

Key Points
Bone marrow-specific deletion of Abi1 in mice results in MPN-like phenotype and is linked to hyperactive SFKs/STAT3/NF-κB signaling.
ABI1 is downregulated in hematopoietic stem/progenitor cells and granulocytes from patients with PMF.
Abstract
Although the pathogenesis of primary myelofibrosis (PMF) and other myeloproliferative neoplasms (MPNs) is linked to constitutive activation of the JAK-STAT pathway, JAK inhibitors have neither curative nor MPN-stem cell-eradicating potential, indicating that other targetable mechanisms are contributing to the pathophysiology of MPNs. We previously demonstrated that Abelson interactor 1 (Abi-1), a negative regulator of Abelson kinase 1, functions as a tumor suppressor. Here we present data showing that bone marrow-specific deletion of Abi1 in a novel mouse model leads to development of an MPN-like phenotype resembling human PMF. Abi1 loss resulted in a significant increase in the activity of the Src family kinases (SFKs), STAT3, and NF-κB signaling. We also observed impairment of hematopoietic stem cell self-renewal and fitness, as evidenced in noncompetitive and competitive bone marrow transplant experiments. CD34+ hematopoietic progenitors and granulocytes from patients with PMF showed decreased levels of ABI1 transcript as well as increased activity of SFKs, STAT3, and NF-κB. In aggregate, our data link the loss of Abi-1 function to hyperactive SFKs/STAT3/NF-κB signaling and suggest that this signaling axis may represent a regulatory module involved in the molecular pathophysiology of PMF.
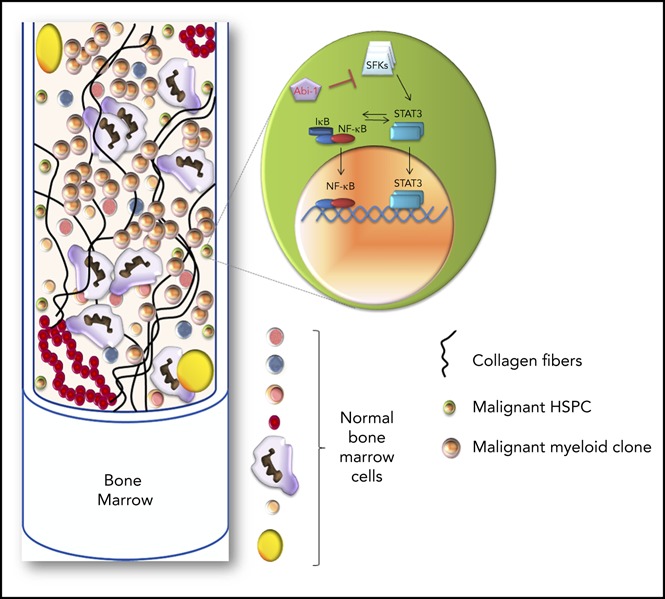
Visual Abstract
Introduction
The phenotype of primary myelofibrosis (PMF) is characterized by progressive bone marrow fibrosis, organomegaly, extramedullary hematopoiesis, thromboembolism, and ultimately, marrow failure or transformation to acute myeloid leukemia (AML).1-3 Median survival in PMF varies between 1 and 15 years, depending on risk factors, and treatment options are limited.3,4 Identification of JAK2-activating mutations as major drivers in myeloproliferative neoplasms (MPNs) prompted clinical development of JAK2 inhibitors.5,6 Ruxolitinib, an ATP-mimetic JAK1/2 inhibitor, induces symptomatic improvement in PMF, but exacerbates associated cytopenias and does not have curative potential, and responses occur regardless of presence of JAK2 mutations.7-11 Therefore, a major need remains to identify other targetable mechanisms contributing to the pathogenesis of PMF and related MPNs, polycythemia vera (PV), and essential thrombocythemia (ET).
Abelson Interactor 1 (Abi-1) is a negative regulator of Abl1 kinase,12-15 involved in regulation of cell proliferation.16,17 By forming a complex with Wiskott-Aldrich syndrome protein family member 2 (WAVE2),18,19 Wiskott-Aldrich Syndrome protein (WASP), or Diaphanous (Dia) formin,16,18-23 Abi-1 affects actin remodeling, cell adhesion, and migration. Abi-1 also interacts with integrin α4 and is involved in integrin β1 signaling.24-26 Abi-1-deficient mice uniformly die in utero with lethal defects of the heart and placenta.19,24 The role of Abi-1 in carcinogenesis is controversial, as both loss or overexpression were implicated in cancer.27-30 Its involvement in malignant hematopoiesis, although reported by us and others, remains unclear.31-35
Here, we present evidence for direct involvement of Abi-1 in homeostasis of hematopoietic system. We found that conditional deletion of Abi1 in murine bone marrow results in impairment of hematopoietic stem cell self-renewal, progressive anemia, megakaryocytosis and myeloid hyperplasia, with resulting PMF-like phenotype characterized by marrow fibrosis and splenomegaly. Furthermore, Abi-1 protein and mRNA levels are decreased in hematopoietic progenitors from patients with PMF, but not from those with ET or PV. Mechanistically, loss of Abi-1 leads to upregulation of Src family kinases (SFKs), STAT3, and NF-κB signaling, suggesting that the Abi-1/SFKs/STAT3/NF-κB axis may represent a new regulatory module involved in the pathophysiology of MPNs.
Materials and methods
Patient samples
CD34+ cells were isolated from the bone marrow of patients with PMF or from healthy marrow purchased from AllCells (Alameda, CA). Granulocytes were isolated from peripheral blood (PB) of patients with ET, PV, primary or secondary (PV- or ET-derived) myelofibrosis, and healthy donors (supplemental Table 1, available on the Blood Web site). CD34 MicroBeads (Miltenyi Biotec, San Diego, CA) and gradient centrifugation were used for CD34+ and granulocyte isolation, respectively. Human subject participation was conducted with informed consent and approved by local ethics committees.
Transgenic mice
Conditional Abi1(fl/fl) mice19 were crossed with B6.Cg-Tg(Mx1-cre+1)Cgn/J strain (#003556, JAX, Bar Harbor, ME)36 to generate Abi1(fl/fl);Tg(Mx1-cre[+/−]) mice. These animals were back-crossed to B6.SJL-PtprcAPepcB/BoyJ(#002014, JAX)(CD45.1) background. Abi1(fl/fl);Tg(Mx1-cre[−]), Abi1(fl/wt);Tg(Mx1-cre[+]), or Abi1(fl/fl);Tg(Mx1-cre[+]) mice were subjected to polyinosinic:polycytidylic acid [poly(I:C)]-induced (Invivo Gen, San Diego, CA) activation of the Cre recombinase under the control of Mx1 promoter to obtain animals with an Abi1(fl/fl);Tg(Mx1-cre[−]), Abi1(−/wt);Tg(Mx1-cre[−]), or Abi1(−/−);Tg(Mx1-cre[+]) genotype, designated as Abi-1WT, Abi-1HET, or Abi-1KO, respectively. Recombination of Abi1floxed allele was confirmed by polymerase chain reaction (PCR). We evaluated 76 Abi-1WT, 41 Abi-1HET, and 85 Abi-1KO animals. Animal experiments were approved by the Institutional Animal Care and Use Committee.
Murine hematopoietic stem/progenitor cells isolation
A biotin-conjugated antibody cocktail containing anti-TER119, CD127, CD8a, Ly-6G, CD11b, CD4, and CD45R was used to stain lineage-committed cells. BUV395-Streptavidin, anti-CD34-FITC, CD117/c-Kit-APC, Ly-6A/E/Sca-1-PE-Cy7 (or -BV605), CD135/Flt3-PE, and CD16/CD32-PE were used to stain hematopoietic stem/progenitors. Cells were sorted using Legacy MoFlo High-Speed cell sorter.
Bone marrow transplantation
For noncompetitive bone marrow transplantation, 5 × 106 marrow cells isolated by flushing from poly(I:C)-uninduced Abi1(fl/fl);Tg(Mx1-cre[+]) or Abi1(fl/fl);Tg(Mx1-cre[−]; CD45.1) mice were injected via tail vein into lethally irradiated (2x475cGy) C57BL/6 wild-type mice recipients (CD45.2; #000664, JAX). Four weeks posttransplant, Abi1 inactivation was performed by poly(I:C) induction. Donor chimerism was evaluated in PB every 4 weeks for 24 weeks. After 24 weeks, marrow was harvested from primary recipients and 5 × 106 cells were transplanted into CD45.2 secondary recipients conditioned as earlier. Donor chimerism in PB was evaluated as earlier. For competitive repopulation assays, marrow cells were isolated via flushing from poly(I:C)-induced Abi-1KO or Abi-1WT (CD45.1) mice. After confirming Abi1 inactivation, donor cells (1 × 106 Abi-1KO or Abi-1WT) were mixed with competitor cells (1 × 106; 1:1, CD45.1:CD45.2) and injected via tail vein into lethally irradiated CD45.2 recipients. Donor chimerism in PB was evaluated as earlier.
Cytokine levels assay
Levels of interleukin (IL)-1α, IL-1β, IL-6, IL-10, IL-12p70, IL-17A, IL-23, IL-27, monocyte chemoattractant protein-1 (MCP-1/CCL2), interferon (IFN)-β, IFN-γ, tumor necrosis factor α (TNF α), and granulocyte-macrophage colony-stimulating factor were detected in plasma of Abi-1WT (n = 11) or Abi-1KO (n = 11) 14-week-old animals, using LEGENDplex Mouse Inflammation Panel (Biolegend, San Diego, CA).
ABI1 silencing in human CD34+ cells
Healthy CD34+ cells (n = 3) were expanded for 48 hours in the presence of thrombopoietin, Flt3/Flk-2 (Fms-like tyrosine kinase 3/fetal liver kinase-2) ligand, stem cell factor, IL-3, IL-6, and granulocyte-macrophage colony-stimulating factor (StemCell Technologies, Cambridge, MA) and incubated for additional 48 hours with 15 μM ABI1-silencing antisense 2′-deoxy-2′fluoro-β-d-arabinonucleotides (FANA-ABI1-ASO; AUM BioTech, Philadelphia, PA). After 48 hours culture with FANA-ABI1-ASO, Abi-1 protein levels were evaluated by immunoblotting, and cell cycle status was determined by 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay.
Liquid chromatography-tandem mass spectrometry
Bone marrow was isolated from 20-week-old Abi-1WT (n = 3) or Abi-1KO (n = 3) animals by flushing. Cell pellets were lysed, and 100 µg protein per sample was subjected to tryptic digestion. Tryptic peptides were subjected to liquid chromatography-tandem mass spectrometry, using an automated proteomic technology platform.37,38
Gene microarrays
Gene array analysis of lineage−, Sca-1+, c-Kit+ (LSK)-enriched cells from 14-week-old Abi-1KO (n = 4) or Abi-1WT (n = 5) mice was done using the Affymetrix-WT Pico Expression Kit (Affymetrix, Louisville, KY) and Affymetrix-3000 7G gene scanner. Partek Genomics Suite v6.6 was used for quality control and data analysis. Microarray data are deposited in GEO as GSE83288.
Abi-1HET bone marrow transplant model of MPLW515L-mediated MPN
MSCV-MPLWT-IRES-GFP and MSCV-MPLW515L-IRES-GFP retroviral vectors were generously provided by Ross Levine (Memorial Sloan Kettering Cancer Center). Marrow was isolated from 14-week-old Abi-1WT or Abi-1HET animals, and Abi1 recombination was verified by PCR. Retroviral transduction and murine marrow transplant assay were performed as previously described.39,40 Abi-1WT/MPLWT, Abi-1HET/MPLWT, Abi-1WT/MPLW515L, and Abi-1HET/MPLW515L transplantation groups were used.
Statistics
Two-tailed unpaired t-tests or log-rank tests were used for between-group comparisons, using Bonferroni correction where appropriate. P < .05 was considered statistically significant. Throughout, *P < .05, **P < .01, and ***P < .001. Two-tailed unpaired t-test and q-values for multiple hypothesis tests, using the R package QVALUE,41 were used to select peptides with significant change in paired analyses.
Additional materials and methods are described in supplemental Information.
Results
Downregulation of ABI1 transcripts in human PMF samples
ABI1 transcript levels in CD34+ cells isolated from bone marrow were decreased by approximately 40% in PMF (n = 5) compared with controls (n = 5; Figure 1A). Gene expression profiles (GEO/GSE53482)42 of CD34+ cells isolated from PB also showed significant downregulation of ABI1 in PMF (n = 42) with mutations in JAK2 or CALR relative to controls (n = 31; supplemental Figure 1A-C). Furthermore, granulocytes from patients with PMF (n = 36), or secondary myelofibrosis post-PV (n = 9), showed a 40% to 60% decrease in ABI1 mRNA relative to control patients (n = 16; Figure 1B). Downregulation of ABI1 in PMF granulocytes was observed regardless of JAK2 or CALR mutation status (Figure 1C). Notably, no significant changes in granulocyte ABI1 transcript levels were noted in ET (n = 15), PV (n = 20), or post-ET myelofibrosis (n = 4; Figure 1B). Consistent with these findings, examination of gene expression profiles of CD34+ cells from bone marrow of ET (n = 24) and PV (n = 26) patients (GEO/GSE103176)43 showed no downregulation of ABI1 (supplemental Figure 1D), indicating that reduced ABI1 expression may be specific to PMF.
Figure 1.

ABI1 is downregulated in hematopoietic progenitor cells and granulocytes obtained from patients with MPNs. (A) Reverse transcription (RT)-PCR analysis of ABI1 transcript levels in CD34+ cells isolated from the bone marrow aspirates of patients with PMF (n = 5) or healthy control patients (n = 5). ABI1 gene expression was normalized to RPP14 (RNaseP) and ACTB. (B) RT-PCR analysis of ABI1 gene expression in peripheral blood granulocytes from patients with PMF (n = 36), ET (n = 15), PV (n = 20), myelofibrosis post-ET (n = 5), and post-PV (n = 9) or from healthy controls (n = 16), normalized to RPP14. (C) RT-PCR analysis of ABI1 gene expression in peripheral blood granulocytes from patients with PMF with JAK2 (JAK2 V617F) or CALR mutations (Del52 or Ins5), normalized to RPP14. Clinical details of the analyzed samples are presented in supplemental Table 1. Boxes represent the interquartile range that contains 50% of the subjects, and the horizontal line in the box indicates the median. *P < .05, ***P < .001.
Inactivation of Abi1 in bone marrow induces leukocytosis, thrombocytosis, anemia, splenomegaly, and decreased survival
To determine the role of Abi-1 in the homeostasis of the hematopoietic system, we phenotyped transgenic Abi-1KO mice carrying bone marrow-selective knockout of Abi1. We first confirmed both inducible inactivation of the Abi1flox allele and loss of Abi-1 protein in the marrow (Figure 2A-C; supplemental Figure 2A-B). Abi-1 levels were also reduced in spleen and liver, but not in heart, lung, or kidney (supplemental Figure 2C). Abi-1KO mice showed femur pallor and splenomegaly that progressed with age, but no change in liver size (Figure 2D-E). Blood count analysis at 14, 35, and 56 weeks indicated progressive leukocytosis, mild anemia, and thrombocytosis in the Abi-1KO, which were absent in Abi-1WT animals (Figure 2F; supplemental Table 2). Survival of Abi-1KO mice decreased 20 weeks post-poly(I:C) injection, with significant weight loss after 60 weeks and death in all animals by 66 weeks of age (Figure 2G; supplemental Figure 2D), likely because of progressive disease. We observed no changes in gross pathology, blood counts, or survival in 56-week-old Abi-1HET animals. Therefore, in subsequent analyses, we focused on characterization of Abi-1KO mice. Our findings suggest that marrow-specific inactivation of Abi1 disrupts normal hematopoiesis.
Figure 2.

Transgenic Abi-1KOmice show splenomegaly, leukocytosis, thrombocytosis, and decreased survival. (A) Schematic representation of the experimental design of deleting Abi1flox allele in Abi1(fl/fl);Tg(Mx1-cre)(+) mice by poly(I:C) induction. Analyses were conducted on animals at 14, 35, or 56 weeks of age (4, 25, or 46 weeks after recombination, respectively). (B) Genomic PCR analysis of Abi1flox deletion efficiency in tail (top) and bone marrow (bottom) DNA. PCR amplification of the mutated, LoxP site containing Abi1 allele produces a 359-nt band, recombined Abi1 allele produces no band, and nonmutated wild type Abi-1 allele produces a 179-nt band. A locus map is presented in supplemental Figure 2A. Tissue from 3 different Abi-1WT, Abi-1HET, and Abi-1KO animals was used. (C) Western blot analysis of Abi-1 protein levels in the bone marrow of Abi-1WT, Abi-1HET, and Abi-1KO mice. Tissue from 3 different animals per group was used. (D) Representative gross anatomy of femurs and spleens of 14-, 35-, and 56-week-old Abi-1WT or Abi-1KO animals. (E) Average spleen and liver sizes of Abi-1WT and Abi-1KO mice. Relative organ weight was calculated as an absolute organ weight (g)/body weight on sacrifice day (g) ×100. Organs from n = 12 sex-matched animals were evaluated per age group. (F) Average white blood cells count, hematocrit values, and platelet count of the peripheral blood obtained from 14-, 35-, and 56-week-old Abi-1WT or Abi-1KO animals performed using automated hematology analyzer. Peripheral blood from at least 12 sex-matched mice was analyzed per each age group. *P < .05, **P < .01, ***P < .001. (G) Survival of Abi-1WT (n = 48) or Abi-1KO (n = 60) animals monitored from birth for 67 weeks (log-rank P = .003).
Abi-1 loss results in myeloid hyperplasia, megakaryocytosis, and fibrosis
As Abi-1KO animals aged, the fraction of myeloid (CD11b+/Gr-1+) cells in their PB increased at the expense of lymphoid B220 cells, whereas the CD3+ lymphoid fraction remained unchanged (Figure 3A). Abi-1KO erythrocytes demonstrated polychromasia, anisopoikilocytosis, and teardrop-shaped forms (Figure 3B; supplemental Figure 3A). Giant platelets were observed in 2/10 14-week-old, 4/10 36-week-old, and 5/10 56-week-old or older Abi-1KO mice. Bone marrow showed hypercellularity with myeloid hyperplasia, erythroid hypoplasia, and megakaryocytosis (Figure 3C-E; supplemental Figure 3B-C). Bone marrow cellularity at the distal aspect of femur in the medial and lateral condyles was 95% for Abi-1WT and 100% for 56-week-old Abi-1KO mice (supplemental Figure 3D). Spleen histology showed expansion of red pulp, with numerous islands of megakaryocytic, myeloid, and erythroid infiltration (Figure 3C-D; supplemental Figure 3B-C). Clusters of megakaryocytes were commonly observed in marrow and spleen of 56-week-old Abi-1KO mice (supplemental Figure 3C). Abi-1KO marrow samples and spleens showed progressive deficits of stainable iron on Prussian blue staining (Figure 3D and 3F; supplemental Figure 4A-B), possibly linked to observed anemia (supplemental Table 2; Figure 2F). Silver stain and semiquantitative grading44 showed progressive increase in reticulin fibrosis in Abi-1KO marrow, from grade 1 (out of 3) at 14 weeks to grade 2 to 3 at 56 weeks (Figure 3G; supplemental Figure 4C). Increased thickness and density of reticulin fibers was present in Abi-1KO spleen, from grade 1 at 14 weeks to grade 2 at 56 weeks (Figure 3G; supplemental Figure 4C). Progressive splenomegaly, megakaryocytosis, and marrow and spleen fibrosis in Abi-1KO animals meet Mouse Models of Human Cancers Consortium criteria for MPN.45
Figure 3.

Abi-1 deficiency results in age progressive increase in circulating granulocytes, anisopoikilocytosis, megakaryocytosis, loss of stainable iron, and fibrosis. (A) Fluorescence-activated cell sorter analysis of CD11b+/Gr-1+, B220+, and CD3+ populations in the peripheral blood of Abi-1WT and Abi-1KO mice, performed at 14, 35, or 56 weeks of age, 20 Abi-1WT and 20 Abi-1KO sex-matched mice were evaluated at each point. (B) Wright staining of representative blood smears obtained from Abi-1KO and Abi-1WT mice at 56 weeks of age; teardrop cells are marked with arrows. Similar results were observed in blood smears of at least 12 sex-matched Abi-1WT or Abi-1KO mice. Magnification ×100, bars correspond to 20 μm. Images were obtained using a Zeiss Axiophot microscope and Zeiss Pan-Apochromat 100×/1.40 oil lens. Representative (C) hematoxylin and eosin or (D) Prussian blue stains of bone marrow from femurs and spleen sections of 56-week-old Abi-1WT or Abi-1KO animals. Similar results were observed in at least 12 sex-matched Abi-1WT or Abi-1KO mice. Bars correspond to 100 μm. Images were obtained using a Zeiss Axiophot microscope with Zeiss Pan-Apochromat 20×/1.0 lens. (E) Average number of megakaryocytes per high power field (hpf) and (F) percentage of macrophages with hemosiderin in the bone marrow and spleen/red pulp evaluated in 14-, 35-, and 56-week-old Abi-1WT or Abi-1KO animals; 6 sex-matched animals were evaluated per age group per genotype. Representative (G) Gomori reticulin staining of the bone marrow from femurs and spleen sections of 56-week-old Abi-1WT or Abi-1KO animals (black stain). Similar results were obtained for at least 12 sex-matched animals per group. Bars correspond to 100 μm. Images were obtained using Zeiss Axiophot microscope with Zeiss Pan-Apochromat 20×/1.0 lens. *P < .05.
Abi-1 deficiency results in increased frequency and cell cycle activity of hematopoietic stem/progenitor cells
We assessed the frequency of hematopoietic stem and progenitor cells in the marrow of 14-week-old Abi-1WT or Abi-1KO mice (supplemental Figure 5A). Deletion of Abi1 resulted in no change in Lin- fractions. However, there were increased frequencies of LSKs (from 2.3% [±0.4%] to 4.6% [±0.8%]) and LK progenitors (from 4.9% [±0.9%] to 17.6% [±1.1%]; Figure 4A-B), a 2.6-fold increase in LT-HSCs, no change in ST-HSCs, a 47% reduction in common myeloid progenitors (CMPs), and a 25% increase in granulocyte-monocyte progenitors (GMPs) (Figure 4C; supplemental Figure 5B). Evaluation of the absolute number of stem/progenitor cells confirmed these trends with the exception of CMP, which showed an increased number in Abi-1KO mice (supplemental Figure 5C).
Figure 4.

Expansion, increased cell cycle activity, decreased engraftability and inflammatory signature in Abi-1 deficient hematopoietic stem/progenitor cells. (A) Frequencies of Lin- cells in 5 × 105 bone marrow. (B) Frequencies of LSK or LK cells in lineage-negative fractions of Abi-1WT or Abi-1KO bone marrow. (C) Frequencies of long-term (LT)- and short-term (ST)-HSCs and multipotent progenitors in LSK fractions were determined by sorting. Sorting was performed using bone marrow obtained from 6 sex-matched 14-week-old Abi-1WT or Abi-1KO mice. Sorting strategy is presented in supplemental Figure 5A. Average percentages of (D) EdU-positive LSK cells in G0/G1, S, and G2/M phase and (E) EdU-positive LT-HSCs in G0/G1 and S/G2/M phase of the cell cycle. Data from 4 experiments each using bone marrow from 14-week-old sex-matched Abi-1WT or Abi-1KO animals are shown as mean. Noncompetitive bone marrow primary (F) and secondary (G) transplant. Bone marrow cells isolated from Abi-1WT or Abi-1KO mice (before poly[I:C] injection) were transplanted into lethally irradiated recipient C57BL/6 wild-type mice in the absence of competitor cells. Four weeks after the transplantation, Abi-1 loss was induced by poly(I:C) administration. Twenty-four weeks posttransplant, bone marrow cells were harvested from primary recipients and transplanted into the secondary recipients. Average donor chimerism in primary and secondary recipients was monitored every 4 weeks for 24 weeks posttransplantation; n = 4 Abi-1WT or n = 4 Abi-1KO donor mice (CD45.1) were used for primary or secondary transplants, and 10 recipient lethally irradiated (CD45.2) mice per Abi-1WT orAbi-1KO transplant group were used. (H) Western blot analysis of Abi-1 protein levels and (I) average percentage of G0/G1, S or G2/M human CD34+ cells (n = 3) exposed to FANA ABI1 silencing antisense oligonucleotide (KD) or scrambled control (Ctrl) for 48 hours. (J) Heat map showing differently expressed transcripts in LSK-enriched cells isolated from the bone marrow of n = 4 Abi-1KO and n = 5 Abi-1WT 14-week-old animals. False discovery rate was < 0.05. (K) Cytokine levels detected in the plasma of 14-weeks old sex-matched Abi-1WT (n = 12) and Abi-1KO (n = 12) animals. *P < .05, **P < .01.
Given this evidence for expansion of HSCs, we evaluated the cell cycle activity of LSK and LT-HSC cells. Abi-1KO mice at 14 weeks showed a higher proportion of LSK cells in S-phase, and 46% more LT-HSCs in the S/G2/M phases relative to Abi-1WT animals (Figure 4D-E; supplemental Figure 5D-E). Transcript expression analysis of genes regulating cell cycle progression in LT-HSCs confirmed these observations (supplemental Figure 5F).
To determine the effect of Abi-1 loss on hematopoietic progenitors, we performed colony-forming unit assays. Results showed a significant decrease in burst forming unit-erythroid and increase in GMP colonies derived from Abi-1KO marrow isolated from 14-week-old mice, consistent with anemia and leukocytosis (supplemental Figure 5G). We further observed a 3-fold increase in megakaryocyte colonies, consistent with the observed megakaryocytosis in Abi-1KO mice (supplemental Figure 5H). We found no indication of growth factor independence or hypersensitivity.
To examine the effect of Abi-1 loss on long-term engraftment and self-renewal of HSCs, we transplanted bone marrow cells isolated from Abi-1WT or Abi-1KO mice (before poly[I:C] injection), into lethally irradiated recipient C57BL/6 wild-type mice in the absence of competitor cells. Four weeks after the transplantation, Abi-1 loss was induced by poly(I:C)-induction. Whereas initial engraftment remained comparable (∼95%) for primary recipients of Abi-1WT or Abi-1KO transplants, 12 weeks postpoly(I:C)-induction, Abi-1KO-transplanted mice showed progressive loss of chimerism (Figure 4F). These mice also exhibited significant weight loss at 24 weeks posttransplant, with an average weight of 22.2 g (±1.8 g) compared with 28.6 g (±3.1 g) for Abi-1WT marrow recipients (P = .04). Bone marrow cells were harvested from primary recipients 24 weeks posttransplant and transplanted into secondary recipients. A further decrease in donor chimerism and significantly reduced initial engraftment of Abi-1KO bone marrow cells were seen in secondary recipients (Figure 4G).
In competitive repopulation assays, C57BL/6 wild-type mice transplanted with the whole bone marrow isolated from Abi-1KO mice 4 weeks post-poly(I:C)-induction (after PCR confirmed recombination) showed progressive decrease in donor chimerism relative to Abi-1WT marrow recipients, reaching significance 4 months after the transplant (supplemental Figure 5I). Of note, initial engraftment at 4 weeks posttransplant was comparable between recipients of Abi-1KO or Abi-1WT marrow (46% and 45% chimerism, respectively).
To confirm a direct link between Abi-1 loss and cell cycle activity in hematopoietic stem/progenitor cells, we performed EdU incorporation assays using CD34+ cells isolated from the bone marrow of healthy donors (n = 3) and exposed to ABI1-silencing FANA antisense oligonucleotides (FANA-ABI1-ASO). More than 50% efficiency of ABI1 silencing after 48 hours incubation with FANA-ABI1-ASO was noted for CD34+ cells (Figure 4H; supplemental Figure 6A). Cellular penetrance of FANA oligos was confirmed by confocal microscopy and fluorescence-activated cell sorting (supplemental Figure 6B-C). EdU incorporation assays showed that 48-hour exposure to FANA-ABI1-ASO resulted in a nearly 2-fold increase in CD34+ cells in S-phase (Figure 4I; supplemental Figure 6D). Overall, our data suggest that deletion of Abi-1 leads to abnormal cell cycle activity, impaired HSC self-renewal, and defective reconstitution of hematopoiesis.
Activation of NF-κB pathway and pro-inflammatory signaling in Abi-1KO mice
To determine factors contributing to the abnormal expansion of hematopoietic stem/progenitor cells in Abi-1-deficient animals, we performed a genome-wide expression analysis of LSK cells from 14-week-old Abi-1KO and Abi-1WT mice. We found significant overexpression of genes regulated by or involved in regulation of the NF-κB pathway (Figure 4J; supplemental Table 3). MetaCore Pathway Analysis indicated that the NF-κB pathway was first of the top 10 dysregulated pathways in the Abi-1KO LSK cells (supplemental Figure 6E). Ingenuity Pathway Analysis identified 3 overlapping networks in which NF-κB activation was predicted on the basis of increased expression of known NF-κB targets (Cd69, Cxcl10, Cxcl2, Hp, and Nfkbia) and regulators (Myd88, Chuk, and Ikbkb; supplemental Figure 6F).46,47 Of note, gene expression profiling of CD34+ cells from patients with PMF (GEO/GSE53482) showed upregulation of the same NF-κB human target genes CXCL10, CD69, and HP (supplemental Figure 7A), as well as genes involved in leukocyte migration and recruitment (FCER1A, FCER1G, FCGR2A, TPSAB1, and TPSB2; supplemental Figure 7B).42
We also assessed levels of 13 pro-inflammatory cytokines, finding a nearly 2-fold increase in IL-1B, IL-12, IL-17, IL-23, IL-27, and MCP-1/CCL2; a roughly 10-fold increase in IFN-γ; and no significant changes in IL-1A, IL-6, IL-10, TNF α, or IFN-β in Abi-1KO plasma (Figure 4K). Overall, these data link upregulation of NF-κB pathway and inflammatory signaling to Abi-1 deficiency in hematopoietic system.
Abi-1-deficient bone marrow cells exhibit increased activity of SFKs, STAT3, and NF-κB
On the basis of our gene expression data indicating active NF-κB signaling in Abi-1KO marrow, we evaluated major components of this pathway by immunoblotting. We found increased levels of IκB accompanied by increased phosphorylation of NF-κB (Figure 5A; supplemental Figure 8A). Surprisingly, Abi-1KO bone marrow showed only modest increase in JAK2 and STAT5 activities, whereas the phosphorylation of STAT3, Akt, and Erk1/2 increased significantly (Figure 5B; supplemental Figure 8B).
Figure 5.

Abi-1 loss in the bone marrow results in instability of WAVE2 complex, and is accompanied by increased phosphorylation of STAT3, SFKs, and NF-κB. Western blot evaluation of the activity status of (A) NF-κB and its inhibitor IκB, (B) JAK2, STAT5, STAT3, Erk1/2, Akt, and (C) SFKs using phospho-site-specific antibodies, and (D) stability of WAVE2 complex components (WAVE2, Nap1, Abi-1, and Sra-1) in Abi-1-deficient bone marrow. Bone marrow from 3 different Abi-1WT and Abi-1KO sex-matched 20-week-old animals was used. (E) Volcano plot of fold change (log 2) vs q-value (−log10) of peak-area for 12 103 peptides identified in Abi-1KO (n = 3) respective to Abi-1WT (n = 3) bone marrow isolated from sex-matched 20-week-old animals. Green represents number of peptides with 2-fold decrease in abundancies, peptides with 2-fold increase in abundancies are presented in red. (F) Heat map showing significantly changed peptides derived from target proteins known to be associated with MPNs identified in liquid chromatography-tandem mass spectrometry analysis performed on Abi-1WT or Abi-1KO bone marrow samples. (G) Immunoblotting assessment of the levels of Abi-1 and activity status of SFKs, STAT3, and NF-κB in CD34+ cells isolated from the bone marrow of patients with PMF and compared with sex- and age-matched healthy control patients (sample details are presented in supplemental Table 1). Whole-cell lysates were used for immunoblotting analyses. (H) A hypothetical schematic depicting the effect of Abi-1 on SFKs, STAT3, and NF-κB signaling. Abi-1 acts as a negative regulator of SFKs activity. Its absence leads to overactive SFKs signaling to STAT3, which becomes activated and cross-activates NF-κB. Overactivity of STAT3 and NF-κB results in cells acquiring inflammatory molecular signature, positively affecting proliferation, survival and ultimately leading to transformation.
On the basis of our previous work showing high-affinity binding between Abi-1 and the Src homology domain 2 of SFKs, we assessed the activity of SFKs.48,49 We found increased phosphorylation of the Src auto-inhibitory Tyr-527 site. However, pan-phospho-Src family antibody showed increased phosphorylation on Tyr-416, consistent with activation of other SFKs (Figure 5C; supplemental Figure 8C). We did not detect phosphorylation of c-Abl on Tyr-412 in Abi-1KO bone marrow cells (supplemental Figure 8D). Immunoblotting showed an Abi-1-loss-associated decrease in the stability of WAVE2 complex components (WAVE2, Nap1, and Sra-1; Figure 5D; supplemental Figure 9A), consistent with our previous findings.19
To obtain unbiased insight into the mechanistic effects of Abi-1 loss, we performed label-free, intensity-based quantitative proteomic analysis of bone marrow from 20-week-old Abi-1KO and Abi-1WT mice. The analysis yielded 12 103 peptides derived from 2464 unique proteins. When quantified, there were significant changes in the abundance of 318 peptides representing 226 unique proteins in the Abi-1KO samples (supplemental Table 4; supplemental Figure 9B). A volcano plot showed that among peptides with a more than 2-fold difference, 25.1% showed lower and 74.9% higher abundance (Figure 5E). Among peptides showing a greater than 2-fold increase in abundance in Abi-1KO samples, we detected peptides derived from calreticulin, CD177, CD97, haptoglobin, Mac-1, myeloperoxidase, STAT1, STAT3, and SFKs Hck and Fgr. We interpreted these data as confirming not only activation of SFKs and STAT3 signaling but also an increase in proteins previously associated with MPN. Within the group of peptides showing a greater than 2-fold decrease in abundance, we detected peptides derived from WAVE2, Sra-1, and Nap1 (Figure 5F).
Finally, immunoblotting analysis performed on CD34+ or CD34− cells isolated from PMF patients showed decreases of 80% and 50% in Abi-1 protein levels, respectively. Similar to Abi-1-depleted murine marrow, we also found significant upregulation of SFKs and NF-κB activities (Figure 5G; supplemental Figure 9C-D). These data indicate that SFKs/STAT3/NF-κB signaling operating in Abi-1-deficient marrow may be involved in pathogenesis of the observed PMF-like phenotype. A model of Abi-1/SFKs/STAT3/NF-κB cross talk is presented in Figure 5H.
Inactivation of 1 copy of Abi-1 accelerates development of MPLW515L-mediated MPN-like disease
To further explore the role of Abi-1 in the pathogenesis of MPNs, we assessed involvement of Abi-1 in 1 of the established models of the disease. Such models are based on expression of mutated JAK2, CALR, or MPL. Because of less pronounced ABI1 loss in granulocytes bearing CALR mutation, and no significant overactivity of JAK2-STAT5 in Abi-1KO marrow, we decided to use the transplant model of MPLW515L-mediated MPN.39 As lethality and penetrance of the MPLW515L model is variable in C57/BL6 wild-type mice recipients (in contrast to full penetrance in tumor growth-permissive Balb/C background),39,50 and homozygous loss of Abi1 allele is sufficient to produce MPN phenotype, we decided to use Abi-1HET animals to achieve controllable manifestation of the disease in double-mutant (Abi-1HET/MPLW515L) animals.
Bone marrow isolated from Abi-1WT (n = 3) and Abi-1HET (n = 3) animals was transduced by retrovirus encoding MPLWT or MPLW515L. Transduction efficiency was higher than 60%. Abi-1WT marrow cells expressing MPLWT or MPLW515L (Abi-1WT/MPLWT or Abi-1WT/MPLW515L), or Abi-1HET marrow cells expressing MPLWT or MPLW515L (Abi-1HET/MPLWT or Abi-1HET/MPLW515L) were transplanted into lethally irradiated C57/BL6 mice (n = 6 per group) (Figure 6A). Analyses performed 60 days posttransplant showed no femur pallor or significant changes in spleen or liver sizes in the Abi-1WT/MPLWT, Abi-1WT/MPLW515L, or Abi-1HET/MPLWT groups. Conversely, Abi-1HET/MPLW515L animals showed 3-fold and 1.7-fold increases in spleen and liver size, respectively, as well as femur pallor (Figure 6B-C). Furthermore, we noted pronounced leukocytosis, thrombocytosis, and polycythemia in Abi-1HET/MPLW515L animals compared with Abi-1WT/MPLW515L (Figure 6D). We also observed a significant increase in Mac-1+/Gr-1+, CD41, TER119+/CD71+, TER119+/CD71−, and B220+ cells in both marrow and blood, and a decrease in CD3+ cells in the marrow of the Abi-1HET/MPLW515L animals (Figure 6E; supplemental Figure 10A-B). Staining for reticulin fibers in the Abi-1HET/MPLW515L mice showed significant (3+) thickening in both marrow and spleen (Figure 6F). Overall, these data suggest that loss of 1 copy of Abi1 markedly accelerates the MPLW515L-mediated MPN in mice.
Figure 6.

Loss of 1 copy of Abi-1 accelerates disease development in the bone marrow transplantation model of MPLW515L-mediated myeloproliferative neoplasm. (A) Schematic representation of Abi-1HET/MPLW515L model. Bone marrow was obtained from 14-week-old sex-matched Abi-1WT (n = 3) or Abi-1HET (n = 3) animals and transduced with retrovirus encoding Abi-1WT or W515L mutated GFP-tagged MPL. Infected bone marrows representing 4 experimental groups Abi-1WT/MPLWT, Abi-1HET/MPLWT, Abi-1WT/MPLW515L, or Abi-1HET/MPLW515L were next transplanted to C57BL/6J females (n = 6 per experimental group). Analyses were conducted 60 days posttransplant. (B). Representative gross pathology images of femurs, spleens, and livers of the animals from each experimental group are shown. (C) Average spleen and liver sizes of Abi-1WT/MPLWT, Abi-1HET/MPLWT, Abi-1WT/MPLW515L, or Abi-1HET/MPLW515L mice. Relative organ weight was calculated as an absolute organ weight (g)/body weight on sacrifice day (g) ×100. Organs from 6 animals per group were evaluated. (D) Average white blood cell count and hematocrit and platelet count of Abi-1WT/MPLWT, Abi-1HET/MPLWT, Abi-1WT/MPLW515L, or Abi-1HET/MPLW515L mice. Peripheral blood from 6 animals per group was analyzed. (E) Average frequencies of Mac1+/Gr-1+, CD41+, CD42+, CD71+/Ter119+ or CD71+/Ter119− as well as B220+ and CD3+ cells in the bone marrow or peripheral blood obtained from Abi-1WT/MPLWT, Abi-1HET/MPLWT, Abi-1WT/MPLW515L, or Abi-1HET/MPLW515L mice. Samples from 6 animals per group were analyzed. (F) Gomori reticulin staining of the bone marrow from femurs and spleen sections of Abi-1WT/MPLW515L or Abi-1HET/MPLW515L animals (black stain). Bars correspond to 100 μm. Images were obtained using Zeiss Axiophot microscope with Zeiss Pan-Apochromat 20×/1.0 lens. *P < .05
Discussion
Extensive evidence implicates dysregulated JAK/STAT signaling in the pathophysiology of MPNs. Nonetheless, JAK2 inhibitors do not have salutary disease-modifying effects at the stem cell level.7-10 The cross talk between JAK/STAT, MAPK, and Akt pathways, and the potential of therapeutic targeting of these pathways, has received much less attention.39,51-53 Inflammation signaling and cytokine involvement in MPNs, particularly PMF, is widely acknowledged; however, the specific role of NF-κB signaling in MPNs was highlighted only recently.54
Abi-1 is an adapter protein involved in regulation of Abl1, PI3K, and Ras signaling, with a previously unknown role in hematopoiesis.12,17,49 To address this gap, we generated bone marrow-specific murine knockout of Abi1. Loss of Abi-1 proved sufficient to induce a MPN-like phenotype that replicates many features of human myelofibrosis. Myeloid hyperplasia, megakaryocytosis, progressive fibrosis, splenomegaly, and extramedullary hematopoiesis observed in Abi-1KO mice resemble other murine models of PMF.6,39,55-58 Notably, we observed no transformation to acute leukemia, yet Abi-1KO animals had shorter lifespan and evidence of cytokine dysregulation. Although there was no significant phenotypic effect of heterozygous Abi-1 loss, given that PMF typically develops in patients older than 55 years (equivalent to 18 months in mice), we continue observation of the effect of heterozygous Abi1 loss in mice beyond 72 weeks of age.
Molecular analyses of the MPN-like phenotype in Abi-1KO mice showed unexpectedly only modest activation of JAK2-STAT5 signaling, with hyperactivation of SFKs, STAT3, and NF-κB. Upregulation of Mpo, integrin αMβ2, Runx1, Hck, Fgr, STAT3, and NF-κB–haptoglobin, seen in proteomic analysis, are consistent with increase in myeloid cells and upregulated SFKs, STAT3, and NF-κB signaling in Abi-1KO mice. Further work will be needed to elucidate how overactive SFKs/STAT3/NF-κB signaling relates to established MPN models. JAK2, MPL, or CALR mutations are frequent events in MPNs,5,6,39,59 and murine models incorporate these variants.39,51,56,57,60-62 In contrast to JAK2V617F models that progress to secondary myelofibrosis via PV-like stage,51,60,61,63 our model resembles the thrombopoietin -driven models expressing MPLW515L or CALRdel52, in which fibrosis develops after a period of primary thrombocytosis.39,56,57,62 To explore the association between Abi-1 deficiency and established pathogenetic mechanisms in MPNs, we assessed how heterozygous loss of Abi-1 affected the MPLW515L-mediated mouse model. Results showed accelerated MPN-like disease in Abi-1HET/MPLW515L animals. These data support a contributory role of Abi-1 to the pathophysiology of MPNs. In addition to these findings, proteomics and immunoblotting (supplemental Figure 9E) indicated upregulation of calreticulin in Abi-1KO. Calreticulin has an established role in MPN pathophysiology, and somatic CALR mutations occur in 30% of PMF.59 At this point, the contribution of increased calreticulin to the Abi-1 loss-dependent phenotype remains unclear. Strikingly, Abi-1 deficiency in hematopoietic stem/progenitor cells results in upregulation of genes controlled, directly or indirectly, by NF-κB. Genome-wide expression profiling of Abi-1KO LSK cells highlighted overexpression of pro-inflammatory pathways associated with NF-κB activation. Upregulation of pro-inflammatory cytokines in patients with PMF,64 as well as TLR activation,65 may contribute to marrow fibrosis. Our data support a mechanism in which Abi-1 loss results in upregulation of SFKs, which in turn activate STAT3, ultimately modulating the NF-κB pathway. Notably, STATs can be activated by SFKs,66,67 and STAT3 and NF-κB pathways are linked.68,69 Interestingly, Ser-32-phosphorylated IκB is not degraded and remains highly expressed in Abi-1KO marrow. IκB phosphorylation on Tyr allows NF-κB activation without IκB degradation.70 Elucidation of how Abi-1 loss might lead to increased NF-κB signaling and the role of IκB phosphorylation in this process will require further research.
Abi-1 loss induced significant impairment of the hemostasis of HSCs. Increased cell cycle activity of Abi-1KO hematopoietic stem/progenitor cells is possibly leading to their expansion, displacement from the marrow, and progressive exhaustion, and is reflected in their impaired self-renewal capacities. Similarly, increased cycling and impaired self-renewal of HSCs was noted in animal models of hematopoietic tissue-specific overactivation of Akt or NF-κB.71,72 Given the activity of Mx1 promotor-driven Cre in mesenchymal stromal cells, it is reasonable to expect that changes in homeostasis of Abi-1KO HSCs may prove to be both intrinsic and nonautonomous in future experiments.
Analyses of ABI1 transcripts in MPNs indicated that its loss may be specific to PMF. Mechanisms responsible for loss of ABI1 transcripts in PMF are however not clear. Notably, single nucleotide polymorphism arrays performed on 151 MPN samples showed microdeletions in the 10p12 region encompassing ABI1 locus.73 Sequencing of ABI1 is warranted to provide mutational profile of the gene in MPNs.
In aggregate, our data provide the first evidence for a role of Abi-1 in the pathogenesis of PMF, and a link to upregulation of the SFKs/STAT3/NF-κB signaling axis. To our knowledge, this is the first model of MPN mechanistically uncoupled from JAK2-STAT5 hyperactivity. Full elucidation of the cross-communication between JAK/SFKs/STAT3/NF-κB signaling may provide insight into the association between proliferative and pro-inflammatory features that characterize MPNs, particularly PMF. Targeting Abi-1 and associated pathways may offer a new therapeutic strategy for human PMF.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Mandy Pereira, Laura Goldberg, Mark Dooner, Elaine Pappa, Michael DelTatto, Joan Boylan, Aleksander F. Sikorski, Paola Guglielmelli, and Loren Fast for helpful suggestions and technical assistance, and Eric Milner for his editorial help. The authors thank Matthew Adler and Veenu Aishwarya from AUM BioTech, LLC and Chris Gould from Biolegend for technical assistance and helpful suggestions. The authors also thank John Crispino and Qiang Jeremy Wen (Northwestern University) for critical comments, discussion, and technical assistance. The authors thank Ross Levine (MSKCC) for MSCV-MPL WT-IRES-GFP and MSCV-MPL W515L-IRES-GFP retroviral vectors.
This work was supported by Brown Biomed Division Dean's Award grant (P.M.D., E.O.), University of Medicine Foundation Integration Funding (P.M.D.), Rhode Island Foundation grant (P.M.D.), a pilot grant from the Lifespan Center of Biomedical Research Excellence for Cancer Research Development (National Institutes of Health/National Institute of General Medical Sciences 1P30GM110759 to P.M.D. and 1P20GM119943 to P.J.Q. and P.M.D.) and Savit Foundation (P.J.Q., P.M.D.). Work in Florence was supported by a grant from Associazione Italiana per la Ricerca sul Cancro (AIRC, Milan, Italy), Special Program Molecular Clinical Oncology 5x1000 to AIRC-Gruppo Italiano Malattie Myeloproliferative project #1005. A.H.-J. and L.K. were supported by National Institutes of Health/National Cancer Institute R01CA161018 (L.K.). A.J.O. is supported by the American Society of Hematology Research Scholar award.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.C., J.M., N.A., M.P., N.K., Y.C., K.L., C.S., X.Y., R.Z., A.P., A.T., J.C., A.H.-J. and O.L. performed experiments, acquired and analyzed data, and contributed to manuscript writing; N.A., D.O.T., A.J.O., T.C.Z., E.O., J.L.R., L.K., P.J.Q., P.A.G., R.M., and A.M.V. analyzed data and contributed to manuscript writing; and P.M.D. designed research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Patrycja M. Dubielecka, Warren Alpert Medical School of Brown University, Signal Transduction Laboratory, Rhode Island Hospital, 1 Hoppin St, Coro West, Suite 5.01, Providence, RI 02903; e-mail: patrycja_dubielecka-szczerba@brown.edu.
REFERENCES
- 1.Tefferi A. Primary myelofibrosis: 2014 update on diagnosis, risk-stratification, and management. Am J Hematol. 2014;89(9):915-925. [DOI] [PubMed] [Google Scholar]
- 2.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937-951. [DOI] [PubMed] [Google Scholar]
- 3.Barbui T, Thiele J, Gisslinger H, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 2018;8(2):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392-397. [DOI] [PubMed] [Google Scholar]
- 5.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779-1790. [DOI] [PubMed] [Google Scholar]
- 6.Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tefferi A, Pardanani A. Serious adverse events during ruxolitinib treatment discontinuation in patients with myelofibrosis. Mayo Clin Proc. 2011;86(12):1188-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787-798. [DOI] [PubMed] [Google Scholar]
- 10.Cervantes F, Vannucchi AM, Kiladjian JJ, et al. ; COMFORT-II investigators. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122(25):4047-4053. [DOI] [PubMed] [Google Scholar]
- 11.Geyer HL, Mesa RA. Therapy for myeloproliferative neoplasms: when, which agent, and how? Hematology Am Soc Hematol Educ Program. 2014;2014:277-286. [DOI] [PubMed] [Google Scholar]
- 12.Shi Y, Alin K, Goff SP. Abl-interactor-1, a novel SH3 protein binding to the carboxy-terminal portion of the Abl protein, suppresses v-abl transforming activity. Genes Dev. 1995;9(21):2583-2597. [DOI] [PubMed] [Google Scholar]
- 13.Xiong X, Cui P, Hossain S, et al. Allosteric inhibition of the nonMyristoylated c-Abl tyrosine kinase by phosphopeptides derived from Abi1/Hssh3bp1. Biochim Biophys Acta. 2008;1783(5):737-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brehme M, Hantschel O, Colinge J, et al. Charting the molecular network of the drug target Bcr-Abl. Proc Natl Acad Sci USA. 2009;106(18):7414-7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikeguchi A, Yang HY, Gao G, Goff SP. Inhibition of v-Abl transformation in 3T3 cells overexpressing different forms of the Abelson interactor protein Abi-1. Oncogene. 2001;20(36):4926-4934. [DOI] [PubMed] [Google Scholar]
- 16.Scita G, Nordstrom J, Carbone R, et al. EPS8 and E3B1 transduce signals from Ras to Rac. Nature. 1999;401(6750):290-293. [DOI] [PubMed] [Google Scholar]
- 17.Innocenti M, Tenca P, Frittoli E, et al. Mechanisms through which Sos-1 coordinates the activation of Ras and Rac. J Cell Biol. 2002;156(1):125-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Innocenti M, Gerboth S, Rottner K, et al. Abi1 regulates the activity of N-WASP and WAVE in distinct actin-based processes. Nat Cell Biol. 2005;7(10):969-976. [DOI] [PubMed] [Google Scholar]
- 19.Dubielecka PM, Ladwein KI, Xiong X, et al. Essential role for Abi1 in embryonic survival and WAVE2 complex integrity. Proc Natl Acad Sci USA. 2011;108(17):7022-7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sini P, Cannas A, Koleske AJ, Di Fiore PP, Scita G. Abl-dependent tyrosine phosphorylation of Sos-1 mediates growth-factor-induced Rac activation. Nat Cell Biol. 2004;6(3):268-274. [DOI] [PubMed] [Google Scholar]
- 21.Campa F, Machuy N, Klein A, Rudel T. A new interaction between Abi-1 and betaPIX involved in PDGF-activated actin cytoskeleton reorganisation. Cell Res. 2006;16(9):759-770. [DOI] [PubMed] [Google Scholar]
- 22.Beli P, Mascheroni D, Xu D, Innocenti M. WAVE and Arp2/3 jointly inhibit filopodium formation by entering into a complex with mDia2. Nat Cell Biol. 2008;10(7):849-857. [DOI] [PubMed] [Google Scholar]
- 23.Innocenti M, Zucconi A, Disanza A, et al. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat Cell Biol. 2004;6(4):319-327. [DOI] [PubMed] [Google Scholar]
- 24.Ring C, Ginsberg MH, Haling J, Pendergast AM. Abl-interactor-1 (Abi1) has a role in cardiovascular and placental development and is a binding partner of the alpha4 integrin. Proc Natl Acad Sci USA. 2011;108(1):149-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4(3):207-214. [DOI] [PubMed] [Google Scholar]
- 26.Sun X, Li Y, Yu W, Wang B, Tao Y, Dai Z. MT1-MMP as a downstream target of BCR-ABL/ABL interactor 1 signaling: polarized distribution and involvement in BCR-ABL-stimulated leukemic cell migration. Leukemia. 2008;22(5):1053-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiong X, Chorzalska A, Dubielecka PM, et al. Disruption of Abi1/Hssh3bp1 expression induces prostatic intraepithelial neoplasia in the conditional Abi1/Hssh3bp1 KO mice. Oncogenesis. 2012;1(9):e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sowalsky AG, Sager R, Schaefer RJ, et al. Loss of Wave1 gene defines a subtype of lethal prostate cancer. Oncotarget. 2015;6(14):12383-12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tod J, Hanley CJ, Morgan MR, et al. Pro-migratory and TGF-β-activating functions of αvβ6 integrin in pancreatic cancer are differentially regulated via an Eps8-dependent GTPase switch. J Pathol. 2017;243(1):37-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinestel K, Brüderlein S, Lennerz JK, et al. Expression and Y435-phosphorylation of Abelson interactor 1 (Abi1) promotes tumour cell adhesion, extracellular matrix degradation and invasion by colorectal carcinoma cells. Mol Cancer. 2014;13(1):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Juskevicius D, Lorber T, Gsponer J, et al. Distinct genetic evolution patterns of relapsing diffuse large B-cell lymphoma revealed by genome-wide copy number aberration and targeted sequencing analysis. Leukemia. 2016;30(12):2385-2395. [DOI] [PubMed] [Google Scholar]
- 32.Chorzalska A, Salloum I, Shafqat H, et al. Low expression of Abelson interactor-1 is linked to acquired drug resistance in Bcr-Abl-induced leukemia. Leukemia. 2014;28(11):2165-2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maruoka M, Sato M, Yuan Y, et al. Abl-1-bridged tyrosine phosphorylation of VASP by Abelson kinase impairs association of VASP to focal adhesions and regulates leukaemic cell adhesion. Biochem J. 2012;441(3):889-899. [DOI] [PubMed] [Google Scholar]
- 34.Yu W, Sun X, Clough N, Cobos E, Tao Y, Dai Z. Abi1 gene silencing by short hairpin RNA impairs Bcr-Abl-induced cell adhesion and migration in vitro and leukemogenesis in vivo. Carcinogenesis. 2008;29(9):1717-1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eto K, Nishikii H, Ogaeri T, et al. The WAVE2/Abi1 complex differentially regulates megakaryocyte development and spreading: implications for platelet biogenesis and spreading machinery. Blood. 2007;110(10):3637-3647. [DOI] [PubMed] [Google Scholar]
- 36.Kühn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427-1429. [DOI] [PubMed] [Google Scholar]
- 37.Yu K, Salomon AR. HTAPP: high-throughput autonomous proteomic pipeline. Proteomics. 2010;10(11):2113-2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahsan N, Belmont J, Chen Z, Clifton JG, Salomon AR. Highly reproducible improved label-free quantitative analysis of cellular phosphoproteome by optimization of LC-MS/MS gradient and analytical column construction. J Proteomics. 2017;165:69-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99(1):310-318. [DOI] [PubMed] [Google Scholar]
- 41.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100(16):9440-9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Norfo R, Zini R, Pennucci V, et al. ; Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative Investigators. miRNA-mRNA integrative analysis in primary myelofibrosis CD34+ cells: role of miR-155/JARID2 axis in abnormal megakaryopoiesis. Blood. 2014;124(13):e21-e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zini R, Guglielmelli P, Pietra D, et al. ; AGIMM (AIRC Gruppo Italiano Malattie Mieloproliferative) investigators. CALR mutational status identifies different disease subtypes of essential thrombocythemia showing distinct expression profiles. Blood Cancer J. 2017;7(12):638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90(8):1128-1132. [PubMed] [Google Scholar]
- 45.Kogan SC, Ward JM, Anver MR, et al. ; Hematopathology subcommittee of the Mouse Models of Human Cancers Consortium. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood. 2002;100(1):238-245. [DOI] [PubMed] [Google Scholar]
- 46.Kfoury A, Virard F, Renno T, Coste I. Dual function of MyD88 in inflammation and oncogenesis: implications for therapeutic intervention. Curr Opin Oncol. 2014;26(1):86-91. [DOI] [PubMed] [Google Scholar]
- 47.Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30(1):43-52 [DOI] [PubMed] [Google Scholar]
- 48.Machida K, Thompson CM, Dierck K, et al. High-throughput phosphotyrosine profiling using SH2 domains. Mol Cell. 2007;26(6):899-915. [DOI] [PubMed] [Google Scholar]
- 49.Dubielecka PM, Machida K, Xiong X, et al. Abi1/Hssh3bp1 pY213 links Abl kinase signaling to p85 regulatory subunit of PI-3 kinase in regulation of macropinocytosis in LNCaP cells. FEBS Lett. 2010;584(15):3279-3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kleppe M, Kwak M, Koppikar P, et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015;5(3):316-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akada H, Yan D, Zou H, Fiering S, Hutchison RE, Mohi MG. Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood. 2010;115(17):3589-3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khan I, Huang Z, Wen Q, et al. AKT is a therapeutic target in myeloproliferative neoplasms. Leukemia. 2013;27(9):1882-1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wiśniewska-Chudy E, Szylberg Ł, Dworacki G, Mizera-Nyczak E, Marszałek A. pSTAT5 and ERK exhibit different expression in myeloproliferative neoplasms. Oncol Rep. 2017;37(4):2295-2307. [DOI] [PubMed] [Google Scholar]
- 54.Fisher DAC, Malkova O, Engle EK, et al. Mass cytometry analysis reveals hyperactive NF Kappa B signaling in myelofibrosis and secondary acute myeloid leukemia. Leukemia. 2017;31(9):1962-1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vannucchi AM, Bianchi L, Cellai C, et al. Development of myelofibrosis in mice genetically impaired for GATA-1 expression (GATA-1(low) mice). Blood. 2002;100(4):1123-1132. [DOI] [PubMed] [Google Scholar]
- 56.Yan XQ, Lacey D, Hill D, et al. A model of myelofibrosis and osteosclerosis in mice induced by overexpressing thrombopoietin (mpl ligand): reversal of disease by bone marrow transplantation. Blood. 1996;88(2):402-409. [PubMed] [Google Scholar]
- 57.Marty C, Pecquet C, Nivarthi H, et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood. 2016;127(10):1317-1324. [DOI] [PubMed] [Google Scholar]
- 58.Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood. 2006;107(11):4274-4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391-2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144-1148. [DOI] [PubMed] [Google Scholar]
- 61.Tiedt R, Hao-Shen H, Sobas MA, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111(8):3931-3940. [DOI] [PubMed] [Google Scholar]
- 62.Chachoua I, Pecquet C, El-Khoury M, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood. 2016;127(10):1325-1335. [DOI] [PubMed] [Google Scholar]
- 63.James C, Mazurier F, Dupont S, et al. The hematopoietic stem cell compartment of JAK2V617F-positive myeloproliferative disorders is a reflection of disease heterogeneity. Blood. 2008;112(6):2429-2438. [DOI] [PubMed] [Google Scholar]
- 64.Hasselbalch HC. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013;24(2):133-145. [DOI] [PubMed] [Google Scholar]
- 65.Micera A, Balzamino BO, Di Zazzo A, Biamonte F, Sica G, Bonini S. Toll-like receptors and tissue remodeling: the pro/cons recent findings. J Cell Physiol. 2016;231(3):531-544. [DOI] [PubMed] [Google Scholar]
- 66.Silva CM. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene. 2004;23(48):8017-8023. [DOI] [PubMed] [Google Scholar]
- 67.Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6(10):587-595. [DOI] [PubMed] [Google Scholar]
- 68.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21(1):11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Imbert V, Rupec RA, Livolsi A, et al. Tyrosine phosphorylation of I kappa B-alpha activates NF-kappa B without proteolytic degradation of I kappa B-alpha. Cell. 1996;86(5):787-798. [DOI] [PubMed] [Google Scholar]
- 71.Kharas MG, Okabe R, Ganis JJ, et al. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood. 2010;115(7):1406-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood. 2017;130(15):1693-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stegelmann F, Bullinger L, Griesshammer M, et al. High-resolution single-nucleotide polymorphism array-profiling in myeloproliferative neoplasms identifies novel genomic aberrations. Haematologica. 2010;95(4):666-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.