

Abstract
Serotonin receptors modulate a variety of physiological processes ranging from perception, cognition and emotion to vascular and smooth muscle contraction, platelet aggregation, gastrointestinal function and reproduction. Drugs that interact with serotonin receptors effectively treat diseases as diverse as migraine headaches, depression, and obesity. Here, we present four structures of a prototypical serotonin receptor—the human 5-HT2B receptor—in complex with chemically and pharmacologically diverse drugs, including methysergide, methylergonovine, lisuride, and LY266097. A detailed analysis of these structures complemented by comprehensive interrogation of signaling illuminates key structural determinants essential for activation. Additional structure-guided mutagenesis experiments revealed binding pocket residues essential for agonist-mediated biased signaling and β-arrestin2 translocation. Given the importance of serotonin receptors for a large number of therapeutic indications, insights derived from these studies should accelerate the design of safer and more effective medications.
Introduction
The human genome encodes 13 distinct serotonin (5-HT) G protein-coupled receptors (GPCRs). Drugs targeting 5-HT GPCRs are approved treatments for a diverse array of indications including obesity, migraine headaches, schizophrenia, anxiety and depression1,2. 5-HT receptors also frequently mediate serious drug side-effects via unanticipated ‘off-target’ actions1,3. A notorious example is that of the now banned appetite suppressant fenfluramine, which exerts its potent anti-obesity actions by activating 5-HT2C receptors4. Fenfluramine was ultimately withdrawn from the market because of a high incidence of drug-induced valvular heart disease (VHD), which occurs due to the ‘off-target’ activation by fenfluramine and its active metabolite norfenfluramine at the closely related 5-HT2B receptor (5-HT2BR)5,6.
Several other medications, including anti-migraine drugs methysergide and ergotamine5, anti-parkinsonian medications pergolide, and cabergoline7, and drugs that treat pituitary adenomas, also have potent off-target actions at 5-HT2BR, and have also been withdrawn or their use severely restricted due to drug-induced VHD1,5. Importantly, both the valvular heart disease and fibrosis associated with carcinoid syndrome have been linked to 5-HT2BR activation8. Consequently, candidate medications are routinely screened for 5-HT2B agonist activity before progressing to clinical trials9,10. Not surprisingly, 5-HT2B antagonists have been proposed as potential therapeutics for VHD and other fibrotic disorders including carcinoid syndrome11. Thus, understanding drugs action at 5-HT2B receptors is clearly important for future drug development.
The 5-HT2BR is a member of the 5-HT2 sub-family of 5-HT receptors, which includes 5-HT2A and 5-HT2C receptors. 5-HT2BR activation via Gq/11 induces phospholipase C activation, inositol phosphate (IP) accumulation, intracellular calcium release and protein kinase C activation1,2. The 5-HT2BR also recruits β-arrestin2 (also known as arrestin-3, hereafter referred to β-arrestin2) and downstream effector activation9,12,13. Drugs like lysergic acid diethylamide (LSD) and ergotamine (ERG) prefer arrestin recruitment and are considered ‘arrestin-biased agonists’9,12,13.
Over the past few years there has been an explosion in available GPCR structural information, which has provided a molecular understanding into ligand recognition14, receptor dynamics and activation 15, and ligand-mediated biased signaling16. To date, three 5-HT receptors have been solved by X-ray crystallography: the 5-HT1B17,18, 5-HT2B12, and 5-HT2C19 receptors, all in complex with the VHD-inducing antimigraine drug ERG. For the 5-HT2B receptor, LSD and ERG structures are available and reveal that ergot ligands engage a presumed orthosteric binding pocket (OBP), which is likely shared with the endogenous ligand 5-HT12,13,17. Indeed, ERG and LSD engage regions outside this OBP, which we have termed the extended binding pocket (EBP). The OBP of 5-HT receptors shares certain features with the OBP exemplified by the β2 adrenergic receptor (β2AR)20,21, and includes highly conserved and critical ligand contacts between the amine nitrogen of ligands and a highly conserved aspartate in transmembrane (TM) domain 3 (e.g. Asp3.32 in Ballesteros-Weinstein numbering scheme22), as well as polar and aromatic contacts in TM5 and 6, respectively. These interactions are thought to facilitate the stabilization of active23 and G protein-bound conformational states24.
Little is known, however, regarding the 5-HT receptor EBP, which encompasses extracellular portions of TM3 and 7, and has been proposed as a potential structural feature that may facilitate biased signaling2. Support for this hypothesis comes from the analysis of the structure of LSD bound to the 5-HT2B receptor, where LSD stereo-selectively engages TM3 and TM7 to evoke potent β-arrestin2 recruitment13. Additional insights into the mechanisms of biased agonism have recently emerged revealing a key ligand interaction between LSD and Leu209 in extracellular loop 2 (EL2), which increases ligand residence time at the receptor contributing to enhanced time-dependent β-arrestin2 recruitment13. Additionally, both TM5 and the EL2 regions have recently been exploited for biased ligand design at aminergic GPCRs25, it is unclear how biased activation occurs via contact with these regions of the receptor.
Going forward, a clearer understanding how ligand interactions with key binding pocket residues lead to the stabilization of active or inactive states will facilitate agonist, biased agonist, and antagonist drug design. Unfortunately, for most crystallized GPCRs, a single ligand (e.g. an agonist or antagonist), has only been solved, precluding such analysis. In instances where agonist and antagonist structures are available, the ligand chemotypes are structurally diverse (e.g. β2AR, adenosine 2A, μ and κ opioid receptors, and others16,26). Consequently, without such information for a given GPCR, it is difficult to leverage existing GPCR structural information for structure-guided drug design.
Here, we identify key residues responsible for 5-HT2BR activation by comparing the binding modes of chemically similar agonists and antagonists at the 5-HT2BR. Importantly, we were able to compare the binding mode of methylergonovine with its parent antimigraine drug, methysergide, which differs by a single methyl substituent. To illuminate antagonism we compared the binding mode of the antagonist lisuride to the agonist LSD, which differ only by stereochemistry and an additional nitrogen atom. Finally, we elucidate biased signaling and subtype-selectivity by clarifying the binding mode of the selective 5-HT2BR antagonist, LY266097. These insights should accelerate the design of safer and more effective medications.
Results
Structural insights into a 5-HT2B activation mechanism
Methysergide is rapidly N-demethylated in vivo to methylergonovine, which is the major active metabolite mediating methysergide’s anti-migraine actions in vivo 27. Unlike methysergide, which is a 5-HT2BR antagonist, methylergonovine is a potent 5-HT2BR agonist6 responsible for the methysergide-induced VHD5. As methysergide differs from methylergonovine by a single –CH3 (Fig. 1a), the pair represents a key ligand set to gain insight into the ligand-based structural features responsible for 5-HT2BR efficacy. Other unsubstituted N(1)-H ligands such as methylergonovine, LSD, and ERG are Gq partial agonists (Fig. 1b). By contrast, the N(1)-methyl or alkyl ergoline ligands methysergide (Fig. 1b) and LY215840 (Supplementary Fig. 1a) are 5-HT2BR antagonists.
Figure 1. Structural insights into a 5-HT2B activation mechanism.
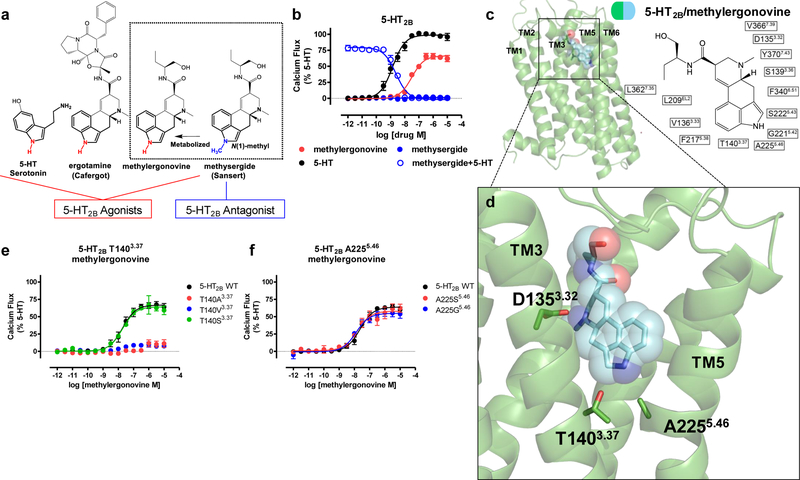
Identification of an 5-HT2BR activation mechanism by the 5-HT2BR-methylergonovine structure a) Structure-activity-relationship of 5-HT2BR ergoline ligands comparing unsubstituted N(1)-H ligands (red) such as 5-HT, ergotamine, and methylergonovine to N(1)-methyl methysergide (blue) b) 5-HT2BR Gq calcium flux activity by 5-HT (black, EC50 = 1.4 nM, Emax = 100%), methylergonovine (red, EC50 = 31 nM, Emax = 66%) and lack of agonist activity by methysergide (blue, closed circles). Methysergide instead acts as a competitive antagonist (blue, open circles, IC50 = 2.4 nM) in response to 5-HT. c) Structure of methylergonovine (blue) at 5-HT2BR (green) with 2D ligand plots of nearby residues (PDB code: 6DRY). d) Close up of methylergonovine binding pose in the binding pocket highlighting D1353.32 interacting with the charged nitrogen of methysergide, and the indole N(1)-H interacting with both T1403.37 and A2255.46 in the orthosteric binding pocket. e) Methylergonovine Gq-mediated calcium flux agonist activity at T140A3.37 (red), T140V3.37 (blue), T140S3.37 (green, EC50 = 18 nM, Emax = 64%) relative to 5-HT2BR wild-type (WT, black, EC50 = 19 nM, Emax = 66%). f) Methylergonovine Gq-mediated calcium flux agonist activity at A225S5.46 (red, EC50 = 15 nM, Emax = 58%), A225G5.46 (blue, EC50 = 12 nM, Emax = 55%) relative to 5-HT2BR wild-type (WT, black, EC50 = 23 nM, Emax = 64%). Data in panels b, e, and f represent mean and S.E.M from three independent experiments (N=3) performed in triplicate.
To identify residues critical for methylergonovine’s agonism, we obtained the crystal structure of the 5-HT2BR-methylergonovine complex at 2.9 Å resolution (Fig. 1c, Table 1). Methylergonovine forms a salt bridge with D1353.32 in the presumed orthosteric site, and the ergoline ring system forms an edge-to-face π- π stack with residues F3406.51 and F3416.52 in TM6—interactions commonly observed in aminergic20,23 and 5-HT structures13,17,19. The binding mode of methylergonovine when compared with LSD and ergotamine bound to 5-HT2BR reveals a subtly different positioning of the indole N(1)-H towards TM5 residues, with ERG the deepest toward A2255.46 and LSD the shallowest toward the backbone of G2215.42 (Supplementary Fig. 1b). This differential positioning is likely caused by a rotation around the axis of the ionic interaction between D3.32 and the protonated ergoline amine group13. Similar to ERG, the N(1)-H of methylergonovine points toward residues A2255.46 and T1403.37 (Fig. 1d, Supplementary Fig. 1c). Although the density of the N(1)-H of methylergonovine is not resolved at a 2.9 Å resolution, this hydrogen likely resides between residues A2255.46 and T1403.37.
Table 1.
Data collection and refinement statistics
| h5-HT2BR-BRIL-2 methylergonovine (6DRY)b | h5-HT2BR-A225G5.46-BRIL-3 methysergide (6DRZ)c | h5-HT2BR-BRIL-1 lisuride (6DRX)d | h5-HT2BR-BRIL-1 LY266097 (6DS0)e | |
|---|---|---|---|---|
| Data collection | ||||
| Space group | C2221 | C2221 | C2221 | C2221 |
| Cell dimensions | ||||
| a, b, c (Å) | 59.7 ,119.5, 171.0 | 59.1, 119.3, 172.6 | 59.4, 118.6, 168.2 | 59.5 120.1 169.5 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 30.0–2.9 (2.99–2.90)a | 30–3.1 (3.19–3.10) | 30.0–3.1 (3.17–3.10) | 30.0–3.2 (3.29–3.20) |
| Rmerge (%) | 15.7 (71.8) | 16.3 (84.5) | 12.2 (95.0) | 15.9 (128.0) |
| I/σ(I) | 7.7 (1.4) | 8.7 (1.2) | 10.9 (1.1) | 9.8 (1.2) |
| CC1/2 | 98.0 (56.0) | 98.0 (61.2) | 99.6 (46.2) | 99.4 (39.1) |
| Completeness (%) | 91.8 (81.1) | 93.4 (94.4) | 97.1 (97.1) | 93.5 (96.5) |
| Redundancy | 2.6 (2.1) | 3.3 (3.3) | 4.4 (4.3) | 3.8 (3.8) |
| Refinement | ||||
| Resolution (Å) | 2.9 | 3.1 | 3.1 | 3.2 |
| No. reflections | 12436 | 10595 | 10679 | 9725 |
| Rwork / Rfree | 23.5 / 27.1 | 22.5 / 26.3 | 24.3 / 28.6 | 22.3 / 26.4 |
| No. atoms | ||||
| 5-HT2BR | 2200 | 2174 | 2144 | 2177 |
| BRIL | 733 | 622 | 506 | 637 |
| Ligand | 25 | 26 | 25 | 26 |
| Lipid and other | 82 | 79 | 53 | 54 |
| B factors | ||||
| 5-HT2BR | 72.5 | 73.8 | 81.7 | 82.7 |
| BRIL | 87.1 | 91.1 | 135.3 | 146.5 |
| Ligand | 70.2 | 57.7 | 77.1 | 81.7 |
| Lipid and other | 90.5 | 84.7 | 84.6 | 100.0 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.003 | 0.004 | 0.002 | 0.002 |
| Bond angles (°) | 0.49 | 0.61 | 0.76 | 0.48 |
Values in parentheses are for highest-resolution shell.
Number of crystals is 11.
Number of crystals is 13.
Number of crystals is 11.
Number of crystals is 8.
To determine if residues T1403.37 and A2255.46 are involved in methylergonovine’s agonism, we mutated residues T1403.37 and A2255.46. T140A3.37 and T140V3.37 mutations significantly diminished methylergonovine’s agonism (Fig. 1e) and binding affinity (Supplementary Table 1), despite similar surface expression levels relative to wild-type (Supplementary Fig. 1d). Although there is not an optimal angle for a hydrogen bond between the N(1) hydrogen and residue T1403.37 in the 5-HT2BR-methylergonovine structure (Supplementary Fig. 1e), the close distance suggests that at least an electrostatic interaction (van der Waals, vdW) between residue T1403.37 and the N(1)-H may occur during activation. Indeed, 5-HT, which also contains an N(1)-H on the indole, displayed weak Gq activity at T140A3.37 and T140V3.37 mutants (Supplementary Fig. 1f), indicating that T1403.37 is essential for receptor activation. To recapitulate a favorable electrostatic interaction, we created the T140S3.37 mutant receptor and found that methylergonovine’s Gq agonism (Fig. 1e) and binding affinity were spared (Supplementary Table 1). By contrast, mutations A225S5.46 and A225G5.46 did not substantially affect methylergonovine’s Gq agonism (Fig. 1f) nor binding affinity (Supplementary Table 1), and spared 5-HT Gq activity (Supplementary Fig. 1g). Thus, methylergonovine’s interaction with T1403.37 is critical for 5-HT2BR activation.
Structure of a 5-HT2BR-A225G5.46 mutant designed to be activated by methysergide
We next sought to understand how the N(1)-methyl of methysergide causes 5-HT2BR antagonism. We hypothesized that methysergide’s N(1)-methyl would either lack interaction with T1403.37 and/or cause a steric clash with A2255.46. Unfortunately, methysergide failed to demonstrate any measureable Gq agonist activity at a T140V3.37 mutant, where it would be expected that the valine and N(1)-methyl could form additional vdW interactions leading to activation (Fig. 2a). By contrast, methysergide demonstrated potent Gq partial agonist activity (Fig. 2a, Supplementary Fig. 2a) at the A225G5.46 mutant, which we hypothesized to introduce more bulk tolerance for methysergide’s N(1)-methyl—a notion consistent with TM5 engagement appearing to be important for G protein-dependent agonism25.
Figure 2. Structure of a 5-HT2BR-A225G5.46 mutant designed to be activated by methysergide.
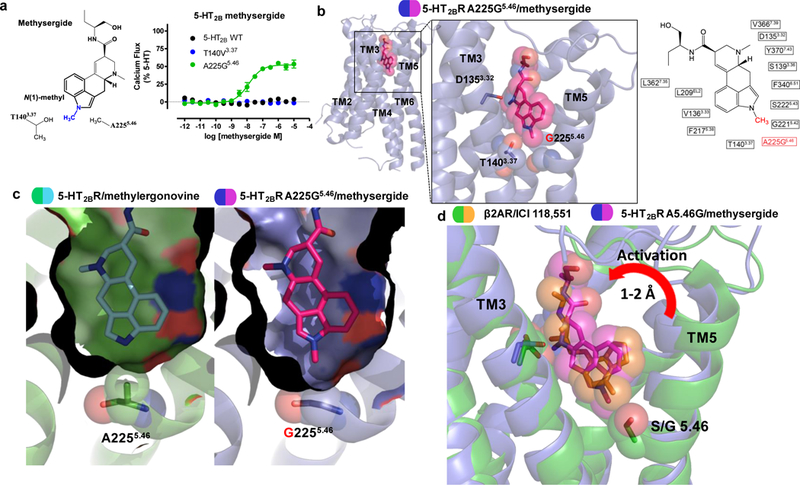
Design and structure of a 5-HT2BR mutant to convert methysergide into an agonist. a) Design of mutation to convert methysergide into an agonist by accommodating the N(1)-methyl (blue) with vdW interaction by T140V3.37 or with space by A225G5.46. 5-HT2BR methysergide Gq-mediated calcium flux activity at 5-HT2BR wild-type (black), T140V3.37 (blue) and A225G5.46 (green, EC50 = 12 nM, Emax = 54%). Data represent mean and S.E.M from three independent experiments (N=3) performed in triplicate. b) Structure of the 5-HT2BR A225G5.46 mutant (blue) in complex with methysergide (purple) indicating that the N(1)-methyl is positioned toward residues T1403.37 and A2255.46 in the orthosteric binding pocket, which is also illustrated in 2D ligand plot of nearby residues (PDB code: 6DRZ). c) Space-filling comparison of the 5-HT2BR-methylergonovine and the 5-HT2BR- A225G5.46-methysergide structures indicating that the A225G5.46 mutant creates space to accommodate methysergide’s N(1)-methyl to achieve a similar pose as seen in the 5-HT2BR-methylergonovine structure. d) Comparison of the binding poses of the 5-HT2BR- A225G5.46-methysergide structure to the β2AR (green) in complex with the inverse agonist ICI 118,551 (orange) indicating a 1–2 Å shift difference in TM5 commonly observed in active versus inactive structures.
To test this hypothesis, we obtained a 5-HT2BR-A225G5.46-methysergide complex structure to 3.1 Å (Fig. 2b, Supplementary Fig. 2b,c, Table 1). Similar to the 5-HT2BR-methylergonvine complex structure, methysergide makes conserved contacts with D1353.32, F3406.51 and F3416.52. Importantly, methysergide’s N(1)-methyl s pointed down into the space at the G2255.46 mutation. As predicted, removing a methyl substituent from A2255.46 to form the glycine G2255.46 mutant creates ‘space’ (e.g. bulk tolerance) for the methysergide’s N(1)-methyl at TM5 (Fig. 2c).
To obtain further insights into methysergide’s engagement at TM5, we compared the 5-HT2BR- A225G5.46-methysergide structure to the inactive state β2-adrenergic receptor (β2AR) structure bound to the antagonist ICI-118,55128. ICI-118,551, like methysergide, also contains a methyl substitution on its core scaffold that appears to sterically ‘push’ against residue S2075.46 in TM5. Alignment of both structures reveals a 1–2 Å shift in TM5, where the extracellular side of TM5 moves closer toward methysergide in the 5-HT2BR-A225G5.46-methysergide structure, where ICI-118,551 is precluding TM5 movement via its methyl substituent in the β2- ICI-118,551 structure (Fig. 2d). Importantly, this TM5 shift is also observed in the nanobody-stabilized active state of β2AR23. Alignment of the β2AR-ICI-118,551 complex structure with the nanobody-stabilized active state of β2AR, reveals that ICI-118,551 precludes this inward TM5 movement via the methyl substituent (Supplementary Fig. 2d). Our results thus reveal that residues T1403.37 and A2255.46 are essential for activation.
Structural basis for a 5-HT2B activation mechanism via the extended binding pocket
We next focused on the role of the EBP in receptor activation. Previously, we showed that LSD’s diethylamide, which is key for LSD’s potent hallucinogenic effects29, contacts TM3 and TM7 within the EBP13. Furthermore, we found that LSD’s recognition in this region is stereo-selective, as LSD’s potent agonism was recapitulated by only the (S,S)-azetidide stereoisomer, a conformationally-restricted diethylamide stereoisomer of LSD13. Less clear, however, is the effect of opposing stereochemistry at the C8 position on antagonist versus agonist recognition. Here we sought to examine other ergoline ligands, such as lisuride, which shares the same ergoline core scaffold as LSD yet possesses an (S)-diethylurea (Fig. 3a) and is a potent 5-HT2BR antagonist30 (Fig. 3b).
Figure 3. Structural basis for a 5-HT2B activation mechanism via the extended binding pocket.
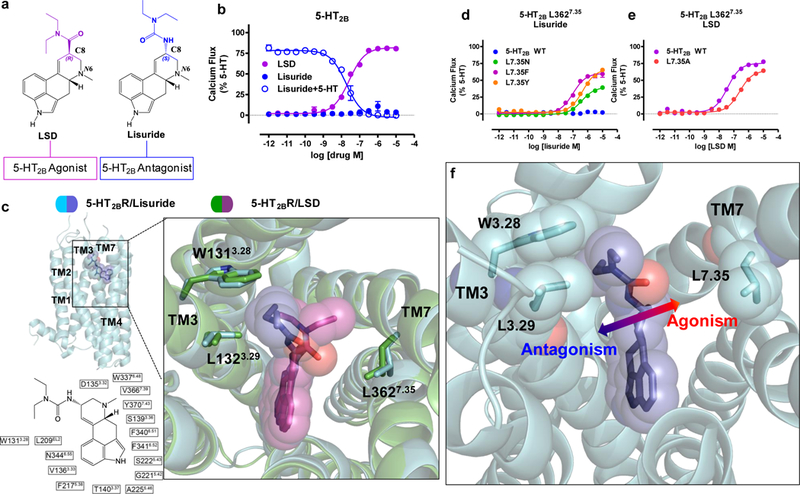
Identification of an activation mechanism via the extended binding pocket illuminated by the 5-HT2BR-lisuride structure. a) Comparison of the chemical structures of LSD to lisuride indicating LSD’s (R) diethylamide (purple) and lisuride’s (S)-diethylurea (blue) lead to either 5-HT2BR agonism or antagonism, respectively. b) 5-HT2BR Gq-mediated calcium flux activity indicating LSD partial Gq agonist activity (purple, EC50 = 40 nM, Emax = 82%), and lack of lisuride agonist activity (blue, closed circles), and instead demonstrating competitive antagonism by lisuride (blue, open circles, IC50 = 25 nM). c) Structure of lisuride (blue) at the 5-HT2BR (light blue) comparing LSD (purple) bound to 5-HT2BR (green) showing lisuride’s (S)-diethylurea wedged between TM3 residues W1313.28 and L1323.29 and making no contact with TM7 L3627.35 (PDB code: 6DRX). d) Lisuride Gq-mediated calcium flux activity at L362N7.35 (green, EC50 = 395 nM, Emax = 41%), L362Y7.35 (orange, EC50 = 465 nM, Emax = 69%), and L362F7.35 (purple, EC50 = 77 nM, Emax = 60%). e) LSD Gq-mediated calcium flux activity at L362A7.35 (red, EC50 = 340 nM, Emax = 67%) relative to 5-HT2BR wild-type (WT, purple, EC50 = 37 nM, Emax = 79%). Data in panels b, d, and e represent mean and S.E.M from three independent experiments (N=3) performed in triplicate. f) Schematic illustrating ligand contact with residue L3627.35 in TM7 in the extended binding pocket leads to 5-HT2BR activation.
To identify the structural basis for lisuride’s antagonism, we solved the 5-HT2BR-lisuride complex structure to a resolution of 3.1 Å (Fig. 3c, Supplementary Fig. 3a,b, Table 1). Alignment of 5-HT2BR-LSD and 5-HT2BR-lisuride structures show similar binding poses in the OBP, where the indole N(1)-H of both LSD and lisuride form a hydrogen bond with the carbonyl backbone at residue G2215.42 in TM5 (Supplementary Fig. 3c). Comparison of these poses at the OBP cannot entirely explain the difference in pharmacological activity between LSD and lisuride. By contrast, comparison of the EBP poses reveals that the (S)-diethylurea of lisuride is exclusively wedged between residues W1313.28 and L1323.29 in a hydrophobic stack making minimal contact with TM7 residues. This results in 1.2 Å less contraction of the binding pocket between residues W1313.28 and L3627.35 compared to the LSD structure. By contrast, LSD’s binding pose, where the diethylamide contacts residue L3627.35 in TM7 (Fig. 3c) exhibits a more contracted binding pocket. This unexpected difference in binding pose likely explains lisuride’s lack of agonism at the 5-HT2BR, where contact with TM7 residue L3627.35 in the EBP appears essential for LSD’s agonism.
To test the hypothesis that ligand contact with TM7 in EBP facillitates 5-HT2BR activation, we mutated residue L3627.35, which is the closest residue in proximity to the lisuride’s diethylurea in TM7, in order to facilitate an interaction between lisuride and TM7. Consistent with our hypothesis, mutation of L3627.35 to asparagine (L362N7.35), which we predicted will form a hydrogen bond with the backbone of lisuride’s diethylurea moiety, resulted in Gq partial agonism (EC50 = 395 nM, Emax = 41%, Fig. 3d). We also mutated L3627.35 to either phenylalanine (L362F7.35) or tyrosine (L362Y7.35), which could facilitate either a hydrophobic-aromatic interaction (F,Y) or hydrogen bond (Y) with the diethylurea of lisuride. At both mutants, lisuride is a potent partial agonist, with EC50 values as low as 77 nM (Fig. 3d) at the L7.35F mutant, a potency comparable to LSD at the 5-HT2BR wild-type (EC50 = 40 nM, Emax = 82%). Mutation of L3627.35 to alanine (L362A7.35) impaired LSD’s Gq agonist potency by 10-fold (EC50 = 401 nM, Emax = 79%, Fig.3e), without altering surface expression levels (Supplementary Fig. 3d). Taken together, our results showing that LSD and lisuride occupy the OBP in a similar fashion, yet exhibit different poses in the EBP indicates that ligand engagement with TM7, specifically with L3627.35, leads to an auxiliary mechanism of agonist activation via the EBP (Fig. 3f).
Divergent actions on β-arrestin recruitment by OBP versus EBP mutations
We previously reported that ergolines, such as ERG and LSD, display preference for β-arrestin2 recruitment over Gq-mediated calcium flux at the 5-HT2BR12, and that LSD’s β-arrestin2 recruitment appears to be time-dependent and a product of its slow off-rate from the 5-HT2BR13. A key structural motif identified for LSD’s potent β-arrestin2 recruitment is EL2; however other regions of the binding pocket that lead to β-arrestin2 recruitment at this receptor remain unexplored. Here, we examined the roles of EBP versus OBP regions for 5-HT2BR β-arrestin2 recruitment.
First, we examined the OBP mutants, T1403.37 and A2255.46, that were critical for Gq activity for methylergonovine and methysergide, respectively. At the T140A3.37 mutation, methylergonovine failed to recruit β-arrestin2 (Fig. 4a) indicating the T140A3.37 mutation disrupts both Gq and β-arrestin2 agonism. Similarly, at the A225G5.46mutation, which restores methysergide’s Gq agonism, also restored methysergide’s β-arrestin2 recruitment (Fig. 4b). These results indicate that OBP activation via T1403.37 and A2255.46 leads to equal contributions for Gq activation and β-arrestin2 recruitment, as observed for the endogenous ligand 5-HT (Supplementary Fig. 4a,b).
Figure 4. Divergent actions on β-arrestin2 recruitment by OBP versus EBP mutations.
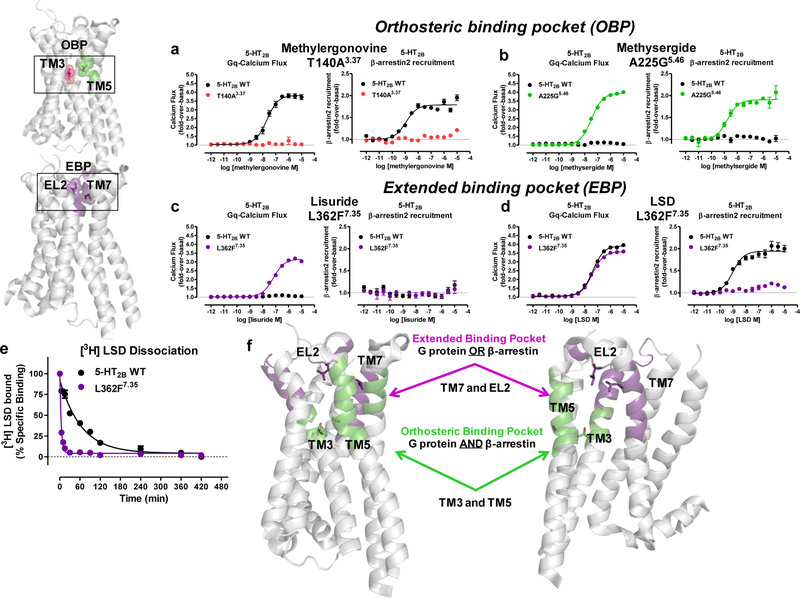
Examination of β-arrestin2 recruitment activity at OBP T140A3.37 and A225G5.46 mutations versus EBP mutation L362F7.35. a) Methylergonovine Gq-mediated calcium flux (left panel) comparing T140A3.37 (red) to 5-HT2BR WT (black, EC50 = 21 nM). β-arrestin2 recruitment (right panel) comparing T140A3.37 (red) to 5-HT2BR WT (black, EC50 = 1.2 nM). b) Methysergide Gq-mediated calcium flux (left panel) comparing A225G5.46 (green, EC50 = 33 nM) to 5-HT2BR WT (black). β-arrestin2 recruitment (right panel) comparing A225G5.46 (green, EC50 = 1.7 nM) to 5-HT2BR WT (black). c) Lisuride Gq-mediated calcium flux (left panel) comparing L362F7.35 (purple, EC50 = 65 nM) to 5-HT2BR WT (black). β-arrestin2 recruitment (right panel) comparing L362F7.35 (purple) to 5-HT2BR WT (black). d) LSD Gq-mediated calcium flux (left panel) comparing L362F7.35 (purple, EC50 = 40 nM) to 5-HT2BR WT (black, EC50 = 42 nM). β-arrestin2 recruitment (right panel) comparing L362F7.35 (purple) to 5-HT2BR WT (black, EC50 = 0.97 nM). Data in panels a-d are expressed as fold-over-basal and represent mean and S.E.M from three independent experiments (N=3) performed in triplicate. e) LSD dissociation comparing 5-HT2BR wild-type (black, koff = 0.015 min−1) to the L362F7.35 mutant (purple, koff = 0.240 min−1). Data represent percent specific binding indicating mean and S.E.M from three independent experiments (N=3) performed in duplicate. f) Schematic comparing the location of the EBP residues L209EL2 and L3627.35 (purple), which can result in either Gq or β-arrestin2 recruitment preference, to the location of OBP residues T1403.37 and A2255.46 (green), which result in equal contributions to Gq activity and β-arrestin2 recruitment.
Surprisingly, the EBP mutant L362F7.35, which restored lisuride’s Gq agonism, did not restore lisuride’s β-arrestin recruitment agonism (Fig. 4c). Although the L362F7.35 mutation did not affect LSD’s Gq agonism, it abolished LSD’s β-arrestin recruitment (Fig. 4d). Interestingly, the impairment of β-arrestin2 recruitment by the L3627.35 mutation appeared to be dependent on type of mutation—L362F7.35 showed the weakest recruitment for β-arrestin with some restoration by L362Y7.35 (Supplementary Fig. 4c).
Further analysis of L362F7.35 revealed no deficits in Gq function as measured by PI hydrolysis (Supplementary Fig. 4d) or by Gq/γ1 dissociation measured by bioluminescence resonance energy transfer (BRET, Supplementary Fig. 4e). By contrast, L362F7.35 attenuated LSD’s time-dependent recruitment of β-arrestin (Supplementary Fig. 4f), as observed previously for the EL2 L209A mutation13. Accordingly, we directly measured LSD’s dissociation rate at the L362F7.35 mutant and measured greater than 10-fold faster dissociation rate compared to 5-HT2BR WT (Fig. 4e). Thus, the L362F7.35 mutation decreases β-arrestin2 recruitment by accelerating dissociation, thereby contributing to β-arrestin2 recruitment. Taken together, ligand recognition in the OBP results in equivalent Gq and β-arrestin2 activity, whereas ligand recognition in the EBP, specifically at TM7, results in either Gq or β-arrestin2 recruitment activity and divergent effects on ligand bias (Fig. 4f).
Structure of 5-HT2BR-LY266097 reveals TM7 as a trigger for biased signaling
Finally, we explored non-ergoline antagonists with distinct scaffolds to determine if they display similar binding modes shared with other 5-HT2BR antagonists. Although lisuride is a 5-HT2BR antagonist, it lacks selectivity for 5-HT2BR, as is commonly observed for many ergolines19. LY266097, however, is a purported selective 5-HT2BR antagonist31 that contains a distinct tetrahydro-beta-carboline pharmacophore, and determining the binding mode of LY266097 could illuminate novel structural determinants 5-HT2BR selectivity.
Accordingly, we crystallized LY266097 in the BRIL-fused 5-HT2BR and solved the 5-HT2BR-LY266097 structure at a 3.2 Å resolution (Fig. 5a, Supplementary Fig. 5a,b, Table 1). Analysis of LY266097’s binding pose reveals that the tetrahydro-beta-carboline core scaffold is oriented in the OBP with the charged nitrogen engaging D1353.32 in TM3, and tetrahydro-beta-carboline core engaging residues F3406.51 and F3416.52 in a π- π aromatic stack. Unexpectedly, when we compared the binding modes of LY266097 and lisuride in the EBP, we observed that LY266097’s 2-chloro-3,4-dimethoxybenzyl substitution is oriented much closer toward TM7 compared to lisuride (Fig. 5b,c). Based on our previous finding that TM7 appears to be important for biased agonism, we hypothesized that LY266097 should show preference for either Gq or β-arrestin2 activity. We found LY266097 to be a modest Gq partial agonism without detectable β-arrestin2 activity (Fig. 5d), and confirmed LY266097’s Gq partial agonism in an additional assay of Gq function via PI hydrolysis (Supplementary Fig. 5c). We also tested LY266097 in an orthologonal β-arrestin2 recruitment assay using BRET, which measures β-arrestin2 recruitment in real-time monitored across specific time points. At no tested time point did LY266097 display β-arrestin2 recruitment (Supplementary Fig. 5d), and instead, displayed potent β-arrestin2 antagonism (Supplementary Fig. 5e). These results indicate that LY266097 displays Gq-bias.
Figure 5. Structure of 5-HT2BR-LY266097 reveals TM7 as a trigger for biased signaling.
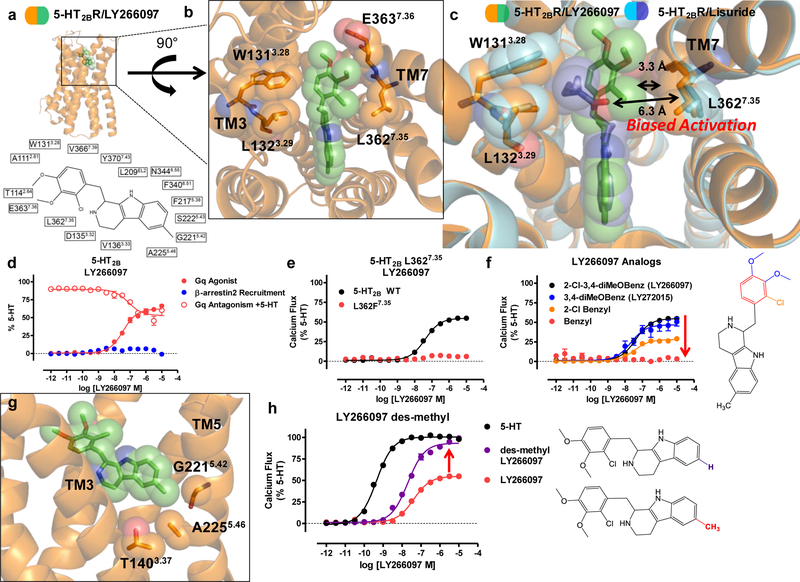
Structure of 5-HT2BR in complex with LY266097 reveals determinants of ligand bias via TM7 a) 5-HT2BR (orange) in complex with LY266097 (green) with 2D ligand plot of nearby residues (PDB code: 6DS0). b) View from the top of the receptor showing 2-chloro-3,4-dimethoxybenzyl substitution of LY266097 is oriented in close proximity to residue L3627.35 in TM7. c) Alignment of the 5-HT2BR-LY266097 and 5-HT2BR-lisuride structures showing that LY266097’s 2-chloro-3,4-dimethoxybenzyl substitution is within 3.3 Å from L3627.35, whereas lisuride’s (S)-diethylurea is further away at 6.3 Å from L3627.35. d) Profiling of LY266097 for ligand bias showing partial Gq agonist activity (red closed circles, EC50 = 37 nM, Emax = 62%) and partial antagonist activity (red open circles, IC50 = 78 nM), but no β-arrestin2 recruitment activity (blue). e) LY266097 Gq-mediated calcium flux activity comparing L3627.35 (red) to 5-HT2BR wild-type (WT, black, EC50 = 41 nM, Emax = 54%). f) Gq-mediated calcium flux activity of benzyl substituted LY266097 analogs, 3,4-diMeOBenzyl (blue, EC50 = 24 nM, Emax = 46%), 2-ClBenzyl (orange, EC50 = 35 nM, Emax = 28%), and unsubstituted Benzyl (red). g) 5-HT2BR-LY266097 structure shows methyl substitution on the tetrahydro-beta-carboline scaffold interacts with the 5-HT2-specific residue G2215.42. h) Gq-mediated calcium flux activity of des-methyl LY266097 analog (purple, EC50 = 20 nM, Emax = 93%) results in near full agonist activity compared to 5-HT (black, EC50 = 0.7 nM) and LY266097 (red, EC50 = 41 nM, Emax = 54%). Data in panels d, e, f, and h represent mean and S.E.M from three independent experiments (N=3) performed in triplicate.
Based on our previous results with lisuride at the L362F7.35 mutation, we hypothesized that the extent of Gq agonism by LY266097 is determined by ligand contact between the 2-chloro-3,4-dimethoxybenzyl and L3627.35. To confirm that LY266097’s contact with TM7 results in selective Gq activation, we tested LY266097 at the L362F7.35 mutation, which was previously found to restore lisuride’s Gq agonist activity. Expectedly, LY266097’s Gq agonist activity was abolished at the L362F7.35 mutation (Fig. 5e), suggesting that the bulkiness of the phenylalanine in the L362F7.35 mutant sterically clashes with the 2-chloro-3,4-dimethoxybenzyl moiety resulting in lack of agonism. To provide additional evidence that TM7 influences activation via strict steric requirements, we synthesized analogs of LY266097 that lack either 2-chloro or 3,4-dimethoxy substitutions to the benzyl moiety, which would be expected to show less TM7 interaction. We find a substitution-dependent decrease in Gq-mediated agonist potency with the unsubstituted benzyl analog showing no detectible Gq agonist activity, indicating that the substitution pattern to LY266097’s scaffold confers Gq agonism (Fig. 5f).
Similar to methysergide, LY266097 contains a methyl substitution to the tetrahydro-beta-carboline scaffold that appears to ‘push’ on TM5 at position G2215.42 (Fig. 5g), a key residue implicated in conferring 5-HT2 subtype-selectivity19. To determine if LY266097’s methyl substitution is partly responsible for Gq-partial agonism, we synthesized the des-methyl analog of LY266097 finding that it is nearly a full agonist (EC50 = 20 nM Emax = 93%, Fig. 5h). These results suggest that the methyl substitution on LY266097 and its interaction with TM5 impair agonism.
Discussion
Here leveraged by four new 5-HT2B crystal structures, including the structure of a mutant receptor A225G5.46 designed to switch a ligand’s efficacy, we provide a comprehensive structural analysis of ligand-specific contacts that lead to GPCR activation. Importantly, this study not only identifies mechanisms of serotonin GPCR activation via the orthosteric site, but also identifies important determinants of β-arrestin recruitment via the extended binding pocket, thereby illuminating determinants of ligand bias that could apply to other GPCRs.
Our results with the 5-HT2BR-methylergonovine and the 5-HT2BR- A225G5.46-methysergide structures illuminate an orthosteric activation mechanism occurring via ligand engagement with residues T1403.37 and A2255.46. Although residue 5.46 has been implicated as an important determinant of activation in β adrenergic receptors21,32, the role of the highly conserved Thr3.37 is less understood. In fact, crystallographic evidence that shows direct ligand engagement with residue 3.37 in TM3 is scarce. In rhodopsin, however, the β-ionone ring of retinal, which is critical for receptor activation, is directly engaged with residue Glu1223.37. Retinal interaction with Glu1223.37 has been posited to lead to disruption of a hydrogen bond between Glu1223.37 and His2115.46, which causes an inward rotation of TM5 transducing the disruption of ionic lock between the intracellular portions of TM3 and TM633. Although here ligand engagement with T1403.37 appears to involve either a hydrogen bond or an electrostatic interaction, it remains to be seen if this ligand contact occurs frequently with other ligands or if its role is more indirect (and in concert with residue 5.46) with the activation process, especially considering that residue Thr3.37 is highly conserved across aminergic GPCRs. Our results do emphasize that ligand engagement with residue 5.46 and TM5 movement is important for an orthosteric mechanism of class A aminergic GPCRs—a region highly conserved in aminergic GPCRs that we have previously exploited to design biased ligands25.
To our knowledge, our results with the 5-HT2BR-lisuride structure are the first to reveal a mechanism of activation occurring via the EBP, specifically via ligand contact with TM7 at residue L3627.35. This extended binding region of the receptor is less conserved allowing for ligand-specific engagement of non-conserved residues, and likely explains the diverse pharmacological action that results from ligand-specific substituents that project from the core scaffold, as in the case of LSD where the diethylamide projects away from the core ergoline scaffold. Interestingly, the same residues, 3.28, 3.29, and 7.35, which are also involved in recognition of the LSD’s diethylamide and the lisuride’s diethylurea, compose part of the allosteric site of muscarinic receptors34. This suggests that ergoline ligands, such as LSD, access a potential allosteric site leading to a diverse pharmacological profile (e.g. antagonist, biased agonist). However, it remains to be seen if bona fide allosteric modulators can selectively target the EBP of serotonin receptors, leading to bias.
Further study of the structural dynamics that lead to activation, either via the OBP or EPB, is needed to clarify trigger motifs (e.g. NPxxY, P-I-F, DRY) involved in either balanced or biased activation. Although BRIL fusion 5-HT2BR structures adopt the “active intermediate” state, more ‘active-like’ 5-HT2BR structures will likely shed light on key trigger motifs involved in G protein versus β-arrestin2 recruitment. Indeed, current inactive state structures of 5-HT2C19 and 5-HT1B18, which have both been co-crystallized with inverse agonists, implicate the conserved P-I-F trigger as motifs essential for inactivation and biased signaling.
In this study, L3627.35 in TM7 appears to be an important determinant of preference for G protein or β-arrestin2 recruitment, as was also implicated for the kappa opioid receptor biased signaling37. In the case of LY266097, which appears to make no specific vdW or hydrogen bond contacts with residues in TM7, our results with L3627.35 mutants suggest that ligand engagement with TM7 may operate under strict steric constraints, as similarly observed in the 5-HT2BR-A225G5.46-methysergide structure. Importantly, hydrophobic contact with L3627.35, as in the case of LSD, leads to enhanced ligand residence time, and contributions from both TM7 and EL2 serve to stabilize ligand residence time. In fact, increased ligand residence time has been shown to lead to increased β-arrestin2 recruitment over time13, and appears to be a hallmark of β-arrestin biased ligands at aminergic GPCRs25. Further study, however, is required to illuminate subsequent signaling after arrestin recruitment, especially as it relates to G protein-dependence, as shown in one recent study8. Nevertheless, our study illuminates receptor recognition of small substitutions to core ligand scaffolds directly modulates G protein versus β-arrestin recruitment preferences.
Identification of critical residue(s) for 5-HT2BR agonist versus antagonist recognition and efficacy encoding is important for drug design strategies, which aim to avoid 5-HT2BR activation and VHD. Based on our previous structural and functional studies with the 5-HT2BR, it is clear that the 5-HT2BR represents an attractive receptor template for clarifying the structural features necessary for biased signaling as they apply to other class A GPCRs. Insights into the structural basis of agonist versus antagonist action at GPCRs are essential for the design of safer and more effective medications.
Online Methods
Generation of 5-HT2B receptor crystallization constructs
5-HT2BR crystal constructs were based on previously published 5-HT2BR constructs fusing thermostabilized apocytochrome b562 RIL (M7W, H102I, R106L) from E. coli – referred to as BRIL - into ICL3 of the receptor38,39. The 5-HT2BR-LY266097 and 5-HT2BR-lisuride complexes were crystallized using a previously crystallized construct (h5-HT2BR-BRIL-1) that lacks N-terminal residues 1–35 and C-terminal residues 406–481, contains a thermostabilizing M144W3.41 mutation40, and possessed residues A1-L106 of BRIL in place of receptor residues Y249-V313 of ICL341. The 5-HT2BR-methylergonovine complex was crystallized using a different previously crystallized construct39 (h5-HT2BR-BRIL-2) that includes V313, but is otherwise identical to h5-HT2BR-BRIL-1. The 5-HT2BR A225G5.46-methysergide complex (h5-HT2BR-BRIL-3) was generated by QuikChange (Agilent) PCR mutating alanine 2255.46 to glycine in the h5-HT2BR-BRIL-2 construct. All constructs also contain a haemagglutinin (HA) signal sequence followed by a FLAG tag at the N terminus, and a PreScission protease site followed by a 10× His tag at the C-terminus to enable purification by immobilized metal affinity chromatography.
Expression and purification of the 5-HT2B crystal constructs.
High-titer recombinant baculovirus (>109 viral particles per ml) was generated using the Bac-to-Bac Baculovirus Expression System (Invitrogen). Recombinant baculovirus was obtained by transfecting ~5 µg of recombinant bacmid into 5×105 settled Spodoptera frugiperda (Sf9) cells (Expression Systems) in a 12 well plate (Corning) using 3 µl Cellfectin II Reagent (Invitrogen). After 5–12 hrs, media was exchanged for 1 mL Sf-900 II SFM media (Invitrogen) and incubated for 4–6 days at 27 °C. P0 viral stock was harvested as the supernatant and used to generate high-titer baculovirus stock by infection of 40–1000 mls of 2×106 Sf9 cells per ml and incubation for three days. Viral titers were determined by flow-cytometric analysis of cells stained with gp64-PE antibody (Expression Systems)41. Expression of 5-HT2BR was carried out by infection of Sf9 cells at a cell density of 2–3×106 cells/ml in ESF921 media (Expression Systems) with P1 virus at a MOI (multiplicity of infection) of 3–5. Cells were harvested by centrifugation at 48 h post infection, washed in PBS, and stored at −80 °C until use. Cells were disrupted by thawing frozen cell pellets in a hypotonic buffer (10 mM HEPES, 10 mM MgCl2, 20 mM KCl, pH 7.5) containing protease inhibitors (500 µM AEBSF, 1 µM E-64, 1 µM Leupeptin, 150 nM Aprotinin). Membranes were purified by repeated centrifugation in a high osmotic buffer ( containing 10 mM HEPES, 1.0 M NaCl, 10 mM MgCl2, 20 mM KCl, pH 7.5) to remove soluble and membrane associated proteins. Purified membranes were directly flash-frozen in liquid nitrogen and stored at −80 °C until protein purification.
Purified membranes were resuspended in buffer containing 10 mM HEPES, 10 mM MgCl2, 20 mM KCl, 150 mM NaCl, pH 7.5, protease inhibitors and incubated for 1 hour at room temperature with either 50 µM methylergonovine (Sigma Cat#M2776), methysergide (Sigma Cat#M137), lisuride (Tocris Cat#4052), or LY266097 (Tocris Cat#4081). After 30 min incubation in the presence of 2 mg/ml iodoacetamide (Sigma), membranes were solubilized in 10 mM HEPES, 150 mM NaCl, pH 7.5, 1% (w/v) n-dodecyl-β-D-maltopyranoside (DDM, Anatrace), 0.2% (w/v) cholesteryl hemisuccinate (CHS, Sigma), 25 µM indicated complex ligand, and protease inhibitors for 2 h at 4 °C. Unsolubilized material was removed by centrifugation at 150,000 × g for 30 min, and 15 mM imidazole was added to the supernatant. Proteins were bound to TALON IMAC resin (Clontech) overnight at 4 °C using approximately 750 µl resin for protein purified from 2 L of cells. The resin was then washed with 10 column volumes (cv) of Wash Buffer I (50 mM HEPES, 800 mM NaCl, pH 7.5, 0.1% (w/v) DDM, 0.02% (w/v) CHS, 20 mM imidazole, 10% (v/v) glycerol, and 20 µM indicated complexing ligand, followed by 10 cv of Wash Buffer II (25 mM HEPES, 150 mM NaCl, pH 7.5, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 10% (v/v) glycerol, and 20 µM indicated complexing ligand). Proteins were eluted in 2.5 cv of Wash Buffer II + 250 mM imidazole, concentrated in a 100 kDa molecular weight cut-off Vivaspin 20 concentrator (Sartorius Stedim) to 500 µl, and imidazole was removed by desalting the protein over PD MiniTrap G-25 columns (GE Healthcare). The C-terminal 10× His-tag was removed by addition of His-tagged PreScission protease (GenScript) and incubation overnight at 4 °C. Protease, cleaved His-tag and uncleaved protein were removed by passing the suspension through equilibrated TALON IMAC resin (Clontech) and collecting the flow-through. 5-HT2BR-ligand complexes were then concentrated to ~30 mg/ml with a 100 kDa molecular weight cut-off Vivaspin 500 centrifuge concentrator (Sartorius Stedim). Protein purity and monodispersity were tested by analytical size-exclusion chromatography.
Lipidic cubic phase crystallization.
5-HT2BR-ligand complexes were reconstituted into lipidic cubic phase (LCP) by mixing protein and a monoolein/cholesterol mixture in a ratio of 40%:54%:6% using the twin-syringe method5. Crystallization was done on 96-well glass sandwich plates (Marienfeld GmbH) in 50 nL LCP drops dispensed from a 10 uL gas-tight pipette (Hamilton) using a handheld dispenser (Art Robbins Instruments) and overlaid with 1 µL of precipitant solution. Upon optimization, crystals were obtained in 100 mM Tris-HCl pH 8.0, 100 mM sodium formate, 30% v/v PEG400 (h5-HT2BR-BRIL-1-LY266097), 100 mM Tris-HCl pH 7.4–7.7, 30–50 mM ammonium tartrate dibasic, 30% v/v PEG400 (h5-HT2BR-BRIL-1-lisuride), 100 mM Tris-HCl pH 7.2–8.0, 170–190 mM potassium phosphate monobasic, 30% v/v PEG400 (h5-HT2BR-BRIL-2-methylergonovine), 100 mM Tris-HCl pH 7.3–7.5, 40–100 mM MgCl2, 30% v/v PEG400 (h5-HT2BR-A225G5.46-BRIL-3-methysergide). All crystals grew to a maximum size of ~70 µm ×30 µm ×20 µm within three days and were harvested directly from the LCP matrix using MiTeGen micromounts before flash-freezing and storage in liquid nitrogen.
Data collection, structure solution and refinement.
X-ray data were collected at the 23ID-B and 23ID-D beamline (GM/CA CAT) at the Advanced Photon Source, Argonne, IL using a 10 µm minibeam at a wavelength of 1.0330 Å and either a MarMosaic 300 CCD or Eiger-16m detector (Dectris). Diffraction data were collected by exposing the crystals for 1–3 s to unattenuated beam using 1° oscillation. Full datasets for each complex were assembled from several crystals due the rapid onset of radiation decay at such high doses. Data were indexed, integrated, scaled, and merged using HKL300043, and initial phases were obtained by molecular replacement in PHASER44 using the 5-HT2BR coordinates from PDB ID: 4IB4 with the ligand, extracellular loops, and orthosteric residues removed. Refinement was performed with PHENIX45 and REFMAC, and included extensive use of simulated annealing to remove model bias. Density was manually examined and coordinates were rebuild in COOT46 using |2Fo| - |Fc|, |Fo| - |Fc|, and omit maps. After refinement, we do not observe any ramachandran outliers in any of the structures: 96.34% and 3.66% (5-HT2BR-methylergonovine), 95.29% and 4.71% (5-HT2BR - A225G5.46-methysergide), 96.7% and 3.3% (5-HT2BR -lisuride), as well as 97.47% and 2.53% (5-HT2BR -LY266097) of residues are in favored and allowed regions as defined by ramachandran statistics. We further observe low molprobity clashscores of 4.48, 4.94, 3.77, and 3.97 for 5-HT2BR -methylergonovine, 5-HT2BR - A225G5.46-methysergide, 5-HT2BR -Lisuride, and 5-HT2BR -LY266097, respectively.
Gq-mediated Calcium Flux FLIPR Assay.
Stable cell lines were generated for wild-type 5-HT2BR and all 5-HT2BR mutants using the Flp-In 293 T-Rex Tetracycline inducible system (Invitrogen, mycoplasma-free). Cell lines were maintained in DMEM containing 10% FBS, 10 µg/mL Blasticidin (Invivogen), and 100 µg/mL Hygromycin B (KSE Scientific). The day before the FLIPR assay, tetracycline-induced cells were seeded into 384-well poly-L-lysine-coated black plates at a density of 10,000 cells/well in DMEM containing 1% dialyzed FBS. On the day of the assay, the cells were incubated with Fluo-4 Direct dye (Invitrogen, 20 µl/well) for 1 h at 37 °C, which was reconstituted in drug buffer (20 mM HEPES-buffered HBSS, pH 7.4) containing 2.5 mM probenecid. After dye load, cells were allowed to equilibrate to room temperature for 15 minutes, and then placed in a FLIPRTETRA fluorescence imaging plate reader (Molecular Dynamics). Drug dilutions were prepared at 5X final concentration in drug buffer (20 mM HEPES-buffered HBSS, pH 7.4) containing 0.1% BSA and 0.01% ascorbic acid, final concentration. Drug dilutions were aliquoted into 384-well plastic plates and placed in the FLIPRTETRA for drug stimulation. Drug solutions used for FLIPR assay were exactly the same as used for the Tango assay. Fluorescence for the FLIPRTETRA were programmed to read baseline fluorescence for 10 s (1 read/s), and afterward 5 µl of drug per well was added and read for a total of 3 min (1 read/s). Fluorescence in each well was normalized to the average of the first 10 reads for baseline fluorescence, and then the maximum-fold increase was determined and calculated as fold-over-basal. Fold-over-basal was plotted as a function of drug concentration, and data were normalized to percent 5-HT stimulation. Data was plotted and non-linear regression was performed using “log(agonist) vs. response” in Graphpad Prism 5.0 to yield Emax and EC50 parameter estimates.
β-Arrestin2 Recruitment Tango Assay.
Measurement of β-Arrestin2 recruitment at 5-HT2BR and mutants utilized constructs that contain the TEV cleavage site and the tetracycline transactivator (tTA) fused to the C terminus of the receptor. Assays were designed and performed as previously described47. HTLA cells expressing TEV fused-β-Arrestin2 (derived from HEK cells and kindly provided by Dr. Richard Axel) were grown and maintained in DMEM containing 10% FBS and 5 µg/mL Puromycin and 100 µg/mL Hygromycin B. On the day of transfection, media was removed from HTLA cells, washed with PBS, and DMEM containing 10% dialyzed FBS and no antibiotics were added. After at least one hour, cells were transfected with the 5-HT2BR or mutant constructs using a calcium phosphate precipitation method48. After at least 24 hours, media was decanted, washed with PBS, and detached using trypsin. Cells were collected via centrifugation, resuspended in DMEM containing 1% dialyzed FBS, and plated into poly-L-lysine coated 384-well white clear bottom cell culture plates at a density of 7,000–10,000 cells per well in a total of 40 µL. The cells were incubated at 37°C and 5% CO2 for at least 6 hours before receiving drug stimulation. Drug solutions were prepared in drug buffer (20 mM HEPES-buffered HBSS, pH 7.4) containing 0.1% BSA and 0.01% ascorbic acid, final concentration. Drug stimulation was performed using the FLIPRTETRA by dispensing 10 µL per well. Drug solutions used for the Tango assay were exactly the same as used for the FLIPR assay, which were performed on the same day. Plates were incubated at 37°C and 5% CO2 for at least 20–22 hours. Afterwards, media was decanted, plates were blotted for excess media, and 20 µl per well of BrightGlo reagent (Promega, diluted 1:20 in drug buffer) was added. The plate was incubated for 20 min at room temperature in the dark before being counted using a Wallac Trilux MicroBeta luminescence counter. Relative luminescence units (RLU) were plotted as a function of drug concentration, normalized to % 5-HT, and analyzed using “log(agonist) vs. response” in GraphPad Prism 5.0.
Phosphoinositide Hydrolysis Assay.
Phosphoinositide (PI) hydrolysis assays measuring Gq-mediated inositol phosphate (IP) accumulation were performed using a scintillation proximity assay (SPA)49,50. On the day before the assay, cells were seeded into 96-well poly-lysine coated white plates at a density of 40–50,000 cells/well in 200 µL inositol-free DMEM (Caisson Labs) containing 1 µCi/well (final concentration) of [3H]-myo-inositol (PerkinElmer) and incubated overnight for 16–18 hours at 37°C and 5% CO2. Next day, label media was siphoned off and replaced with 200 µL inositol-free DMEM. Drug stimulation was initiated with addition of 50 µL of 5X concentrated drug dilutions made up in drug buffer (20 mM HEPES-buffered HBSS, pH 7.4) containing 0.1% BSA and 0.01% ascorbic acid, final concentration. To capture IP accumulation, lithium chloride (10 µL/well, 15 mM final concentration) was added 15 minutes before lysis. The assay was terminated by decanting media and adding 40 µL of 50 mM cold formic acid. After overnight incubation at 4°C, 10 µL of lysates were added to 96-well flexible, clear microplates (PerkinElmer) containing 75 µL of 0.2 mg/well RNA binding yttrium silicate beads (PerkinElmer), and incubated for one to two hours wrapped in foil on a shaker at room temperature. Afterwards, plates were centrifuged at 300xg for 1 minute, and radioactivity was measured using a Wallac MicroBeta Trilux plate reader (PerkinElmer) using SPA paralux setting. IP accumulation data were plotted as counts per minute (CPM) as a function of drug concentration, normalized to % 5-HT stimulation, and analyzed using “log(agonist) vs. response” in GraphPad Prism 5.0.
Bioluminescence Resonance Energy Transfer (BRET) Arrestin Assay.
To measure 5-HT2BR-mediated β-Arrestin2 recruitment as measured by BRET1, HEK293T cells (ATCC CRL-11268; 59587035; mycoplasma-free) in DMEM supplemented with 10% dialyzed FBS were co-transfected in a 1:1:15 ratio with human 5-HT2BR containing C-terminal fused renilla luciferase (RLuc8), GRK2, and Venus-tagged N-terminal β-arrestin2 in DMEM supplemented with 10% dialyzed FBS using TransiT-2020 (Mirus). To measure 5-HT2BR-mediated Gq activation via Gq/γ1 dissociation as measured by BRET2, HEK293T cells in DMEM supplemented with 10% dialyzed FBS were co-transfected in a 1:1:1:1 ratio with RLuc8-fused at residue 121 of human Gαq (Gαq- RLuc8), a GFP2-fused to the C-terminus of human Gγ1(Gγ1-GFP2), human Gβ1, and either 5-HT2BR WT or mutant using TransiT-2020. After at least 18–24 hours, transfected cells were plated in poly-lysine coated 96-well white clear bottom cell culture plates in DMEM containing 1% dialyzed FBS at a density of 30–40,000 cells in 200 µl per well and incubated overnight. The next day, media was decanted and cells were washed with 60 µL of drug buffer (1× HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4), then 60 µL of drug buffer was added per well. For kinetic experiments, plates were incubated at 37°C at least 20 minutes prior to receiving drug stimulation. Afterwards, 30 µL of drug (3X) was added per well and incubated for designated time points. For β-Arrestin2 recruitment assays, 10 µL of the RLuc substrate, coelenterazine h (Promega, 5 µM final concentration) was added per well, incubated an additional 15 minutes to allow for substrate diffusion, and plates were immediately read for both luminescence at 485 nm and fluorescent eYFP emission at 530 nm for 1 second per well using a Mithras LB940. The ratio of eYFP/RLuc was calculated per well and the net BRET1 ratio was calculated by subtracting the eYFP/RLuc per well from the eYFP/RLuc ratio in wells without Venus-β-Arrestin2 present. For Gq/γ1 dissociation assays, 10 µL of the RLuc substrate coelenterazine 400a (Nanolight Technology, 5 µM final concentration) was added per well, incubated an additional 10 minutes to allow for substrate diffusion, and plates were immediately read for both luminescence at 400 nm and fluorescent GFP2 emission at 510 nm for 1 second per well using a Mithras LB940. BRET2 ratios were calculated as a the ratio of GFP2 fluorescence over Either the net BRET1 or BRET2 ratios were plotted as a function of drug concentration using Graphpad Prism 5 (Graphpad Software Inc., San Diego, CA). Data were normalized to % 5-HT stimulation and analyzed using nonlinear regression “log(agonist) vs. response” in Graphpad Prism 5.0.
Radioligand Binding Assays.
Binding assays were performed using stable Flp-In 293 T-Rex Tetracycline-inducible cell lines for 5-HT2BR or mutants. Binding assays were set up in 96-well plates in standard binding buffer (50 mM Tris, 10 mM MgCl2, 0.1 mM EDTA, 0.1% BSA, 0.01% ascorbic acid, pH 7.4) using [3H]-LSD (PerkinElmer, specific activity = 76–82.2 Ci/mmol) as the radioligand. For competitive binding assays, all assays were performed with various concentrations of cold unlabeled ligands (1 pM to 10 µM), [3H]-LSD (0.2 −0.7 nM), and resuspended membranes in a total volume of 225 µL. Competition assays were incubated for 4 hours at 37°C in a humidified incubator until harvesting. For [3H]-LSD dissociation assays, assays utilized at least 2–3 radioligand concentrations (0.3–2.0 nM) and were pre-incubated for 2 hours at 37°C in a humidified incubator in a total of 200 µL volume per well. To initiate dissociation, 10 µL of cold excess LSD (10 µM) was added per well at specific time points ranging from 2 minutes to 7 hours, and immediately at time = 0 min plates were harvested. For all assays, non-specific activity was defined as the addition of 10 µM SB 206553. Plates were harvested using vacuum filtration onto 0.3% polyethyleneimine pre-soaked 96 well Filtermat A (PerkinElmer) and washed three times 250 µL per well of cold wash buffer (50 mM Tris, pH 7.4). Filters were dried and scintillation cockatil (Meltilex, PerkinElmer) was melted and allowed to cool to room temperature. Afterwards, filter plates were placed in cassettes and read using a Wallac Trilux Microbeta counter (PerkinElmer). Data were analyzed either using “One-site-homologous” to yield Kd, “One-site-Fit Ki” to yield Ki or “Dissociation-One phase exponential decay” to yield estimates of koff using Graphpad Prism 5.0.
Chemistry General Procedures.
HPLC spectra for all compounds were acquired using an Agilent 1200 Series system with DAD detector. Chromatography was performed on a 2.1×150 mm Zorbax 300SB-C18 5 μm column with water containing 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.4 mL/min. The gradient program was as follows: 1% B (0−1 min), 1−99% B (1−4 min), and 99% B (4−8 min). High-resolution mass spectra (HRMS) data were acquired in positive ion mode using an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. Nuclear Magnetic Resonance (NMR) spectra were acquired on a Bruker DRX-600 spectrometer (600 MHz 1H, 150 MHz 13C) or a Varian Mercury spectrometer (400 MHz 1H, 100 MHz 13C). Chemical shifts are reported in ppm (δ). Preparative HPLC was performed on Agilent Prep 1200 series with UV detector set to 254 nm. Samples were injected into a Phenomenex Luna 75 × 30 mm, 5 μm, C18 column at room temperature. The flow rate was 40 mL/min. A linear gradient was used with 10% (or 50%) of MeOH (A) in H2O (with 0.1 % TFA) (B) to 100% of MeOH (A). HPLC was used to establish the purity of target compounds. All final compounds had > 95% purity using the HPLC methods described above. Synthesis schemes and compound purification details can be found in Supplementary Note 1.
Supplementary Material
Acknowledgements
This work was supported by NIH Grant R01MH61887 (B.L.R.), the NIMH Psychoactive Drug Screening Program Contract (B.L.R.), R01NS100930 (J.J.), U19MH82441 (J.J. and B.L.R.), the Michael Hooker Distinguished Chair of Pharmacology (B.L.R.), and F31-NS093917 (R.H.J.O). We thank J. Smith, R. Fischetti and the staff of APS GM/CA for assistance in the development and use of the minibeam and beamtime at GM/CA-CAT beamline 23-ID at the Advanced Photon Source, which is supported by National Cancer Institute grant Y1-CO-1020 and National Institute of General Medical Sciences grant Y1-GM-1104. Use of the Advanced Photon Source was supported by the Office of Science of the U.S. Department of Energy.
Footnotes
Competing financial interests
The authors declare no competing financial interests
Data availability
Macromolecular structure data is available via the worldwide protein data bank (wwPDB): 5-HT2BR-methylergonovine (PDB code: 6DRY), 5-HT2BR-A225G5.46-methysergide (PDB code: 6DRZ). 5-HT2BR-lisuride (PDB code: 6DRX), 5-HT2BR-LY266097 (PDB code: 6DS0). Generated and analyzed data sets that support the findings of this study are available in the source data files or from the corresponding authors upon reasonable request.
References
- 1.Berger M, Gray JA & Roth BL The expanded biology of serotonin. Annu Rev Med 60, 355–66 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCorvy JD & Roth BL Structure and function of serotonin G protein-coupled receptors. Pharmacol Ther 150, 129–42 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen JA & Roth BL Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu Rev Pharmacol Toxicol 51, 117–44 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Vickers SP, Clifton PG, Dourish CT & Tecott LH Reduced satiating effect of d-fenfluramine in serotonin 5-HT(2C) receptor mutant mice. Psychopharmacology (Berl) 143, 309–14 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Roth BL Drugs and valvular heart disease. N Engl J Med 356, 6–9 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Rothman RB et al. Evidence for possible involvement of 5-HT2B receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation 102, 2836–2841 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Zanettini R et al. Valvular heart disease and the use of dopamine agonists for Parkinson’s disease. N Engl J Med 356, 39–46 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Gustafsson BI, Hauso O, Drozdov I, Kidd M & Modlin IM Carcinoid heart disease. Int J Cardiol 129, 318–24 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Huang XP et al. Parallel functional activity profiling reveals valvulopathogens are potent 5-hydroxytryptamine(2B) receptor agonists: implications for drug safety assessment. Mol Pharmacol 76, 710–22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papoian T et al. Regulatory Forum Review*: Utility of in Vitro Secondary Pharmacology Data to Assess Risk of Drug-induced Valvular Heart Disease in Humans: Regulatory Considerations. Toxicologic Pathology 45, 381–388 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Hauso O et al. Long-term serotonin effects in the rat are prevented by terguride. Regul Pept 143, 39–46 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Wacker D et al. Structural features for functional selectivity at serotonin receptors. Science 340, 615–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wacker D et al. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 168, 377–389 e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katritch V, Cherezov V & Stevens RC Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci 33, 17–27 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Latorraca NR, Venkatakrishnan AJ & Dror RO GPCR Dynamics: Structures in Motion. Chem Rev 117, 139–155 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Wacker D, Stevens RC & Roth BL How Ligands Illuminate GPCR Molecular Pharmacology. Cell 170, 414–427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C et al. Structural basis for molecular recognition at serotonin receptors. Science 340, 610–4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin W et al. Crystal structure of the human 5-HT1B serotonin receptor bound to an inverse agonist. Cell Discov 4, 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng Y et al. 5-HT2C Receptor Structures Reveal the Structural Basis of GPCR Polypharmacology. Cell 172, 719–730 e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherezov V et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258–65 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ring AM et al. Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ballesteros JA & Weinstein H Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. in Methods in Neurosciences Vol. 25 (ed. Sealfon SC) 366–428 (Academic Press, 1995). [Google Scholar]
- 23.Rasmussen SG et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469, 175–80 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rasmussen SG et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCorvy JD et al. Structure-inspired design of beta-arrestin-biased ligands for aminergic GPCRs. Nat Chem Biol 14, 126–134 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stevens RC et al. The GPCR Network: a large-scale collaboration to determine human GPCR structure and function. Nat Rev Drug Discov 12, 25–34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bredberg U, Eyjolfsdottir GS, Paalzow L, Tfelt-Hansen P & Tfelt-Hansen V Pharmacokinetics of methysergide and its metabolite methylergometrine in man. Eur J Clin Pharmacol 30, 75–7 (1986). [DOI] [PubMed] [Google Scholar]
- 28.Wacker D et al. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc 132, 11443–5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nichols DE, Monte A, Huang X & Marona-Lewicka D Stereoselective pharmacological effects of lysergic acid amides possessing chirality in the amide substituent. Behav Brain Res 73, 117–9 (1996). [DOI] [PubMed] [Google Scholar]
- 30.Hofmann C et al. Lisuride, a dopamine receptor agonist with 5-HT2B receptor antagonist properties: absence of cardiac valvulopathy adverse drug reaction reports supports the concept of a crucial role for 5-HT2B receptor agonism in cardiac valvular fibrosis. Clin Neuropharmacol 29, 80–6 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Audia JE et al. Potent, selective tetrahydro-beta-carboline antagonists of the serotonin 2B (5HT2B) contractile receptor in the rat stomach fundus. J Med Chem 39, 2773–80 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Sato T et al. Pharmacological Analysis and Structure Determination of 7-Methylcyanopindolol-Bound beta1-Adrenergic Receptor. Mol Pharmacol 88, 1024–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahuja S & Smith SO Multiple switches in G protein-coupled receptor activation. Trends Pharmacol Sci 30, 494–502 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Kruse AC et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nygaard R et al. The dynamic process of beta(2)-adrenergic receptor activation. Cell 152, 532–42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Staus DP et al. Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 535, 448–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Che T et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 172, 55–67 e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wacker D et al. Structural features for functional selectivity at serotonin receptors. Science 340, 615–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wacker D et al. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 168, 377–389 e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roth CB, Hanson MA & Stevens RC Stabilization of the human beta2-adrenergic receptor TM4-TM3-TM5 helix interface by mutagenesis of Glu122(3.41), a critical residue in GPCR structure. J Mol Biol 376, 1305–19 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chun E et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–76 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caffrey M & Cherezov V Crystallizing membrane proteins using lipidic mesophases. Nat Protoc 4, 706–31 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minor W, Cymborowski M, Otwinowski Z & Chruszcz M HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62, 859–66 (2006). [DOI] [PubMed] [Google Scholar]
- 44.McCoy AJ et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kroeze WK et al. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat Struct Mol Biol 22, 362–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jordan M, Schallhorn A & Wurm FM Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res 24, 596–601 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bourdon DM, Wing MR, Edwards EB, Sondek J & Harden TK Quantification of isozyme-specific activation of phospholipase C-beta2 by Rac GTPases and phospholipase C-epsilon by Rho GTPases in an intact cell assay system. Methods Enzymol 406, 489–99 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Huang XP et al. Parallel functional activity profiling reveals valvulopathogens are potent 5-hydroxytryptamine(2B) receptor agonists: implications for drug safety assessment. Mol Pharmacol 76, 710–22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.