

ABSTRACT
Renal osteodystrophy (ROD) is the bone component of chronic kidney disease mineral and bone disorder (CKD‐MBD). ROD affects bone quality and strength through the numerous hormonal and metabolic disturbances that occur in patients with kidney disease. Collectively these disorders in bone quality increase fracture risk in CKD patients compared with the general population. Fractures are a serious complication of kidney disease and are associated with higher morbidity and mortality compared with the general population. Furthermore, at a population level, fractures are at historically high levels in patients with end‐stage kidney disease (ESKD), whereas in contrast the general population has experienced a steady decline in fracture incidence rates. Based on these findings, it is clear that a paradigm shift is needed in our approach to diagnosing and managing ROD. In clinical practice, our ability to diagnose ROD and initiate antifracture treatments is impeded by the lack of accurate noninvasive methods that identify ROD type. The past decade has seen advances in the noninvasive measurement of bone quality and strength that have been studied in kidney disease patients. Below we review the current literature pertaining to the epidemiology, pathology, diagnosis, and management of ROD. We aim to highlight the pressing need for a greater awareness of this condition and the need for the implementation of strategies that prevent fractures in kidney disease patients. Research is needed for more accurate noninvasive assessment of ROD type, clinical studies of existing osteoporosis therapies in patients across the spectrum of kidney disease, and the development of CKD‐specific treatments. © 2018 The Authors. JBMR Plus published by Wiley Periodicals, Inc. on behalf of the American Society for Bone and Mineral Research.
Keywords: CKD, ESKD, CKD‐MBD, FRACTURES, OSTEOPOROSIS, BONE DISEASE
Introduction
Renal bone disease or renal osteodystrophy (ROD) is a complex disorder of bone in patients with chronic kidney disease (CKD).1, 2, 3 Progressive kidney disease results in metabolic and hormonal disturbances that impair bone quality and is characterized by abnormal remodeling (low, normal or high turnover) with or without abnormalities in mineralization. Altered bone and mineral metabolism in kidney disease is part of a broader systemic disorder defined by the Kidney Disease Improving Global Outcomes (KIDGO) as CKD‐mineral and bone disorder (CKD‐MBD).4 CKD‐MBD is manifested by either one or a combination of: 1) abnormalities of calcium, phosphate, parathyroid hormone (PTH), or vitamin D metabolism; 2) abnormalities of bone turnover, mineralization, volume or strength, and linear growth; and 3) vascular or soft tissue calcification. Manifestations of CKD‐MBD begin early in CKD, with near‐normal kidney function, and the severity of CKD‐MBD and its clinical outcomes of increased fracture risk increase in parallel with declining renal function.5, 6, 7
In practical terms, our ability to confidently diagnose ROD type in CKD and to initiate strategies that could prevent fractures remains limited by the lack of accurate and noninvasive diagnostic tools. The measurement of calcium, phosphate, vitamin D, and PTH identify metabolic abnormalities associated with CKD‐MBD, but these remain poor markers of ROD and have insufficient discrimination of ROD turnover type and mineralization.8, 9 Bone turnover markers (BTMs), routinely used in non‐CKD patients to monitor fracture risk and osteoporosis therapy, lack validation in CKD and end‐stage chronic kidney disease (ESKD) and are consequently used infrequently. Transiliac crest bone biopsy remains the gold standard tool to assess bone quality in metabolic bone diseases; however, bone biopsy is invasive, expensive, painful, requires time‐consuming measurements, and is available at only a few centers worldwide.
Over the past decade, advancements in the field of metabolic bone diseases have the potential to alter the paradigm of how we approach renal bone disease. In 2017, KDIGO updated its guidelines to recommend risk classification of patients with kidney disease for fracture by measurement of areal bone mineral density (BMD) by dual‐energy X‐ray absorptiometry (DXA).10 Noninvasive measurement of cortical and trabecular microarchitecture is now possible with both high‐resolution imaging techniques and novel algorithms that reinterpret grayscale variation in DXA images. The diagnostic utility of BTMs in kidney disease is also being refined and fracture risk assessment tools based on clinical risk factors alone have been developed. Below we review the state of the field pertaining to the epidemiology, pathology, diagnosis, and management of renal bone disease in patients with CKD 3‐5D. Furthermore, we provide our personal interpretation of the current issues and advocate for a greater clinical awareness of this condition and the pressing need to develop and test strategies to prevent fractures in these patients.
Pathophysiology of Renal Osteodystrophy
ROD is the bone component of CKD‐MBD and is defined as alterations in bone morphology associated with progressive CKD that can be quantified by bone histomorphometry.(4) It is important to note that although CKD is defined as an estimated glomerular filtration rate (eGFR) <60 mL/min/1.73m2, changes of CKD‐MBD are present even with mild renal impairment (eGFR 60–90 mL/min/1.73m2).11, 12, 13 The historically complex nomenclature pertaining to ROD was simplified by the KDIGO working group and aligned with the standard nomenclature recommended by the American Society for Bone and Mineral Research.4, 14 Classification of ROD type was based on the turnover (high, normal, or low), mineralization (normal or abnormal), and volume (high, normal, or low) of bone (TMV classification system, Table 1). The aim of the revised classification was to encompass the most significant bone abnormalities in kidney disease that would inform management decisions.
Table 1.
Bone Turnover, Mineralization, and Volume (TMV) Classification System for Renal Osteodystrophya
| Turnover | Mineralization | Volume |
|---|---|---|
| Low | Low | |
| Normal | ||
| Normal | Normal | |
| Abnormal | ||
| High | High |
Reprinted with permission from aMoe et al.4
Historically, bone abnormalities in patients with kidney disease were attributed to alterations in PTH and 1,25 dihydroxyvitamin D [1,25(OH)2D]. The characterization of circulating and bone‐derived hormones and the changes that occur with progressive kidney disease have fundamentally altered our understanding of the changes of CKD‐MBD and the pathogenesis of ROD. A detailed discussion of these changes is beyond the scope of this review. However, progressive CKD is generally associated with increased levels of PTH, fibroblast growth factor 23 (FGF‐23), osteoprotegerin, sclerostin, and Dickkopf‐related protein 1 (DKK1) and reductions in α‐Klotho, serum 25‐hydroxyvitamin D [25(OH)D], and [1,25(OH)2D].12, 13, 15, 16, 17, 18, 19, 20, 21, 22 The expression of bone turnover such as bone‐specific alkaline phosphatase (BSAP), procollagen type 1 N‐terminal (P1NP), C‐terminal telopeptide of type 1 collagen (CTX), and tartrate‐resistant acid phosphatase (TRAP‐5b) is more variable and dependent not only on the degree of renal impairment but also bone turnover.23 Together, these changes impact bone formation, resorption, and mineralization through their effects on osteoblast and osteocyte function.
Expression of skeletal proteins such as FGF‐23, dentin matrix protein 1, and matrix extracellular phosphoglycoprotein is also altered in CKD. In a study by Periera and colleagues,24 FGF‐23 and DMP1 expression was increased across all stages of CKD (compared with healthy controls), whereas there was no difference in MEPE expression. FGF‐23 and DMP1 were inversely related to osteoid accumulation, whereas MEPE was inversely related to bone volume, suggesting a role for FGF‐23 and DMP1 in bone mineralization and MEPE in the regulation of bone volume. The Wnt/β‐catenin pathway is essential for normal osteoblast differentiation and function and therefore normal bone formation. Sclerostin and Dickkopf‐1 (DKK1) are two circulating inhibitors of this pathway; these inhibit lipoprotein receptor‐related protein 5/6 activation of Wnt signaling and impair normal osteoblast differentiation.21, 25, 26, 27 Sclerostin is primarily expressed in skeletal tissue, and its expression is maintained during aging; in contrast, expression of DKK1 is more general and decreases in bone with age.28, 29, 30 Inhibition of sclerostin and DKK1 leads to increased bone formation in humans and animal models.31, 32, 33, 34, 35 Furthermore, animal studies suggest that DKK1 overexpression negatively impacts bone healing, suggesting a role for DKK1 inhibition during the fracture repair process.36, 37 Sclerostin and DKK1 levels are elevated in CKD, with sclerostin levels increased early in CKD, and generally preceding the rise of FGF‐23 and β‐catenin.27, 38 In a study of ESKD patients, sclerostin levels were inversely associated with reduced bone formation and bone loss over a 1‐year period.39, 40 These studies highlight the complex endocrine and paracrine bone‐renal signaling pathways and suggest that the pathogenesis of ROD is driven by changes within osteocytes that occur early in CKD.
In 2001, the National Institutes of Health defined osteoporosis as a skeletal disorder characterized by compromised bone strength, predisposing to an increased risk of fracture.41 Bone strength is a combined measure that reflects both bone density and quality. Bone density can be determined by DXA; however, bone quality is more broadly defined and pertains to bone material properties, such as bone remodeling, microdamage, microarchitecture, and collagen and mineral characteristics.42 Disorders of bone quality accumulate with age and directly affect the mechanical properties of bone and therefore fracture risk. In health, damaged areas of bone are constantly targeted for ongoing remodeling and repair. In CKD, some or all aspects of bone quality may be impaired (Table 2).43, 44, 45, 46, 47, 48 This includes defective mineralization (osteomalacia), abnormal remodeling (low‐ or high‐turnover bone disease), increased microarchitectural impairment (cortical porosity), and accumulation of microdamage and advanced glycation end products (AGE) cross‐linking.43, 44
Table 2.
Bone Changes Associated With Hormonal and Metabolic Changes of End‐Stage Kidney Disease
| Decreased bone density |
|---|
| Alterations in bone microarchitecture |
| • Cortical porosity |
| • Cortical thinning and trabecularization |
| • Trabecular thinning and dropout |
| • Disruption in balance and orientation of newly formed and mature bone |
| Decreased bone quality |
| • Mineralization (osteomalacia) |
| • Abnormal remodeling (loss of normal repair processes) |
| о Adynamic bone disease |
| о Low turnover |
| о High turnover |
| • Microdamage accumulation |
| о Reduced resistance to impact |
| • Advanced glycation end products cross‐linking |
| о Loss of elasticity and tissue embrittlement |
Kidney disease patients have both traditional and CKD‐specific risk factors for bone disease and fractures (Table 3). For example, older age, low body weight, postmenopausal status, a history of previous fractures, increased risk of falls, and the use of immunosuppressive medications that promote bone loss are all common in CKD cohorts.49, 50, 51, 52 Metabolic disturbances that occur due to CKD, including decreased levels of nutritional and activated vitamin D, disordered calcium and phosphorous metabolism, premature hypogonadism, hyperparathyroidism, and metabolic acidosis all contribute to abnormalities in bone strength.4, 53, 54, 55, 56 Patients with kidney disease are also more likely to have reduced physical activity, postural hypotension, and decreased muscle mass, which increase susceptibility to falls and fractures.57, 58 The age‐specific risk of fracture associated with CKD is higher in younger age groups, but the absolute risk of fracture increases with age, suggesting an interaction between CKD‐specific and traditional risk factors for fracture in older CKD and ESKD patients.7, 59, 60
Table 3.
General and CKD‐Specific Risk Factors for Bone Loss and Fractures
| General risk factors | CKD‐specific |
|---|---|
| Patient‐related (non‐modifiable) | • Hyperparathyrodism |
| • Age | • Low nutritional and activated vitamin D |
| • Sex | • Disordered mineral metabolism |
| • Ethnicity | • Chronic inflammation |
| • Past history of fracture | • Metabolic acidosis |
| • Premature hypogonadism | |
| General (modifiable) | • Medications |
| • Low physical activity | о Steroids |
| • Smoking | о Phosphate binders (eg, aluminium) |
| • Alcohol | о CNI |
| • Medications (eg, streoids) | • Dietary restriction |
| • Diabetes | • Dialysis‐related amyloidosis |
| • Sarcopenia | |
| • Chronic inflammatory disorders | • Higher prevalence of general risk factors for osteoporosis |
CKD = chronic kidney disease; CNI = calcineurin inhibitor.
Bone biopsy studies in CKD patients have provided important insights into the patterns of ROD observed in patients across the spectrum of CKD. In patients with CKD stages 3 to 5 (non‐dialysis), some data suggest that up to three‐quarters of patients have histologic evidence of renal osteodystrophy.1, 61, 62, 63, 64, 65, 66 Depending on the cohort studied, there is considerable variation in the prevalence of ROD types, and findings include a predominance of high or adynamic bone disease and even normal bone. In ESKD, recent large bone biopsy studies have characterized tissue‐level impairments in bone quality that reflect current CKD‐MBD management strategies. In a seminal study, Malluche and colleagues used the TMV classification system to evaluate 630 bone biopsies from adult hemodialysis patients from Europe and the United States.9 For turnover, 58%, 25%, and 18% of patients had low, high, and normal turnover, respectively. There were clear racial differences in turnover: low bone turnover predominated in whites (62%) and normal or high turnover predominated in blacks (68%). For mineralization, defects were uncommon (3% of patients). For volume, low, normal, or high cancellous bone volume was equally distributed among whites, but high volume predominated in blacks. Furthermore, blacks had normal cortical thickness with higher porosity, but whites had an equal distribution of low or normal thickness with high or normal porosity. Trabecular microarchitecture was also examined, with trabecular thickness being low in 37% of patients, normal in 40% of patients, and high in 13% of patients. Trabecular separation was normal in most (78%) of patients. Interestingly, both black and white patients with high bone turnover had increased porosity, and more than 80% of patients with low cancellous bone volume had thin trabeculae. These findings were supported in a recent study, where bone histomorphometric assessment of turnover (bone formation rate/bone surface [BFR/BS]) was performed in 492 dialysis patients.8 Low turnover was the dominant lesion being present in 59% (n = 289), whereas high turnover was present in 17% (n = 83). In a smaller study of 35 hemodialysis patients, those with low bone turnover had more microstructural abnormalities (lower cancellous bone volume and trabecular thickness) than those with high or normal turnover.67 Conversely, those with high turnover had reduced mineral content and reduced stiffness.
These data suggest that early CKD is characterized by high bone turnover. In ESKD, low‐turnover disease is the dominant lesion; however, high‐turnover lesions are present in a significant proportion of patients, whereas mineralization defects are comparably low in both groups. Importantly, the studies above highlight that ROD is more than bone turnover but a global disorder of bone quality and strength. It has been proposed that ROD and fractures related to kidney disease be considered as a subtype of osteoporosis (analogous to steroid‐induced osteoporosis), as bone quality and strength are impaired to a greater extent than age‐related osteoporosis.68 Although intentionally provocative, this definition highlights the need for a more practical and easily translatable definition of ROD. This should not only incorporate bone biopsy findings but surrogate parameters of bone strength (such as BTMs, DXA) that are easily accessible in daily clinical practice, facilitate diagnosis and inclusion in therapeutic trials, and inform treatment decisions.
Fracture Epidemiology in CKD and ESKD
The incidence and prevalence of fractures increases with CKD stage and has been reported to be 2‐ to 17‐fold higher in CKD patients compared with the general population.7, 59, 69, 70, 71, 72 Recent studies have provided important insights into secular trends of fracture epidemiology in ESKD, highlighting differences in fracture rates between general population and ESKD cohorts and in fracture incidence rates at the central and peripheral skeleton.
Longitudinal studies using USRDS and US Medicare data have compared the incidence of hip fractures in ESKD and non‐ESKD cohorts.73, 74 In general, hip fracture rates in ESKD increased steadily from the early 1990s until the mid 2000s (an increase of 43%), with a nonsignificant reduction in incidence after 2004 (Fig. 1).73 More recent data from the US Nationwide Inpatient Sample examined age‐ and sex‐standardized hip fracture rates in ESKD and found a 12.6% decrease between 2003 and 2011.75 However, in 2010, hip fracture rates in ESKD remained 27% higher compared with 1996, and individuals aged 66 years or older with ESKD had a markedly higher incidence of hip fractures compared with those without ESKD (31.9 versus 8.0 per 1000 patient‐years). Furthermore, although these data suggest that hip fracture rates are decreasing, peripheral (arm and leg) fractures have more than doubled over the corresponding period.60 It is important to put these data in perspective, and the small decrease in hip fractures reported must be interpreted in the context of a persistently high overall fracture rate in ESKD patients compared with the general population. All studies report that current fracture rates are significantly higher in ESKD compared with general population cohorts, and fractures remain more prevalent today than they were in 1996.
Figure 1.
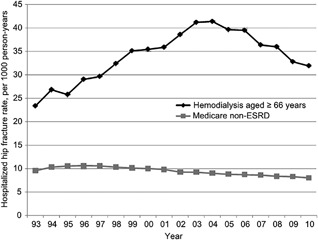
Adjusted hospitalized fracture rates per 1000 person‐years in Medicare point‐prevalent hemodialysis and non‐ESKD patients aged 66 years or older. Reprinted with permission from Arneson et al.73
The morbidity, mortality, and health care costs associated with fractures are higher for patients with kidney disease compared with the general population.68 Patients with CKD and ESKD experience longer hospitalization after a fracture, with reported mortality rates between 16% and 60%.74, 76, 77 Many patients do not return to their premorbid level of function after a hip fracture, with as many as 80% discharged to a long‐term‐care facility after a fracture.5 It was estimated that in 2010 hip fracture‐associated expenses in patients with CKD and ESKD were in excess of $600 million USD,(5) and in the general US population, the costs of fractures and associated morbidity are projected to increase to more than $25 billion USD by 2025.78
Noninvasive Assessment of Bone Quality and Fracture Risk in CKD
Bone biopsy is the gold standard for assessing bone quality in kidney disease and informs treatment options based on bone formation rates and mineralization characteristics. However, its utility as an everyday clinical tool is limited by lack of availability and long duration of time required to process and analyze bone tissue. Therefore, there is great interest in the use of noninvasive approaches to assess bone quality in kidney‐related bone disease so that fracture risk classification and the selection of antifracture treatments can be implemented broadly in the renal clinic. Imaging modalities such as DXA, Trabecular bone score (TBS), conventional Quantitative computed tomography (QCT), High‐resolution peripheral QCT (HR‐pQCT) and micro magnetic resonance imaging (MRI) assess bone density and/or structural aspects of bone quality, whereas PTH and BTMs assess aspects of bone quality that cannot be assessed by imaging, such as bone formation rates and mineralization. We will discuss the role of each of these in CKD.
Bone Density and the Fracture Risk Assessment Tool
DXA is the clinical standard to determine BMD and measure fracture risk in the general population and is integral to the World Health Organization definition of osteoporosis (T‐score ≤–2.5).79 Historically the role of DXA to assess bone health and fracture risk in CKD3‐5D was controversial, as small cross‐sectional studies did not demonstrate that DXA discriminated prevalent fractures, and BMD measurements did not predict type of ROD. However, several recent longitudinal studies in patients across the spectrum of CKD and ESKD have demonstrated that low BMD at the hip and forearm do predict incident fractures.80, 81, 82, 83 These studies reported that the WHO T‐scores perform similarly in patients with and without CKD, with regard to fracture prediction, and resulted in the revised KDIGO recommendation to include BMD measurement in patients with CKD 3‐5D to assess fracture risk.
In the general population, estimates of fracture prediction are improved by the addition of clinical risk factors to BMD measurements. The Fracture Risk Assessment (FRAX) was developed to provide 10‐year absolute risk of major osteoporotic or hip fracture by combining 10 clinical risk factors, with or without femoral neck BMD, into a fracture risk algorithm.51 The clinical relevance of FRAX in CKD remains unclear. In a study from the Canadian Multicentre Osteoporosis Study, 320 individuals with an eGFR <60 mL/min, and 1787 with an eGFR ≥60 mL/min were followed for a mean of 4.8 years.81 This study showed that FRAX did not differ in its ability to predict major osteoporotic fractures, despite differences in underlying renal function. It is important to note that the incidence of fractures in the CKD patients was low and most patients did not have evidence of CKD‐MBD. In a cross‐sectional study of 353 patients with CKD (mean eGFR of 28 mL/min), approximately 30% had prevalent fractures; FRAX with femoral neck BMD discriminated those with and without fractures but was not superior to femoral neck BMD alone.84 All three study groups (FRAX alone, FRAX with BMD, BMD alone) were better discriminants of fracture than age alone. In a study of 485 Japanese hemodialysis patients, FRAX did not predict increased fracture risk over a 3.3‐year median follow‐up.80 It is important to note the relatively short follow‐up of this study, along with concerns about the accuracy of the fracture risk assessment data. In a recent study of 718 hemodialysis patients who were followed for a period of 2 years, the area under the curve (AUC) for FRAX was 0.76 (95% confidence interval [CI] 0.69–0.82) for major fractures and 0.70 (95% CI 0.69–0.84) for hip fractures, and FRAX discriminated fractures better than individual elements in the FRAX algorithm, although this did not include BMD.85 These studies suggest that more research is needed to determine the usefulness of FRAX in CKD and ESKD. More specifically, some of the clinical factors included in current FRAX algorithms may not be relevant in these patients and CKD‐specific fracture assessment tools need to be developed. These should incorporate predictors of fracture specific to kidney disease, for example, bone alkaline phosphatase (BSAP) or PTH.86
Assessment of Bone Microarchitecture—High‐Resolution Imaging
An important limitation of DXA is that it does not assess the 3‐dimensional structure of bone. Therefore, high‐resolution imaging methods such as QCT, HR‐pQCT, and micro‐MRI have been developed to provide 3‐dimensional imaging of bone density and microarchitectural aspects of bone quality, including cortical and trabecular volumetric BMD, geometry, microarchitecture, and strength. The TBS, although not a high‐resolution imaging modality, has also emerged as an important tool in the assessment of bone microarchitecture and will be discussed below. Micro‐MRI assessment of bone microarchitecture has been evaluated in general and kidney disease cohorts; however, studies of fracture discrimination are lacking, as such no further discussion has been included in this review.87, 88, 89, 90, 91, 92, 93
Conventional QCT has a resolution of 300 μm3 and quantifies volumetric BMD and cortical and trabecular geometry. In CKD and ESKD, studies utilizing QCT have shown that cortical deficits predominate and these could discriminate and predict future fractures.94, 95, 96 HR‐pQCT has a higher nominal resolution (60 to 82 μm3), which allows for quantification of trabecular number, thickness, and separation (Fig. 2). Finite element analysis (FEA) has been used in biomechanics to determine the mechanical behavior (and therefore strength) of bone.97 The advent of high‐resolution imaging has allowed FEA to be used in the assessment of bone strength, stiffness, and failure load, either in its entirety or in individual (cortical or trabecular) compartments.98, 99 Recent developments in HR‐pQCT processing methods have been developed to characterize cortical porosity and trabecular rod and plate structure, which have been strongly associated with bone strength.100, 101, 102
Figure 2.
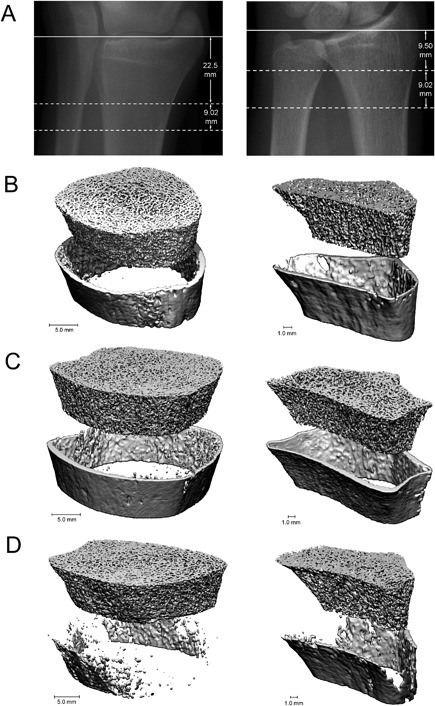
HR‐pQCT provides detailed images of bone microarchitecture at the radius (left) and tibia (right). Scout view (A) reference line position (solid line) and the measurement site (dotted line). Images from healthy, postmenopausal white female (B). Images from female with CKD, no fractures (C). Images from female with CKD and prevalent fractures (D). Reprinted with permission from Nickolas et al.103
The ability of HR‐pQCT analysis of bone microarchitecture at the distal radius and tibia to assess fracture risk and bone quality has been evaluated in kidney disease. Specifically, HR‐pQCT was reported to discriminate and predict fractures, define abnormalities in bone quality that adversely affect bone strength, and identify microstructural abnormalities that account for reduced bone density as measured by DXA.103, 104, 105, 106, 107, 108 Measures from DXA and HR‐pQCT were associated with prevalent fractures in patients with CKD.103, 104 Patients with fractures had lower BMD by DXA and lower cortical and trabecular volumetric BMD and thinner cortices and trabeculae by HR‐pQCT. These abnormalities were more severe with longer duration and severity of CKD. A study of 211 men and women with CKD 3–5 assessed the ability of areal BMD by DXA and volumetric BMD by HR‐pQCT to discriminate fractures.105 Both tests discriminated fracture status and the addition of HR‐pQCT measures to BMD by DXA did not improve discrimination. In a study of 74 prevalent hemodialysis patients, those with fractures had reduced cortical and trabecular microarchitecture compared with those without fractures.106 Changes to bone microarchitecture and calculated bone strength were assessed in 33 ESKD patients and 33 age‐matched controls.107 Cortical and trabecular bone microarchitecture and calculated bone strength were altered in ESKD patients; these changes were more pronounced in females. HR‐pQCT has also been used to determine the microarchitectural mechanisms of bone loss in CKD. In a longitudinal study of 54 patients with CKD 2‐5D, the mean annualized loss of bone density by DXA at the radius was 2.9%.108 With HR‐pQCT, this was characterized by loss of cortical area, density, and thickness and a significant increase in cortical porosity.
The use of HR‐pQCT in the clinic is not currently practical because of cost and limited availability. Thus, methods to assess bone microarchitecture that can be implanted in the clinic have high potential to assist with the diagnosis and management of patients with metabolic bone diseases. TBS was developed to assess trabecular microarchitecture (ie, bone quality) by using software analysis that measures grayscale homogeneity from lumbar DXA images.109 In early studies, TBS has been shown to correlate with trabecular microarchitecture as measured by micro‐computed tomography, HR‐pQCT, and iliac crest bone biopsy.110, 111, 112 TBS was also shown to predict fractures independently of clinical risk factors and areal BMD by DXA113, 114, 115 and has been incorporated into international fracture risk guidelines and predictive algorithms such as FRAX.116 TBS has also been investigated in patients with kidney disease. In a recent study of 50 patients with CKD 3‐5D, TBS was associated with trabecular bone volume, width, and spacing, as well as cortical width as measured by bone biopsy.117 Luckman and colleagues112 reported that TBS measures correlated with both cortical and trabecular microarchitecture by HR‐pQCT in ESKD patients before kidney transplantation and that changes in TBS measurements reflected changes in trabecular microarchitecture and failure load but not cortical microarchitecture 12 months after transplantation. In a Canadian cohort of 1476 patients, those with an eGFR <60 mL/min and a TBS value below the median (<1.277) had a higher 5‐year fracture probability that was independent of BMD and clinical risk factors for fracture (hazard ratio [HR] = 1.62 per SD decrease in TBS).118 Further verification and qualification of TBS as a fracture prediction tool in patients with kidney disease is needed.
Bone Turnover Markers
In patients without CKD, BTMs can be used to assess fracture risk and monitor osteoporosis therapy. In patients with CKD and ESKD, they provide mild to moderate accuracy in the noninvasive assessment of bone turnover and mineralization. In some cases, BTMs can be used instead of bone biopsy to inform antifracture strategies, although the utility of BTMs to improve patient‐related outcomes such as fractures remains unproven. Markers of bone formation (osteoblast function) include BSAP, osteocalcin, and P1NP. Bone resorption markers (osteoclast function) include TRAP‐5b and CTX.
In kidney disease, PTH and BSAP are the most widely tested BTMs to assess bone turnover, and BSAP, vitamin D, calcium, and phosphorus are used to assess mineralization.8, 9, 10, 67, 119 Generally, extremes of PTH and BSAP identify bone turnover type based on bone biopsy finding in CKD and ESKD. In a study of 132 patients with CKD 3–5, plasma PTH measurements effectively distinguished patients with and without low bone turnover, AUC 0.96 for CKD stages 3 and 4, and 0.86 for CKD stage 5.119 In a study of 141 hemodialysis patients, an intact PTH < 420 pg/mL increased the positive predictive value (PPV) for low bone turnover from 74% to 90% in white patients, whereas an intact PTH < 340 pg/mL increased the PPV for low bone turnover from 48% to 90% in black patients.120 In 492 ESKD patients, the AUC for discriminating low versus non‐low bone turnover was 0.701 for PTH, 0.757 for BSAP, and 0.718 for PTH and BSAP in combination.8 In another study of patients with ESKD, PTH values within the middle range (150 to 300 pg/mL) less reliably identified underlying histology.121 In age‐related osteoporosis, BSAP reflects bone formation and correlates with bone histology and other BTMs.122, 123, 124 In a study of 42 ESKD patients, BSAP and PTH were compared with bone biopsy.122 BSAP levels were higher in patients with high compared with low turnover (66.9 versus 10.8 ng/mL, p < 0.0005), were more strongly correlated with bone formation than PTH, and levels > 20 ng/mL reliably excluded adynamic bone disease. Total ALP has sometimes been used as a surrogate marker of BSAP, particularly in the absence of liver disease. However, ALP has high biological variation (>20%), and a high BSAP can still result in a normal ALP level.125, 126
PTH and BTMs may also have clinical utility in predicting bone loss and fractures. In a cross‐sectional study of 82 CKD patients, higher levels of PTH and BTMs were associated with lower cortical and trabecular density and increased cortical and trabecular thinning.104 In the same study, higher levels of PINP, osteocalcin, and TRAP discriminated fracture. In a prospective 2‐year study of 89 hemodialysis patients, baseline BSAP was strongly associated with loss of BMD cortical mass and volume.127 In prospective studies of patients with CKD before (n = 52) and after renal transplantation (n = 47), the microarchitectural and biochemical mechanisms of bone loss were examined by BTMs, DXA, and HR‐pQCT.108, 128 Higher levels of PTH, BSAP, osteocalcin, PINP, TRAP, and CTX predicted the loss of cortical area, density, and thickness, increase in cortical porosity, and decreased bone strength. The ability of PTH and BTMs to predict fractures was assessed in a prospective study of 485 ESKD patients. Incident fracture was associated with PTH levels either <150 pg/mL or >300 pg/mL compared with 150 to 300 pg/mL (p < 0.01).80 In the same study, BSAP was a very useful surrogate marker for any type of incident fracture risk (AUC = 0.766, p < 0.0001).
These data suggest that PTH and BTMs may have greater clinical utility in assessing bone quality and fracture risk in CKD and ESKD, in particular when used in conjunction with bone imaging methods. For example, fracture risk can be estimated by DXA. However, deciding on a treatment (vitamin D, calcimimetic, antiresorptive, anabolic) will also require consideration of bone turnover. PTH and BTMs can potentially be used to inform which pharmacologic agent is most appropriate. However, before BTMs can be widely used to manage renal bone disease, significant barriers need to be overcome. BSAP lacks a readily available automated assay, and there are concerns of cross‐reactivity with the liver iso‐enzyme,129 there are no validated reference ranges of BTMs in patients with CKD and ESKD, many BTMs are cleared by the kidney (monomeric P1NP, osteocalcin, and CTX) and BTMs have high intra‐ and interassay variability and biological variability.130
Treatment of ROD and Preventing Fractures in CKD
Management of kidney‐associated bone disease for fracture risk reduction is controversial. First, until the release of the 2017 KDIGO guidelines, fracture risk classification in CKD3‐5D was not recommended. Second, there are no antifracture treatments that have been developed specifically for patients with CKD‐MBD. However, since emerging data and anecdotal experience with existing antifracture agents suggest that they are safe, we expect that these agents will be more widely used in patients with CKD 3‐5D, especially as more patients undergo DXA screening. In this section, we will briefly review the antifracture treatments that are in current clinical use and their potential application to patients with kidney disease.
Management of CKD‐MBD
Management of the abnormalities associated with CKD‐MBD must occur before initiating specific antifracture therapy in patients with CKD 3‐5D. A detailed discussion of this is comprehensively articulated in the updated KIDGO guidelines and associated publications.10, 53, 131 In brief, supplementing with vitamin D (nutritional and/or active), lowering phosphate, initiating calcimimetics, and deciding on the need for parathyroidectomy are critically important to treating kidney‐bone disease and can have some antifracture benefits.132, 133, 134, 135, 136, 137 In our opinion, after optimized management of CKD‐MBD, one should consider additive treatment with an agent demonstrated to have antifracture efficacy in the general population (Fig. 3). The updated 2017 KDIGO CKD‐MBD Guidelines clearly endorse the use specific antifracture therapies in CKD 1–2 and to a lesser extent in CKD stage 3 in the absence of abnormalities of CKD‐MBD (recommendation 4.3.1 and 4.3.2).10 However, In patients with CKD 3–5 and evidence of CKD‐MBD, the use of osteoporosis therapies is not directly addressed (recommendation 4.3.3): “In patients with CKD G3a–G5D with biochemical abnormalities of CKD‐MBD and low BMD and/or fragility fractures, we suggest that treatment choices take into account the magnitude and reversibility of the biochemical abnormalities and the progression of CKD, with consideration of a bone biopsy (2D).” It is noteworthy that the KDIGO update no longer mandates that a bone biopsy should be obtained before starting osteoporosis treatment, in part because of the increasing experience with the use of osteoporosis medications in CKD patients.
Figure 3.
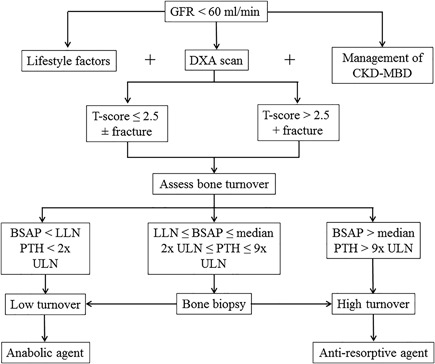
Algorithm for fracture risk screening and initiation of antifracture strategies in patients with CKD. DXA = dual‐energy X‐ray absorptiometry; CKD‐MBD = chronic kidney disease‐mineral and bone disorder; PTH = parathyroid hormone; BSAP = bone‐specific alkaline phosphatase; LLN = lower limit of the normal reference range; ULN = upper limit of the normal reference range. Lifestyle factors include weight‐bearing exercise, cessation of smoking, adequate nutrition, moderate alcohol intake, and fall prevention strategies. Management of CKD‐MBD includes phosphate lowering, vitamin D supplementation (nutritional and active), calcimimetics, and parathyroidectomy. Anabolic agents include teriparatide and abaloparatide. Antiresorptive agents include bisphosphonate and denosumab.
Antiresorptive Agents
Bisphosphonates and denosumab are commonly used in the treatment of senile and glucocorticoid‐induced osteoporosis; these agents lower remodeling rates and may be helpful in preventing bone loss and fracture in normal and high‐turnover bone disease. These should be avoided in patients with adynamic bone disease, but to date there are no studies in CKD that definitively demonstrate that bisphosphonates cause adynamic bone disease. There are no primary safety and efficacy data on the use of antiresorptive therapies in patients with CKD‐MBD, but there are post hoc analyses of the registration trials in patients with mild to moderate CKD (without biochemical manifestations of CKD‐MBD). These agents can therefore be used in CKD patients who have similar characteristics to the inclusion criteria of the registration trials, but in CKD 3‐5D patients with evidence of CKD‐MBD, further evaluation of the underlying ROD type needs to be undertaken before the use of these agents for fracture prevention.
Bisphosphonates
Bisphosphonates are selectively taken by osteoclasts, where they inhibit farnesyl pyrophosphate synthase and the synthesis of isoprenoid compounds, which are essential for osteoclast activity, thereby reducing osteoclast‐mediated bone resorption. Oral bisphosphonates have poor bioavailability (<1%); around 50% of the drug is not taken up by osteoclasts and cleared by the kidney. The actual amount of bisphosphonate retained in bone depends on the remodeling space, the GFR, and bone turnover rate.52 Given the lack of clinical trial data, oral bisphosphonates are not recommended in patients with eGFR < 30 mL/min because of concerns about excessive accumulation in bone and long‐term suppression of bone remodeling.
However, data from recent fracture intervention trials suggest that these drugs can be safely used in patients with reduced eGFR. In a post hoc analysis of 9 randomized, double‐blind trials comparing risedronate to placebo, 8996 females were identified as having kidney impairment, with the majority having a creatinine clearance of 30 to 80 mL/min.138 Risedronate effectively preserved BMD and reduced the incidence of vertebral fractures, and overall and renal function‐related adverse effects were similar in both groups and independent of renal function. Bone biopsy data of 57 patients with renal impairment showed no evidence of adynamic bone disease or mineralization defects. In a secondary analysis of the Fracture Intervention Trial, 581 women (9.9%) had a GFR < 45 mL/min.139 Women with a reduced eGFR had a 5.6% increase in total hip BMD, and alendronate increased BMD regardless of eGFR. Treatment with alendronate reduced clinical and spine fractures to a similar degree in those with and without renal impairment. There were no differences in adverse outcomes by renal function. A recent systematic review by Wilson and colleagues140 compared bisphosphonate to placebo in six studies (n = 1013) of patients with CKD (four of these were in renal transplant recipients).139, 141, 142, 143, 144, 145 Given the heterogeneity of the studies included, the use of bisphosphonates was associated with a nonsignificant reduction in fracture risk and moderate evidence that bisphosphonates may reduce loss of lumbar spine BMD but not femoral neck BMD. A recent post hoc analysis of three Japanese RCTs146, 147, 148 compared risedronate to placebo in 852 patients with an eGFR from 30 to >90 mL/min (with 228 having an eGFR <60).149 Patients were analyzed according to eGFR level ≥90, 60 to <90, and ≥30 to <60 mL/min/1.73 m2, and lumbar BMD, BTMs (CTX, P1NP, and BSAP) were evaluated at 48 weeks. The increase in lumbar BMD (p < 0.001) and inhibition of BTMs (p < 0.001) with the use of risedronate did not differ in the eGFR subgroups. There are no data on the optimal duration of bisphosphonate therapy in CKD or ESKD. General population data suggest 3 to 5 years of therapy, with a lack of evidence on fracture reduction beyond 5 years and concerns about atypical femoral fractures.150, 151, 152
Denosumab
Denosumab is a humanized monoclonal antibody against the Receptor activator of nuclear factor kappa‐Β ligand (RANKL), reducing bone turnover by inhibiting osteoclast proliferation and development. The Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) Trial recruited 7808 postmenopausal women and demonstrated that denosumab administered twice yearly for 36 months was associated with a reduction in the risk of vertebral (68%), nonvertebral (20%), and hip fractures (40%) compared with placebo.153 Denosumab is not cleared by the kidney, so unlike bisphosphonates, there is no risk of excess drug accumulation. Its effectiveness and safety in CKD (without evidence of CKD‐MBD) were assessed in a secondary analysis of patients from the FREEDOM trial, where treatment effect was compared across CKD categories (CKD 4 = 73, CKD 3 = 2817, CKD 2 = 4069).154 There was no difference in treatment efficacy and adverse effects by renal function, and denosumab increased BMD at the spine and hip and showed a 62% reduction in incident vertebral fractures.
The secondary analysis of the FREEDOM trial did not describe an association between hypocalcemia and decreased renal function. However, case reports and anecdotal experience suggest that this may occur in patients with CKD, hyperparathyroidism, and low vitamin D. In a case series of 8 patients with ESKD and 6 patients with CKD treated with denosumab, 8 patients developed severe hypocalcemia.155 In two studies of single‐dose denosumab in CKD and ESKD, hypocalcemia was the most common side effect; however, appropriate pretreatment with calcium and calcitriol was protective of clinically significant hypocalcemia.156, 157 These and other clinical data suggest that denosumab may have a role in patients with CKD 3‐5D, with appropriate calcium, vitamin D supplementation, and monitoring for hypocalcemia. Denosumab is metabolized in the reticuloendothelial system, and its clinical effect wanes after 6 months; frequency and duration of treatment in CKD 3‐5D remain unclear.
The increasing off‐label use of antiresorptive therapies in kidney disease patients reflects not only the decreasing concern regarding their short‐term safety but also the lack of specific antifracture treatments for patients with CKD‐MBD. No doubt further observational data will continue to guide and refine clinical practice, but ultimately prospective trials are needed to better define safety and antifracture efficacy of these drugs in patients with CKD‐MBD and an eGFR of <30 mL/min/1.73m2.
Osteoanabolic Agents
Osteoanabolic agents are forms of recombinant PTH, and their use in CKD remains controversial. In CKD, high‐baseline PTH levels promote cortical bone loss; therefore, these agents should not be used in patients with high‐turnover bone disease. However, they may increase bone turnover and bone density in patients with adynamic bone disease.
Teriparatide is a recombinant peptide of the first 34 amino acids of human PTH. In health, its antifracture efficacy is caused by an increase in osteoblast number, leading to increased bone formation and thickening of trabecular and cortical bone.158 Its efficacy in primary osteoporosis was demonstrated in two large clinical trials.159, 160 A post hoc analysis of Fracture Prevention Trial examined the safety and efficacy of teriparatide in 736 women with CKD (majority GFR 50 to 79 mL/min, and none <30 mL/min).161 Compared with placebo, teriparatide significantly increased lumbar spine and femoral neck BMD and had similar efficacy in preventing vertebral and nonvertebral fractures across all stages of CKD. The main adverse effects were hypercalcemia and hyperuricemia and these were more common with reduced eGFR. In a separate post hoc analysis from the same trial, improvements in BMD were correlated with improvements in trabecular microarchitecture.162 Teriparatide has also been used in patients with adynamic bone disease, where it improved BMD at the lumbar spine but not the femoral neck.163 A prospective, 48‐week study examined the effect of once‐weekly teriparatide in 22 hemodialysis patients with low BMD and hypoparathyroidism.164 Over the treatment period, there was an increase in lumbar spine but not femoral neck BMD. The observed increase in BSAP strongly correlated with improvements in BMD. There was no clinically significant hypercalcemia, and hypotension was the most common adverse event.
Abaloparatide is a newer parathyroid hormone‐related protein (PTHrP) analog, with stronger affinity for the transient state of the PTH1/PTH receptor and a more anabolic profile (compared with teriparatide) given less bone resorption and hypercalcemia.165, 166 In a study of 222 postmenopausal women, daily abaloparatide (20, 40, or 60 μg) was compared with teriparatide 20 μg or placebo.167 BMD was increased at the lumbar spine, femoral neck in a dose‐dependent manner, and to a greater extent than with teriparatide. Abaloparatide was compared with placebo and open‐label teriparatide in a study of 2461 postmenopausal women.168 New vertebral fractures occurred less frequently in the active treatment groups, and BMD increases were greater with abaloparatide than placebo. The incidence of hypercalcemia was lower with abaloparatide compared with teriparatide. In a secondary analysis of changes on bone histomorphometry, there was no evidence of abnormal mineralization, bone marrow abnormalities, or presence of excess osteoid.169 Patients treated with abaloparatide had lower eroded surface on histomorphometry versus placebo but similar increase in cortical porosity compared with teriparatide. This is supported by a smaller increase in CTX (a resorption marker) with abaloparatide compared with teriparatide. Based on the available data, abaloparatide has the ability to increase bone mass and formation, with less bone resorption and hypercalcemia. It has the potential to become an ideal agent for the treatment of patients with CKD and low to normal bone turnover; however, that data is currently lacking.
Sclerostin is a protein encoded by the SOST gene and secreted by osteocytes. Loss‐of‐function SOST mutations result in a high bone mass phenotype through increased bone formation, and sclerostin has predominantly anti‐anabolic effects on bone through inhibition of the Wnt‐signaling pathway. Inhibition of sclerostin is therefore of great interest in patients with CKD 3‐5D, particularly those with low bone mass and low bone turnover where antiresorptive therapies are contraindicated. In trials of postmenopausal women with osteoporosis, romosozumab increased BMD to a greater extent than existing anabolic agents and decreased vertebral and nonvertebral fractures.170, 171, 172 In another study, comparing romosozumab to teriparatide, there was a greater increase in cortical BMD (compared with trabecular BMD) in the romosozumab arm compared with a reduction in cortical BMD in the teriparatide group.173 Increased cardiovascular events were reported in one (but no other) romosozumab study.171 As such, it remains unclear whether romosozumab increases cardiac risk, but given the large CVD burden in patients with kidney disease, further study of romosozumab in CKD and ESKD should be suspended until these issues are clarified in future studies. Inhibition of DKK1 is another target for potential novel anabolic agents, although the clinical development of these drugs lags behind that of sclerostin inhibitors. In animal models, inhibition of DKK1 by monoclonal antibodies (DKK1‐ab) generally increased bone formation and mass. In a mouse model, changes of CKD‐MBD including ROD were ameliorated after the administration of DKK1‐ab (in combination with phosphate binder therapy).174 In patients with multiple myeloma, administration of DKK1‐ab reduced bone resorption and reduced bone formation.34 Finally, the dual inhibition of sclerostin and DKK‐1 leads to synergistic bone formation in rodents and non‐human primates.175 These studies have important implications for patients with kidney disease, and clinical studies of DKK1‐ab are needed in CKD and ESKD cohorts.
Take‐Home Messages and Rethinking Bone Disease in CKD
In CKD patients, fracture rates are more than 10‐fold higher compared with age‐ and sex‐matched individuals without CKD. Although fracture incidence in the general population has fallen over the last two decades, it has increased in patients with ESKD. In the clinic, treatment of CKD‐MBD is focused on the correction of abnormalities associated with parathyroid hormone and phosphate, a strategy that has not mitigated the effects of CKD on fracture risk. Despite advances that have improved our ability to assess fracture risk in the clinic, we continue to lack noninvasive tools that predict turnover and diagnose ROD type, which would inform fracture prevention strategies. Furthermore, despite an ever‐increasing choice of antifracture treatments in the general population, we lack data on their safety and efficacy in CKD 3‐5D. Therefore, it is time to rethink bone disease in patients with kidney disease.
Kidney‐related bone disease is complex and multifaceted, as such any gains are likely to be incremental rather than revolutionary. A starting point could be implementation of the successful fracture screening, prevention, and treatment programs used in the general population to CKD 3‐5D. The 2017 KDIGO guidelines justify the use of DXA, a readily available and inexpensive fracture risk screening tool. Lifestyle measures such as smoking cessation, weight‐bearing exercise, alcohol moderation, and improved nutrition all have proven antifracture efficacy in the general population and could be argued as being even more important in CKD. Vitamin D deficiency is common in CKD and should be routinely supplemented given its skeletal benefits and low risk of adverse consequences. The role of calcium supplementation is less well defined in CKD and ESKD and cannot be recommended in these patients.
Our ability to initiate specific, mostly antiresorptive osteoporosis treatments in CKD 3‐5D remains limited by our inability to effectively assess bone turnover and a historical fear of propagating low turnover and atypical femoral fractures. In the clinic setting, the interpretation and treatment of the current markers of CKD‐MBD should not occur in isolation but in view of the broader endocrine and skeletal disturbances. Although imperfect, PTH and BSAP improve our ability to discriminate bone turnover and some of the microstructural derangements. These should be used with DXA and be further studied with novel algorithms such as TBS that can inform on bone microarchitecture, with only minimal modifications to existing imaging infrastructure. Finally, fracture prediction algorithms such as FRAX should be broadly utilized to increase awareness of fractures; however, these need further validation and refinement to improve their accuracy in patients with kidney disease. Many, if not all, of these measures are feasible and could be readily implemented in most developed countries.
Ongoing work in improving diagnosis of ROD type is needed. Bone biopsy will remain the gold standard; however, given the required expertise, its role will always be confined to specialized tertiary institutions. High‐resolution imaging techniques provide a glimpse of the future, where imaging could obviate the need for a physical biopsy. However, today, even the most advanced high‐resolution imaging techniques remain inadequate at evaluating bone turnover and mineralization, both of which remain essential to informing treatment decisions. We have no doubt that high‐resolution imaging will continue to evolve and there are countless examples in medicine where technology has diminished the need for invasive diagnostic tests but seldom have these been superseded. Therefore, we believe that bone biopsy will remain an important diagnostic and research tool, such as in validating studies of high‐resolution imaging techniques. As such, its availability should be fostered and rationally coordinated across regions and health care networks.
CKD patients are historically disadvantaged by their exclusion from large general population clinical trials, and this is clearly the case in osteoporosis. Much of the current data come from secondary analyses of large osteoporosis trials, where patients were excluded if they had evidence of CKD‐MBD. The safety and efficacy of existing and novel agents in CKD 3‐5D remain unclear, and studies of these drugs are urgently needed in these cohorts. Future studies also need to target patients at various stages in the evolution of renal bone disease; for example, prevention of bone loss in CKD, reduction of fractures in ESKD, as well as any effects on the systemic aspects of CKD‐MBD. For this to occur, stronger advocacy from kidney societies and a paradigm shift from drug companies around the world are required. As physicians, we need to recognize the burden of fractures and bone disease in our patients and work tirelessly to promote better awareness and utilization of available diagnostic and therapeutic resources, such as greater off‐label use of osteoporosis medications in CKD 3‐5D. To this end, collaborations with our endocrinology colleagues should be fostered in both the research and clinical settings. In the words of Lao‐Tzu, “A journey of a thousand miles begins with a single step.” No doubt the road ahead is long, harboring many challenges and false starts; however, to accept the status quo is simply not an option.
Disclosures
MD has nothing to declare. TN has participated in a Scientific Advisory Board for Amgen. He has received grant support from Amgen for an Investigator Sponsored Study.
Acknowledgments
Authors’ roles: MJD and TLN contributed equally to the development and writing of this manuscript.
References
- 1. Hamdy NA, Kanis JA, Beneton MN, et al. Effect of alfacalcidol on natural course of renal bone disease in mild to moderate renal failure. BMJ. 1995; 310 (6976): 358–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Slatopolsky E, Delmez JA. Renal osteodystrophy. In: Avioli LV, Krane SM, editors. Metabolic bone disease and clinically related disorders. 3rd ed San Diego: Academic Press; 1998. pp. 443–64. [Google Scholar]
- 3. Slatopolsky E, Gonzalez E, Martin K. Pathogenesis and treatment of renal osteodystrophy. Blood Purif. 2003; 21 (4–5): 318–26. [DOI] [PubMed] [Google Scholar]
- 4. Moe S, Drüeke T, Cunningham J, et al. Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006; 69 (11): 1945–53. [DOI] [PubMed] [Google Scholar]
- 5. Kim SM, Long J, Montez‐Rath M, Leonard M, Chertow GM. Hip fracture in patients with non‐dialysis‐requiring chronic kidney disease. J. Bone Miner Res. 2016; 31 (10): 1803–9. [DOI] [PubMed] [Google Scholar]
- 6. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu C‐Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004; 351(13):1296–305. [DOI] [PubMed] [Google Scholar]
- 7. Naylor KL, McArthur E, Leslie WD, et al. The three‐year incidence of fracture in chronic kidney disease. Kidney Int. 2014; 86 (4): 810–8. [DOI] [PubMed] [Google Scholar]
- 8. Sprague SM, Bellorin‐Font E, Jorgetti V, et al. Diagnostic accuracy of bone turnover markers and bone histology in patients with CKD treated by dialysis. Am J Kidney Dis. 2016; 67 (4): 559–66. [DOI] [PubMed] [Google Scholar]
- 9. Malluche HH, Mawad HW, Monier‐Faugere M‐C. Renal osteodystrophy in the first decade of the new millennium: analysis of 630 bone biopsies in black and white patients. J Bone Miner Res. 2011; 26 (6): 1368–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ketteler M, Block GA, Evenepoel P, et al. Executive summary of the 2017 KDIGO Chronic Kidney Disease‐Mineral and Bone Disorder (CKD‐MBD) Guideline Update: what's changed and why it matters. Kidney Int. 2017; 92 (1): 26–36. [DOI] [PubMed] [Google Scholar]
- 11. National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis. 2002; S1–266. [PubMed] [Google Scholar]
- 12. Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int. 2007; 71 (1): 31–8. [DOI] [PubMed] [Google Scholar]
- 13. Isakova T, Xie H, Yang W, et al. Fibroblast growth factor 23 and risks of mortality and end‐stage renal disease in patients with chronic kidney disease. JAMA. 2011; 305 (23): 2432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parfitt AM, Drezner MK, Glorieux FH, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987; 2 (6): 595–610. [DOI] [PubMed] [Google Scholar]
- 15. Kuro‐o M. A phosphate‐centric paradigm for pathophysiology and therapy of chronic kidney disease. Kidney Int Suppl (2011). 2013; 3 (5): 420–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barker SL, Pastor J, Carranza D, et al. The demonstration of αKlotho deficiency in human chronic kidney disease with a novel synthetic antibody. Nephrol Dial Transplant. 2015; 30 (2): 223–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dhayat NA, Ackermann D, Pruijm M, et al. Fibroblast growth factor 23 and markers of mineral metabolism in individuals with preserved renal function. Kidney Int. 2016; 90 (3): 648–57. [DOI] [PubMed] [Google Scholar]
- 18. Kuro‐o M. Phosphate and Klotho. Kidney Int. 2011; 79121 (121): S20–3. [DOI] [PubMed] [Google Scholar]
- 19. Jüppner H. Phosphate and FGF‐23. Kidney Int Suppl. 2011; 79 (121): S24–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Komaba H, Lanske B. Role of Klotho in bone and implication for CKD. Curr Opin Nephrol Hypertens. 2018; 27 (4): 298–304. [DOI] [PubMed] [Google Scholar]
- 21. Ke HZ, Richards WG, Li X, Ominsky MS. Sclerostin and Dickkopf‐1 as therapeutic targets in bone diseases. Endocr Rev. 2012; 33 (5): 747–83. [DOI] [PubMed] [Google Scholar]
- 22. Damasiewicz MJ, Toussaint ND, Polkinghorne KR. Fibroblast growth factor 23 in chronic kidney disease: new insights and clinical implications. Nephrology (Carlton). 2011; 16 (3): 261–8. [DOI] [PubMed] [Google Scholar]
- 23. Nickolas TL. The utility of circulating markers to predict bone loss across the CKD spectrum. Clin J Am Soc Nephrol. 2014; 9 (7): 1160–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pereira RC, Jüppner H, Azucena‐Serrano CE, et al. Patterns of FGF‐23, DMP1, and MEPE expression in patients with chronic kidney disease. Bone. 2009; 45 (6): 1161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baron R, Rawadi G. Targeting the Wnt/beta‐catenin pathway to regulate bone formation in the adult skeleton. Endocrinology. 2007; 148 (6): 2635–43. [DOI] [PubMed] [Google Scholar]
- 26. Johnson ML, Kamel MA. The Wnt signaling pathway and bone metabolism. Curr Opin Rheumatol. 2007; 19 (4): 376–82. [DOI] [PubMed] [Google Scholar]
- 27. Schiavi SC. Sclerostin and CKD‐MBD. Curr Osteoporos Rep. 2015; 13 (3): 159–65. [DOI] [PubMed] [Google Scholar]
- 28. Li J, Sarosi I, Cattley RC, et al. Dkk1‐mediated inhibition of Wnt signaling in bone results in osteopenia. Bone. 2006; 39 (4): 754–66. [DOI] [PubMed] [Google Scholar]
- 29. Poole KES, van Bezooijen RL, Loveridge N, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB.J. 2005; 19 (13): 1842–4. [DOI] [PubMed] [Google Scholar]
- 30. Krupnik VE, Sharp JD, Jiang C, et al Functional and structural diversity of the human Dickkopf gene family. Gene. 1999; 238 (2): 301–13. [DOI] [PubMed] [Google Scholar]
- 31. Li X, Ominsky MS, Warmington KS, et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009; 24 (4): 578–88. [DOI] [PubMed] [Google Scholar]
- 32. Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single‐dose, placebo‐controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011; 26(1):19–26. [DOI] [PubMed] [Google Scholar]
- 33. Pinzone JJ, Hall BM, Thudi NK, et al. The role of Dickkopf‐1 in bone development, homeostasis, and disease. Blood. 2009; 113 (3): 517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yaccoby S, Ling W, Zhan F, Walker R, Barlogie B, Shaughnessy JD. Antibody‐based inhibition of DKK1 suppresses tumor‐induced bone resorption and multiple myeloma growth in vivo. Blood. 2007; 109 (5): 2106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Diarra D, Stolina M, Polzer K, et al. Dickkopf‐1 is a master regulator of joint remodeling. Nat Med. 2007; 13 (2): 156–63. [DOI] [PubMed] [Google Scholar]
- 36. Chen Y, Whetstone HC, Lin AC, et al. Beta‐catenin signaling plays a disparate role in different phases of fracture repair: implications for therapy to improve bone healing. PLoS Med. 2007; 4 (7): e249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim J‐B., Leucht P, Lam K, et al. Bone regeneration is regulated by wnt signaling. J. Bone Miner Res. 2007; 22 (12): 1913–23. [DOI] [PubMed] [Google Scholar]
- 38. Graciolli FG, Neves KR, Barreto F, et al. The complexity of chronic kidney disease‐mineral and bone disorder across stages of chronic kidney disease. Kidney Int. 2017; 91(6):1436–46. [DOI] [PubMed] [Google Scholar]
- 39. Cejka D, Herberth J, Branscum AJ, et al. Sclerostin and Dickkopf‐1 in renal osteodystrophy. Clin J Am Soc Nephrol. 2011; 6 (4): 877–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Malluche HH, Davenport DL, Cantor T, Monier‐Faugere M‐C. Bone mineral density and serum biochemical predictors of bone loss in patients with CKD on dialysis. Clin J Am Soc Nephrol. 2014; 9(7):1254–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA. 2001; 285(6):785–95. [DOI] [PubMed] [Google Scholar]
- 42. Seeman E, Delmas PD. Bone quality—the material and structural basis of bone strength and fragility. N Engl J Med. 2006; 354(21):2250–61. [DOI] [PubMed] [Google Scholar]
- 43. Vashishth D, Gibson GJ, Khoury JI, Schaffler MB, Kimura J, Fyhrie DP. Influence of nonenzymatic glycation on biomechanical properties of cortical bone. Bone. 2001; 28 (2): 195–201. [DOI] [PubMed] [Google Scholar]
- 44. Akkus O, Adar F, Schaffler MB. Age‐related changes in physicochemical properties of mineral crystals are related to impaired mechanical function of cortical bone. Bone. 2004; 34 (3): 443–53. [DOI] [PubMed] [Google Scholar]
- 45. Burr DB. The complex relationship between bone remodeling and the physical and material properties of bone. Osteoporos Int. 2015; 26(3):845–7. [DOI] [PubMed] [Google Scholar]
- 46. Bala Y, Seeman E. Bone's material constituents and their contribution to bone strength in health, disease, and treatment. Calcif Tissue Int. 2015; 97 (3): 308–26. [DOI] [PubMed] [Google Scholar]
- 47. Schaffler MB, Choi K, Milgrom C. Aging and matrix microdamage accumulation in human compact bone. Bone. 1995; 17(6):521–5. [DOI] [PubMed] [Google Scholar]
- 48. Reilly GC, Currey JD. The effects of damage and microcracking on the impact strength of bone. J Biomech. 2000; 33 (3): 337–43. [DOI] [PubMed] [Google Scholar]
- 49. Cummings SR, Melton LJ. Epidemiology and outcomes of osteoporotic fractures. Lancet. 2002; 359(9319):1761–7. [DOI] [PubMed] [Google Scholar]
- 50. Damasiewicz MJ, Ebeling PR. Management of mineral and bone disorders in renal transplant recipients. Nephrology (Carlton). 2017; 22 Suppl 2: 65–9. [DOI] [PubMed] [Google Scholar]
- 51. Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int. 2008; 19(4):385–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Miller PD. Bone disease in CKD: a focus on osteoporosis diagnosis and management. Am J Kidney Dis. 2014; 64 (2): 290–304. [DOI] [PubMed] [Google Scholar]
- 53.Kidney Disease Improving Global Outcomes KDIGO CKD MBD Work Group. KDIGO Clinical Practice Guideline for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease‐Mineral and Bone Disorder (CKD‐MBD). Kidney Int Suppl. 2009; 76 (Suppl 113):1–130. [DOI] [PubMed] [Google Scholar]
- 54. Jadoul M, Albert JM, Akiba T, et al. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study. Kidney Int. 2006; 70 (7): 1358–66. [DOI] [PubMed] [Google Scholar]
- 55. Miller PD. Unrecognized and unappreciated secondary causes of osteoporosis. Endocrinol Metab Clin North Am. 2012; 41(3):613–28. [DOI] [PubMed] [Google Scholar]
- 56. Hylander B, Lehtihet M. Testosterone and gonadotropins but not SHBG vary with CKD stages in young and middle aged men. Basic Clin Androl. 2015; 25: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. West SL, Jamal SA, Lok CE. Tests of neuromuscular function are associated with fractures in patients with chronic kidney disease. Nephrol Dial Transplant. 2012; 27 (6): 2384–8. [DOI] [PubMed] [Google Scholar]
- 58. Fahal IH. Uraemic sarcopenia: aetiology and implications. Nephrol Dial Transplant. 2014; 29(9):1655–65. [DOI] [PubMed] [Google Scholar]
- 59. Alem AM, Sherrard DJ, Gillen DL, et al. Increased risk of hip fracture among patients with end‐stage renal disease. Kidney Int. 2000; 58 (1): 396–9. [DOI] [PubMed] [Google Scholar]
- 60. Wagner J, Jhaveri KD, Rosen L, Sunday S, Mathew AT, Fishbane S. Increased bone fractures among elderly United States hemodialysis patients. Nephrol Dial Transplant. 2014; 29 (1): 146–51. [DOI] [PubMed] [Google Scholar]
- 61. Malluche HH, Ritz E, Lange HP, et al. Bone histology in incipient and advanced renal failure. Kidney Int. 1976; 9(4):355–62. [DOI] [PubMed] [Google Scholar]
- 62. Mora Palma FJ, Ellis HA, Cook DB, et al. Osteomalacia in patients with chronic renal failure before dialysis or transplantation. Q J Med. 1983; 52(207):332–48. [PubMed] [Google Scholar]
- 63. Coen G, Mazzaferro S, Ballanti P, et al. Renal bone disease in 76 patients with varying degrees of predialysis chronic renal failure: a cross‐sectional study. Nephrol Dial Transplant. 1996; 11 (5): 813–9. [DOI] [PubMed] [Google Scholar]
- 64. Elder G. Pathophysiology and recent advances in the management of renal osteodystrophy. J Bone Miner Res. 2002; 17(12):2094–105. [DOI] [PubMed] [Google Scholar]
- 65. Carvalho C, Magalhães J, Neto R, et al. Cortical bone analysis in a predialysis population: a comparison with a dialysis population. J Bone Miner Metab. 2016; 35 (5): 513–21. [DOI] [PubMed] [Google Scholar]
- 66. Lehmann G, Ott U, Kaemmerer D, Schuetze J, Wolf G. Bone histomorphometry and biochemical markers of bone turnover in patients with chronic kidney disease Stages 3 − 5. Clin Nephrol. 2008; 70(4):296–305. [DOI] [PubMed] [Google Scholar]
- 67. Malluche HH, Porter DS, Monier‐Faugere M‐C., Mawad H, Pienkowski D. Differences in bone quality in low‐ and high‐turnover renal osteodystrophy. J Am Soc Nephrol. 2012; 23 (3): 525–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Moe SM. Renal osteodystrophy or kidney‐induced osteoporosis? Curr Osteoporos Rep. 2017; 15 (3): 194–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Coco M, Rush H. Increased incidence of hip fractures in dialysis patients with low serum parathyroid hormone. Am J Kidney Dis. 2000; 36 (6): 1115–21. [DOI] [PubMed] [Google Scholar]
- 70. Nickolas TL, McMahon DJ, Shane E. Relationship between moderate to severe kidney disease and hip fracture in the United States. J Am Soc Nephrol. 2006; 17(11):3223–32. [DOI] [PubMed] [Google Scholar]
- 71. Ball AM, Gillen DL, Sherrard D, et al. Risk of hip fracture among dialysis and renal transplant recipients. JAMA. 2002; 288 (23): 3014–8. [DOI] [PubMed] [Google Scholar]
- 72. Isakova T, Craven TE, Scialla JJ, et al. Action to Control Cardiovascular Risk in Diabetes (ACCORD) Trial. Change in estimated glomerular filtration rate and fracture risk in the Action to Control Cardiovascular Risk in Diabetes Trial. Bone. 2015; 78:23–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Arneson TJ, Li S, Liu J, Kilpatrick RD, Newsome BB, St Peter WL. Trends in hip fracture rates in US hemodialysis patients, 1993‐2010. Am J Kidney Dis. 2013; 62(4):747–54. [DOI] [PubMed] [Google Scholar]
- 74. Nair SS, Mitani AA, Goldstein BA, Chertow GM, Lowenberg DW, Winkelmayer WC. Temporal trends in the incidence, treatment, and outcomes of hip fracture in older patients initiating dialysis in the United States. Clin J Am Soc Nephrol. 2013; 8(8):1336–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kim SM, Liu S, Long J, Montez‐Rath ME, Leonard MB, Chertow GM. Declining rates of hip fracture in end‐stage renal disease: analysis from the 2003‐2011 nationwide inpatient sample. J Bone Miner Res. 2017; 32(11):2297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Abbott KC, Oglesby RJ, Hypolite IO, et al. Hospitalizations for fractures after renal transplantation in the United States. Ann Epidemiol. 2001; 11 (7): 450–7. [DOI] [PubMed] [Google Scholar]
- 77. Beaubrun AC, Kilpatrick RD, Freburger JK, Bradbury BD, Wang L, Brookhart MA. Temporal trends in fracture rates and postdischarge outcomes among hemodialysis patients. J Am Soc Nephrol. 2013; 24(9):1461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Burge R, Dawson‐Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis‐related fractures in the United States, 2005‐2025. J Bone Miner Res. 2007; 22(3):465–75. [DOI] [PubMed] [Google Scholar]
- 79. Kanis JA, Melton LJ, Christiansen C, Johnston CC, Khaltaev N. The diagnosis of osteoporosis. J Bone Miner Res. 1994; 9 (8): 1137–41. [DOI] [PubMed] [Google Scholar]
- 80. Iimori S, Mori Y, Akita W, et al. Diagnostic usefulness of bone mineral density and biochemical markers of bone turnover in predicting fracture in CKD stage 5D patients—a single‐center cohort study. Nephrol Dial Transplant. 2012; 27(1):345–51. [DOI] [PubMed] [Google Scholar]
- 81. Naylor KL, Garg AX, Zou G, et al. Comparison of fracture risk prediction among individuals with reduced and normal kidney function. Clin J Am Soc Nephrol. 2015; 10 (4): 646–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. West SL, Lok CE, Langsetmo L, et al. Bone mineral density predicts fractures in chronic kidney disease. J Bone Miner Res. 2015; 30 (5): 913–9. [DOI] [PubMed] [Google Scholar]
- 83. Yenchek RH, Ix JH, Shlipak MG, et al. Health, Aging, and Body Composition Study. Bone mineral density and fracture risk in older individuals with CKD. Clin J Am Soc Nephrol. 2012; 7(7):1130–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jamal SA, West SL, Nickolas TL. The clinical utility of FRAX to discriminate fracture status in men and women with chronic kidney disease. Osteoporos Int. 2014; 25 (1): 71–6. [DOI] [PubMed] [Google Scholar]
- 85. Przedlacki J, Buczyńska‐Chyl J, Koźmiński P, et al. The utility of FRAX® in predicting bone fractures in patients with chronic kidney disease on hemodialysis: a two‐year prospective multicenter cohort study. Osteoporos Int. 2018; 29(5):1105–15. [DOI] [PubMed] [Google Scholar]
- 86. Jamal SA, Nickolas TL. Bone imaging and fracture risk assessment in kidney disease. Curr Osteoporos Rep. 2015; 13(3):166–72. [DOI] [PubMed] [Google Scholar]
- 87. Link TM, Majumdar S, Augat P, et al. In vivo high resolution MRI of the calcaneus: differences in trabecular structure in osteoporosis patients. J Bone Miner Res. 1998; 13 (7): 1175–82. [DOI] [PubMed] [Google Scholar]
- 88. Majumdar S, Genant HK, Grampp S, et al. Correlation of trabecular bone structure with age, bone mineral density, and osteoporotic status: in vivo studies in the distal radius using high resolution magnetic resonance imaging. J Bone Miner Res. 1997; 12(1):111–8. [DOI] [PubMed] [Google Scholar]
- 89. Wehrli FW, Hwang SN, Ma J, Song HK, Ford JC, Haddad JG. Cancellous bone volume and structure in the forearm: noninvasive assessment with MR microimaging and image processing. Radiology. 1998; 206 (2): 347–57. [DOI] [PubMed] [Google Scholar]
- 90. Seifert AC, Wehrli FW. Solid‐state quantitative (1)H and (31)P MRI of cortical bone in humans. Curr Osteoporos Rep. 2016; 14(3):77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Techawiboonwong A, Song HK, Leonard MB, Wehrli FW. Cortical bone water: in vivo quantification with ultrashort echo‐time MR imaging. Radiology. 2008; 248 (3): 824–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chang G, Boone S, Martel D, et al. MRI assessment of bone structure and microarchitecture. J Magn Reson Imaging. 2017; 46(2):323–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sharma AK, Toussaint ND, Elder GJ, et al. Magnetic resonance imaging based assessment of bone microstructure as a noninvasive alternative to histomorphometry in patients with chronic kidney disease. Bone. 2018; 114: 14–21. [DOI] [PubMed] [Google Scholar]
- 94. Jamal SA, Gilbert J, Gordon C, Bauer DC. Cortical pQCT measures are associated with fractures in dialysis patients. J Bone Miner Res. 2006; 21 (4): 543–8. [DOI] [PubMed] [Google Scholar]
- 95. Leonard MB. A structural approach to skeletal fragility in chronic kidney disease. Semin Nephrol. 2009; 29 (2): 133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Denburg MR, Tsampalieros AK, de Boer IH, et al. Mineral metabolism and cortical volumetric bone mineral density in childhood chronic kidney disease. J Clin Endocrinol Metab. 2013; 98(5):1930–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Prendergast PJ. Finite element models in tissue mechanics and orthopaedic implant design. Clin Biomech (Bristol, Avon). 1997; 12 (6): 343–66. [DOI] [PubMed] [Google Scholar]
- 98. Pistoia W, van Rietbergen B, Lochmüller E‐M., Lill CA, Eckstein F, Rüegsegger P. Estimation of distal radius failure load with micro‐finite element analysis models based on three‐dimensional peripheral quantitative computed tomography images. Bone. 2002; 30 (6): 842–8. [DOI] [PubMed] [Google Scholar]
- 99. Boutroy S, Van Rietbergen B, Sornay‐Rendu E, Munoz F, Bouxsein ML, Delmas PD. Finite element analysis based on in vivo HR‐pQCT images of the distal radius is associated with wrist fracture in postmenopausal women. J Bone Miner Res. 2008; 23 (3): 392–9. [DOI] [PubMed] [Google Scholar]
- 100. Nishiyama KK, Macdonald HM, Buie HR, Hanley DA, Boyd SK. Postmenopausal women with osteopenia have higher cortical porosity and thinner cortices at the distal radius and tibia than women with normal aBMD: an in vivo HR‐pQCT study. J Bone Miner Res. 2010; 25(4):882–90. [DOI] [PubMed] [Google Scholar]
- 101. Nishiyama KK, Pauchard Y, Nikkel LE, et al. Longitudinal HR‐pQCT and image registration detects endocortical bone loss in kidney transplantation patients. J Bone Miner Res. 2015; 30 (3): 554–61. [DOI] [PubMed] [Google Scholar]
- 102. Liu XS, Stein EM, Zhou B, et al. Individual trabecula segmentation (ITS)‐based morphological analyses and microfinite element analysis of HR‐pQCT images discriminate postmenopausal fragility fractures independent of DXA measurements. J Bone Miner Res. 2012; 27(2):263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nickolas TL, Stein E, Cohen A, et al. Bone mass and microarchitecture in CKD patients with fracture. J Am Soc Nephrol. 2010; 21 (8): 1371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Nickolas TL, Cremers S, Zhang A, et al. Discriminants of prevalent fractures in chronic kidney disease. J Am Soc Nephrol. 2011; 22 (8): 1560–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Jamal SA, Cheung AM, West SL, Lok CE. Bone mineral density by DXA and HR pQCT can discriminate fracture status in men and women with stages 3 to 5 chronic kidney disease. Osteoporos Int. 2012; 23(12):2805–13. [DOI] [PubMed] [Google Scholar]
- 106. Cejka D, Patsch JM, Weber M, et al. Bone microarchitecture in hemodialysis patients assessed by HR‐pQCT. Clin J Am Soc Nephrol. 2011; 6 (9): 2264–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Trombetti A, Stoermann C, Chevalley T, et al. Alterations of bone microstructure and strength in end‐stage renal failure. Osteoporos Int. 2013; 24(5):1721–32. [DOI] [PubMed] [Google Scholar]
- 108. Nickolas TL, Stein EM, Dworakowski E, et al. Rapid cortical bone loss in patients with chronic kidney disease. J Bone Miner Res. 2013; 28 (8): 1811–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Pothuaud L, Carceller P, Hans D. Correlations between grey‐level variations in 2D projection images (TBS) and 3D microarchitecture: applications in the study of human trabecular bone microarchitecture. Bone. 2008; 42(4):775–87. [DOI] [PubMed] [Google Scholar]
- 110. Roux JP, Wegrzyn J, Boutroy S, Bouxsein ML, Hans D, Chapurlat R. The predictive value of trabecular bone score (TBS) on whole lumbar vertebrae mechanics: an ex vivo study. Osteoporos Int. 2013; 24 (9): 2455–60. [DOI] [PubMed] [Google Scholar]
- 111. Muschitz C, Kocijan R, Haschka J, et al. TBS reflects trabecular microarchitecture in premenopausal women and men with idiopathic osteoporosis and low‐traumatic fractures. Bone. 2015; 79:259–66. [DOI] [PubMed] [Google Scholar]
- 112. Luckman M, Hans D, Cortez N, et al. Spine trabecular bone score as an indicator of bone microarchitecture at the peripheral skeleton in kidney transplant recipients. Clin J Am Soc Nephrol. 2017; 12 (4): 644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hans D, Barthe N, Boutroy S, Pothuaud L, Winzenrieth R, Krieg M‐A. Correlations between trabecular bone score, measured using anteroposterior dual‐energy X‐ray absorptiometry acquisition, and 3‐dimensional parameters of bone microarchitecture: an experimental study on human cadaver vertebrae. J Clin Densitom. 2011; 14(3):302–12. [DOI] [PubMed] [Google Scholar]
- 114. Iki M, Tamaki J, Kadowaki E, et al. Trabecular bone score (TBS) predicts vertebral fractures in Japanese women over 10 years independently of bone density and prevalent vertebral deformity: the Japanese Population‐Based Osteoporosis (JPOS) cohort study. J Bone Miner Res. 2014; 29 (2): 399–407. [DOI] [PubMed] [Google Scholar]
- 115. McCloskey EV, Odén A, Harvey NC, et al. A meta‐analysis of trabecular bone score in fracture risk prediction and its relationship to FRAX. J Bone Miner Res. 2016; 31(5):940–8. [DOI] [PubMed] [Google Scholar]
- 116. McCloskey EV, Odén A, Harvey NC, et al. Adjusting fracture probability by trabecular bone score. Calcif Tissue Int. 2015; 96 (6): 500–9. [DOI] [PubMed] [Google Scholar]
- 117. Ramalho J, Marques IDB, Hans D, et al. The trabecular bone score: relationships with trabecular and cortical microarchitecture measured by HR‐pQCT and histomorphometry in patients with chronic kidney disease. Bone. 2018; 116:215–20. [DOI] [PubMed] [Google Scholar]
- 118. Naylor KL, Prior J, Garg AX, et al. Trabecular bone score and incident fragility fracture risk in adults with reduced kidney function. Clin J Am Soc Nephrol. 2016; 11 (11): 2032–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lehmann G, Stein G, Hüller M, Schemer R, Ramakrishnan K, Goodman WG. Specific measurement of PTH (1‐84) in various forms of renal osteodystrophy (ROD) as assessed by bone histomorphometry. Kidney Int. 2005; 68 (3): 1206–14. [DOI] [PubMed] [Google Scholar]
- 120. Herberth J, Branscum AJ, Mawad H, Cantor T, Monier‐Faugere M‐C., Malluche HH. Intact PTH combined with the PTH ratio for diagnosis of bone turnover in dialysis patients: a diagnostic test study. Am J Kidney Dis. 2010; 55 (5): 897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Barreto FC, Barreto DV, Moysés RMA, et al. K/DOQI‐recommended intact PTH levels do not prevent low‐turnover bone disease in hemodialysis patients. Kidney Int. 2008; 73 (6): 771–7. [DOI] [PubMed] [Google Scholar]
- 122. Ureña P, Hruby M, Ferreira A, Ang KS, de Vernejoul MC. Plasma total versus bone alkaline phosphatase as markers of bone turnover in hemodialysis patients. J Am Soc Nephrol. 1996; 7 (3): 506–12. [DOI] [PubMed] [Google Scholar]
- 123. Chiang C. The use of bone turnover markers in chronic kidney disease‐mineral and bone disorders. Nephrology (Carlton). 2017; 22 Suppl 2:11–3. [DOI] [PubMed] [Google Scholar]
- 124. Magnusson P, Larsson L, Magnusson M, Davie MW, Sharp CA. Isoforms of bone alkaline phosphatase: characterization and origin in human trabecular and cortical bone. J Bone Miner Res. 1999; 14 (11):1926–33. [DOI] [PubMed] [Google Scholar]
- 125. Panteghini M, Bonora R, Pagani F. Measurement of pancreatic lipase activity in serum by a kinetic colorimetric assay using a new chromogenic substrate. Ann Clin Biochem. 2001; 38 (Pt 4):365–70. [DOI] [PubMed] [Google Scholar]
- 126. Tibi L, Chhabra SC, Sweeting VM, Winney RJ, Smith AF. Multiple forms of alkaline phosphatase in plasma of hemodialysis patients. Clin Chem. 1991; 37 (6):815–20. [PubMed] [Google Scholar]
- 127. Malluche HH, Monier‐Faugere M‐C, Blomquist G, Davenport DL. Two‐year cortical and trabecular bone loss in CKD‐5D: biochemical and clinical predictors. Osteoporos Int. 2017; 65:235. [DOI] [PubMed] [Google Scholar]
- 128. Iyer SP, Nikkel LE, Nishiyama KK, et al. Kidney transplantation with early corticosteroid withdrawal: paradoxical effects at the central and peripheral skeleton. J Am Soc Nephrol. 2014; 25 (6):1331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Jean G, Souberbielle J‐C, Zaoui E, et al. Total and bone‐specific alkaline phosphatases in haemodialysis patients with chronic liver disease. Clin Biochem. 2012; 45(6):436–9. [DOI] [PubMed] [Google Scholar]
- 130. McNerny EMB, Nickolas TL. Bone quality in chronic kidney disease: definitions and diagnostics. Curr Osteoporos Rep. 2017; 15 (3):207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Isakova T, Nickolas TL, Denburg M, et al. KDOQI US Commentary on the2017 KDIGO Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease‐Mineral and Bone Disorder (CKD‐MBD). Am J Kidney Dis. 2017; 70 (6):737–51. [DOI] [PubMed] [Google Scholar]
- 132. Coen G, Mantella D, Manni M, et al. 25‐hydroxyvitamin D levels and bone histomorphometry in hemodialysis renal osteodystrophy. Kidney Int. 2005; 68 (4):1840–8. [DOI] [PubMed] [Google Scholar]
- 133. Rudser KD, de Boer IH, Dooley A, Young B, Kestenbaum B. Fracture risk after parathyroidectomy among chronic hemodialysis patients. J Am Soc Nephrol. 2007; 18 (8):2401–7. [DOI] [PubMed] [Google Scholar]
- 134. Patel L, Bernard LM, Elder GJ. Sevelamer versus calcium‐based binders for treatment of hyperphosphatemia in CKD: a meta‐analysis of randomized controlled trials. Clin J Am Soc Nephrol. 2016; 11 (2):232–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Jamal SA, Vandermeer B, Raggi P, et al. Effect of calcium‐based versus non‐calcium‐based phosphate binders on mortality in patients with chronic kidney disease: an updated systematic review and meta‐analysis. Lancet. 2013; 382(9900):1268–77. [DOI] [PubMed] [Google Scholar]
- 136. Moe SM, Abdalla S, Chertow GM, et al. Effects of cinacalcet on fracture events in patients receiving hemodialysis: the EVOLVE trial. J Am Soc Nephrol. 2015; 26 (6):1466–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Behets GJ, Spasovski G, Sterling LR, et al. Bone histomorphometry before and after long‐term treatment with cinacalcet in dialysis patients with secondary hyperparathyroidism. Kidney Int. 2015; 87(4):846–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Miller PD, Roux C, Boonen S, Barton IP, Dunlap LE, Burgio DE. Safety and efficacy of risedronate in patients with age‐related reduced renal function as estimated by the Cockcroft and Gault method: a pooled analysis of nine clinical trials. J Bone Miner Res. 2005; 20 (12):2105–15. [DOI] [PubMed] [Google Scholar]
- 139. Jamal SA, Bauer DC, Ensrud KE, et al. Alendronate treatment in women with normal to severely impaired renal function: an analysis of the fracture intervention trial. J Bone Miner Res. 2007; 22 (4):503–8. [DOI] [PubMed] [Google Scholar]
- 140. Wilson LM, Rebholz CM, Jirru E, et al. Benefits and harms of osteoporosis medications in patients with chronic kidney disease: a systematic review and meta‐analysis. Ann Intern Med. 2017; 166(9):649–58. [DOI] [PubMed] [Google Scholar]
- 141. Coco M, Glicklich D, Faugere MC, et al. Prevention of bone loss in renal transplant recipients: a prospective, randomized trial of intravenous pamidronate. J Am Soc Nephrol. 2003; 14 (10):2669–76. [DOI] [PubMed] [Google Scholar]
- 142. Walsh SB, Altmann P, Pattison J, et al. Effect of pamidronate on bone loss after kidney transplantation: a randomized trial. Am J Kidney Dis. 2009; 53 (5):856–65. [DOI] [PubMed] [Google Scholar]
- 143. Toussaint ND, Lau KK, Strauss BJ, Polkinghorne KR, Kerr PG. Effect of alendronate on vascular calcification in CKD stages 3 and 4: a pilot randomized controlled trial. Am J Kidney Dis. 2010; 56(1):57–68. [DOI] [PubMed] [Google Scholar]
- 144. Smerud KT, Dolgos S, Olsen IC, et al. A 1‐year randomized, double‐blind, placebo‐controlled study of intravenous ibandronate on bone loss following renal transplantation. Am J Transplant. 2012; 12 (12):3316–25. [DOI] [PubMed] [Google Scholar]
- 145. Torregrosa J‐V., Fuster D, Gentil M‐A., et al. Open‐label trial: effect of weekly risedronate immediately after transplantation in kidney recipients. Transplantation. 2010; 89 (12):1476–81. [DOI] [PubMed] [Google Scholar]
- 146. Fukunaga M, Kushida K, Kishimoto H, et al. A comparison of the effect of risedronate and etidronate on lumbar bone mineral density in Japanese patients with osteoporosis: a randomized controlled trial. Osteoporos Int. 2002; 13(12):971–9. [DOI] [PubMed] [Google Scholar]
- 147. Kushida K, Fukunaga M, Kishimoto H, et al. A comparison of incidences of vertebral fracture in Japanese patients with involutional osteoporosis treated with risedronate and etidronate: a randomized, double‐masked trial. J Bone Miner Metab. 2004; 22 (5):469–78. [DOI] [PubMed] [Google Scholar]
- 148. Kishimoto H, Fukunaga M, Kushida K, et al. Efficacy and tolerability of once‐weekly administration of 17.5 mg risedronate in Japanese patients with involutional osteoporosis: a comparison with 2.5‐mg once‐daily dosage regimen. J Bone Miner Metab. 2006; 24(5):405–13. [DOI] [PubMed] [Google Scholar]
- 149. Shigematsu T, Muraoka R, Sugimoto T, Nishizawa Y. Risedronate therapy in patients with mild‐to‐moderate chronic kidney disease with osteoporosis: post‐hoc analysis of data from the risedronate phase III clinical trials. BMC Nephrol. 2017; 18 (1):66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Whitaker M, Guo J, Kehoe T, Benson G. Bisphosphonates for osteoporosis—where do we go from here? N Engl J Med. 2012; 366 (22):2048–51. [DOI] [PubMed] [Google Scholar]
- 151. Shane E, Burr D, Abrahamsen B, et al. Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2014; 29(1):1–23. [DOI] [PubMed] [Google Scholar]
- 152. Black DM, Schwartz AV, Ensrud KE, et al. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long‐term Extension (FLEX): a randomized trial. JAMA. 2006; 296 (24):2927–38. [DOI] [PubMed] [Google Scholar]
- 153. Cummings SR, San Martin J, McClung MR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009; 361(8):756–65. [DOI] [PubMed] [Google Scholar]
- 154. Jamal SA, Ljunggren O, Stehman‐Breen C, et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res. 2011; 26(8):1829–35. [DOI] [PubMed] [Google Scholar]
- 155. Dave V, Chiang CY, Booth J, Mount PF. Hypocalcemia post denosumab in patients with chronic kidney disease stage 4‐5. Am J Nephrol. 2015; 41(2):129–37. [DOI] [PubMed] [Google Scholar]
- 156. Block GA, Bone HG, Fang L, Lee E, Padhi D. A single‐dose study of denosumab in patients with various degrees of renal impairment. J Bone Miner Res. 2012; 27 (7):1471–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Chen C‐L., Chen N‐C., Hsu C‐Y., et al. An open‐label, prospective pilot clinical study of denosumab for severe hyperparathyroidism in patients with low bone mass undergoing dialysis. J Clin Endocrinol Metab. 2014; 99 (7):2426–32. [DOI] [PubMed] [Google Scholar]
- 158. Bilezikian JP, Matsumoto T, Bellido T, et al. Targeting bone remodeling for the treatment of osteoporosis: summary of the proceedings of an ASBMR workshop. J Bone Miner Res. 2009; 24 (3):373–85. [DOI] [PubMed] [Google Scholar]
- 159. Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1‐34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001; 344(19):1434–41. [DOI] [PubMed] [Google Scholar]
- 160. Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid‐induced osteoporosis. N Engl J Med. 2007; 357 (20):2028–39. [DOI] [PubMed] [Google Scholar]
- 161. Miller PD, Schwartz EN, Chen P, Misurski DA, Krege JH. Teriparatide in postmenopausal women with osteoporosis and mild or moderate renal impairment. Osteoporos Int. 2007; 18(1):59–68. [DOI] [PubMed] [Google Scholar]
- 162. Chen P, Miller PD, Recker R, et al. Increases in BMD correlate with improvements in bone microarchitecture with teriparatide treatment in postmenopausal women with osteoporosis. J Bone Miner Res. 2007; 22 (8):1173–80. [DOI] [PubMed] [Google Scholar]
- 163. Cejka D, Kodras K, Bader T, Haas M. Treatment of hemodialysis‐associated adynamic bone disease with teriparatide (PTH1‐34): a pilot study. Kidney Blood Press Res. 2010; 33 (3):221–6. [DOI] [PubMed] [Google Scholar]
- 164. Sumida K, Ubara Y, Hoshino J, et al. Once‐weekly teriparatide in hemodialysis patients with hypoparathyroidism and low bone mass: a prospective study. Osteoporos Int. 2016; 27(4):1441–50. [DOI] [PubMed] [Google Scholar]
- 165. Hattersley G, Dean T, Corbin BA, Bahar H, Gardella TJ. Binding selectivity of abaloparatide for PTH‐type‐1‐receptor conformations and effects on downstream signaling. Endocrinology. 2016; 157 (1):141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Jilka RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone. 2007; 40(6):1434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Leder BZ, O'Dea LSL, Zanchetta JR, et al. Effects of abaloparatide, a human parathyroid hormone‐related peptide analog, on bone mineral density in postmenopausal women with osteoporosis. J Clin Endocrinol Metab. 2015; 100 (2):697–706. [DOI] [PubMed] [Google Scholar]
- 168. Miller PD, Hattersley G, Riis BJ, et al. Effect of abaloparatide vs placebo on new vertebral fractures in postmenopausal women with osteoporosis: a randomized clinical trial. JAMA. 2016; 316 (7):722–33. [DOI] [PubMed] [Google Scholar]
- 169. Moreira CA, Fitzpatrick LA, Wang Y, Recker RR. Effects of abaloparatide‐SC (BA058) on bone histology and histomorphometry: the ACTIVE phase 3 trial. Bone. 2017; 97:314–9. [DOI] [PubMed] [Google Scholar]
- 170. McClung MR, Grauer A, Boonen S, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. 2014; 370 (5):412–20. [DOI] [PubMed] [Google Scholar]
- 171. Saag KG, Petersen J, Brandi ML, et al. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med. 2017; 377 (15):1417–27. [DOI] [PubMed] [Google Scholar]
- 172. Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. 2016; 375 (16):1532–43. [DOI] [PubMed] [Google Scholar]
- 173. Langdahl BL, Libanati C, Crittenden DB, et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: a randomised, open‐label, phase 3 trial. Lancet. 2017; 390 (10102):1585–94. [DOI] [PubMed] [Google Scholar]
- 174. Fang Y, Ginsberg C, Seifert M, et al. CKD‐induced wingless/integration1 inhibitors and phosphorus cause the CKD‐mineral and bone disorder. J Am Soc Nephrol. 2014; 25(8):1760–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Florio M, Gunasekaran K, Stolina M, et al. A bispecific antibody targeting sclerostin and DKK‐1 promotes bone mass accrual and fracture repair. Nat Commun. 2016; 7:11505. [DOI] [PMC free article] [PubMed] [Google Scholar]