

Abstract
Background
Loss-of-function mutations in progranulin (GRN) cause frontotemporal dementia. Patients with GRN mutations present with a uniform subtype of TDP-43 pathology at autopsy (FTLD-TDP type A); however, age at onset and clinical presentation are variable, even within families. We aimed at identifying potential genetic factors modifying disease onset and disease risk in GRN mutation carriers.
Methods
In the discovery stage, genome-wide logistic and linear regression analyses were performed to test association of genetic variants with disease risk (case/control status) and age at onset. Suggestive loci (p<10−5) were genotyped in a replication cohort, followed by a meta-analysis. The effect of genome-wide significant variants at the novel GFRA2 locus on expression of GFRA2 was assessed using mRNA expression studies in cerebellar tissue samples from the Mayo Clinic brain bank. The effect of the GFRA2 locus on progranulin protein (PGRN) levels was studied using previously generated ELISA-based expression data. Co-immunoprecipitation experiments in HEK293T cells were performed to test for a direct interaction between GFRA2 and PGRN
Findings
Previously ascertained patients and controls were enrolled in the current study between October 2014 and October 2017. After quality control measures, statistical analyses in the discovery stage included 382 unrelated symptomatic GRN mutation carriers and 1,146 controls free of neurodegenerative disorders collected from 34 research centers located in North America, Australia and Europe. In the replication stage, 210 patients, including 67 symptomatic GRN mutation carriers and 143 pathologically-confirmed non-GRN FTLD-TDP type A patients, and 1,798 controls free of neurodegenerative diseases were recruited from 26 sites, of which 20 sites overlapped with the discovery stage. No genome-wide significant association with age at onset was identified in the discovery, replication or meta-analysis. However, in the case/control analysis, we replicated the previously reported TMEM106B association (meta-analysis:rs1990622, p=3·54×10−16, OR=0·54, 95% CI: 0·46 − 0·63), and identified a novel genome-wide significant locus at GFRA2 on chromosome 8p21.3 associated with disease risk (meta-analysis: rs36196656, p=1·58×10−8, OR=1·49, 95% CI: 1·30 − 1·71). Expression analyses showed that the risk-associated allele at rs36196656 decreased GFRA2 mRNA levels in cerebellar tissue. No effect of rs36196656 on plasma and cerebrospinal fluid PGRN levels was detected by ELISA; however, co-immunoprecipitation experiments in HEK cells did suggest a direct binding of PGRN and GFRA2.
Interpretation
The identification of TMEM106B and GFRA2 as potential modifiers of disease risk in GRN carriers raises the possibility that TMEM106B and GFRA2-related pathways are targets for therapies; yet, the biological interaction between PGRN and these disease modifiers requires further study. These potential genetic modifiers might also provide opportunities to select and stratify patients for future clinical trials and, when more is known about their potential effects, to inform genetic counselling,especially in the context of asymptomatic individuals.
INTRODUCTION
Frontotemporal lobar degeneration (FTLD) represents a collection of neurodegenerative diseases accounting for 5–10% of all dementia patients and 10–20% of patients with an onset of dementia before 65 years.1 Three clinical variants have been described: the behavioral variant of frontotemporal dementia (bvFTD), and two language variants of FTLD including the non-fluent and the semantic variant of primary progressive aphasia (PPA). The most common pathological subtype of FTLD is characterized by aggregates of the TAR DNA-binding protein 43, TDP-43 (FTLD-TDP).2,3 Four different FTLD-TDP pathological subtypes have been defined based on the morphology and anatomical distribution of the TDP-43 pathology (A to D).2
Mutations in progranulin (GRN) are the second most common genetic cause of FTLD-TDP, accounting for 5–20% of FTLD with positive family history.4–6 All currently known heterozygous pathogenic GRN mutations cause disease through a uniform disease mechanism, i.e. the loss of 50% functional progranulin protein (PGRN), leading to haploinsufficiency.4 Additionally, all patients with GRN mutations present with FTLD-TDP type A at autopsy.2 Despite this uniform disease mechanism and pathological presentation, clinical research has made clear that the age at symptom onset and clinical phenotype associated with GRN mutations are highly variable, even within the same family, and the penetrance of GRN mutations is not complete, even at old age.7,8 Importantly, a genome-wide association study performed in 2010 reported variants in the transmembrane protein 106 B locus (TMEM106B) as a risk factor for FTLD-TDP and subsequent studies established TMEM106B as a modifier of disease risk in individuals with GRN mutations.9–11 Identification of additional genetic modifiers of GRN-associated frontotemporal dementia could lead to improved genetic counselling, and could suggest potential new targets for disease-modifying therapies. We therefore aimed to identify additional genetic modifiers in GRN mutation carriers through genome-wide association analyses in the largest collection of unrelated symptomatic GRN patients ascertained to date.
METHODS
Participants
Participants for this study were all Caucasian and recruited at 40 international clinical and/or pathological research centers in Italy, US, France, Spain, UK, Canada, The Netherlands, Sweden, Australia, Denmark, Poland and Germany (appendix p.3, Supplementary Table 1). No restriction in terms of age, sex or race was applied to the initial selection; however statistical analysis only included white individuals (appendix p.3). Identification of GRN mutations, and assessment of TDP-43 pathological subtype, was performed at each individual site. For the discovery stage we obtained DNA from a total of 33 centers from 493 symptomatic GRN carriers from North America, Europe and Australia, and 505 controls from Italy and Spain (Table 1). We also obtained genetic data from 1,986 controls free from neurodegenerative diseases from the Genome-wide association study of Parkinson disease: Genes and Environment from the CIDR consortium (NCBI dbGaP phs000196.v3.p1 NINDS CIDR PD Environment; hereinafter referred to as CIDR dataset and considered one site, Table 1, appendix p.3, Supplementary Table 1 and Supplementary Figure 1). Additional and non-overlapping patients (n=210) and controls free from neurodegenerative diseases (n=1,798) for the replication stage were recruited from 26 centers, 20 overlapping with the discovery stage and 6 newly identified centers (Table 1; Supplementary Table 1). The 210 replication-stage cases included 67 patients with GRN mutations unrelated and independent from the discovery stage and 143 GRN-negative patients with pathologically confirmed FTLD-TDP type A.
Table 1. Demographics of patients and controls included in the study.
The median age at onset, age at death and age at last healthy visit of patients and controls included in the discovery and replication stage are presented. N=number of individuals; IQR=interquartile range. NA=not applicable.
| Discovery | Replication | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Group | Age at onset (IQR) |
Age at death (IQR) |
Age at last healthy visit (IQR) |
% female (N) |
Total | Age at onset (IQR) | Age at death (IQR) |
Age at last healthy visit (IQR) |
% female (N) |
Total |
|
GRN
mutation Carriers |
60.0(55.0 – 66.0) | 66.0(61.0 – 73 0) | NA | 55 2%(211) | 382 | 59.0(55.0 – 65 0) | 65.0(60.8 – 71.0) | NA | 52 2%(35) | 67 |
| Controls | NA | NA | 62.0(56 0 – 67 0) | 55.0%(630) | 1146 | NA | 77.0(64 0 – 81 0) | 62.0(53.0 – 71.0) | 47 5%(853) | 1798 |
|
GRN-negative FTLD-TDP >Type A |
NA | NA | NA | NA | NA | 70.0(62.0 – 76 8) | 79.0(68.0 – 85 0) | NA | 42 7%(61) | 143 |
Age at onset was defined as the age at which first disease symptoms appeared, including initial cognitive dysfunction in judgment, language, memory, or changes in behavior or personality.
Informed consent for genetic studies was given by patients and controls during life, or by next of kin at time of death for autopsy material, with approval of each institution’s Institutional Review Board.
Procedures and statistical analysis
Genotyping and quality control (QC) procedures for the discovery stage are described in detail in appendix (p.3–4). Genome-wide association analyses, using logistic and linear regressions, were performed to test the association of genetic variants with patient/control status (disease risk) and age at onset, respectively, under an additive model for allele effects and adjusting for age, sex, and the first two principal components of genetic variation (PCs) when appropriate (appendix p.4). As exploratory analyses, association of variants with absence or presence of specific first clinical symptoms (memory, behavior or language impairment) or presence of parkinsonism at any time during the course of the disease was tested among patients using logistic regression adjusting for age, sex, and first two PCs (appendix p.4; Supplementary Results). Association of previously reported putative genetic modifier variants in known neurodegenerative diseases genes with disease presentation and age at onset were also determined and reported.
Lead variants or a proxy associated at p<10−5 with disease risk or age at onset in the discovery stage were selected for the replication stage. Genotyping and quality control measures for this stage are described in detail in appendix (p.4–5). Association analyses were performed using logistic or linear regressions to replicate association of genetic variants suggestively associated with disease risk or age at onset, adjusting for age and sex when appropriate under an additive model. Thirty-six variants at 34 loci were analyzed in the replication stage, and thus a Bonferroni-corrected significance threshold of p<1–5×10−3 was employed in this stage. Meta-analyses of the discovery and replication results were performed under a fixed effects model. We also calculated I2 heterogeneity statistics to evaluate the degree of heterogeneity of the effects in the discovery and replication stages, and for SNPs with I2 suggesting moderate or high heterogeneity (I2 >0.3) we also performed a random effects meta-analysis, to verify that conclusions regarding association would not change under this model. Using the discovery data, a test of interaction was performed for the genome-wide significant loci found to modify disease risk in GRN mutation carriers. Specifically, using the top variants from the TMEM106B and GFRA2, a logistic regression model was fit with both variant genotypes and their multiplicative effect as predictors of risk, and a likelihood ratio test of the multiplicative term was performed to assess the effect of the variant interaction on disease risk.
To determine the effect of the lead variant at the GFRA2 locus (rs36196656) on brain GFRA2 mRNA expression levels, quantitative real-time PCR was performed in cerebellar tissue samples of AA and CC carriers (appendix p.5). Effect of rs36196656 on progranulin protein (PGRN) levels in plasma and cerebrospinal fluid (CSF) was assessed by Taqman genotyping of 345 individuals for which levels of PGRN were previously determined by ELISA12, using linear regression adjusting for age and sex. Whole-genome sequence data from 959 control individuals from the Mayo Clinic biobank was used to estimate linkage disequilibrium measures (D’ and r2) between all variants at the GFRA2 locus and rs36196656.
To study the direct interaction between PGRN and GFRA2, HEK293T cells were co-transfected with GFRA2 and PGRN. Cell lysates were collected and subjected to immunoprecipitation (appendix p.6).
RESULTS
In the discovery stage of our study we obtained DNA samples from 493 patients carrying 120 different loss-of-function mutations in GRN (appendix p.3; Supplementary Table 2). Three mutations were identified in more than 20 patients: p.Thr272Serfs*10 (n=97), p.Arg493* (n=35) and c.709–1G> (n=31). Patients had a median age at onset of 60 0 years (interquartile range, IQR 55 0 – 66 0) and 55 2% (n=211) were female (Table 1). Large variability in the age at onset was detected even among patients with the same mutation. Indeed, among patients with the most frequent mutation p.Thr272Serfs*10, ages at onset ranged from 39 to 82 years with a median age at onset at 62 0 years (IQR 56 0 – 66 0). To identify genetic modifiers of disease risk and disease onset in this unique cohort of patients with GRN mutations, we performed a two-stage genome-wide association study. After QC, the discovery stage included 382 unrelated symptomatic GRN mutation carriers and 1,146 unrelated controls. Genome-wide logistic regression analysis identified an expected highly significant association with variants at the GRN locus on chr17q21 (Figure 1). Haplotype analyses using 16 variants around GRN showed that this association was driven by distantly related individuals sharing founder haplotypes corresponding to the most common mutations in our cohort. We estimated the presence of a shared haplotype in 100% (n=22) of patients carrying the p.709–1G>A mutation and in 63 (80 8%) of patients carrying the p.Thr272Serfs*10 mutation, whereas 18 patients with p.Arg493* (60 0%) were estimated to carry one of two founder haplotypes. We also detected the known TMEM106B locus including 93 variants with genome-wide significant association and in strong linkage disequilibrium (LD; D’>0 8, r2>0 6) with the lead variant rs7791726 (p=1–53*10−10 ,OR=0 53, 95% confidence interval CI: 0 44–0 64; Figure 1; Supplementary Figures 2 and 3). In particular, the lead variant rs7791726 is in strong LD with the previously reported TMEM106B variants rs1990622, rs3173615 and rs1990620 (D’=1, r2>0 8). No additional genome-wide significant association signals were detected throughout the genome; however, 29 additional loci showed suggestive association at p<10−5 (Figure 1, Supplementary Table 3). After adjustment with the lead variant on chr17q21 (rs141568868), these suggestive associations did not change substantially suggesting that they are independent events from the chr17q21 locus. In a separate analysis, genome-wide linear regression analysis of onset age within the patient cohort did not identify any genome-wide significant association signals; however, 14 loci showed suggestive association (p<10−5) (Figure 1, Table 3; Supplementary Figure 3 and Supplementary Table 4). Since only the wild-type copy of GRN is expressed in patients with GRN mutations, we analyzed the effect of rs5848 located in the 3’UTR of GRN comparing patients homozygous for the common (C) and rare (T) alleles at this marker; however, no significant association with onset age was observed (p=0 36).
Figure 1: Manhattan plots of the case/control and age at onset analyses.
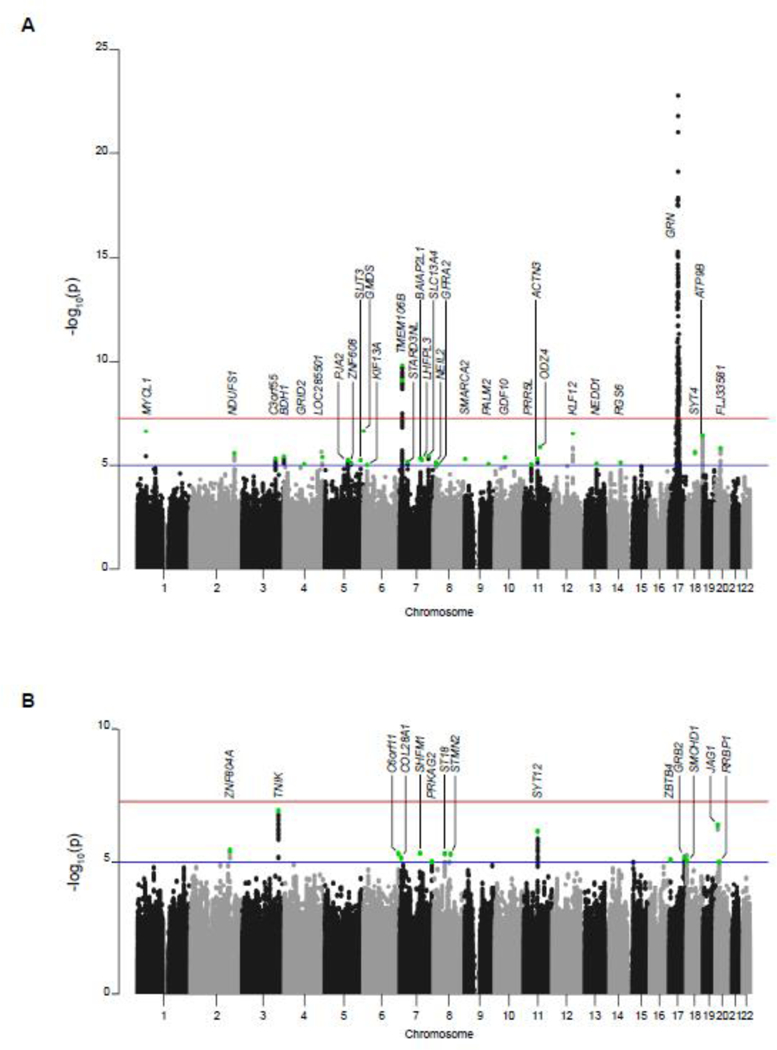
Negative log10- transformed p-values are shown for each variant genotyped on the y axis in function of the chromosomal position on the x axis. The red line represents the genome-wide significant threshold (p=5×10−8). The blue line denotes suggestive associations with p<10-5. Green dots represent the variants that were included in the design for follow-up in the replication stage. (A) case/controls analysis. (B) Age at onset analysis. Please note that at some loci a proxy of the top variant was selected for genotyping in the replication stage.
Table 3. Loci identified in age at onset analysis.
Suggestive variants identified in the discovery stage (p<10−5) and followed-up in the replication stage, as well as the meta-analyses are represented. Variant p-values and beta values were calculated using an additive genetic model. Minor alleles were treated as effect alleles. The locus name is determined by the closest gene to the significant variant. MAF=minor allele frequency; P=p-value.
| Discovery | Replication | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant | Positiona | Major/ minor allele |
Locus name |
MAF patients/controls |
Association | MAF patients/controls |
Association | Meta-analysisb | ||||
| Beta (95%CI) |
P | Beta (95%CI) |
P | Beta (95%CI) |
P | I2 | ||||||
| rs116316277 | 2:185834886 | C/T | ZNF804A | 0·03 | 8·09(4·72 − 11·6) | 3·58×10−6 | 0·08 | −1·03(-6·79 − 4·72) | 7·26 × 10−1 | 5·76(2·85 − 8·67) | 1·04×10−4 | 86·1 |
| rs6809184 | 3:170888198 | C/T | TNIK | 0·05 | −6·78(−9·24 – −4·32) | 1·22×10−7 | 0·09 | −0·54(−5·29 − 4·21) | 8·24 × 10−1 | −5·46(−7·64 – −3·27) | 1·01×10−6 | 80·9 |
| rs12189587 | 6:165332257 | C/T | C6orf11 | 0·11 | −4·05(−5·76 – −2·34) | 4·83×10−6 | 0·13 | 1·4(−2·32 − 5·11) | 4·62 × 10−1 | −3·1(−4·65 – −1·54) | 9·44×10−5 | 85·3 |
| rs6962939 | 7:7524226 | T/A | COL28A1 | 0·04 | −6·02(−8·61 – −3·43) | 7·00×10−6 | 0·06 | −6·15(−12·56 − 0·25) | 6·13 × 10−2 | −6·04(−8·43 – −3·64) | 8·18×10−7 | 0 |
| rs2922921 | 7:96398079 | G/A | SHFM1 | 0·02 | 9·65(5·58 − 13·72) | 4·65×10−6 | 0·06 | −0·54(−7·77 − 6·7) | 8·84 × 10−1 | 7·20(3·65 − 10·75) | 6·93×10−5 | 82·7 |
| rs77466830 | 7:151529171 | C/A | PRKAG2 | 0·32 | 2·91(1·64 − 4·18) | 9·49×10−6 | 0·43 | −0·13(−2·36 − 2·11) | 9·12 × 10−1 | 2·17(1·06 − 3·27) | 1·18×10−4 | 81·3 |
| rs9792144 | 8:53081551 | C/G | ST18 | 0·12 | 3·99(2·30 − 5·68) | 4·88×10−6 | 0·18 | 2·99(−0·26 − 6·24) | 7·28 × 10−2 | 3·78(2·28 − 5·28) | 7·55×10−7 | 0 |
| rs3922636 | 8:80383502 | G/A | STMN2 | 0·19 | 3·28(1·89 − 4·67) | 5·08×10−6 | 0·31 | 0·49(−2·23 − 3·21) | 7·23 × 10−1 | 2·70(1·47 − 3·94) | 1·83×10−5 | 68·4 |
| rs12943707 | 17:73317510 | C/G | GRB2 | 0·29 | −2·8(−4·00 – −1·6) | 6·40×10−6 | 0·4 | −0·41(−2·80 − 1·98) | 7·38 × 10−1 | −2·32(−3·39 – −1·25) | 2·22×10−5 | 67·5 |
| rs1561819 | 18:2712629 | G/A | SMCHD1 | 0·49 | −2·41(−3·46 – −1·36) | 8·96×10−6 | 0·51 | −0·81(−2·97 − 1·35) | 4·61 × 10−1 | −2·11(−3·05 – −1·16) | 1·23×10−5 | 41·5 |
| rs6108746 | 20:10902771 | T/C | JAG1 | 0·19 | 3·54(2·19 − 4·89) | 4·23×10−7 | 0·25 | 1·69(−1·11 − 4·48) | 2·38 × 10−1 | 3·19(1·98 − 4·41) | 2·59×10−7 | 27·4 |
| rs6111609 | 20:17664546 | C/A | RRBP1 | 0·22 | 2·86(1·61 − 4·11) | 9·83×10−6 | 0·22 | 2·88(−0·03 − 5·8) | 5·41 × 10−2 | 2·86(1·71 − 4·01) | 1·05×10−6 | 0 |
Positions are based on the Human Genome version 38 (hg38).
For SNPs with I2 >0.3, a random effects meta-analysis was also performed. The p-values were generally larger in the random effects meta-analysis, and the results were consistent with the fixed effects, showing that none of these SNPs were significantly associated with the outcome.
The replication stage of the association study, which included 210 patients (67 symptomatic GRN mutation carriers and 143 patients with pathologically confirmed FTLD-TDP type A without known mutations) and 1,798 controls (Table 1), identified significant association at the Bonferroni-corrected level of p<1 5×10−3 for two loci nominated by the case-control discovery GWAS (Table 2). None of the loci nominated through the discovery GWAS of age at disease onset withstood Bonferroni correction (Table 3). The strongest signal in the case-control analysis was at the TMEM106B locus with marker rs3173615 (p=8 97×10−8, OR=0 53, 95% CI: 0 47 – 0 63). The lead variant at the second locus was rs36196656 located within intron 3 of the gene encoding GDNF family receptor alpha 2 (GFRA2; MAFpatients=0 44, MAFcontrols=0 35 p=4 35×10−4, OR=1 46, 95% CI: 118 – 1 80). In the meta-analysis of discovery and replication stages, both the TMEM106B and GFRA2 loci reached genome-wide significance (TMEM106B, rs3173615, p=3 78×10−16, OR=0 54, 95% CI: 0 47 – 0 63; GFRA2, p=158×10−8, rs36196656, OR=1 49, 95% CI: 1 30–1 71, Table 2). No other loci showed p<5×10−8 in the meta-analysis. Conditional analysis adjusted for the TMEM106B variant rs3173615 in the discovery stage had no effect on the association at the GFRA2 variant rs36196656 (p=5 80×10−6, OR=1 54, 95% CI: 1–28-1 85). Moreover, tests of interactions between these variants provided no evidence for interaction effects on disease risk (interaction p>0 1), indicating that the effect of the GFRA2 variant on disease risk is not modified by the TMEM106B genotype that a person carries, and vice versa. These results suggest that the associations at TMEM106B and GFRA2 are independent.
Table 2. Loci identified in age at onset analysis.
Suggestive variants identified in the discovery stage (p<10−5) and followed-up in the replication stage, as well as the meta-analyses are represented. Variant p-values and beta values were calculated using an additive genetic model. Minor alleles were treated as effect alleles. The locus name is determined by the closest gene to the significant variant. MAF=minor allele frequency; P=p-value.
| Discovery | Replication | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant | Positiona | Major/ minor allele |
Locus name |
MAF patients/controls |
Association | MAF patients/controls |
Association | Meta-analysisb | ||||
| Beta (95%CI) |
P | Beta (95%CI) |
P | Beta (95%CI) |
P | I2 | ||||||
| rs13393316 | 2:206999339 | A/G | NDUFS1 | 0·10/0·16 | 0·50(0·38 − 0·67) | 2·65×10-6 | 0·12/0·14 | 0·81(0·59 − 1·13) | 2·14×10-1 | 0·62(0·50 − 0·77) | 1·34×10−10 | 78·5 |
| rs4680382 | 3:157324261 | G/A | C3orf55 | 0·59/0·32 | 1·5(1·26 − 1·78) | 4·75×10−10 | 0·35/0·35 | 1·00(0·80 − 1·24) | 9·86×10−10 | 1·28(1·12 − 1·47) | 3·46×10−10 | 87·7 |
| rs13072484 | 3:197136822 | G/A | BDH1 | 0·29/0·21 | 1·54(1·28 − 1·85) | 3·79×10−10 | 0·23/0·22 | 1·03(0·81 − 1·32) | 7·98×10−10 | 1·34(1·15 − 1·55) | 1·08×10−10 | 84·5 |
| rs79095029 | 5:108855306 | C/G | PJA2 | 0·03/0·08 | 0·35(0·23 − 0·55) | 5·72×10−10 | 0·07/0·08 | 0·99(0·66 − 1·49) | 9·64×10−10 | 0·62(0·46 − 0·84) | 2·03×10−10 | 91·0 |
| rs146261599 | 5:123600139 | T/G | ZNF608 | 0·05/0·02 | 2·91(1·82 − 4·64) | 7·64×10−10 | 0·02/0·03 | 1·09(0·54 − 2·17) | 8·14×10−10 | 2·13(1·45 − 3·15) | 1·24×10−10 | 81·3 |
| rs181675566 | 5:168651912 | T/C | SLIT3 | 0·04/0·01 | 3·86(2·15 − 6·90) | 5·72×10−10 | 0·01/0·02 | 0·60(0·24 − 1·53) | 2·88×10−10 | 2·29(1·40 − 3·76) | 1·02×10−10 | 90·8 |
| rs6904835c | 6:17810195 | T/C | KIF13A | 0·32/0·24 | 1·50(1·25 − 1·80) | 9·67×10−10 | 0·29/0·27 | 1·08(0·86 − 1·36) | 5·08×10−10 | 1·32(1·15 − 1·53) | 9·94×10−10 | 79·5 |
| rs3173615cd | 7:12269417 | C/G | TMEM106B | 0·27/0·39 | 0·55(0·45 − 0·66) | 7·81×10−10 | 0·27/0·42 | 0·53(0·42 − 0·67) | 8·97×10−10 | 0·54(0·47 − 0·63) | 3·78×10−10 | 0 |
| rs7791726cd | 7:12283329 | G/C | TMEM106B | 0·26/0·39 | 0·53(0·44 − 0·64) | 1·53×10−10 | 0·28/0·42 | 0·55(0·44 − 0·70) | 4·71×10−10 | 0·54(0·46 − 0·63) | 3·80×10−10 | 0 |
| rs1990622cd | 7:12283787 | A/G | TMEM106B | 0·26/0·39 | 0·53(0·44 − 0·65) | 1·61×10−10 | 0·28/0·42 | 0·55(0·44 − 0·70) | 4·09×10−10 | 0·54(0·46 − 0·63) | 3·54×10−10 | 0 |
| rs62443267 | 7:38153313 | C/T | STARD3NL | 0·19/0·19 | 0·62(0·50 − 0·76) | 6·83×10−10 | 0·25/0·25 | 0·93(0·73 − 1·20) | 5·91×10−10 | 0·74(0·63 − 0·86) | 1·64×10−10 | 84·1 |
| rs141226303 | 7:104251213 | A/G | LHFPL3 | 0·04/0·01 | 3·73 (2·11 − 6·59) | 5·61×10−10 | 0·02/0·01 | 1·06(0·47 − 2·38) | 8·92×10−10 | 2·46(1·55 − 3·93) | 1·47×10−10 | 83·9 |
| rs3110811c | 7:135402648 | A/G | SLC13A4 | 0·29/0·20 | 1·55(1·29 − 1·87) | 3·50×10−10 | 0·21/0·23 | 0·82(0·64 − 1·06) | 1·37×10−10 | 1·25(1·07 − 1·45) | 3·91×10−10 | 93·5 |
| rs10101195c | 8:11623212 | C/A | NEIL2 | 0·18/0·26 | 0·62(0·51 − 0·77) | 7·50×10−10 | 0·20/0·23 | 0·79(0·61 − 1· 02) | 7·06×10−10 | 0·68(0·58 − 0·80) | 3·71×10−10 | 47·9 |
| rs36196656cd | 8:21621247 | C/A | GFRA2 | 0·46/0·37 | 1·51(1·26 − 1·82) | 9·44×10−10 | 0·44/0·35 | 1·46(1·18 − 1·80) | 4·35×10−10 | 1·49 (1·30 − 1·71) | 1·58×10−10 | 0 |
| rs10816848 | 9:112421435 | T/A | PALM2 | 0·42/0·49 | 0·68(0·57 − 0·80) | 8·74×10−10 | 0·46/0·49 | 0·87(0·70 − 1·07) | 1·90×10−10 | 0·69(0·65 − 0·85) | 6·37×10−10 | 69·4 |
| rs78781776 | 11:36466533 | A/G | PRR5L | 0·10/0·05 | 2(1·47 − 2·72) | 8·88×10−06 | 0·09/0·07 | 1·27(0·86 − 1·86) | 2·28×10−10 | 1·68 (1·32 − 2·13) | 2·37×10−10 | 69·8 |
| rs10791882c | 11:66319313 | G/A | ACTN3 | 0·46/0·37 | 1·49(1·26 − 1·77) | 5·01×10−06 | 0·40/0·39 | 1·06(0·85 − 1·31) | 6·17×10−10 | 1·30 (1·14 − 1·49) | 1·06×10−10 | 83·5 |
| rs10860097 | 12:97199656 | A/T | NEDD1 | 0·05/0·02 | 3·43(2·14 − 5·50) | 2·88×10−07 | 0·03/0·02 | 1·29(0·70 − 2·37) | 4·12×10−10 | 2·38(1·64 − 3·45) | 5·15×10−10 | 83·9 |
| rs61965655 | 13:74712915 | T/A | KLF12 | 0·07/0·04 | 2·33(1·61 − 3·39) | 8·52×10−06 | 0·04/0·05 | 0·91(0·55 − 1·52) | 7·31×10−10 | 1·71(1·24 − 2·27) | 4·17×10−10 | 88·2 |
| rs847358 | 14:72780521 | G/A | RGS6 | 0·53/0·44 | 1·46(1·24 − 1·73) | 7·43×10−06 | 0·45/0·46 | 0·97(0·79 − 1·20) | 7·76×10−10 | 1·25(1·10 − 1·42) | 8·39×10−10 | 88·9 |
| rs12605286 | 18:41150167 | G/A | SYT4 | 0·23/0·31 | 0·6(0·48 − 0·74) | 2·37×10−06 | 0·29/0·26 | 1·20(0·95 − 1·51) | 1·28×10−10 | 0·82(0·70 − 0·96) | 1·41×10−10 | 94·6 |
| rs7240419c | 18:76928989 | G/A | ATP9B | 0·31/0·22 | 1·62(1·34 − 1·94) | 3·80×10−07 | 0·25/0·23 | 1·10(0·86 − 1·41) | 4·37×10−10 | 1·41 (1·21 − 1·63) | 5·96×10−10 | 83·0 |
| rs6076187 | 20:24082578 | G/A | FLJ33581 | 0·07/0·03 | 2·47 (1·71 − 3·57) | 1·53×10−06 | 0·04/0·04 | 0·87(0·50 − 1·53) | 6·39×10−10 | 1·80(1·32 − 2·45) | 1·74×10−10 | 89·2 |
Positions are based on the Human Genome version 38 (hg38).
At the 4 SNPs for which association was replicated, the I2 heterogeneity statistic is 0, showing no heterogeneity of effects between the two stages, and suggesting that a fixed effects metaanalysis is appropriate. For SNPs with I2>0.3, a random effects meta-analysis was also performed. The p-values were generally larger in the random effects meta-analysis, and the results were consistent with the fixed effects, showing that none of these SNPs were significantly associated with the outcome.
Variants annotated as eQTL in the GTex database
Variants that are study-wide significant at the replication stage after Bonferroni correction.
At the putative novel GFRA2 locus both patients with GRN mutations and FTLD-TDP type A without known mutations contributed to the observed association in the replication stage (Supplementary Results). While more significant association was detected when only GRN patients were included (p=3 11×10−3, OR=1 69, 95% CI: 1–19-2–40; Supplementary Table 5), the FTLD-TDP type A patients showed a comparable allele frequency and odds ratio at rs36196656 (p=108×10−2, OR 1 40, 95% CI: 1 08–1 82; Supplementary Table 6).
To identify possible functional variants at the newly identified putative GFRA2 locus, we queried publicly available data and whole-genome sequence data from 959 control individuals from the Mayo Clinic biobank which showed two single nucleotide polymorphisms (SNP, rs144692383 and rs150047054) and a 3-bp deletion (rs36144451) in strong linkage disequilibrium (r2>0 8) with the lead variant rs36196656 (Figure 2A, Supplementary Table 7). All four variants are located in close proximity within GFRA2 intronic regions: intron 3 of GFRA2 transcript variant A (NM_001495), intron 2 of GFRA2 transcript variant B (NM_001165038) and intron 1 of GFRA2 transcript variant C (NM_001165039) depending on alternative splicing at the GFRA2 locus (Figure 2A). Several of these variants are predicted to affect transcription factor binding sites and histone marks and they all are expression quantitative loci (eQTL) for GFRA2 in testis (p=1 80×10-14; www.gtexportal.org ). Indeed, GFRA2 RNA expression analyses in cerebellar tissue samples from individuals with rs36196656 ‘CC’ (n=24) and ‘AA’ (n=24) genotypes available from the Mayo Clinic brain bank showed substantial variability in expression among individuals but confirmed a 40% reduction in all GFRA2 transcripts in brains of homozygous carriers of the risk allele (AA) compared to CC carriers, which reached significance when analyzing all GFRA2 variants (p=0 04) or variant A individually (p=0 01) (Figure 2B). GFRA2 transcript variant A was consistently the predominant transcript expressed in cerebellum (Supplementary Figure 4A, B) and no significant difference in the ratio of GFRA2 transcripts (A, B, and C) was observed between AA and CC carriers (data not shown). Since the potential functional variant(s) underlying the observed association could also be less frequent than the lead variant, we further identified all variants with D’>0 8, which resulted in an additional 130 single nucleotide variants, none of which were coding (data not shown).
Figure 2: GFRA2 genetic locus and expression studies.
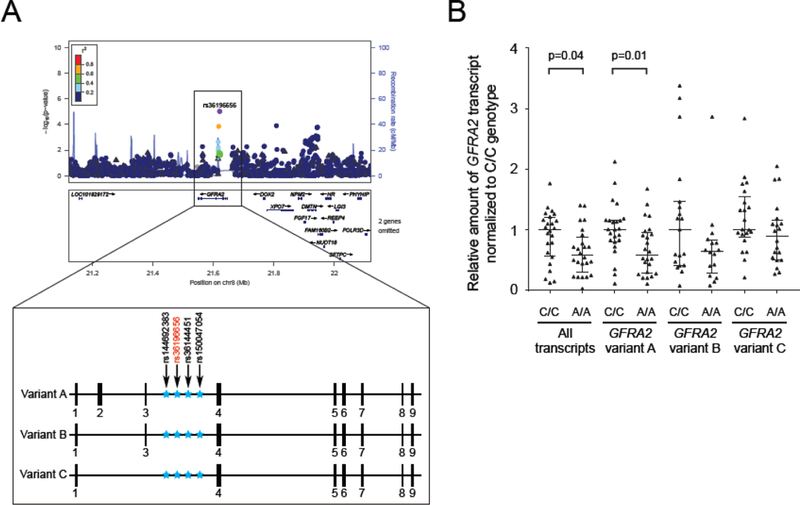
(A) The GFRA2 locus zoom plot is presented on the top panel . Each dot represents a genotyped (triangle) or imputed (circle) variant. The purple dot is the most significant variant (rs36196656) among variants in the region. Dots are colored from red to blue according to the r2 showing their degree of linkage disequilibrium with rs36196656 (grey color indicates an r2 of zero). The blue line shows the estimated recombination rate. The bottom panel presents the GFRA2 gene and its three GFRA2 transcripts. Exons are represented as small black boxes and non-coding regions as straight line. The location of three variants in strong linkage disequilibrium (black arrows) with rs36196656 (red arrow) are represented as blue stars across the different GFRA2 transcripts. (B) Cerebellar mRNA expression level of GFRA2 transcripts stratified by rs36196656 genotype. All values are normalized to two reference genes and within each assay, expression levels are shown normalized to homozygous rs36196656-CC carriers. cM=centimorgan, Mb=megabase.
In order to assess a potential direct effect of GFRA2 markers on PGRN expression levels in plasma and CSF, we performed a linear regression adjusting for age and sex, which showed that rs36196656 is not associated with PGRN levels in both plasma and CSF in 345 individuals (p=061 and p=0 67 respectively; Supplementary Figure 5A and B). We next hypothesized that GFRA2 might directly interact with PGRN and serve as a receptor for PGRN. Indeed, using transient overexpression of untagged PGRN and GFRA2 in HEK293T cells, immunoprecipitation of GFRA2 pulled down PGRN in cell lysates. Reciprocally, immunoprecipitation of PGRN pulled down GFRA2 (Figure 3A and B).
Figure 3: Interaction of PGRN and GFRA2.
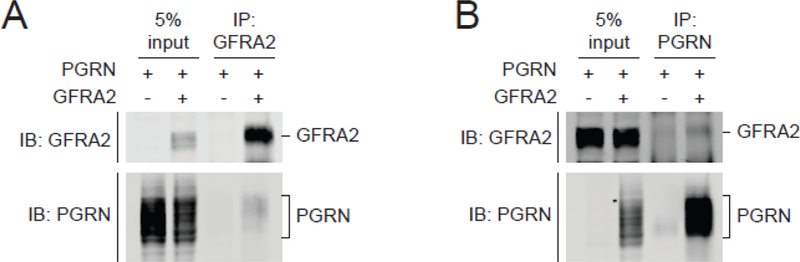
GFRA2 and PGRN immunoblots are displayed after immunoprecipitation with anti-GFRA2 antibody of cell lysates (A) of HEK293T cotransfected with untagged PGRN and untagged GFRA2 or vector control. Similarly, GFRA2 and PGRN immunoblots are displayed after immunoprecipitation with anti-PGRN antibody of cell lysates (B) of HEK293T co-transfected with untagged GFRA2 and untagged PGRN or vector control. IP=immunoprecipitation; IB=immunoblotting; 5% input=5% of the total amount of cell lysates used for immunoprecipitation.
DISCUSSION
Using an unbiased two-stage genome-wide association study in the largest available collection of unrelated FTLD patients with pathogenic GRN mutations, we identified two association signals, one at the known TMEM106B locus and one at a novel putative locus encompassing GFRA2. GRN mutations are a relatively rare cause of FTLD and despite the international nature of our collaboration we were limited by the number of GRN carriers we were able to identify. In the discovery stage, we therefore relied on the uniform loss-of-function disease mechanism associated with pathogenic GRN mutations and combined genetic analysis of patients with 120 distinct mutations. In the replication stage, newly identified GRN mutation carriers were combined with FTLD-TDP type A patients with unknown genetic etiology which are pathologically indistinguishable from GRN carriers and possibly share common pathomechanisms. Using this approach, genome-wide significant associations were detected when symptomatic patients were compared to healthy controls, suggesting that TMEM106B and GFRA2 are able to modify disease risk. Moreover, the allele at the lead GFRA2 variant (rs36196656) associated with reduced disease risk was shown to correlate with increased brain mRNA expression of GFRA2 transcripts.
Our study confirms TMEM106B as the strongest modifier of disease risk in GRN mutation carriers and GRN-negative FTLD-TDP type A patients. Published studies already established that variants associated with the TMEM106B risk haplotype correlate with increased expression of TMEM106B11 and increases in the amount of TMEM106B have been reported to be detrimental to lysosomal health and function.13–15 Among the variants in strong LD, several functional candidates have been reported including rs3173615 encoding TMEM106B p.Thr185Ser and the non-coding variant rs1990620 suggested to affected higher-order chromatin architecture at the TMEM106B locus.15,16 We estimated that GRN carriers of the TMEM106B protective haplotype (tagged by the ‘G’ allele of rs3173615) have 50% lower odds to develop disease symptoms as compared to non-protective haplotype carriers. Indeed, despite a population frequency of 14 2% in our control cohort, only 4 out of 382 (1 0%) unrelated symptomatic patients were homozygous rs3173615 ‘GG’ carriers, suggesting that many GRN mutation carriers who are also homozygous for the protective TMEM106B haplotype never develop symptoms. This is a remarkable finding for a disease gene once thought to be nearly fully penetrant and prompts the important question as to whether TMEM106B genotyping should be performed routinely when GRN genetic testing is requested or should at least be discussed as a crucial component of predictive GRN genetic testing and counselling protocols, especially in asymptomatic individuals.
The GFRA2 locus was identified as a second independent potential modifier of disease risk, which reached significance in the meta-analysis of our combined discovery and replication stages. Both GRN carriers and FTLD-TDP type A patients without mutations contributed to the observed association. Expression data points to a potential disease mechanism in which risk-associated variants at the GFRA2 locus decrease brain mRNA expression of GFRA2. Whether these variants similarly affect GFRA2 protein expression, remains to be tested. GFRA2 is the preferential co-receptor for neurturin (NRTN), one of four members of the glial cell line-derived neurotrophic factor (GDNF) family ligands (GFLs) with an important role in neuronal differentiation, proliferation and surviva.l17 NRTN further requires the transmembrane signaling receptor tyrosine kinase RET to assemble as a multi-component receptor system. Upon binding of NRTN to GFRA2, RET activates downstream signaling pathways including mitogen activated protein kinase (MAPK), extracellular signal-regulated kinase 1/2 (ERK1/2) and AKT. In-vitro, we obtained evidence of a direct binding of PGRN to GFRA2 which could suggest that GFRA2 may be a signaling receptor for PGRN; however, future experiments both in vitro and in vivo will be needed to determine the functional consequences of this interaction. If it is confirmed that GFRA2 indeed serves as a receptor for PGRN, one possible future therapeutic avenue could be to enhance their binding, e.g. by using small molecules or compounds. Another possibility, which is not mutually exclusive, is that PGRN and GFRA2 are part of independent neurotrophic signaling pathways. In this scenario, reduced neurotrophic signaling in GFRA2 risk allele carriers may facilitate the development of symptoms in GRN mutation carriers, which are already vulnerable as a result of reduced neurotrophic PGRN signaling. A loss of neurotrophic GFRA2 signaling may also affect FTLD-TDP type A patients without GRN mutations, especially since GFRA2 expression appears to be enriched in the frontal and motor cortex, highly vulnerable regions in FTLD (Supplementary Figure 4C-E). The observation of impaired behavior and memory deficits in GFRA2 knock-out mice further supports this.18 Excitingly, GDNF (another GFL with preferential binding to GFRA1) and NRTN have already been extensively studied for their neuroprotective potential in Parkinson’s disease (PD) models and clinical trials in PD patients have been performed by delivery of GDNF and NRTN as purified proteins or by means of viral vector mediated gene delivery to the brain.19–21 While none of these studies have yet shown efficacy in clinical trials, the brain delivery of GFLs was found to be safe and provides hope that modified gene-therapy approaches to boost GFRA2/NRTN signaling could be developed and tested in the context of sporadic FTLD and GRN patients.
Our study did not identify genome-wide significant associations with age at disease onset. Variability in the clinical presentation of FTLD and the subjective nature of defining disease onset may have contributed to this, especially since 40 clinical centers contributed data to this study. The focus on unrelated symptomatic patients as opposed to extended families where a more limited number of genetic factors are expected to contribute to disease onset may have further limited our ability to observe significant association. One previous study in 4 large families reported a 13 year decrease in onset age for carriers of the TMEM106B risk allele10; however, no association with age at onset was observed for TMEM106B in our study (rs3173615, p=0 87, Beta=−0 12, 95% CI 1–59-1 35).
Our study also has limitations. First, only symptomatic unrelated GRN mutation carriers were included in the analysis. Individual GRN families were generally small with limited numbers of symptomatic and informative asymptomatic carriers available, limiting the ability to perform family-based studies. Second, since patient samples were collected in various countries, population stratification could bias the results. To address this issue, we combined publically available control genotype data with newly generated genotypes from control individuals ascertained in Italy and Spain, allowing each patient to be matched to 3 geographical controls, followed by standard methodology to correct for any remaining bias. Importantly, detailed analysis at the newly identified putative GFRA2 locus across geographical populations, showed consistent ORs associated with the lead variant (rs36196656) (Supplementary Table 10). Third, FTLD-TDP type A patients without GRN mutations were included in the replication stage. While this broadens the potential impact of TMEM106B and GFRA2 associations to sporadic FTLD patients, our approach likely discounted a number of genetic modifiers specific to GRN mutation carriers. Finally, our functional studies were limited to GFRA2 and thus it remains possible that other genes in addition to GFRA2 may contribute to the observed association on chromosome 8.
In conclusion, this is the first large-scale genome-wide association study focused on genetic modifiers in patients with GRN mutations and the first study in a homogenous cohort of genetically defined FTLD patients. Two loci - TMEM106B and GFRA2 - were shown to harbor genetic variants able to modify the disease risk. These modifiers may inform genetic counselling in families and could aid in future clinical trial designs. More importantly, identification of these modifiers in human subjects supports TMEM106B and GFRA2-related pathways as potential targets for therapies. Accordingly, improving lysosomal function and/or increasing GFRA2 expression or signaling in FTLD-relevant brain areas may be viable treatment options and important areas for future research which could complement the current translational research efforts focused on increasing GRN levels.22–24
RESEARCH IN CONTEXT
Evidence before this study
Mutations in the progranulin gene (GRN) are an important cause of frontotemporal lobar degeneration with TDP-43 pathology (FTLD-TDP). Pathogenic mutations are heterozygous and cause disease through a uniform mechanism leading to 50% loss of functional progranulin protein (PGRN). We searched for the terms “GRN” OR “PGRN” AND “onset age variability” in PubMed on January 30th 2018 including all publications from the database inception and identified seven publications reporting large age at onset variability among GRN mutation carriers, suggesting that genetic modifiers may be in part responsible for the phenotypic presentation. We also searched PubMed with the terms “GRN” OR “PGRN” AND “Genome-wide association study” for reports published on January 30th 2018, without restriction on language of publication and including all publications from the database inception and identified one previous study focused on FTLD-TDP which included 80 GRN mutation carriers in a genome-wide association analyses. That study identified TMEM106B as a risk factor in FTLD-TDP patients, with a particular strong effect in GRN mutation carriers, suggesting an effect of TMEM106B variants on disease penetrance in individuals with GRN mutations. No other genome-wide association studies in GRN patients have been performed prior to the current study.
Added value of this study
Through international collaborations we were able to use a 5-fold larger cohort of patients with GRN mutations compared to the previous genome-wide association study. Importantly, using a two-stage association study, we confirmed the TMEM106B locus as the most important modifier of disease risk in GRN mutation carriers and we were able to estimate that GRN carriers of the TMEM106B protective haplotype (tagged by the ‘G’ allele of rs3173615) have 50% lower odds to develop disease symptoms as compared to non-protective haplotype carriers. We also newly identified the GFRA2 locus on chromosome 8p21.3 as a potential genome-wide significant modifier of disease risk in patients with GRN mutations. The lead variant at the GFRA2 locus (rs36196656) is located within GFRA2 intron 3 and was shown to affect the expression profile of GFRA2.
Functional studies also showed that PGRN binds to GFRA2 in vitro.
Implications of all available evidence
The identification of genetic variants in TMEM106B and GFRA2 as modifiers of the disease risk in patients with GRN mutations provides new avenues towards biomarker discovery and the development of therapeutic approaches for FTLD patients. These genetic variants might further inform genetic counselling in families and could aid in future clinical trial designs.
Supplementary Material
ACKNOWLEDGMENTS
We thank all colleagues and staff at the participating centers for their help with recruitment of patients. Specifically we thank Masood Manoochehri, Chan Foong, M.S., Huei-Hsin Chiang Ph.D., Andrew King, Ivy Trinh, Jeffrey Metcalf, Silvana Archetti, Pheth Sengdy, Alice Fok, Ewa Narożańska, Ph.D., David Lacomis, MD, Prof. Nick Fox, Prof. Martin Rossor, Prof. Jason Warren, Virginia Phillips, Linda Rousseau, Monica Casey-Castanedes, Michael DeTure, Ph.D., Rosanna Colao, MD, Gianfranco Puccio, M.D., Sabrina A.M. Curcio, Ph.D., Livia Bernardi, Ph.D., Eric M. Wassermann, M.D., Martin Farlow, M.D., Ann Hake, M.D., Dimitrios I. Kapogiannis M.D., Keiji Yamaguchi, M.D., Matthew Hagen, M.D., Ph.D., Jose Bonnin, M.D., Melissa Gener, M.D., Lina Riedl, M.D., and The French research network on FTD and FTD/ALS for clinical, pathological and genetic characterization of patients.
This work was supported by NIH grants from the NIA: P30 AG019610 (MNS, EMR, NGR-R, DWD); P30 AG012300 (CLW, BME); P50 AG025688 (JG, MG); P50 AG08702 (LSH); P30 AG013854 (EHB, MMM, SW, CG); P50 AG005133 (JK, OL); P30 AG019610 (TGB); P01 AG003991 and P50 AG005681 (NJC and CC); R01 AG044546, RF1 AG053303, P30 AG10124 (JQT), P01 AG017586 (VMY-L) and P01 AG026276 (CC); U01 AG045390, UO1 AG006786 and RO1 AG041797 (BFB); R01 AG037491 (KAJ); P50 AG016574 (RCP, BFB, RR, DWD, DSK, NRG-R); R01 AG051848, R21 AG051839, P50 AG005131 (RAR); RF AG051504, U01 AG046139 (NE-T); P50 AG023501, P01 AG019724 (BS). The study was in also in part supported by the following NIH grants from NINDS: R35 NS097261 (RR); R01 NS076837 (EDH); P30 NS055077 (MG); U54 NS092089 (BFB, ALB); P01 NS084974 (DWD); U24 NS072026 (TGB); R01 NS080820 (N.E-T); P50 NS072187 (ZKW). In addition this work was also supported by NIH grants from the NIDCD: R01 DC008552 (SW, MMM) and from the VA: I01 BX003040 (RAR).
This work was further supported by grants from the Consortium for Frontotemporal dementia (RR), the Bluefield Project to Cure FTD (XZ), the Mayo Clinic Dorothy and Harry T. Mangurian Jr. Lewy Body Dementia Program and the Little Family Foundation (BFB); the Carl B. and Florence E. King Foundation (BME, CLW); the McCune Foundation and the Winspear Family Center for Research on the Neuropathology of Alzheimer Disease (CLW); the University of Pittsburgh Brain Institute (JK); and the Arizona Department of Health Services contract 211002, Arizona Biomedical Research Commission contracts 4001, 0011, 05– 901, and 1001; the Michael J. Fox Foundation for Parkinson’s Research; GHR Foundation (RCP, TGB), Mayo Clinic Foundation and the Sun Health Foundation (TGB); and the Arizona Department of Health Services (EMR); The Tau Consortium and the Consortium for Frontotemporal Dementia Resesarch (BS).
We thank the CIHR for the following supports provided for this study: MOP 13129 (SEB, MM); M0P137116 (MM); #179009 (RG-Y, IRM); #327387 (ECF); as well as the Canadian Consortium on Neurodegeneration in Aging (SEB, ER), in particular from CCNA #137794 (RG-Y, IRM); and the LC Campbell Foundation (SEB). In addition, this study was supported by the Ricerca Corrente, Italian Ministry of Health (GR, RG, LB, GB, DG, FT); and specifically by the Ministry of Health Finalizzata 2011 ref 14GRSB and Fondazione Cassa di Risparmio di Firenze ref 2015–0722 (BN, CF, SB, SS), by AIRAlzh onlus - ANCC-COOP (IP) as well as the Telethon Foundation and Ricerca Finalizzata, Italian Ministry of Health (FT).
Part of the tissue samples were supplied by The London Neurodegenerative Diseases Brain Bank, which receives funding from the Medical Research Council UK and as part of the Brains for Dementia Research programme, jointly funded by Alzheimer’s Research UK and Alzheimer’s Society (CT). SP-B was supported by the MRC grant G0701441. The Dementia Research Centre at UCL is supported by Alzheimer’s Research UK, Brain Research Trust, and The Wolfson Foundation. This work is further supported by the NIHR Queen Square Dementia Biomedical Research Unit, the NIHR UCL/H Biomedical Research Centre and the Leonard Wolfson Experimental Neurology Centre (LWENC) Clinical Research Facility. JH received funding from the Wellcome /MRC Centre on Parkinson’s. RF was supported by the Alzheimer’s Society Grant 284 and RM by The office of the Dean of the School of Medicine, Department of Internal Medicine, at Texas Tech University Health Sciences Center. JDR was supported by an MRC Clinician Scientist Fellowship (MR/M008525/1) and has received funding from the NIHR Rare Disease Translational Research Collaboration (BRC149/NS/MH). ILB, FP and DH received funding from the Program “Investissements d’avenir” ANR-10-IAIHU-06 and the PHRC FTLDexome (promotion AP-HP). GMH, JBK, JRH and OP are part of The ForeFront Brain and Mind project team, a large collaborative research group dedicated to the study of neurodegenerative diseases funded by the National Health and Medical Research Council of Australia (NHMRC) Program Grant (#1037746) and Project Grant (#1062539), Dementia Research Team Grant (#1095127) and NeuroSleep Centre of Research Excellence (#1060992), as well as the Australian Research Council Centre of Excellence in Cognition and its Disorders Memory Program (#CE110001021), and the Sydney Research Excellence Initiative 2020. OP is supported by a NHMRC Senior Research Fellowship (APP1103258). GMH is supported by a NHMRC Senior Principal Research Fellowship (630434). CG is supported by grants provided by Swedish Research Council (Dnr 521–2010-3134, 529–2014-7504, 2015–02926), Alzheimer foundation Sweden, Brain Foundation Sweden, Swedish FTD Initiative, Swedish Brain Power, Karolinska Institutet doctoral funding, Gamla tjanarinnor, Stohnes foundation, Dementia foundation Sweden and the Stockholm County Council (ALF project). MN is funded by German Helmholtz Association, Nomis Foundation and German Research Foundation. MS is funded by the Else Kröner Fresenius Stiftung. JDS is supported by German Federal Ministry of Education and Research (FTLDc 01GI1007A). RS-V is supported by Fundació Marató de TV3, Barcelona, Spain (grant no. 20143810). JCvS is funded by Memorabel 2013 Presympt FTD #70–73305-98–105, JPND RiMod #733051024 and Alzheimer Nederland, de Ruiter #WE.15–2014-08. Finally, JS is supported by Ministry of Health, Medical University of Gdansk.
Funding
National Institute on Aging, National Institute of Neurological Disorders and Stroke, Canadian Institutes of Health Research, Italian Ministry of Health, UK National Institute for Health Research, National Health and Medical Research Council of Australia and the French National Research Agency.
Role of the funding source
The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Footnotes
DECLARATION OF INTEREST
MNS reports grants from Avid Radiopharmaceuticals, grants and personal fees from Axovant Sciences, grants and personal fees from Biogen, grants from Genentech, personal fees from Grifols, grants and personal fees from Eli Lilly, grants from Merck, grants from Pfizer, grants from Roche, personal fees from Sanofi, grants from Suven Life Sciences, grants and personal fees from vTv Therapeutics, and holds stock in Brain Health, Muses Labs, and Versanum, outside the submitted work.
ALB reports grants from US National Institutes of Health, grants from Bluefield Project to Cure Frontotemporal Dementia, grants from CBD Solutions, grants from the Tau Consortium, personal fees from AbbVie, grants from Biogen, grants from Bristol-Myers Squibb, grants from C2N Diagnostics, personal fees from Delos Pharmaceuticals, grants and non-financial support from Eli Lilly, personal fees from Denali Therapeutics, personal fees from Alector, grants from FORUM Pharmaceuticals, grants from Genentech, personal fees from Janssen Pharmaceutica, personal fees from Celgene, grants from Roche, grants from TauRx Therapeutics, personal fees from Merck, personal fees from Novartis, personal fees from Toyama Chemical, personal fees from UCB, grants from the Association for Frontotemporal Degeneration, outside the submitted work.
GMH reports grants from the Australian National Health and Medical Research Council (grant numbers 1037747, 1079679), during the conduct of the study.
ZKW reports grants from the NIH/ National Institute of Neurological Disorders and Stroke (grant number P50 NS072187) during the conduct of the study and RCP reports personal fees from Roche, personal fees from Merck, personal fees from Genentech, personal fees from Biogen, during the conduct of the study.
All other authors declare no conflict of interest related to this study.
REFERENCES
- 1.Graff-Radford NR, Woodruff BK. Frontotemporal dementia. Semin Neurol 2007; 27(1): 48–57. [DOI] [PubMed] [Google Scholar]
- 2.Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. ActaNeuropathol 2011; 122(1): 111–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314(5796): 130–3. [DOI] [PubMed] [Google Scholar]
- 4.Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 2012; 8(8): 423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006; 442(7105): 916–9. [DOI] [PubMed] [Google Scholar]
- 6.Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006; 442(7105): 920–4. [DOI] [PubMed] [Google Scholar]
- 7.Rademakers R, Baker M, Gass J, et al. Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C-->T (Arg493X) mutation: an international initiative. Lancet Neurol 2007; 6(10): 857–68. [DOI] [PubMed] [Google Scholar]
- 8.Rademakers R, Rovelet-Lecrux A. Recent insights into the molecular genetics of dementia. Trends Neurosci 2009; 32(8): 451–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finch N, Carrasquillo MM, Baker M, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 2011; 76(5): 467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cruchaga C, Graff C, Chiang HH, et al. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol 2011; 68(5): 581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Deerlin VM, Sleiman PM, Martinez-Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 2010; 42(3): 234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicholson AM, Finch NA, Thomas CS, et al. Progranulin protein levels are differently regulated in plasma and CSF. Neurology 2014; 82(21): 1871–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brady OA, Zheng Y, Murphy K, Huang M, Hu F. The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. HumMol Genet 2013; 22(4): 685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen-Plotkin AS, Unger TL, Gallagher MD, et al. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci 2012; 32(33): 11213–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicholson AM, Rademakers R. What we know about TMEM106B in neurodegeneration. Acta Neuropathol 2016; 132(5): 639–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallagher MD, Posavi M, Huang P, et al. A Dementia-Associated Risk Variant near TMEM106B Alters Chromatin Architecture and Gene Expression. Am J Hum Genet 2017; 101(5): 643–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 2002; 3(5): 383–94. [DOI] [PubMed] [Google Scholar]
- 18.Voikar V, Rossi J, Rauvala H, Airaksinen MS. Impaired behavioural flexibility and memory in mice lacking GDNF family receptor alpha2. Eur J Neurosci 2004; 20(1): 308–12. [DOI] [PubMed] [Google Scholar]
- 19.Bartus RT, Weinberg MS, Samulski RJ. Parkinson’s disease gene therapy: success by design meets failure by efficacy. Mol Ther 2014; 22(3): 487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindholm D, Makela J, Di Liberto V, et al. Current disease modifying approaches to treat Parkinson’s disease. Cell Mol Life Sci 2016; 73(7): 1365–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirik D, Cederfjall E, Halliday G, Petersen A. Gene therapy for Parkinson’s disease: Disease modification by GDNF family of ligands. Neurobiol Dis 2017; 97(Pt B): 179–88. [DOI] [PubMed] [Google Scholar]
- 22.Cenik B, Sephton CF, Dewey CM, et al. Suberoylanilide hydroxamic acid (vorinostat) up-regulates progranulin transcription: rational therapeutic approach to frontotemporal dementia. J Biol Chem 2011; 286(18): 16101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sha SJ, Miller ZA, Min SW, et al. An 8-week, open-label, dose-finding study of nimodipine for the treatment of progranulin insufficiency from GRN gene mutations. Alzheimers Dement (N Y) 2017; 3(4): 507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Capell A, Liebscher S, Feilerer K, et al. Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J Neurosci 2011; 31(5): 1885–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.