

Abstract
Azobenzene is a prototypical molecular switch that can be reversibly photoisomerized between the nearly planar and apolar trans form, and the distorted, polar cis form. Most studies related to azobenzene derivatives have focused on planar adsorbed molecules. We present herein the study of a three‐dimensional shape‐persistent molecular architecture consisting of four tetrahedrally arranged azobenzene units that is adsorbed on a Ag(111) surface. While the azobenzenes of the tripod in contact with the surface lost their switching ability, different isomers of the upright standing arm of the tetramer were obtained reversibly and efficiently by illumination at different wavelengths, revealing time constants of only a few minutes. Diffusion on the surface was dependent on the isomeric state—trans or cis—of the upright oriented azobenzene group. Hence, molecular mobility can be modulated by its isomeric state, which suggests that molecular growth processes could be controlled by external stimuli.
Keywords: azo compounds, molecular switches, photoisomerization, scanning probe microscopy, surface diffusion
Molecular photoswitches1, 2 are not only at the basis of fundamental processes in nature, but also of interest for applications that include responsive materials and surfaces, information processing and storage, catalysis and separation, molecular machines, energy conversion, and drug design, for example, for switchable surfaces3 or as conductance switches.4 The photoisomerization of azobenzene (Figure 1 a) has been extensively studied in the gas phase, in solution, in polymer matrices, in liquid crystals, in thin films,1, 5 on nanoparticle surfaces,6, 7 and, more recently, in crystalline porous materials.8
Figure 1.
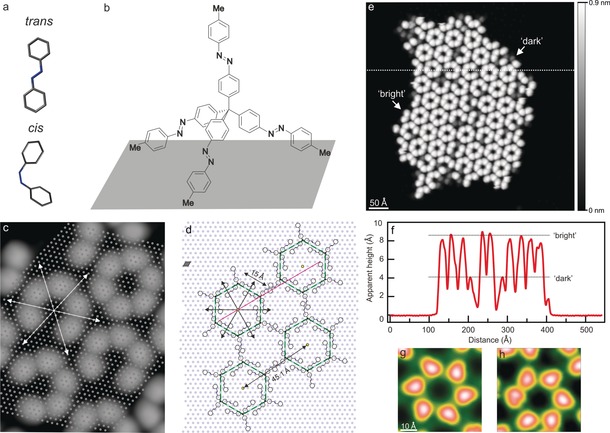
Tetra‐azo islands on Ag(111). a) Molecular structure of the trans and cis isomers of azobenzene (calculated by HyperChem). b) Chemical formula of the tetra(azobenzene)methane species, adsorbed on an atomically flat surface, with all its four azobenzene units in the trans configuration. c) STM image of several self‐assembled hexamers with respect to the underlying Ag(111) lattice (indicated by gray dots), as identified by imaging a small area near the cluster with atomic resolution (see Figure S4). d) Sketch of a possible assembly. The Ag lattice unit cell is plotted in gray. Surface symmetry directions are shown in (c,d) by arrows. e) STM image (544×544 Å2, 1 V, 6 pA) of a tetra‐azo island consisting of bright and dark lobes (see Figure S5 for details). f) Height profile (along the dashed line in (e)) revealing the characteristic apparent heights of bright and dark lobes. g,h) STM images (50×50 Å2, 0.5 V, 5 pA) of hexamers of both chiralities.
The adsorption of azobenzene derivatives on a surface is advantageous since it confines the molecules to two dimensions and allows imaging of single molecules in real space by scanning probe microscopy.9, 10, 11, 12 The surface can have a strong influence on the isomerization process, that is, on the switching behavior,13 and subtle variations in the molecular interaction with the surface can be used to control molecular properties.14 Azobenzene adsorption on metal surfaces typically results in quenching of their light‐induced isomerization, owing to electronic coupling to the substrate,15 which can be weakened by adding bulky side groups to the azobenzene unit, thus reducing the interaction with the surface.16
Another strategy to enhance the photomechanical activity of molecular switches on a surface is to integrate them into molecular architectures that are inherently rigid and three‐dimensional, such as tripod structures. For example, reversible photoisomerization was observed for (tetra)molecular architectures consisting of three adamantane moieties17 or thiols18, 19 anchored to the surface and a fourth branch with an azobenzene group. This approach ensures stable adsorption through the tripod linker and an upright arrangement of the molecular switch unit, and thus electronic decoupling from the surface. When integrating two20 or three21, 22 azobenzene groups into one and the same molecule, the flexible structure of these molecules resulted in flat conformations on the surface, as all azobenzene moieties were adsorbed planar. Hence, a vertical orientation and/or the electronic decoupling of the switching units from the surface was not achieved, and molecular architectures that contain several switching units and maintain their three‐dimensional conformation upon adsorption at a surface were not realized so far.
Herein we present a study of tetra(azobenzene)methane molecules (named tetra‐azo in the following), which consist of four azobenzene moieties converging at a central tetrahedral carbon atom, on a metal surface.8 The idea was that the molecules would adsorb with three azobenzene units in contact with the surface and the fourth one pointing upright, owing to their rigid tetrahedral shape (Figure 1 b).
In this regard, tetra‐azo can be viewed as a nanoscale caltrop. When tetra‐azo molecules were deposited (see the Experimental Section) in the all‐trans configuration onto the Ag(111) surface, which was kept at room temperature during preparation, they were found to assemble in ordered islands (Figure 1 c–e; see also Figure S1 in the Supporting Information). Such islands appear as six‐lobed objects compatible with hexameric aggregates, as sketched in Figure 1 d. The lobes were present in two different apparent heights of (4.19±0.24) and (8.87±0.12) Å (approximately constant over various bias voltages; see Figure S2), as determined from height profiles (Figure 1 f; see also Figure S11). They are named “dark” and “bright”, respectively, in the following.
The hexamer units are chiral superstructures, both enantiomorphs of which are observed (Figure 1 g,h; see also Figure S3). Note that the tetra‐azo molecules are originally achiral (in the gas phase), but apparently form chiral structures when assembling on the surface, similar to other molecules.23, 24 Each hexamer is typically composed of one handedness only, probably owing to steric hindrance. Hexamers with an undefined chirality, which represent a non‐uniform appearance of the individual molecules, are also observed, but only rarely (see Figure S3). The precise adsorption site and geometry of the molecules was determined with the help of atomically resolved images of the Ag(111) surface (Figure 1 c; see also Figure S4). By symmetrically arranging the threefold tripod arms (the fourth arm protrudes vertically off the surface), in analogy to the tetrahedral arrangement proposed previously,17, 19 we determined the molecular adsorption geometry, which is in agreement with the STM images (Figure 1 d).
On the basis of the three‐dimensional structure of a tetra‐azo molecule (Figure 1 b), it seems reasonable to associate the “bright” and “dark” molecules with different isomers of the azobenzene unit standing upright (see Figure S12). This interpretation is in agreement with scanning probe microscopy studies of self‐assembled monolayers (SAMs)25, 26 or tripod molecules,18, 19 resulting in either large or small apparent heights in the images if the upright azobenzene group is in the trans or cis state, respectively. We therefore assigned the “bright” and “dark” lobes on the surface to the trans and cis configuration, respectively, of the upwards pointing arm of the tetramer. In other words: the trans isomers appear bright, owing to their rather linear structure pointing approximately vertically off the surface, and the cis isomers appear darker. The other three arms (see Figure S5), which are adsorbed at the surface, are most likely always in the trans state.21, 27 This assignment is on one hand based on previous studies, in which the trans state of azobenzene derivatives is the energetically most stable form in the gas phase,1 in the solid state,8 and on various metal surfaces9, 10, 14, 27 after deposition at room temperature. Importantly, also tri‐azobenzene systems adsorb on Ag(111) with all azobenzene arms in the trans state,21 a configuration that is probably similar to the azo‐tripod linker in the present case. On the other hand, the molecular islands appear very regular—both in the lateral and vertical direction (see Figure S11)—which seems unlikely if one or more surface‐adsorbed legs of an azo‐tetrapod molecule were in the cis state. The threefold symmetry observed for individual molecules (see Figure 1 c) is consistent with this assignment of the surface‐adsorbed arms to the trans state.
While the majority (about 80 %) of the molecules in islands are in the “bright” and thus trans state after deposition (with the sample kept at room temperature during deposition), a fraction of 20 % are found in the cis state. Hence, we assign the small fraction of cis isomers to the adsorption of these species that do not relax into the trans isomer upon adsorption, owing to their lifting from the surface in the tetramer geometry.15, 27
To prove our assignment of trans (bright lobes) and cis isomers (dark lobes) on the surface and to study the isomerization properties of the tetra‐azo molecules in the clusters, we illuminated them with laser light of different wavelengths. Starting with the pristine surface (i.e., after molecule deposition; Figure 2 a), the sample was illuminated with 532 nm light. STM imaging of precisely the same surface area before and after light exposure (Figure 2 a,b, respectively) was then used to determine how the molecules in the “bright” and “dark” states evolved. After illumination for 30 min, the number of “bright” molecules decreased strongly (Figure 2 b). Note that the shape and apparent height of new dark and bright lobes were equivalent to the initial surface (see Figure S6).
Figure 2.
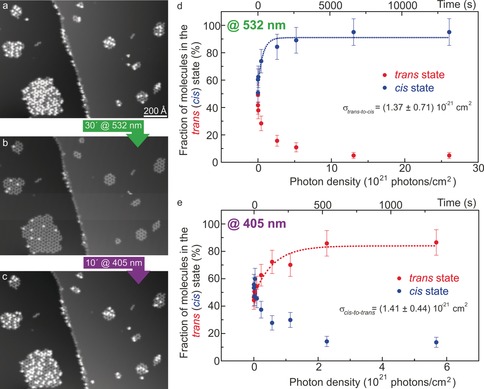
Reversible photoisomerization. a–c) STM images of the same molecular clusters (0.9 V, 7.9 pA) in their original state (a), after illumination at 532 nm for 30 min (b), and after illumination at 405 nm for 10 min (c; see Figure S8 for magnified images). d,e) Fraction of molecules in the “bright” (“dark”) state as a function of the photon flux and exposure time upon exposure to laser light at 532 (d) and 405 nm (e). Data points (in d,e) were fitted with the exponential function and the time constants τ=457 s (d) and τ=154 s (e).
Hence, the 532 nm light causes trans→cis isomerization of individual molecules. This result indicates that only the trans‐azobenzene units can absorb photons of 532 nm and/or the cis→trans isomerization is much less efficient. This wavelength is longer than the absorption range of all‐trans tetra‐azo molecules in solution,8 but it is known that surface adsorption of trans‐azobenzene derivatives can enable red‐shifted photoreactivity.28 The reversibility of the process29 was explored by illuminating the “dark”‐enriched surface (Figure 2 b) for 10 min with 405 nm light, upon which the population of “bright” molecules increased significantly (Figure 2 c). Hence, reversible photoisomerization (trans→cis→trans) was achieved.
By fitting the data points with an exponential function that describes the solution of a first‐order rate master equation with time as a variable (top axis in Figure 2 d,e), we found that isomerization occurs rather fast with time constants of 457 s for trans→cis isomerization and an even smaller value of 154 s for cis→trans switching (Figure 2 d,e). For both processes, the total number of molecules at the surface remained constant. A more precise measure for the photoinduced switching, referring to the photon density (bottom axis in Figure 2 d,e) and thus including the spot size on the surface (see the Supporting Information), are the cross‐sections σ(trans→cis)=(1.37±0.71)×10−21 cm2 (Figure 2 d) and σ(cis→trans)=(1.41±0.44)×10−21 cm2 (Figure 2 e). These values are larger than for azobenzene derivatives adsorbed flat on a metal surface (about 10−22 cm2),30 in agreement with the relatively small time constant, thus revealing that the upright standing azobenzene is partially decoupled from the metallic surface. On the other hand, they are smaller than those for azobenzene derivatives on an insulator surface (10−18 and 10−20 cm2)15 and in solution (about 10−19 cm2),13 thus indicating only partial decoupling. Other tripod systems have shown more efficient decoupling with cross‐sections between 2.5×10−19 cm2 and 4×10−18 cm2 for a (p‐cyano)azobenzene‐tripod system17 or about 10−18[31] and 10−17 cm2 32 for azobenzene chains in self‐assembled monolayers on Au(111), where the distance between the azobenzene unit and the metal surface is larger. Besides photoswitching, isomerization could also be induced in a controlled fashion by STM manipulation, and reversible cycles trans→cis→trans were possible, resulting in exactly the same molecular appearances (see Figure S9). About 25 % of the molecules did not switch, either with light or by STM manipulation, and remained always in the dark state (see the Supporting Information).
Individual tetramer molecules were also present, typically consisting of three lobes of low apparent height arranged in a tripod geometry and a bright lobe in the center with an apparent height of about 4 Å (Figure 3 a). This height corresponds to that of the “dark” molecules in the islands (see Figure 1 f), and we therefore conclude that the upwards pointing azobenzene arm is in the cis state for this species. On the other hand, another type of single molecule shows a much brighter lobe in the center. Figure 3 b shows a dimer that contains both types: The molecule at the bottom is in the “dark” state, whereas the molecule at the top appears with a height of about 9 Å and is therefore identified as the “bright” species (see Figure 1 f). Hence, we assign the bright lobe to a tetra‐azo molecule in the all‐trans state attached to another molecule in which the upwards pointing arm is in the cis state.
Figure 3.
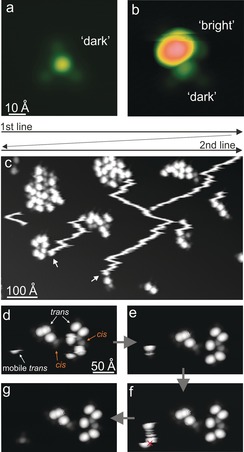
Isomer‐dependent diffusion. a,b) STM images (62×62 Å2 with 0.5 V, 20 pA (a) and −1 V, 1.2 pA (b)) of individual tetra‐azo molecules in both states. c) STM overview image (810×473 Å2, 1 V, 22 pA) of “bright” molecules that diffuse over the surface under the influence of the tip (scanning lines are indicated at the top). Arrows in (c) mark the positions in which the “bright” molecules stop their diffusion during scanning and become stable, because they adsorb near other molecules. d–g) Subsequent STM images of the same surface area with a smaller tunneling current (6 pA and 1 V), with only an isolated trans tetra‐azo molecule diffusing (marked at the left as “mobile trans”).
When following the evolution of a surface area with time, we observed interesting molecular dynamics. Whereas the molecules within the islands remained stable, isolated tetra‐azo species could move over the surface during STM scanning, which is not unusual, as the intermolecular interaction in islands stabilizes the molecules. However, we found different dynamics for the two molecular states: While single tetra‐azo molecules in the “dark” (cis) state were usually left unperturbed at low tunneling currents (i.e., large tip heights; Figure 3 a), “bright” (trans) molecules could detach from island edges and were then mobile as individual entities.
This mobility is visible in STM images as fuzzy stripes (Figure 3 c) that result from the STM scanning direction (horizontal lines from top to bottom of the image as sketched in Figure 3 c). Hence, these lines reflect single molecules that are mobile on the surface. Finally, the mobile trans molecules adsorb close to a molecular cluster and then remain stable, which is visible as the lines end and a bright lobe appears at the terminus (marked by arrows in Figure 3 c). Apparently, the trans molecules are continuously pulled or pushed downwards (line‐by‐line in the scanning direction) by the STM tip during imaging, thus resulting in the characteristic stripes in the images that start at a molecular island from which the molecule detaches and end at another island towards the bottom of the image.
This interpretation was confirmed by imaging at a smaller tunneling current (at the same bias voltage), that is, a larger tip height, under which conditions the detachment of trans tetra‐azo molecules from the islands cannot be observed (Figure 3 d–g). Diffusion of the trans molecules is so efficient that a static “bright” (trans) molecule could not be imaged even at rather large tip–surface distances. Such stripes were never observed for the “dark” (cis) molecules, which reveals the isomer sensitivity of the diffusion, as also confirmed by STM manipulation. Whereas the three cis as well as the trans tetra‐azo molecules in clusters always remained in place, the trans molecule at the left diffused (Figure 3 d,e). However, if isomerization was induced by a voltage pulse (similar to Figure S9) on the trans molecule (in Figure 3 f), diffusion was stopped, and the molecule could be imaged as a motionless entity afterwards, appearing with a smaller height characteristic of the cis state (Figure 3 g).
These observations indicate very different mobilities of isolated molecules with the upwards oriented azobenzene arm in the trans or the cis state. Their diffusion on the surface therefore depends on their isomeric state, which, in turn, can be changed at will by STM manipulation. The change in mobility can be due to a modification of either the molecule–surface or the molecule–tip interaction upon isomerization. The influence of the tip on the molecular motion is apparent from the characteristic top‐to‐bottom direction of motion in the images (Figure 3 c). However, the isomer‐specific difference persists if various tip heights are used (by changing the tunneling current between 1 and 25 pA at a constant bias voltage; see the Supporting Information). Since the molecule–tip interaction is substantially modified in this range, but the effect remains qualitatively unchanged, we believe that the molecule–surface interaction is mainly responsible for the observed effect. Accordingly, the change in mobility is tentatively assigned to a modification of the molecule–surface coupling, which is reduced in the case of molecules in the “bright” (trans) state, owing to the protruding trans‐azobenzene arm that interacts less with the Ag(111) surface, thereby lowering the diffusion barrier. The difference in the dipole moment of the two isomers (0 D for the trans and 3.2 D for the cis state in the gas phase)33 might also play a role.
In summary, we adsorbed three‐dimensional shape‐persistent molecules comprising four azobenzene switching units on a surface, which resulted in ordered hexamers. The four azo arms, which are chemically and structurally identical, become functionally non‐equivalent upon adsorption: Only the upright standing unit maintains its ability to photoisomerize. Light‐induced switching between the trans and cis state was found to be reversible, as followed for precisely the same molecules at the surface, and efficient as compared with planar azobenzene adsorption on a metal surface. Finally, we found isomer‐dependent diffusion of single molecules on the surface, and showed that the diffusive motion can be turned on and off in a single molecule by tip‐induced isomerization. This unprecedented behavior could open up new routes for controlling molecular mobility on a surface, either locally with the scanning probe tip or remotely by light.
Experimental Section
Tetra‐azo molecules were synthesized and characterized according to a previously published procedure.8 The Ag(111) sample was cleaned by repeated neon ion sputtering (1.3 keV) and subsequent annealing at 720 K. Tetra(azobenzene)methane molecules (named tetra‐azo) were deposited from a Knudsen cell (sublimation temperature of 543 K) onto Ag(111) held at room temperature. STM images were recorded in constant‐current mode at 7.5 K with the bias voltage referring to the sample with respect to the grounded STM tip. Since molecules at the island edges can readily be dislocated if small tip heights are used, imaging was carried out at rather small tunneling currents of a few picoamperes.
CW diode lasers with wavelengths of 405 and 532 nm, a spot size of about 0.22 mm2 on the surface, and an average intensity of 1.85 W cm−2 were used for illumination. During light exposure, the STM tip was retracted by macroscopic distances of about 6–7 mm to avoid shadowing and field‐enhancement effects, whereas the lateral shift upon reapproach did not exceed 100 nm, thus allowing retrieval of exactly the same area on the sample surface.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the European Commission for financial support through the “Mechanics with Molecules” project MEMO (FET open project No.: 766864) and the “Leaps” project (ERC Advanced Grant No.: 692981).
C. Nacci, M. Baroncini, A. Credi, L. Grill, Angew. Chem. Int. Ed. 2018, 57, 15034.
Contributor Information
Dr. Christophe Nacci, http://www.nanograz.com.
Dr. Massimo Baroncini, http://www.credi-group.it.
Prof. Dr. Alberto Credi, Email: alberto.credi@unibo.it.
Prof. Dr. Leonhard Grill, Email: leonhard.grill@uni-graz.at.
References
- 1. Rau H., Photochromism—Molecules and Systems, Elsevier, Amsterdam, 2003, p. 165. [Google Scholar]
- 2. Feringa B. L., Browne W. R., Molecular switches, Wiley, New York, 2011. [Google Scholar]
- 3. Liu Y., Mu L., Liu B., Kong J., Chem. Eur. J. 2005, 11, 2622. [DOI] [PubMed] [Google Scholar]
- 4. Jan van der Molen S., Liljeroth P., J. Phys. Condens. Matter 2010, 22, 133001. [DOI] [PubMed] [Google Scholar]
- 5. Hugel T., Holland N. B., Cattani A., Moroder L., Seitz M., Gaub H. E., Science 2002, 296, 1103. [DOI] [PubMed] [Google Scholar]
- 6. Manna D., Udayabhaskararao T., Zhao H., Klajn R., Angew. Chem. Int. Ed. 2015, 54, 12394; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12571. [Google Scholar]
- 7. Lee J.-W., Klajn R., Chem. Commun. 2015, 51, 2036. [DOI] [PubMed] [Google Scholar]
- 8. Baroncini M., d'Agostino S., Bergamini G., Ceroni P., Comotti A., Sozzani P., Bassanetti I., Grepioni F., Hernandez T. M., Silvi S., Venturi M., Credi A., Nat. Chem. 2015, 7, 634. [DOI] [PubMed] [Google Scholar]
- 9. Alemani M., Peters M. V., Hecht S., Rieder K.-H., Moresco F., Grill L., J. Am. Chem. Soc. 2006, 128, 14446. [DOI] [PubMed] [Google Scholar]
- 10. Choi B.-Y., Kahng S.-J., Kim S., Kim H., Kim H. W., Song Y. J., Ihm J., Kuk Y., Phys. Rev. Lett. 2006, 96, 156106. [DOI] [PubMed] [Google Scholar]
- 11. Henzl J., Mehlhorn M., Gawronski H., Rieder K.-H., Morgenstern K., Angew. Chem. Int. Ed. 2006, 45, 603; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 617. [Google Scholar]
- 12. Comstock M. J., Levy N., Cho J., Berbil-Bautista L., Crommie M. F., Poulsen D. A., Frechet J. M. J., Appl. Phys. Lett. 2008, 92, 123107. [Google Scholar]
- 13. Tegeder P., J. Phys. Condens. Matter 2012, 24, 394001. [DOI] [PubMed] [Google Scholar]
- 14. Dri C., Peters M. V., Schwarz J., Hecht S., Grill L., Nat. Nanotechnol. 2008, 3, 649. [DOI] [PubMed] [Google Scholar]
- 15. Jaekel S., Lindner R., Richter A., Bechstein R., Nacci C., Hecht S., Kühnle A., Grill L., ACS Nano 2018, 12, 1821. [DOI] [PubMed] [Google Scholar]
- 16. McNellis E. R., Bronner C., Meyer J., Weinelt M., Tegeder P., Reuter K., Phys. Chem. Chem. Phys. 2010, 12, 6404. [DOI] [PubMed] [Google Scholar]
- 17. Wagner S., Leyssner F., Kördel C., Zarwell S., Schmidt R., Weinelt M., Rück-Braun K., Wolf M., Tegeder P., Phys. Chem. Chem. Phys. 2009, 11, 6242. [DOI] [PubMed] [Google Scholar]
- 18. Takamatsu D., Fukui K.-I., Aroua S., Yamakoshi Y., Org. Biomol. Chem. 2010, 8, 3655. [DOI] [PubMed] [Google Scholar]
- 19. Takamatsu D., Yamakoshi Y., Fukui K.-I., J. Phys. Chem. B 2006, 110, 1968. [DOI] [PubMed] [Google Scholar]
- 20. Mielke J., Selvanathan S., Peters M., Schwarz J., Hecht S., Grill L., J. Phys. Condens. Matter 2012, 24, 394013. [DOI] [PubMed] [Google Scholar]
- 21. Scheil K., Gopakumar T. G., Bahrenburg J., Temps F., Maurer R. J., Reuter K., Berndt R., J. Phys. Chem. Lett. 2016, 7, 2080. [DOI] [PubMed] [Google Scholar]
- 22. Gopakumar T. G., Davran-Candan T., Bahrenburg J., Maurer R. J., Temps F., Reuter K., Berndt R., Angew. Chem. Int. Ed. 2013, 52, 11007; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11213. [Google Scholar]
- 23. Schöck M., Otero R., Stojkovic S., Hümmeling F., Gourdon A., Laegsgaard E., Stensgaard I., Joachim C., Besenbacher F., J. Phys. Chem. B 2006, 110, 12835. [DOI] [PubMed] [Google Scholar]
- 24. Gopakumar T. G., Matino F., Schwager B., Bannwarth A., Tuczek F., Kröger J., Berndt R., J. Phys. Chem. C 2010, 114, 18247. [Google Scholar]
- 25. Pace G., Ferri V., Grave C., Elbing M., von Hänisch C., Zharnikov M., Mayor M., Rampi M. A., Samorì P., Proc. Natl. Acad. Sci. USA 2007, 104, 9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. El Garah M., Palmino F., Cherioux F., Langmuir 2010, 26, 943. [DOI] [PubMed] [Google Scholar]
- 27. Alemani M., Selvanathan S., Ample F., Peters M. V., Rieder K.-H., Moresco F., Joachim C., Hecht S., Grill L., J. Phys. Chem. C 2008, 112, 10509. [Google Scholar]
- 28. Hagen S., Kate P., Leyssner F., Nandi D., Wolf M., Tegeder P., J. Chem. Phys. 2008, 129, 164102. [DOI] [PubMed] [Google Scholar]
- 29. Bandara H. M. D., Burdette S. C., Chem. Soc. Rev. 2012, 41, 1809. [DOI] [PubMed] [Google Scholar]
- 30. Wolf M., Tegeder P., Surf. Sci. 2009, 603, 1506. [Google Scholar]
- 31. Moldt T., Przyrembel D., Schulze M., Bronsch W., Boie L., Brete D., Gahl C., Klajn R., Tegeder P., Weinelt M., Langmuir 2016, 32, 10795. [DOI] [PubMed] [Google Scholar]
- 32. Krekiehn N. R., Müller M., Jung U., Ulrich S., Herges R., Magnussen O. M., Langmuir 2015, 31, 8362. [DOI] [PubMed] [Google Scholar]
- 33. Piyanzina I., Minisini B., Tayurskii D., Bardeau J.-F., J. Mol. Model. 2015, 21, 34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary