrated to HRD1 LKO mice (Fig 6A). CREBH was dramatically decreased after AAV‐shCrebh administration in the HRD1 LKO mice (Fig 6B). As expected, FGF21 mRNA and protein levels were repressed by AAV‐shCrebh administration in the HRD1 LKO mice (Fig 6C and D). Body height and tibia length retardation by HRD1 ablation were rescued by AAV‐shCrebh administration (Fig 6E). Accordingly, the growth hormone JAK‐STAT5 target genes, including Igf1, Cyp2d9, cyp4a12, were also rescued by AAV‐shCrebh administration (Fig 6F). Female infertility and estrous cycle of HRD1 LKO mice were also rescued by AAV‐shCrebh administration (Fig 6G and H). These results demonstrate that HRD1 represses FGF21 expression through CREBH degradation.
Figure 6. CREBH ablation rescues the phenotypes induced by hepatic HRD1 deletion.
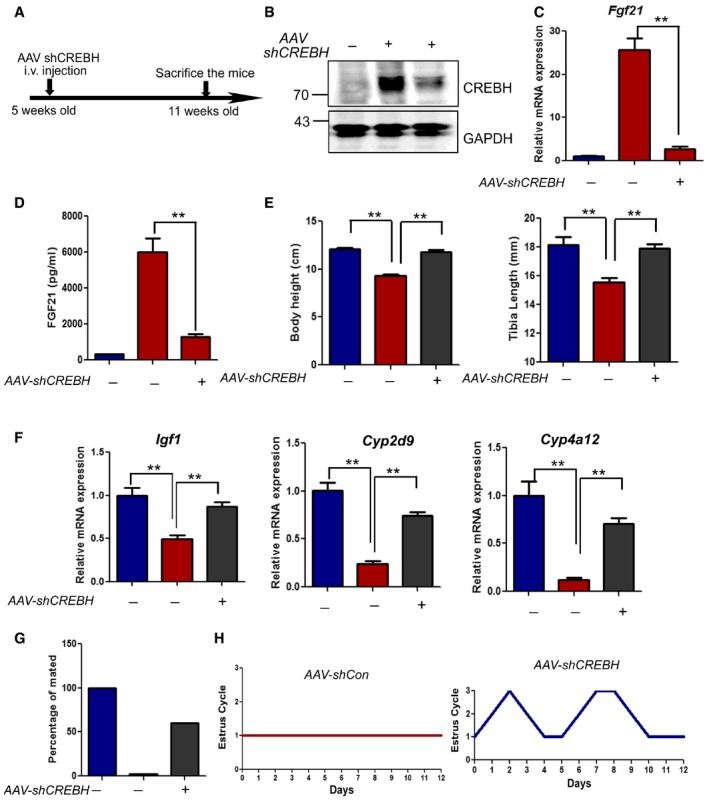
-
AFlowchart of the study design for the knockdown CREBH in vivo.
-
BHepatic CREBH protein levels 5 weeks after AAV‐shCrebh injection (n = 5 for each group).
-
CHepatic Fgf21 mRNA in the WT and L‐HRD1 KO mice 5 weeks after AAV‐shCrebh injection (n = 5 for each group).
-
DSerum FGF21 protein levels in the WT and L‐HRD1 KO mice 5 weeks after AAV‐shCrebh injection (n = 5 for each group).
-
EBody height and tibia length of the WT and L‐HRD1 KO mice 5 weeks after AAV‐shCrebh injection (n = 5 for each group).
-
FHepatic Igf1, Cyp2d9, and Cyp4a12 mRNA in the WT and L‐HRD1 KO mice 5 weeks after AAV‐shCrebh injection (n = 5 for each group).
-
G, HPercentage of the mated and estrous cycle of the WT and L‐HRD KO 5 weeks after AAV‐shCrebh injection.
We then determined whether the hepatic HRD1 regulates FGF21 gain‐of‐function‐like phenotypes through CREBH‐FGF21 axis by generating Hrd1 f/f Fgf21 f/fAlb‐Cre+ mice (HRD1/FGF21 double KO, DKO mice). Notably, further FGF21 deletion largely rescued the growth retardation of HRD1 LKO mice because the DKO animals exhibited a comparable body height and tibia length compared with HRD LKO mice (Fig 7A–D). Body weight and the growth hormone JAK‐STAT5 target genes, including Hsd3b5, Cyp2d9, cyp4a12, were also partially rescued by FGF21 ablation (Fig 7B and E). Importantly, the onset of the estrous cycle of the DKO mice but not HRD1 LKO mice could be observed at the age of 4 months (Fig 7F). In addition, in contrast to the fact that HRD1 LKO mice exhibited attenuated rhythm including consuming a higher percentage of daily food intake, water intake, and activity to 40% during the rest (light) period, both the food and water intake and activity of the HRD1/FGF21 DKO mice decreased to a level comparable to WT mice (Fig 7G). Therefore, our results indicated that HRD1 regulated growth, female reproduction, and the diurnal circadian behavior in an FGF21‐dependent manner.
Figure 7. Hepatic FGF21 ablation rescues the phenotypes induced by hepatic HRD1 deletion.
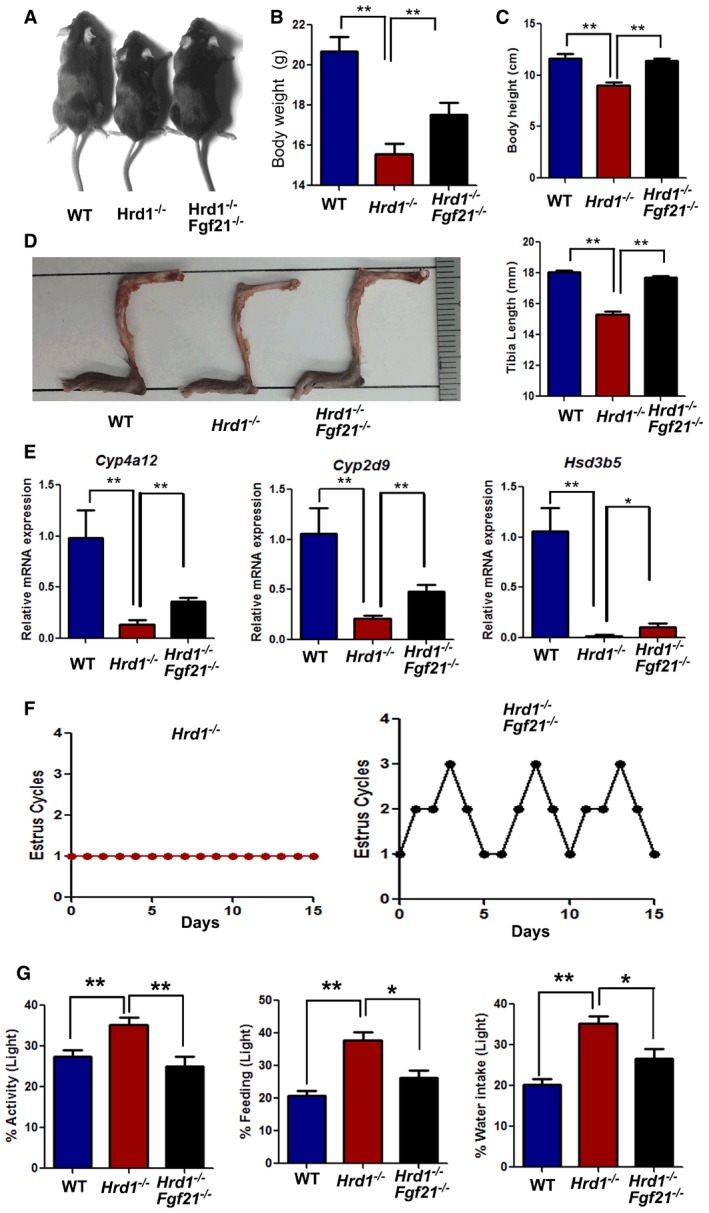
-
AExamples of the body sizes of the WT, L‐HRD KO, and HRD1/FGF21 DKO mice.
-
B, CBody weight and body height of the WT, L‐HRD KO, and HRD1/FGF21 DKO mice (n = 5 for each group).
-
DTibia lengths of the WT, L‐HRD KO, and HRD1/FGF21 DKO mice fed with normal chow diet (n = 5 for each group).
-
EHepatic Hsd3b5, Cyp2d9, and Cyp4a12 mRNA in the WT, L‐HRD1 KO, and HRD1/FGF21 DKO mice (n = 5 for each group).
-
FEstrous cycle of L‐HRD KO and HRD1/FGF21 DKO mice at the age of 4 months.
-
GPercentage of the activity, feeding, and water intake of the WT, L‐HRD KO, and HRD1/FGF21 DKO mice (n = 5 for each group).
Discussion
Our study demonstrates that the hepatic HRD1‐ERAD catalyzes polyubiquitination of CREBH possibly via K27‐linked chains to suppress FGF21 transcription in liver to control liver crosstalk with multiple distal organs including brain, bone, and ovary (Fig 8 and Appendix Fig S10). HRD1‐ERAD is the first line of defense against the accumulation of misfolded proteins in the ER lumen to control the protein quality (Chen et al, 2013; Sun et al, 2015). It has been shown that ERAD targets its substrates for degradation through K11 and K48‐linked polyubiquitination (Locke et al, 2014). However, our study demonstrated that liver HRD1 catalyzes CREBH ubiquitination possibly in a K27‐linked manner, defining a novel type of polyubiquitin chain for ERAD. More interestingly, this putative K27‐linked polyubiquitination catalyzed by HRD1 is predominantly conjugated at the lysine 294 of CREBH, which can be acetylated by the PCAF acetyltransferase (Kim et al, 2015). It will be interesting to further study whether and how the competitive regulation by ubiquitination and acetylation at the same lysine residue controls CREBH transcription activity in regulating liver FGF21 production. While the protein expression levels of both full‐length and activated forms of CREBH are increased, HRD1 could only interact with the full‐length CREBH to catalyze its ubiquitination. Therefore, it is likely that the increased active form of CREBH is a consequence of the diminished degradation of its pre‐spliced full‐length protein in HRD1‐null liver. In addition to CREBH, our genome‐wide proteomic and RNA‐seq analysis identified more than 700 proteins increased in HRD1‐null liver without mRNA expression changes, implying additional substrates may exist for HRD1. Indeed, further FGF21 deletion only partially rescued the body weight gain in HRD1 LKO mice, and our laboratory is currently investigating the FGF21‐independent HRD1 functions and the underlying molecular mechanisms in liver.
Figure 8. Working model of how the HRD1 regulates the circadian behavior, growth, and female reproduction through CREBH‐FGF21 axis.
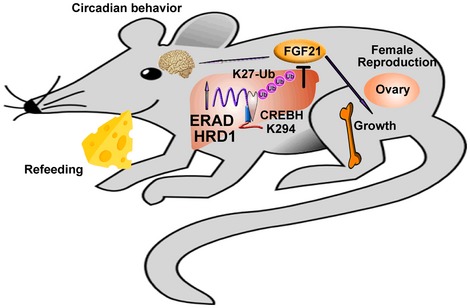
During the long‐term fasting, hepatokine FGF21 is upregulated to directly target adipose tissue to facilitate fat utilization and activate the ketogenesis signaling (Badman et al, 2009; BonDurant et al, 2017; Fisher et al, 2012; Huang et al, 2017). In addition, FGF21 overexpression results in a variety of abnormalities including female infertility, circadian behavior disruption and bone loss (Bookout et al, 2013; Owen et al, 2013; Wei et al, b). FGF21 expression is positively regulated by the ER stress‐associated transcriptional factors ATF4, CREBH, and PPARα during metabolic stress including fasting, excises, and chronic ER stress (Galman et al, 2008; Kim et al, 2014; Wan et al, 2014). Our studies demonstrate that HRD1 controls FGF21 expression by catalyzing ubiquitination‐mediated degradation of the transcription factor CREBH but not ATF4. It has been well established that PPARα is involved in FGF21 transcription (Galman et al, 2008). Therefore, further studies are needed to determine whether HRD1 regulates FGF21 expression through PPARα signaling, such as the crosstalk between CREBH and PPARα.
Materials and Methods
Mice and metabolic measurements
HRD1 floxed mice were used as previously described (Kong et al, 2016; Xu et al, 2016; Yang et al, 2014). Albumin‐Cre (catalogue no. 016832) and FGF21 floxed mice (catalogue no. 022361) were purchased from the Jackson Laboratory (Potthoff et al, 2009). All mice were bred in a specific pathogen‐free facility, and all animal experiments were approved by the Institutional Animal Care and Use Committees (IACUC) at Northwestern University.
Measurements of food intake, activity, and water intake were taken using a Comprehensive Lab Animal Monitoring System from the core facility of the Northwestern University. Animals were maintained on a standard chow diet under 12‐h light and dark cycles beginning at 5:00 a.m. and 5:00 p.m., respectively. The high‐fat diet (45 kcal% fat) is from the Research Diet Inc.
Plasmids and antibodies
The HRD1, CREBH, CREBH K294R, and the HA‐ubiquitin expression plasmids were constructed as reported previously (Zheng et al, 2016). K6 (22900), K11(22901), K27(22902), K29(22903), K33(17607), K48(17605), and K63(17606) ubiquitin plasmids were purchase from addgene. K27R ubiquitin plasmid was kindly provided by Dr. Hongquan Zhang (Wei et al, 2017). Antibodies to Flag (F1804) and Flag‐HRP (A8592) were purchased from Sigma (St. Louis, MO, USA), and antibodies to Myc, ubiquitin, Myc (9E10), Myc (9E10)‐HRP, and HA (F7)‐HRP were purchased from Santa Cruz (Santa Cruz, CA, USA). Phospho‐STAT5 (9359), STAT5 (94205), GAPDH (14C10), and ATF4 (11815) were purchased from Cell Signaling. Nuclear Matrix Protein p84 antibody (GTX102919) was purchased from GeneTex. Polyclonal anti‐CREBH antibody was raised by immunizing rabbits with a mouse CREBH protein fragment spanning N‐terminal amino acids 75–250 of mouse CREBH protein (Zhang et al, 2006). All antibody working concentration for immunoblotting is 1 μg/ml.
Cell culture, transfection, and immunoprecipitation
HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum. Transfections were performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. HEK293 cells were transfected with the indicated expression vectors. After 24 h, the cells were lysed in lysis buffer supplemented with protease (Roche, Basel, Switzerland) and phosphatase inhibitor cocktails. Co‐immunoprecipitation and immunoblotting analyses were performed as described previously (Wei et al, 2012a; Xu et al, 2016).
Genome‐wide RNA sequencing and real‐time RT‐PCR
WT and HRD1 LKO mice were fasted overnight and refed for 4 h. Genome‐wide RNA sequencing was performed as described previously. Counts of reads and RPKM (reads per kilobase pair per million reads mapped) values for each gene were determined using ERANGE software and were tested for differential expression in R using the limma package and the IBMT (intensity‐based moderated t‐statistic) method.
Quantitative PCR was run on a Bio‐Rad IQ2 PCR machine, and each PCR mixture contained 40 ng of cDNA template and 10 nM primers in 15 μl of SYBR Green reaction mix (Bio‐Rad). Expression values were normalized to those that were obtained with the control actb (encoding beta‐actin). Expression was measured using the primers as mentioned previously (Wei et al, 2014), and changes in gene expression levels were calculated by the 2−ΔΔCt method (Wei et al, 2014).
Protein extraction and trypsin digestion
500 μl 8 M urea (Sigma U5128, in 0.1 M Tris–HCl pH 8.5) was added to 10 mg mouse liver tissues, with high‐speed oscillation for 2 min at 70 Hz to extract proteins by high‐throughput tissue grinder. The supernatants were saved after centrifugation at 14,000 g for 20 min at 4°C. The protein concentration was measured, 4 μl of 1 M DTT were added to 200 μg protein and incubated for 1 h at 37°C, and then 20 μl of 1 M IAA were added and incubated at room temperature for 30 min in the dark. The sample was transferred to microcon YM‐10 (Millipore, Cat. No 42407) and then centrifuged at 14,000 g for 20 min. The filter units were washed with 8 M urea and 50 mM NH4HCO3 solution. The filter units were transferred to new collection tubes. Add 4 μg trypsin (trypsin:protein = 1:50) and incubate overnight at 37°C. The peptides were extracted with H2O and dried in a Savant Speed‐Vac.
HPLC fractionation
The sample was then fractionated into fractions by high pH reverse‐phase HPLC using a Waters XBridge BEH300 C18 column (5 μm particles, 4.6 mm ID, 250 mm length). Briefly, peptides were first separated with a gradient of 5–98% acetonitrile in 10 mM ammonium bicarbonate pH 10 over 45 min into 32 fractions. Then, the peptides were combined into six fractions and dried by vacuum centrifuging.
LC‐MS/MS analysis
The peptides were resolved in 0.1% formic acid and were analyzed using an ultra‐performance LC‐MS/MS platform. The LC separation was performed on an Ultimate 3000 (Thermo Fisher Scientific, San Jose, CA, USA) with an in‐house packed capillary column (150 μm I.D. × 12 cm) with 1.9 μm C18 reverse‐phase fused‐silica (Michrom Bioresources, Inc., Auburn, CA, USA). The sample was eluted with a 90‐min nonlinear gradient ramped from 6 to 40% mobile phase B (phase A: 0.1% formic acid in water, phase B: 0.08% formic acid in 80% acetonitrile) at a 0.6 μl/min flow rate. Eluted peptides were analyzed using an Q‐Exactive HF orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). The MS1 was analyzed over a mass range of 300–1,400 Da with a resolution of 120,000 at m/z 200. The isolation width was 1.6 m/z for precursor ion selection. The automatic gain control (AGC) was set to 3 × 106, and the maximum injection time (MIT) was 80 ms. The normalized collision energy was 27 ev. Top 20 MS2 spectra were acquired with a resolution of 15,000 at m/z 200, the AGC was set at 2 × 104, and the MIT was 19 ms. The dynamic exclusion was set at 12 s.
The proteins with more than one unique peptides were kept for further analysis. Differential expressed proteins were analyzed by using R package, ProstaR and DAPAR. The missing values of protein intensity were replaced with minimum value of each replicate. Then, the data were logarithmic‐transformed. Differentially expressed proteins were detected using a limma‐moderated t‐test. P‐values were adjusted by Benjamini–Hochberg procedure. Proteins were regarded as significantly differentially expressed between two groups with P‐value < 0.01 and log2 fold change < −2 or > 2. The functional classification of the differentially expressed proteins with Gene Ontology was carried out by the DAVID webtool.
Histological and estrous cycle analysis
H&E staining was performed as described previously (Wei et al, 2014). Briefly, ovary tissues from mice were fixed overnight in 10% neutral formalin and embedded in paraffin. Paraffin‐embedded tissues were cut into sections and stained with hematoxylin and eosin. The stage of the estrous cycle was determined by the vaginal cytology as described (Byers et al, 2012).
Blood chemistry analysis
Secreted mouse FGF21 in serum were determined using commercially available ELISA kits (mf2100, R&D, USA), followed by analysis with Multiskan MK3 (Thermo Scientific). Blood glucose, serum cholesterol, and TG levels were determined according to the manufacturer's instructions (Abcam, USA).
AAV‐mediated gene delivery
shRNA sequences against mouse Crebh encoding the hairpin shRNA, and control luciferase (GTTGCGCGGAGGAGTTGTG) were cloned into pAAV8‐D(+)‐U6‐siRNA‐CMV‐GFP vector via BamHI and EcoRI restriction sites. AAV was generated from these plasmids at the Harvard Children's Hospital Virus Core (Boston, MA) on a fee for service basis. AAV8 was injected once into 5‐ to 6‐week‐old mice via the tail vein at the dose of 5–10 × 1011 viral genome copies/mouse. Blood was collected via a small nick in the tail for subsequent ELISA analysis of circulating FGF21 levels after 2 or more weeks postinjection.
Statistical analyses
Statistical analysis was performed using Student's t‐test, and P < 0.05 was considered significant. Data are expressed as the mean ± s.d.
Author contributions
JW, LC, WX, FL, YY, YW, YZ, YX, BG, ZY, JM‐C, and CJ performed the experiments and analyzed the data. YY, BG, and KZ contributed critical reagent and to experimental design. RMG, HP, FH, JW, and DF designed the study, analyzed the data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This work was supported by grants from the Special Funds for Major State Basic Research of China (2014CBA02001) to F. H and J.W. and the National Institutes of Health (NIH) R01 grants (AI079056, AI108634, AR006634, and CA232347), and the National Natural Science Foundation Grant of China (81773061) to D.F. and NIH grant DK090313 to K.Z.
The EMBO Journal (2018) 37: e98942
See also: https://doi.org/10.15252/embj.201899277 (November 2018) and https://doi.org/10.15252/embj.2018100676 (November 2018)
Contributor Information
Hui Pan, Email: panhui20111111@163.com.
Jian Wang, Email: wangjian@bmi.ac.cn.
Fuchu He, Email: hefc@bmi.ac.cn.
Kezhong Zhang, Email: kzhang@med.wayne.edu.
Deyu Fang, Email: fangd@northwestern.edu.
References
- Badman MK, Koester A, Flier JS, Kharitonenkov A, Maratos‐Flier E (2009) Fibroblast growth factor 21‐deficient mice demonstrate impaired adaptation to ketosis. Endocrinology 150: 4931–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- BonDurant LD, Ameka M, Naber MC, Markan KR, Idiga SO, Acevedo MR, Walsh SA, Ornitz DM, Potthoff MJ (2017) FGF21 regulates metabolism through adipose‐dependent and ‐independent mechanisms. Cell Metab 25: 935–944.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookout AL, de Groot MH, Owen BM, Lee S, Gautron L, Lawrence HL, Ding X, Elmquist JK, Takahashi JS, Mangelsdorf DJ, Kliewer SA (2013) FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat Med 19: 1147–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA (2000) Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 103: 1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers SL, Wiles MV, Dunn SL, Taft RA (2012) Mouse estrous cycle identification tool and images. PLoS One 7: e35538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PH, Chen X, Lin Z, Fang D, He X (2013) The structural basis of R‐spondin recognition by LGR5 and RNF43. Genes Dev 27: 1345–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW, Kopito RR (2011) Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol 14: 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos‐Flier E, Spiegelman BM (2012) FGF21 regulates PGC‐1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev 26: 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holland WL, Kharitonenkov A, Bumol T, Schilske HK, Moller DE (2013) The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab 18: 333–340 [DOI] [PubMed] [Google Scholar]
- Galman C, Lundasen T, Kharitonenkov A, Bina HA, Eriksson M, Hafstrom I, Dahlin M, Amark P, Angelin B, Rudling M (2008) The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARalpha activation in man. Cell Metab 8: 169–174 [DOI] [PubMed] [Google Scholar]
- Gao B, Lee SM, Chen A, Zhang J, Zhang DD, Kannan K, Ortmann RA, Fang D (2008) Synoviolin promotes IRE1 ubiquitination and degradation in synovial fibroblasts from mice with collagen‐induced arthritis. EMBO Rep 9: 480–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Rine J (1994) Regulated degradation of HMG‐CoA reductase, an integral membrane protein of the endoplasmic reticulum, in yeast. J Cell Biol 125: 299–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Zhong L, Lee JTH, Zhang J, Wu D, Geng L, Wang Y, Wong CM, Xu A (2017) The FGF21‐CCL11 axis mediates beiging of white adipose tissues by coupling sympathetic nervous system to type 2 immunity. Cell Metab 26: 493–508.e4 [DOI] [PubMed] [Google Scholar]
- Inagaki T, Lin VY, Goetz R, Mohammadi M, Mangelsdorf DJ, Kliewer SA (2008) Inhibition of growth hormone signaling by the fasting‐induced hormone FGF21. Cell Metab 8: 77–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumida Y, Yahagi N, Takeuchi Y, Nishi M, Shikama A, Takarada A, Masuda Y, Kubota M, Matsuzaka T, Nakagawa Y, Iizuka Y, Itaka K, Kataoka K, Shioda S, Niijima A, Yamada T, Katagiri H, Nagai R, Yamada N, Kadowaki T, et al (2013) Glycogen shortage during fasting triggers liver‐brain‐adipose neurocircuitry to facilitate fat utilization. Nat Commun 4: 2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim DH, Hur KY, Kim HK, Ko T, Han J, Kim HL, Kim J, Back SH, Komatsu M, Chen H, Chan DC, Konishi M, Itoh N, et al (2013a) Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 19: 83–92 [DOI] [PubMed] [Google Scholar]
- Kim KH, Kim SH, Min YK, Yang HM, Lee JB, Lee MS (2013b) Acute exercise induces FGF21 expression in mice and in healthy humans. PLoS One 8: e63517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Mendez R, Zheng Z, Chang L, Cai J, Zhang R, Zhang K (2014) Liver‐enriched transcription factor CREBH interacts with peroxisome proliferator‐activated receptor alpha to regulate metabolic hormone FGF21. Endocrinology 155: 769–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Mendez R, Chen X, Fang D, Zhang K (2015) Lysine acetylation of CREBH regulates fasting‐induced hepatic lipid metabolism. Mol Cell Biol 35: 4121–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong S, Yang Y, Xu Y, Wang Y, Zhang Y, Melo‐Cardenas J, Xu X, Gao B, Thorp EB, Zhang DD, Zhang B, Song J, Zhang K, Zhang J, Zhang J, Li H, Fang D (2016) Endoplasmic reticulum‐resident E3 ubiquitin ligase Hrd1 controls B‐cell immunity through degradation of the death receptor CD95/Fas. Proc Natl Acad Sci USA 113: 10394–10399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan T, Morgan DA, Rahmouni K, Sonoda J, Fu X, Burgess SC, Holland WL, Kliewer SA, Mangelsdorf DJ (2017) FGF19, FGF21, and an FGFR1/beta‐Klotho‐activating antibody act on the nervous system to regulate body weight and glycemia. Cell Metab 26: 709–718.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston CM, Ifrim MF, Cowan AE, Weller SK (2009) Virus‐Induced Chaperone‐Enriched (VICE) domains function as nuclear protein quality control centers during HSV‐1 infection. PLoS Pathog 5: e1000619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke M, Toth JI, Petroski MD (2014) Lys11‐ and Lys48‐linked ubiquitin chains interact with p97 during endoplasmic‐reticulum‐associated degradation. Biochem J 459: 205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhi H, Kaufman RJ (2011) Endoplasmic reticulum stress in liver disease. J Hepatol 54: 795–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13: 1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen BM, Bookout AL, Ding X, Lin VY, Atkin SD, Gautron L, Kliewer SA, Mangelsdorf DJ (2013) FGF21 contributes to neuroendocrine control of female reproduction. Nat Med 19: 1153–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff MJ, Inagaki T, Satapati S, Ding X, He T, Goetz R, Mohammadi M, Finck BN, Mangelsdorf DJ, Kliewer SA, Burgess SC (2009) FGF21 induces PGC‐1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci USA 106: 10853–10858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi M, Fujisaka S, Cai W, Winnay JN, Konishi M, O'Neill BT, Li M, Garcia‐Martin R, Takahashi H, Hu J, Kulkarni RN, Kahn CR (2017) Adipocyte dynamics and reversible metabolic syndrome in mice with an inducible adipocyte‐specific deletion of the insulin receptor. Cell Metab 25: 448–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solon‐Biet SM, Cogger VC, Pulpitel T, Heblinski M, Wahl D, McMahon AC, Warren A, Durrant‐Whyte J, Walters KA, Krycer JR, Ponton F, Gokarn R, Wali JA, Ruohonen K, Conigrave AD, James DE, Raubenheimer D, Morrison CD, Le Couteur DG, Simpson SJ (2016) Defining the nutritional and metabolic context of FGF21 using the geometric framework. Cell Metab 24: 555–565 [DOI] [PubMed] [Google Scholar]
- Sun S, Shi G, Sha H, Ji Y, Han X, Shu X, Ma H, Inoue T, Gao B, Kim H, Bu P, Guber RD, Shen X, Lee AH, Iwawaki T, Paton AW, Paton JC, Fang D, Tsai B, Yates JR III, et al (2015) IRE1alpha is an endogenous substrate of endoplasmic‐reticulum‐associated degradation. Nat Cell Biol 17: 1546–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar S, Zhou Y, Li D, Rossulek M, Dong J, Somayaji V, Weng Y, Clark R, Lanba A, Owen BM, Brenner MB, Trimmer JK, Gropp KE, Chabot JR, Erion DM, Rolph TP, Goodwin B, Calle RA (2016) A long‐acting FGF21 molecule, PF‐05231023, decreases body weight and improves lipid profile in non‐human primates and type 2 diabetic subjects. Cell Metab 23: 427–440 [DOI] [PubMed] [Google Scholar]
- Wan XS, Lu XH, Xiao YC, Lin Y, Zhu H, Ding T, Yang Y, Huang Y, Zhang Y, Liu YL, Xu ZM, Xiao J, Li XK (2014) ATF4‐ and CHOP‐dependent induction of FGF21 through endoplasmic reticulum stress. Biomed Res Int 2014: 807874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wei W, Krzeszinski JY, Wang Y, Wan Y (2015) A liver‐bone endocrine relay by IGFBP1 promotes osteoclastogenesis and mediates FGF21‐induced bone resorption. Cell Metab 22: 811–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Yuan Y, Jin C, Chen H, Leng L, He F, Wang J (2012a) The ubiquitin ligase TRAF6 negatively regulates the JAK‐STAT signaling pathway by binding to STAT3 and mediating its ubiquitination. PLoS One 7: e49567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Dutchak PA, Wang X, Ding X, Wang X, Bookout AL, Goetz R, Mohammadi M, Gerard RD, Dechow PC, Mangelsdorf DJ, Kliewer SA, Wan Y (2012b) Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator‐activated receptor gamma. Proc Natl Acad Sci USA 109: 3143–3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Wei C, Wang M, Qiu X, Li Y, Yuan Y, Jin C, Leng L, Wang J, Yang X, He F (2014) The GTPase‐activating protein GIT2 protects against colitis by negatively regulating Toll‐like receptor signaling. Proc Natl Acad Sci USA 111: 8883–8888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Wang X, Zhan J, Chen Y, Fang W, Zhang L, Zhang H (2017) Smurf1 inhibits integrin activation by controlling Kindlin‐2 ubiquitination and degradation. J Cell Biol 216: 1455–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Zhao F, Qiu Q, Chen K, Wei J, Kong Q, Gao B, Melo‐Cardenas J, Zhang B, Zhang J, Song J, Zhang DD, Zhang J, Fan Y, Li H, Fang D (2016) The ER membrane‐anchored ubiquitin ligase Hrd1 is a positive regulator of T‐cell immunity. Nat Commun 7: 12073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Qiu Q, Gao B, Kong S, Lin Z, Fang D (2014) Hrd1‐mediated BLIMP‐1 ubiquitination promotes dendritic cell MHCII expression for CD4 T cell priming during inflammation. J Exp Med 211: 2467–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ (2006) Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 124: 587–599 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xie Y, Berglund ED, Coate KC, He TT, Katafuchi T, Xiao G, Potthoff MJ, Wei W, Wan Y, Yu RT, Evans RM, Kliewer SA, Mangelsdorf DJ (2012) The starvation hormone, fibroblast growth factor‐21, extends lifespan in mice. Elife 1: e00065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Kim H, Qiu Y, Chen X, Mendez R, Dandekar A, Zhang X, Zhang C, Liu AC, Yin L, Lin JD, Walker PD, Kapatos G, Zhang K (2016) CREBH couples circadian clock with hepatic lipid metabolism. Diabetes 65: 3369–3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6