

Abstract
HGF/c-Met signalling pathway plays an important role in the development of cancers. A series of 6,7-dimethoxy-4-anilinoquinolines possessing benzimidazole moiety were synthesised and identified as potent inhibitors of the tyrosine kinase c-Met. Their in vitro biological activities against three cancer cell lines (A549, MCF-7, and MKN-45) were also evaluated. Most of these compounds exhibited moderate to remarkable potency. Among them, compound 12n showed the most potent inhibitory activity against c-Met with IC50 value of 0.030 ± 0.008 µM and it also showed excellent anticancer activity against the tested cancer cell lines at low micromolar concentration. Molecular docking verified the results and revealed the possible binding mode of the most promising compound 12n into the ATP-binding site of c-Met kinase.
Keywords: c-Met; inhibitor; 6,7-Dimethoxy-4-anilinoquinoline; synthesis; antitumour
1. Introduction
The cellular mesenchymal to epithelial transition factor (c-Met), also termed as Met or hepatocyte growth factor receptor (HGFR), is a member of receptor tyrosine kinase which encoded by c-Met proto-oncogene1–3. Hepatocyte growth factor (HGF), also known as scatter factor (SF), is the only known high-affinity natural ligand of c-Met. Binding of HGF to c-Met results in receptor homodimerization and phosphorylation and then c-Met is able to recruit several adaptor proteins that in turn activate a number of pathways, including RAS-RAF-MEK-ERK axis, PI3K-AKT-mTOR cascade, SRC, and STAT34–6.
Under normal physiological condition, HGF/c-Met signalling pathway is involved in cell proliferation, survival, migration, scattering, motility and invasion, and plays important roles in mammalian embryonic development, tissue homeostasis, and wound healing7–10. However, when deregulated, the c-Met/HGF pathway leads to tumorigenesis and metastasis7. Abnormal activation of c-Met signalling due to gene amplification, rearrangement or mutation, transcriptional regulation as well as autocrine or paracrine ligand stimulation, has been involved in various types of human cancers, such as head, neck, thyroid, lung, gastric, oesophageal, breast, ovarian, pancreatic, prostatic, and colorectal carcinomas11–19. Additionally, aberrant c-Met activation has been demonstrated to be associated with the acquired resistance of tumour cells during approved therapies13,20,21. As a result, c-Met has emerged as a promising target for cancer treatment.
In the past few years, different strategies have been pursued to inhibit abnormal c-Met signalling pathway, including HGF and c-Met biological antagonists22,23, antibodies against HGF or c-Met24,25, and small-molecule c-Met inhibitors26–28. Among them, ATP-competitive small-molecule c-Met kinase inhibitor has obtained remarkable achievements within the pharmaceutical industry, leading to the marketing of crizotinib (1) and cabozantinib (5), and dozens of candidates currently under clinical trials, such as capmatinib (2), volitinib (3), AMG 337 (4), foretinib (6), and BMS 777607 (7) (Figure 1)8. All these small-molecule c-Met inhibitors can basically be categorised into two classes (classes I and II) according to their structures and binding modes with c-Met. Class I c-Met inhibitors (1–4) bind to the entrance of ATP-binding site in a U-shaped conformation around Met1211, while class II inhibitors (5–7) bind to c-Met in an extended conformation that stretches from the ATP-binding site to the deep hydrophobic pocket (Figure 2).
Figure 1.

Structures of the representative small-molecule c-Met kinase inhibitors.
Figure 2.

(A) Binding mode of class I inhibitor crizotinib with c-Met. (B) Binding mode of class II inhibitor foretinib with c-Met.
In our previous studies, a series of N-(2-phenyl-1H-benzo[d]imidazol-5-yl)quinazolin-4-amine derivatives were discovered as potent c-Met inhibitors29. In an ongoing effort to pursue novel potent c-Met inhibitors, we modified the previous structures by replacing of the quinazolin fragment with the 6,7-dimethoxyquinolone moiety from cabozantinib (5) (Figure 3). All the target compounds were synthesised and evaluated for their inhibitory activities against c-Met kinase and antiproliferative activities against three human cancer cell lines including A549 (human lung cancer), MCF-7 (human breast cancer), and MKN-45 (human gastric cancer). In addition, the structure-activity relationships and docking studies are also presented in this paper.
Figure 3.

Design strategy for the target compounds.
2. Experimental
2.1. Chemistry
All reagents and solvents were purchased from commercial sources and used as received without further purification. Reactions were monitored by thin-layer chromatography (TLC) in silica gel and the TLC plates were visualised by exposure to ultraviolet light (254 and 365 nm). Compounds were purified using flash column chromatography over silica gel (200–300 mesh). Melting points (uncorrected) were determined on a RY-1 MP apparatus. 1H NMR spectra were recorded on a Bruker AV-300 spectrometer at room temperature with tetramethylsilane (TMS) as an internal standard. Chemical shifts were reported in ppm (δ). ESI-MS spectra were recorded on an Agilent/HP 1100 Series LC/MSD Trap SL mass spectrometer. High resolution mass spectrometry (HRMS) was obtained on a Q-tof high resolution mass spectrometer.
2.1.1. General procedure for the preparation of the 5-nitro-2-phenyl-1H-benzo[d]imidazole derivatives (10a-s)
As described in our previous study29, a mixture of 4-nitro-o-phenylenediamine (21 mmol) and substituted benzoic acid 9a–s (20 mmol) in PPA (40 ml) was stirred at 120–150 °C for 5 h. The reaction was quenched with water and the pH was adjusted to 6 with saturated NaOH. The filter cake was washed with water and recrystallized from ethyl acetate to give corresponding compounds 10a–s.
2.1.2. General procedure for the preparation of the 2-phenyl-1H-benzo[d]imidazol-5-amine derivatives (11a-s)
As described in our previous study29, two methods were applied to prepare the intermediates 11a–s.
Method A: A suspension of 10 (5.0 mmol, R=CH3, OCH3, CH2CH3, C(CH)3) and 10% Pd/C (0.13 g) in methanol (30 mL) was hydrogenated under normal pressure for 5 h at room temperature. Filtration and evaporation gave corresponding compound 11.
Method B: Compound 10 (5.0 mmol, R=F, Cl, Br, I) and iron (1.1 g, 20 mmol) were suspended in aqueous ethanol (120 mL, 70% v/v) containing acetic acid (2 mL, 30 mmol) and heated at reflux for 2 h. The reaction mixture was cooled to room temperature. Filtration and evaporation gave corresponding compound 11.
2.1.3. General procedure for the preparation of the 6,7-dimethoxy-N-(2-phenyl-1H-benzo[d]imidazol-6-yl)quinolin-4-amine derivatives (12a-s)
A mixture of 4-chloro-6,7-dimethoxyquinoline (5 mmol) and appropriate amount of substituted anilines (11a–s, 6 mmol) in isopropanol (40 mL) was stirred at reflux for 5 h. The reaction mixture was concentrated under reduced pressure and the solid residue was purified by column chromatography on silica gel, eluting with CH2Cl2/CH3OH (10/1) to furnish the target compounds 12a–s.
2.1.3.1. N-(2–(2-fluorophenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12a)
Yellow powder, yield: 54%, mp: 247–249 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 10.65 (s, 1H), 8.33–8.23 (m, 2H), 8.09 (s, 1H), 7.83 (d, J = 8.5 Hz, 1H), 7.73 (d, J = 1.8 Hz, 1H), 7.63–7.58 (m, 1H), 7.52–7.43 (m, 2H), 7.40 (d, J = 5.3 Hz, 2H), 7.32 (dd, J = 8.6, 2.0 Hz, 1H), 6.69 (d, J = 7.0 Hz, 1H), 4.01 (s, 3H), 3.99 (s, 3H). HRMS (ESI+): calcd C24H20FN4O2 (M + H+) = 415.1570, found = 415.1572.
2.1.3.2. N-(2–(3-fluorophenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12b)
Yellow powder, yield: 55%, mp: 286–288 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 11.54 (s, 1H), 8.85 (s, 1H), 8.33 (s, 1H), 8.13–8.07 (m, 4H), 7.81 (d, J = 9.1 Hz, 1H), 7.69 (s, 2H), 7.48 (d, J = 6.8 Hz, 1H), 7.34 (d, J = 4.3 Hz, 1H), 4.03 (s, 3H), 4.01 (s, 3H). HRMS (ESI+): calcd C24H20FN4O2 (M + H+) = 415.1570, found = 415.1576.
2.1.3.3. N-(2–(4-fluorophenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12c)
Yellow powder, yield: 60%, mp: 312–315 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 10.56 (s, 1H), 8.15–8.19 (m, 3H), 8.01 (s, 1H), 7.69 (d, J = 8.5 Hz, 1H), 7.59 (s, 1H), 7.38–7.28 (m, 3H), 7.19 (dd, J = 8.4, 2.0 Hz, 1H), 6.58 (d, J = 7.0 Hz, 1H), 3.91 (s, 3H), 3.89 (s, 3H). HRMS (ESI+): calcd C24H20FN4O2 (M + H+) = 415.1570, found = 415.1572.
2.1.3.4. N-(2–(2-chlorophenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12d)
Yellow powder, yield: 51%, mp: 287–290 °C. 1H-NMR (300 MHz; DMSO-d6): δ (ppm) 10.77 (s, 1H), 8.33 (d, J = 6.6 Hz, 1H), 8.15 (s, 1H), 7.97 (dd, J = 6.9, 2.2 Hz, 1H), 7.88 (d, J = 8.6 Hz, 1H), 7.79 (d, J = 1.7 Hz, 1H), 7.73 (d, J = 7.4 Hz, 1H), 7.61 (td, J = 7.1, 1.8 Hz, 2H), 7.42–7.39 (m, 2H), 6.71 (d, J = 6.9 Hz, 1H), 4.02 (s, 3H), 4.00 (s, 3H). HRMS (ESI+): calcd C24H20ClN4O2 (M + H+) = 431.1275, found = 431.1284.
2.1.3.5. N-(2–(3-chlorophenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12e)
Yellow powder, yield: 46%, mp: 250–252 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 10.67 (s, 1H), 8.35–8.28 (m, 2H), 8.19 (d, J = 6.7 Hz, 1H), 8.10 (s, 1H), 7.82 (d, J = 8.6 Hz, 1H), 7.73 (s, 1H), 7.63 (d, J = 5.3 Hz, 2H), 7.38 (s, 1H), 7.33 (d, J = 8.4 Hz, 1H), 6.69 (d, J = 7.0 Hz, 1H), 4.01 (s, 3H), 4.00 (s, 3H). HRMS (ESI+): calcd C24H20ClN4O2 (M + H+) = 431.1275, found = 431.1279.
2.1.3.6. N-(2–(4-chlorophenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12f)
Yellow powder, yield: 58%, mp: 330–332 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 13.89 (s, 1H), 10.67 (s, 1H), 8.33 (s, 1H), 8.23 (d, J = 8.3 Hz, 2H), 8.11 (s, 1H), 7.79 (d, J = 8.5 Hz, 1H), 7.68 (d, J = 8.9 Hz, 3H), 7.38 (s, 1H), 7.30 (d, J = 8.7 Hz, 1H), 6.68 (d, J = 7.0 Hz, 1H), 4.01 (s, 3H), 3.99 (s, 3H). HRMS (ESI+): calcd C24H20ClN4O2 (M + H+) = 431.1275, found = 431.1276.
2.1.3.7. N-(2–(4-bromophenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12g)
Yellow powder, yield: 64%, mp: 280–282 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 10.70 (s, 1H), 8.34 (d, J = 7.0 Hz, 1H), 8.20–8.14 (m, 3H), 7.82 (d, J = 8.2 Hz, 3H), 7.72 (s, 1H), 7.42 (d, J = 3.6 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H), 6.70 (d, J = 6.9 Hz, 1H), 4.03 (s, 3H), 4.01 (s, 3H). HRMS (ESI+): calcd C24H20BrN4O2 (M + H+) = 475.0770, found = 475.0773.
2.1.3.8. N-(2–(2-iodophenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12h)
Brown powder, yield: 53%, mp: 207–209 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 12.69 (s, 1H), 8.76 (s, 1H), 8.24 (s, 1H), 8.07 (d, J = 7.8 Hz, 1H), 7.73 (s, 1H), 7.66 (d, J = 6.6 Hz, 2H), 7.57 (t, J = 7.5 Hz, 2H), 7.24 (s, 3H), 6.77 (s, 1H), 3.95 (s, 3H), 3.91 (s, 3H). HRMS (ESI+): calcd C24H20IN4O2 (M + H+) = 523.0631, found = 523.0639.
2.1.3.9. N-(2–(3-iodophenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12i)
Brown powder, yield: 60%, mp: 201–203 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 13.76 (s, 1H), 10.60 (s, 1H), 8.58 (s, 1H), 8.33 (s, 1H), 8.23 (d, J = 8.0 Hz, 1H), 8.07 (s, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.80 (d, J = 8.4 Hz, 1H), 7.70 (s, 1H), 7.40–7.29 (m, 3H), 6.68 (d, J = 7.0 Hz, 1H), 4.00 (s, 3H), 4.00 (s, 3H). HRMS (ESI+): calcd C24H20IN4O2 (M + H+) = 523.0631, found = 523.0636.
2.1.3.10. 6,7-Dimethoxy-N-(2-(o-tolyl)-1H-benzo[d]imidazol-6-yl)quinolin-4-amine (12j)
Yellow powder, yield: 48%, mp: 174–175 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 10.87 (s, 1H), 8.37 (s, 1H), 8.21 (s, 1H), 7.91 (d, J = 8.6 Hz, 1H), 7.83 (d, J = 6.9 Hz, 2H), 7.55–7.43 (m, 5H), 6.75 (d, J = 6.9 Hz, 1H), 4.04 (s, 3H), 4.01 (s, 3H), 2.65 (s, 3H). HRMS (ESI+): calcd C25H23N4O2 (M + H+) = 411.1821, found = 411.1828.
2.1.3.11. 6,7-Dimethoxy-N-(2-(m-tolyl)-1H-benzo[d]imidazol-6-yl)quinolin-4-amine (12k)
Yellow powder, yield: 67%, mp: 339–342 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 13.91 (s, 1H), 10.66 (s, 1H), 8.35 (s, 1H), 8.08 (d, J = 4.5 Hz, 2H), 8.02 (d, J = 7.7 Hz, 1H), 7.82 (d, J = 8.5 Hz, 1H), 7.73 (s, 1H), 7.51 (t, J = 7.7 Hz, 1H), 7.42–7.34 (m, 3H), 6.70 (d, J = 7.0 Hz, 1H), 4.01 (s, 3H), 4.00 (s, 3H), 2.44 (s, 3H). HRMS (ESI+): calcd C25H23N4O2 (M + H+) = 411.1821, found = 411.1819.
2.1.3.12. 6,7-Dimethoxy-N-(2-(p-tolyl)-1H-benzo[d]imidazol-6-yl)quinolin-4-amine (12l)
Yellow powder, yield: 75%, mp: 316–319 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 10.70 (s, 1H), 8.35 (d, J = 6.2 Hz, 1H), 8.15–8.11 (m, 3H), 7.83 (d, J = 8.6 Hz, 1H), 7.74 (d, J = 1.9 Hz, 1H), 7.45 (d, J = 8.0 Hz, 2H), 7.39 (s, 1H), 7.36 (d, J = 8.8 Hz, 1H), 6.71 (d, J = 7.0 Hz, 1H), 4.01 (s, 3H), 3.99 (s, 3H), 2.41 (s, 3H). HRMS (ESI+): calcd C25H23N4O2 (M + H+) = 411.1821, found = 411.1827.
2.1.3.13. N-(2–(4-ethylphenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12m)
Yellow powder, yield: 42%, mp: 210–212 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 10.66 (s, 1H), 8.34 (s, 1H), 8.15 (d, J = 8.0 Hz, 2H), 8.10 (s, 1H), 7.82 (d, J = 8.5 Hz, 1H), 7.72 (s, 1H), 7.47 (d, J = 8.0 Hz, 2H), 7.38–7.36 (m, 2H), 6.70 (d, J = 7.0 Hz, 1H), 4.01 (s, 3H), 4.00 (s, 3H), 2.72 (q, J = 7.8 Hz, 2H), 1.25 (t, J = 7.6 Hz, 3H). HRMS (ESI+): calcd C26H25N4O2 (M + H+) = 425.1978, found = 425.1980.
2.1.3.14. N-(2–(4-(tert-butyl)phenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12n)
Yellow powder, yield: 38%, mp: 257–259 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 10.79 (s, 1H), 8.35 (s, 1H), 8.21 (d, J = 8.1 Hz, 2H), 8.16 (s, 1H), 7.86 (d, J = 8.6 Hz, 1H), 7.78 (s, 1H), 7.68 (d, J = 8.2 Hz, 2H), 7.42 (s, 2H), 6.73 (d, J = 7.0 Hz, 1H), 4.02 (s, 3H), 3.99 (s, 3H), 1.35 (s, 9H). HRMS (ESI+): calcd C28H29N4O2 (M + H+) = 453.2291, found = 453.2294.
2.1.3.15. 6,7-Dimethoxy-N-(2–(2-methoxyphenyl)-1H-benzo[d]imidazol-6-yl)quinolin-4-amine (12o)
Brown powder, yield: 74%, mp: 164–166 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 8.79–8.75 (m, 1H), 8.35–8.32 (m, 1H), 8.25 (s, 1H), 7.75 (s, 1H), 7.67 (d, J = 8.6 Hz, 1H), 7.58 (s, 1H), 7.47 (d, J = 8.6 Hz, 1H), 7.26 (d, J = 8.9 Hz, 2H), 7.19–7.10 (m, 1H), 6.80 (s, 1H), 4.04 (s, 3H), 3.95 (s, 3H), 3.91 (s, 3H). HRMS (ESI+): calcd C25H23N4O3 (M + H+) = 427.1770, found = 427.1776.
2.1.3.16. N-(2–(2,6-difluorophenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12p)
Yellow powder, yield: 55%, mp: 208–210 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 8.77 (s, 1H), 8.23 (d, J = 5.3 Hz, 1H), 7.74–7.62 (m, 3H), 7.54 (s, 1H), 7.34 (t, J = 8.4 Hz, 2H), 7.23 (d, J = 7.5 Hz, 2H), 6.72 (d, J = 5.4 Hz, 1H), 3.95 (s, 3H), 3.91 (s, 3H). HRMS (ESI+): calcd C24H19F2N4O2 (M + H+) = 433.1476, found = 433.1482.
2.1.3.17. N-(2–(2,6-dichlorophenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12q)
Yellow powder, yield: 57%, mp: 254–257 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 8.76 (s, 1H), 8.24 (d, J = 5.2 Hz, 1H), 7.72–7.52 (m, 6H), 7.24–7.21 (m, 2H), 6.73 (d, J = 5.3 Hz, 1H), 3.95 (s, 3H), 3.91 (s, 3H). HRMS (ESI+): calcd C24H19Cl2N4O2 (M + H+) = 465.0885, found = 465.0890.
2.1.3.18. N-(2–(3,4-dichlorophenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12r)
Yellow powder, yield: 52%, mp: 336–338 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 10.73 (s, 1H), 8.48 (d, J = 2.0 Hz, 1H), 8.33 (d, J = 6.5 Hz, 1H), 8.22 (dd, J = 8.4, 2.0 Hz, 1H), 8.13 (s, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.83 (d, J = 8.5 Hz, 1H), 7.75–7.69 (m, 1H), 7.42 (d, J = 3.9 Hz, 1H), 7.35 (dd, J = 8.5, 1.9 Hz, 1H), 6.69 (d, J = 7.0 Hz, 1H), 4.01 (s, 3H), 3.99 (s, 3H). HRMS (ESI+): calcd C24H19Cl2N4O2 (M + H+) = 465.0885, found = 465.0884.
2.1.3.19. N-(2–(2-bromo-5-fluorophenyl)-1H-benzo[d]imidazol-6-yl)-6,7-dimethoxyquinolin-4-amine (12s)
Brown powder, yield: 48%, mp: 284–286 °C. 1H-NMR (300 MHz; DMSO- d6): δ (ppm) 10.79 (s, 1H), 8.33 (t, J = 6.6 Hz, 1H), 8.18 (s, 1H), 7.94–7.85 (m, 2H), 7.78–7.73 (m, 2H), 7.48–7.37 (m, 3H), 6.70 (d, J = 7.0 Hz, 1H), 4.02 (s, 3H), 3.99 (s, 3H). HRMS (ESI+): calcd C24H19BrFN4O2 (M + H+) = 493.0675, found = 493.0685.
2.2. c-Met Inhibition assay
A Caliper motility shift assay was applied to test the potency of synthesised compounds against c-Met kinase. Briefly, the compounds were dissolved in DMSO (Sigma) at 0.5 mM concentration, and then were diluted 50× to the final desired highest inhibitor concentration (10 μM) in reaction by DMSO. For all compounds, 100 μL of the diluted compound solution in tubes was transferred to a well on a 96-well plate (Corning) and was serially diluted 10× by transferring 10 μL dilution to 90 μL DMSO in the next well. 100 μL DMSO was added to two empty wells in the same 96-well plate for no compound control and no enzyme control. The plate was marked as source plate. 10 μL of compound solution from source plate was transferred to a new 96-well plate as the intermediate plate. Then, 90 μL of 1× kinase base buffer (50 mM HEPES, pH 7.5, 0.0015% Brij-35, 10 mM MgCl2, 2 mM DTT) was added to each well of the intermediate plate and mixed for 10 min on a shaker. After that, 5 μL solution from each well of the intermediate plate was transferred to a 384-well plate (Corning) as the assay plate in duplicates.c-Met kinase (Carna) was added in 1× kinase base buffer to prepare 2.5× enzyme solution, and FAM-labled peptide (GL Biochem) and ATP (Sigma) were added in 1× kinase base buffer to prepare 2.5× peptide solution. 10 μL of 2.5× enzyme solution was added to each well of the 384-well assay plate that already contained 5 μL of compound in 10% DMSO, and then incubated at room temperature for 10 min. Subsequently, 10 μL of 2.5× peptide solution was added to each well and incubated at 28 °C for specified period of time. After that, 25 μL of stop buffer (100 mM HEPES, pH 7.5, 0.015% Brij-35, 0.2% Coating Reagent, 50 mM EDTA) was added to stop reaction. The data were collected on Caliper and the conversion data were copied from Caliper program.
Inhibition values were obtained according to the following formula: percent inhibition = (max-conversion)/(max-min) × 100, in which “max” stands for DMSO control while “min” stands for no enzyme activity control. IC50 values were calculated from three independent experiments by fitting the data in XLfit with the following equation: Y = Bottom + (Top-Bottom)/(1 + (IC50/X)^HillSlope).
2.3. Cell proliferation assay
The anti-proliferative activities of the prepared compounds (12a-12s) were evaluated against A549, MCF-7 and MKN-45 cell lines by MTT assay in vitro. Briefly, tumour cells were cultured to log phase in RPMI 1640 medium supplemented with 10% foetal bovine serum (FBS). After diluting to 2 × 104 cells mL−1 with the complete medium, 100 µL of the obtained cell suspension was added to each well of 96-well culture plates. Subsequently, incubation was performed at 37 °C in 5% CO2 for 24 h before the anti-proliferative assessment. Tested samples at pre-set concentrations were added to 6 wells with cabozantinib as a positive reference. After 48 h exposure period, 40 µL of PBS containing 2.5 mg mL−1 of MTT was added to each well. Four hours later, 100 µL extraction solution (10% SDS-5% isobutyl alcohol-0.010M HCl) was added. After an overnight incubation at 37°C, the absorbance was measured at a wavelength of 570 nm with an ELISA microplate reader. All compounds were tested three times in each tumour cell lines. The IC50 values were the averages of three determinations and calculated by concentration-response curve fitting method.
2.4. Molecular docking
Molecular docking of compounds 12a–s into the three dimensional X-ray structure of c-Met kinase (PDB code: 3CD8)30 was carried out using the Discovery Stutio (version 4.0)31 as implemented through the graphical user interface Discovery Studio CDOCKER protocol. The three-dimensional structures of the compounds 12a–s were constructed using ChemBio 3 D Ultra 14.0 software [Chemical Structure Drawing Standard; Cambridge Soft corporation, USA (2014)], then they were energetically minimised by using MMFF94. The crystal structures of c-Met kinase were retrieved from the RCSB Protein Data Bank (http://www.rcsb.org). All bound waters and ligands were eliminated from the protein and the polar hydrogen was added. The whole 3CD8 was defined as a receptor and the site sphere was selected based on ATP binding site of 3CD8. Compounds 12a–s were placed during the molecular docking procedure. Types of interactions of the docked protein with ligand were analysed after the end of molecular docking.
3. Results and discussion
3.1. Chemistry
The route employed to synthesise the title 6,7-dimethoxy-4-anilinoquinolones bearing benzimidazole moiety is outlined in Scheme 1, which was illustrated in detail in our previous study29. Briefly, the preparation of the target compounds started from a series of commercially available substituted benzoic acids 9a–s. Condensation of 9a–s with 4-nitrobenzene-1,2-diamine (8) in polyphosphoric acid (PPA) at 120–150 °C for 5 h gave the 5-nitro-2-substituted-phenyl-benzimidazole intermediates 10a–s. Hydrogenation of 5-nitro-2-substituted-phenyl-benzimidazole intermediates 10–s with Pd/C/H2 in methanol under normal pressure at room temperature for 5 h or Fe/AcOH in ethanol under reflux for 2 h provided the 5-amino–2-substituted-phenyl-benzimidazole intermediates 11a–s. Condensation of 11a–s with 4-chloro-6,7-dimethoxyquinoline in isopropanol under reflux for 5 h gave the target compounds 12a–s.
Scheme 1.

General procedure for the synthesis of 6,7-dimethoxy-4-anilinoquinolines. Reagents and conditions: (a) PPA, 120–150 °C, 5 h; (b) Pd/C, H2, CH3OH, r.t., 5 h; (c) Fe, AcOH, EtOH, reflux, 2 h; (d) 4-chloro-6,7-dimethoxyquinoline, isopropanol, reflux, 5 h.
3.2. Biological evaluation
3.2.1. In vitro c-Met kinase assay
All the synthesised 6,7-dimethoxy-N-(2-phenyl-1H-benzo[d]imidazol-6-yl)quinolin-4-amine derivatives 12a–s were evaluated for their inhibitory activity against c-Met kinase using Caliper motility shift assay. The results were summarised in Table 1 with cabozantinib as a positive control.
Table 1.
Chemical structures of target compounds and their c-Met inhibitory activities in vitro. 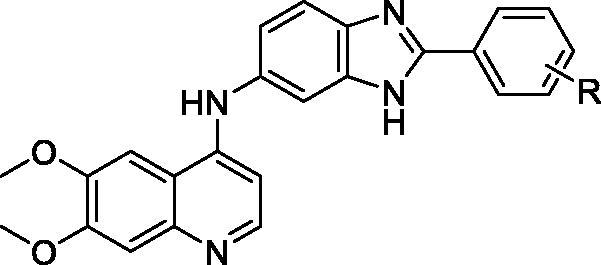
| Compound | R | c-Met inhibition |
|
|---|---|---|---|
| % Inhibition (10 μΜ)a | IC50 (μM)a | ||
| 12a | 2-F | 16.9 ± 3.5 | N.D.b |
| 12b | 3-F | 62.8 ± 1.2 | 1.2 ± 0.4 |
| 12c | 4-F | 89.2 ± 0.3 | 0.11 ± 0.02 |
| 12d | 2-Cl | 17.4 ± 3.7 | N.D. |
| 12e | 3-Cl | 54.5 ± 6.9 | 4.8 ± 0.5 |
| 12f | 4-Cl | 63.8 ± 0.7 | 0.93 ± 0.10 |
| 12g | 4-Br | 54.2 ± 0.1 | 5.5 ± 0.9 |
| 12h | 2-I | 21.6 ± 2.5 | N.D. |
| 12i | 3-I | 66.0 ± 0.8 | 0.54 ± 0.13 |
| 12j | 2-CH3 | 23.0 ± 0.4 | N.D. |
| 12k | 3-CH3 | 52.7 ± 1.6 | 8.5 ± 2.0 |
| 12l | 4-CH3 | 69.0 ± 1.2 | 0.32 ± 0.07 |
| 12m | 4-CH2CH3 | 95.0 ± 0.3 | 0.056 ± 0.012 |
| 12n | 4-C(CH3)3 | 97.1 ± 3.5 | 0.030 ± 0.008 |
| 12o | 2-OCH3 | 24.2 ± 0.1 | N.D. |
| 12p | 2,6-di-F | 25.9 ± 1.5 | N.D. |
| 12q | 2,6-di-Cl | 37.0 ± 4.0 | N.D. |
| 12r | 3,4-di-Cl | 55.4 ± 2.9 | 5.3 ± 1.6 |
| 12s | 2-Br-5-F | 23.0 ± 1.1 | N.D. |
| cabozantinibc | N.D. | 0.0045 ± 0.0006 | |
n = 3 (mean ± SD).
N.D.: not determined.
Used as a positive control.
As illustrated in Table 1, some of these novel 6,7-dimethoxy-4-anilinoquinolones bearing benzimidazole fragment were found to be active against c-Met kinase. Among them, compound 12n exhibited the most potent inhibitory activity against c-Met with IC50 value of 0.030 ± 0.008 μM.
Structure–activity relationships (SARs) were inferred from the data shown in Table 1. Various functional groups were introduced to the phenyl ring attached to the benzimidazole fragment, which played important roles in the optimisation of c-Met inhibitory potency.
Compounds 12a–c with fluoro substituent at different position of the phenyl ring showed distinct inhibitory activity against c-Met. Compound 12c with para-fluoro substituent showed 11-fold more potent activity (IC50 = 0.11 ± 0.02 μM) than compound 12 b with meta-fluoro substituent (IC50 = 1.2 ± 0.4 μM), whereas ortho- fluoro substitution (12a) led to diminished activity. The result suggested that substituent at different positions led to different inhibitory activities in the following order: para- > meta- > ortho-. This rule was also found in other single substituted compounds (12dvs12e and 12f, 12 hvs12i, 12jvs12k and 12l).
The inhibitory activities against c-Met of compounds with different para-substituents on the phenyl ring declined in the following order: compound 12n with para- tert-butyl substituent exhibited the most potent inhibitory activity against c-Met (IC50 = 0.030 ± 0.008 μM); compound 12 m with para-ethyl substituent displayed slightly weaker inhibitory activity (IC50 = 0.056 ± 0.012 μM); compound 12 l with para-methyl substituent showed much weaker inhibitory activity (IC50 = 0.32 ± 0.07 μM). Besides, compounds 12c, 12f and 12 g with different para-halogen substituents on the phenyl ring presented distinguishing inhibitory activities against c-Met: compound 12c with para-fluoro substituent displayed potent activity (IC50 = 0.11 ± 0.02 μM); compound 12f with para-chloro substituent exhibited moderate activity (IC50 = 0.93 ± 0.10 μM), which was about 9-fold lower than compound 12c; compound 12 g with para-bromo substituent showed mild activity (IC50 = 5.5 ± 0.9 μM), which was about 50-fold lower than compound 12c. The above results indicated that introduction of bulk lipophilic substituent is favourable for the activity, probably due to that it can form hydrophobic interaction with the active site of c-Met kinase.
Apart from single substituted compounds (12a–o), we also synthesised several compounds with double halogen substituents on the phenyl ring (12p–s). Compound 12r with two chloro substituents at para and meta positions displayed mild inhibitory activity against c-Met (IC50 = 5.3 ± 1.6 μM). However, as long as one ortho position of the phenyl ring was occupied by any halogen substituent (e.g. 12p, 12q and 12s), it resulted in the loss of activity. The above results also demonstrated that introduction of substituents at ortho position of the phenyl ring was unfavourable for the activity.
3.2.2. In vitro antiproliferation assay
All the synthesised compounds 12a–n were evaluated for their antiproliferation activities against A549 (human lung cancer), MCF-7 (human breast cancer) and MKN-45 (human gastric cancer) cell lines by MTT assay, using cabozantinib as the positive control. The results expressed as IC50 values were presented in Table 2. Among the tested compounds, compound 12n with the most potent c-Met inhibitory activity also displayed the most potent anticancer activities against A549, MCF-7 and MKN-45 with IC50 values of 7.3 ± 1.0 μM, 6.1 ± 0.6 μM, and 13.4 ± 0.5 μM, respectively, which were comparable to the reference drug cabozantinib. Basically, the SARs analysis result of antiproliferation activities of the tested compounds were consistent with that of their inhibitory activities against c-Met kinase, which suggested that the potent anticancer activities of the synthesised compounds were probably related to their c-Met inhibitory activities.
Table 2.
Chemical structures of target compounds and their antiproliferation activities against A549, MCF-7, and MKN-45 cell lines in vitro. 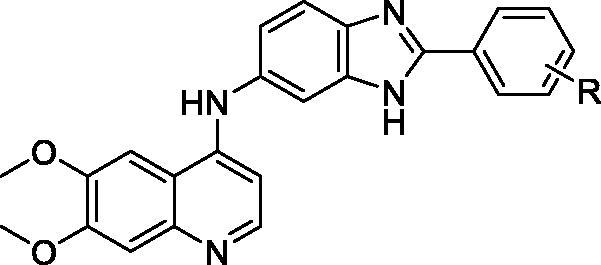
| Compound | R | Proliferative inhibition (IC50, µM)a |
||
|---|---|---|---|---|
| A549 | MCF-7 | MKN-45 | ||
| 12a | 2-F | >100 | >100 | >100 |
| 12b | 3-F | 45.0 ± 3.3 | 48.6 ± 5.2 | 57.5 ± 1.9 |
| 12c | 4-F | 15.6 ± 2.5 | 11.3 ± 1.6 | 21.0 ± 1.8 |
| 12d | 2-Cl | >100 | >100 | >100 |
| 12e | 3-Cl | 53.8 ± 5.0 | 63.5 ± 4.8 | 85.7 ± 3.7 |
| 12f | 4-Cl | 32.4 ± 3.6 | 29.2 ± 1.4 | 39.5 ± 2.5 |
| 12g | 4-Br | 38.5 ± 2.4 | 46.3 ± 3.7 | 55.0 ± 3.4 |
| 12h | 2-I | 92.4 ± 5.1 | >100 | >100 |
| 12i | 3-I | 23.8 ± 2.0 | 30.5 ± 1.6 | 36.1 ± 2.7 |
| 12j | 2-CH3 | >100 | >100 | >100 |
| 12k | 3-CH3 | 72.6 ± 6.5 | 78.3 ± 3.5 | 87.2 ± 6.7 |
| 12l | 4-CH3 | 27.3 ± 1.6 | 24.0 ± 0.9 | 31.8 ± 1.4 |
| 12m | 4-CH2CH3 | 12.5 ± 1.3 | 18.4 ± 2.1 | 14.9 ± 0.7 |
| 12n | 4-C(CH3)3 | 7.3 ± 1.0 | 6.1 ± 0.6 | 13.4 ± 0.5 |
| 12o | 2-OCH3 | >100 | >100 | >100 |
| 12p | 2,6-di-F | >100 | >100 | >100 |
| 12q | 2,6-di-Cl | >100 | >100 | >100 |
| 12r | 3,4-di-Cl | 83.6 ± 4.0 | 79.2 ± 1.7 | >100 |
| 12s | 2-Br-5-F | >100 | 92.7 ± 5.3 | >100 |
| cabozantinibb | 4.5 ± 0.8 | 7.2 ± 0.5 | 11.8 ± 1.4 | |
n = 3 (mean ± SD).
Used as a positive control.
3.3. Molecular docking studies
To further elucidate the interaction between the synthesised compounds and c-Met kinase, molecular docking of compounds 12a–s into the ATP binding site of c-Met kinase (PDB: 3CD8) was performed using the Discovery Studio 4.0/CDOCKER protocol.
The binding model of the most potent compound 12n and c-Met is depicted in Figures 4(A) and (B). Visual inspection of the pose of compound 12n into c-Met ATP-binding site revealed that compound 12n was tightly embedded into the binding pocket via three conventional hydrogen bonds, two π-π stacked interactions, three π-alkyl interactions one π-sulfur interaction, and many Van der Waals interactions. Specifically, the methoxyl oxygen atom at the quinoline forms a hydrogen bond (O…H-O: 2.70 Å, 117.11°) with the hydroxyl hydrogen atom of TYR1230. The phenyl ring of quinoline forms a π-π stacked interaction (distance: 4.04 Å) with the phenyl ring of TYR1230, a π-alkyl interaction with VAL1092 (distance: 5.39 Å), and a π-sulfur interaction with MET1211 (distance: 3.68 Å) as well. In addition, the pyridine ring of quinoline form another π-π hydrophobic interaction (distance: 4.83 Å) with the phenyl ring of TYR1230 and also forms a π-alkyl interaction with MET1211 (distance: 4.93 Å). These results indicated that the quinoline moiety plays an important role in the combination of the receptor and ligand. Besides, the amino hydrogen atom of benzimidazole forms a hydrogen bond (O…H-N: 2.46 Å, 136.32°) with the carbonyl oxygen atom of LYS1161. The phenyl ring of benzimidazole forms a π-alkyl interaction with ILE1084 (distance: 5.24 Å). These results suggested that the introduction of benzimidazole moiety may be favourable for the interaction between compound 12n and c-Met, which might be beneficial for the enhancement of the binding affinity, leading to the increased c-Met inhibition and consequent anticancer activity of compound 12n. Moreover, the hydrogen atom of the amino group which links the qunoline and benzimidazole moieties forms a hydrogen bond with the carbonyl oxygen atom of ILE1084 (O…H-N: 2.75 Å, 116.72°). Furthermore, it is noteworthy that the highly lipophilic tert-butyl group at the phenyl ring occupies the hydrophobic pocket of c-Met and forms hydrophobic interactions with the amino acid residues nearby, which may contribute to the potency increment of compound 12n against c-Met. The binding mode of compound 12 m with comparable activity to 12n was shown in Figures 5(A) and (B). Compound 12 m also formed various types of interactions with c-Met, which may contribute to its potency.
Figure 4.

(A) 2D molecular docking modelling of compound 12n with c-Met kinase. (B) 3D model of the interaction between compound 12n and c-Met ATP-binding site.
Figure 5.

(A) 2D molecular docking modelling of compound 12m with c-Met kinase. (B) 3D model of the interaction between compound 12m and c-Met ATP-binding site.
4. Conclusions
In summary, a series of 6,7-dimethoxy-4-anilinoquinolone derivatives bearing the benzimidazole scaffold have been designed and synthesised as potent inhibitors against c-Met kinase. Their antiproliferation activities against three human cancer cell lines (A549, MCF-7, and MKN-45) have also been evaluated. Compound 12n showed the most potent inhibitory activity against c-Met with IC50 value of 0.030 ± 0.008 μM, and exhibited the strongest inhibitory activities against A549, MCF-7 and MKN-45 cancer cell lines with IC50 values of 7.3 ± 1.0 μM, 6.1 ± 0.6 μM, and 13.4 ± 0.5 μM, respectively. Analysis of SARs indicated that introduction of bulk lipophilic substituent to the phenyl ring linking to the benzimidazole moiety is beneficial for the potency of compounds against c-Met. Molecular docking of the most potent inhibitor 12n into ATP-binding site of c-Met kinase was performed and the result demonstrated that compound 12n could bind tightly with the active site of c-Met by various interactions. All the above results indicated that compound 12n could be a potential anticancer agent and deserves further study.
Funding Statement
This work was supported by Natural Science Foundation of Jiangsu Province [Grant No. BK2012760], College Students Innovation Project for the R&D of Novel Drugs [Grant No. J1310032], National College Students' innovation and entrepreneurship training program [Grant No. 201810316057G], and Fundamental Research Funds for the Central Universities [Grant No. 2632018ZD09].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Bottaro DP, Rubin JS, Faletto DL, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991;251:802–4. [DOI] [PubMed] [Google Scholar]
- 2.Bertotti A, Comoglio PM. Tyrosine kinase signal specificity: lessons from the HGF receptor. Trends Biochem Sci 2003;28:527–33. [DOI] [PubMed] [Google Scholar]
- 3.Galimi F, Brizzi MF, Comoglio PM. The hepatocyte growth factor and its receptor. Stem Cells Suppl 1996;11:22–30. [DOI] [PubMed] [Google Scholar]
- 4.Ponzetto C, Bardelli A, Zhen Z, et al. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994;77:261–71. [DOI] [PubMed] [Google Scholar]
- 5.Furge KA, Zhang YW, Vande Woude GF. Met receptor tyrosine kinase: enhanced signaling through adapter proteins. Oncogene 2000;19:5582–9. [DOI] [PubMed] [Google Scholar]
- 6.Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat Rev Cancer 2002;2:289–300. [DOI] [PubMed] [Google Scholar]
- 7.Birchmeier C, Birchmeier W, Gherardi E, et al. Met, metastasis, motility and more. Nat Rev Mol Cell Biol 2003;4:915–25. [DOI] [PubMed] [Google Scholar]
- 8.Parikh PK, Ghate MD. Recent advances in the discovery of small molecule c-Met Kinase inhibitors. Eur J Med Chem 2018;143:1103–38. [DOI] [PubMed] [Google Scholar]
- 9.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol 2010;11:834–48. [DOI] [PubMed] [Google Scholar]
- 10.Chmielowiec J, Borowiak M, Morkel M, et al. c-Met is essential for wound healing in the skin. J Cell Biol 2007;177:151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim YC, Kang HJ, Moon JH. C-Met pathway promotes self-renewal and tumorigenecity of head and neck squamous cell carcinoma stem-like cell. Oral Oncol 2014;50:633–9. [DOI] [PubMed] [Google Scholar]
- 12.Ruco L, Scarpino S. The pathogenetic role of the HGF/c-Met system in papillary carcinoma of the thyroid. Biomedicines 2014;2:263–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA 2007;104:20932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houldsworth J, Cordon-Cardo C, Ladanyi M, et al. Gene amplification in gastric and esophageal adenocarcinomas. Cancer Res 1990;50:6417–22. [PubMed] [Google Scholar]
- 15.Gunasinghe NP, Wells A, Thompson EW, et al. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev 2012;31:469–78. [DOI] [PubMed] [Google Scholar]
- 16.Tang C, Jardim DL, Falchook GS, et al. MET nucleotide variations and amplification in advanced ovarian cancer: characteristics and outcomes with c-Met inhibitors. Oncoscience 2013;1:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delitto D, Vertes GE, Hughes SJ, et al. c-Met signaling in the development of tumorigenesis and chemoresistance: potential applications in pancreatic cancer. World J Gastroenterol 2014;20:8458–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Humphrey PA, Zhu X, Zarnegar R, et al. Hepatocyte growth factor and its receptor (c-MET) in prostatic carcinoma. Am J Pathol 1995;147:386–96. [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Yu XF, Zou J, et al. Prognostic value of c-Met in colorectal cancer: a meta-analysis. World J Gastroenterol 2015;21:3706–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engelman JA, Zejnullahu K, Mitsudomi T, et al. Met amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039–43. [DOI] [PubMed] [Google Scholar]
- 21.Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012;487:500–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan AM, Rubin JS, Bottaro DP, et al. Identification of a competitive HGF antagonist encoded by an alternative transcript. Science 1991;254:1382–5. [DOI] [PubMed] [Google Scholar]
- 23.Michieli P, Mazzone M, Basilico C, et al. Targeting the tumor and its microenvironment by a dual-function decoy Met receptor. Cancer Cell 2004;6:61–73. [DOI] [PubMed] [Google Scholar]
- 24.Burgess T, Coxon A, Meyer S, et al. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potentialagainst hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res 2006;66:1721–9. [DOI] [PubMed] [Google Scholar]
- 25.Martens T, Schmidt NO, Eckerich C, et al. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin Cancer Res 2006;12:6144–52. [DOI] [PubMed] [Google Scholar]
- 26.Ugolini M, Kenigsberg A, Rak F, et al. Discovery, pharmacokinetic and pharmacological properties of the potent and selective MET kinase inhibitor, 1-{6-[6-(4-Fluoro-phenyl)-[1,2,4]triazolo[4,3-b]pyridazin-3-ylsulfanyl]-benzothiazol-2-yl}-3-(2-morpholin-4-yl-ethyl)-urea (SAR125844). J Med Chem 2016;59:7066–74. [DOI] [PubMed] [Google Scholar]
- 27.Tang Q, Wang L, Duan Y, et al. Discovery of novel 7-azaindole derivatives bearing dihydropyridazine moiety as c-Met kinase inhibitors. Eur J Med Chem 2017;133:97–106. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Zhao J, Zhang B, et al. Discovery of [1,2,4]triazolo[3,4-b][1,3,4]thiadiazole derivatives as novel, potent and selective c-Met kinase inhibitors: Synthesis, SAR study, and biological activity. Eur J Med Chem 2018;150:809–16. [DOI] [PubMed] [Google Scholar]
- 29.Shi L, Wu TT, Wang Z, et al. Discovery of quinazolin-4-amines bearing benzimidazole fragments as dual inhibitors of c-Met and VEGFR-2. Bioorg Med Chem 2014;22:4735–44. [DOI] [PubMed] [Google Scholar]
- 30.Albrecht BK, Harmange JC, Bauer D, et al. Discovery and optimization of triazolopyridazines as potent and selective inhibitors of the c-Met kinase. J Med Chem 2008;51:2879–82. [DOI] [PubMed] [Google Scholar]
- 31.Accelrys Software Inc. Discovery Studio Visualizer, Release 4.0. Accelrys Software Inc., San Diego, CA, USA, 2013. [Google Scholar]