

Abstract
As humans move and alter habitats, they change the disease risk for themselves, their commensal animals and wildlife. Bartonella bacteria are prevalent in mammals and cause numerous human infections. Understanding how this genus has evolved and switched hosts in the past can reveal how current patterns were established and identify potential mechanisms for future cross-species transmission. We analyzed patterns of Bartonella transmission and likely sources of spillover using the largest collection of Bartonella gltA genotypes assembled, including 67 new genotypes. This pathogenic genus likely originated as an environmental bacterium and insect commensal before infecting mammals. Rodents and domestic animals serve as the reservoirs or at least key proximate host for most Bartonella genotypes in humans. We also find evidence of exchange of Bartonella between phylogenetically distant domestic animals and wildlife, likely due to increased contact. Care should be taken to avoid contact between humans, domestic animals and wildlife to protect the health of all.
Author summary
As humans move around the globe they contact new environments, potentially introducing novel diseases to wildlife, domestic animals and humans. Understanding how current infection patterns were established and how humans have likely altered them can help protect human, animal and environmental health. We traced the evolution of and distribution of globally distributed, pathogenic Bartonella, a common and well-studied bacterial genus in wildlife and humans that can cause cat scratch disease, trench fever and other diseases. We showed that humans are likely changing disease risk for themselves and the animals in their environment by moving themselves and domestic animals, as evidenced by large geographic movements of infections or shared infections in distantly related species. Not only does this increase our knowledge about Bartonella, an important emerging pathogen, but our investigation can serve as a model for elucidating the driving role of humans in changing disease landscapes.
Introduction
Human movements and actions have numerous impacts for wildlife disease [1,2]. These impacts are of concern both from a wildlife conservation standpoint [1,2] and from a public health perspective (spillover). Over 60% of emerging infectious diseases in the world are zoonotic, meaning they are transmitted from animals to humans [3]. Despite the fact that zoonosis is an important component of emerging infectious diseases, it is often difficult to trace the ecology and evolution of zoonotic pathogens [4]. Most efforts to identify the source of zoonoses occur after a human has become infected. Because spillover events are rare and often infection prevalence in the reservoir species is low, it can be difficult to trace the origin of potential zoonoses. However, Bartonella bacteria are an exception to this pattern. This genus of bacteria has been found in numerous taxa and is usually at high prevalence [5]. Bartonella is a blood-borne pathogen, found in many animals. It is the cause of cat scratch fever, Carrion’s disease and trench fever as well as a number of incidents of endocarditis in humans and has been hypothesized to be the cause of unexplained febrile illness in a number of cases [5,6]. Therefore, it is an ideal pathogen to focus on in tracing zoonotic potential as well as potential impacts on the native hosts. In this study, we construct some of the largest global phylogenies to date of Bartonella from both 16s rRNA genes and citrate synthase (gltA) to determine the evolutionary history of Bartonella, patterns of host switching and geographic constraint and its spillover into humans or from human commensals into wild species. The citrate synthase gene is known to give high power to discriminate between Bartonella strains and is one of the most commonly sequenced Bartonella genes [7,8]. We also examine 16s as it is the most commonly sequenced locus for metagenomics studies, though it gives low power to discriminate between Bartonella species [8].
Materials and methods
Ethics statement
Research was approved by the Stanford University Administrative Panel on Laboratory Animal Care (protocol 26920) and conducted under the appropriate Costa Rican permits (RT-044-2015-OT-CONAGEBIO, RT-042-2015-OT-CONAGEBIO, 121-2012-SINAC, RT-019-2013-OT-CONAGEBIO, 226-2012-SINAC).
In order to ascertain broader patterns of spillover and Bartonella transmission between species, sequences were downloaded from Genbank on 30 November 2016 using the search term “Bartonella gltA” and again on 21 May 2018 using the search terms “Bartonella” AND (“gltA” OR “GltA” OR “GLTA” OR “glta” OR “citrate synthase”) and limiting the search to sequences uploaded in the previous 900 days in order to update our dataset with the most recently published sequences. A separate search was conducted using the search term “Bartonella 16s” on 1 February 2017. Insect microbiome studies that detected Bartonella were also used [9–15]. Bartonella from Costa Rican bats in a mosaic agricultural landscape, including previously published [16] and 67 new sequences (isolated as in Judson et al. [16]) are also incorporated in this study (Genbank accession numbers MH234314 –MH234380). Metadata were downloaded from Genbank and/ or confirmed by examining the cited publication and are summarized in S1 and S2 Files. When data in Genbank were not associated with a publication, geography was inferred by the host range and/or title information in Genbank. The host of questing ticks was undetermined and therefore denoted as “unknown.” In some cases, genomes of Bartonella strains were published independently from their hosts; in this case we searched other literature to find the source of the strain. Sequences that were not in fact Bartonella gltA were removed manually and sequences were aligned using the Geneious alignment algorithm and refined using MUSCLE in Geneious (version 8.1.9 [17]). Sequences that were significantly redundant (or multiple sequences of the same species of Bartonella) were excluded to reduce the size of the resultant phylogenies. This was especially the case for the published genomes of named Bartonella species; many of these sequences lacked data on when and where they were isolated and were therefore excluded from our analysis. We also excluded some fragments that were too short or had substantial missing data within the alignments, as well as fragments which misaligned significantly at the ends, causing us to doubt the quality of these end base calls. Sequences that contained one or two base pair deletions not found in other sequences were also eliminated as we doubted the quality of the sequences. Sequences with deletions in multiples of three base pairs were retained as these likely represent actual deletions of amino acids. Alignments were manually inspected and corrected. Two alignments were produced, one of 540 bp and one of 277 bp. The first contained 677 sequences in total and the second included 1,060 unique sequences.
In order to test for patterns in host specificity and biogeography we also constructed Bayesian phylogenies using BEAST 2 [18] for the 540 bp fragment and the 277 bp fragment. Alignments were split into three partitions based on the base pair’s position in the codon and run in PartitionFinder to determine the best nucleotide substitution models using AICc [19]. These parameters were then used to configure the parameters for the BEAST 2 run. For both the 540 bp and 277 bp run, PartitionFinder determined that all three positions should be run under the same mode, a GTR+I+G model. As empirical and maximum likelihood estimated base frequencies usually have little impact on the phylogeny, we used observed base frequencies for both sets of nucleotides [19].
We tested two different models for the phylogenetic hypothesis based on the 540 bp fragment. Both analyses were run with a gamma site model with empirical base frequencies, an estimated proportion of invariant sites and all nucleotide transition/transversion frequencies except the CT transition rate estimated. The gamma shape prior was set to an exponential distribution with a mean of 1; the proportion of invariant sites was set to a uniform distribution between 0 and 1; all nucleotide substitution rates were set to a gamma distribution with an alpha of 2 and a beta of 0.5 or 0.25 for transitions and transversions respectively. In all cases Bartonella was constrained to be monophyletic with Brucella melitensis as an outgroup. The first model tested was a strict clock model with a constant population size coalescent model with vague priors as has been used for previous phylogenetic analyses of Bartonella [20,21] with the population size prior set to a 1/X distribution. The second was a birth death model run with a log-normal distributed relaxed molecular clock. The birth rate prior was set to a uniform distribution between 0 and 10,000; the relaxed clock mean prior was set to a uniform distribution between negative infinity and infinity; the relaxed clock standard deviation prior was set to an exponential distribution with a mean of 1; the death rate prior was set to a uniform distribution between 0 and 1. Additionally a clade of Artibeus lituratus and Artibeus watsoni-associated Bartonella was estimated to have evolved at the divergence of the two bat species (KJ816682, MH234319, MH234329, MH234330) and the prior distribution was estimated with a log-normal distribution with a mean of 8.5 mya (SD = 2.73) based on previous estimates [22–27] collated in TimeTree [28]. We used this clade as a calibration point as it was strongly supported in all of our initial analyses, regardless of model and was nested within other Central American bat-associated strains and therefore unlikely to have been impacted by human influence.
For the 277 bp tree we ran our simulations with a GTR distribution, an estimated proportion of invariant sites and a gamma distribution of rates. We tested two models, a strict clock, constant population size coalescent model as described in the first model for the 540 bp alignment and a birth death model with a relaxed log normal clock as described in the second model for the 540 bp alignment. In both models we constrained Brucella melitensis to be an outgroup but no other calibrations were included. All gltA model were run for 2.5 x 107 generations and sampled every 50,000 generations.
All gltA models converged with all parameters showing an effective sample size (ESS) over 100 (with the exception of the inferred relative death rate in the relaxed clock model of the 540bp alignment) and most showing an ESS over 200. The two models for the 540bp alignment were compared using AICM of the likelihood [29] with 1,000 bootstraps implemented through Tracer as model comparison using path sampling was not practical. For the 540bp alignment the best model was the second–a relaxed log normal clock calibrated with host divergence dates (dAICM = 356.48). For the 277bp alignment a strict clock was favored over a relaxed clock (dAICM = 441.86). Maximum clade credibility (MCC) trees were produced using TreeAnnotator, mean heights and a burn in of 10%.
In order to understand the evolutionary origin of Bartonella we constructed a phylogeny using sequences from the 16s rRNA gene. All 450 sequences were aligned and trimmed to the same length (259 bp) in Geneious. We constructed a phylogenetic hypothesis in BEAST 2 using a strict clock and a birth death model with vague priors as described in the birth death models for the gltA genes with Rhizobium leguminosarum as an outgroup. The model was run for 107 generations; most ESS were above 300, though the birth rate and death rate ESS were roughly 100. As we were not concerned with speciation dynamics but rather broad topology, we considered this hypothesis to be sufficiently sampled.
The 277 bp gltA MCC tree was used in an analysis of host specificity and geographic conservation between related Bartonella species. Many nodes did not have good support so we conducted all analyses using only nodes with a Bayesian posterior probability of the likelihood of 0.7 or above. Using the fitDiscrete function in geiger [30], four models of discrete character evolution were fit—one using a lambda transformation, one using a white noise transformation, one using an early burst transformation and one using no transformation to model the evolution of host order (with strains isolated from ectoparasites assigned to the ectoparasite’s host) and broad geographic region of isolation both by continent (all except Antarctica) and by Old World versus New World. Fit of the models was assessed using AICc weights and log-likelihoods.
Host switches and sharing of clades between geographic regions was assessed by manually examining the MCC phylogenetic hypothesis based on a 277bp fragment of gltA, by examining the location of Genbank records with identical genotypes and by searching the literature for the distribution of named Bartonella species. A host switch or geographic shift was inferred so as to capture the minimum number of shifts with Bayesian posterior probabilities of at least 0.7. We also assessed shifts at posterior probabilities of the likelihood of at least 0.9 and 0.95 to ensure our results were robust regardless of our cut-off.
In inferring the influence of humans on Bartonella, we categorized genotypes or monophyletic clades as being found in one or more of the following categories: rats, other rodents (excluding rats), humans, domestic carnivores, wild carnivores, domestic artiodactyls, wild artiodactyls, shrews, and bats. We also noted other wild animals that were rarer in our dataset (e.g. pikas, wild hares and wild primates). Genotypes found in rats, domestic animals and humans were categorized as “human-associated” and the rest as “non-human-associated”. Human-associated Bartonella in this study does not necessarily mean the genotype has been found in a human but rather in a human or a commensal animal or an ectoparasite on a human or commensal animal. Sometimes the metadata contained within Genbank and the publications were insufficient to allow us to distinguish the exact species from which the Bartonella was isolated. All genotypes isolated from Rattus sp., their ectoparasites or an organism denoted “rat” were counted as human-associated rats. Although many species of rodents are commensal with humans (e.g. [31]), all other rodents were counted as not being human-associated to avoid the need to categorize over 100 rodent species, as well as incorporate rodents for which the species was unknown (N = 50). This division therefore renders our analyses of human-associated strains conservative.
Additionally, we determined all instances of Bartonella transferring between host orders represented in our dated, 540bp phylogenetic hypothesis to determine the ages of such transfers and test whether transfers to humans were more recent than other transfers. For these analyses we only used clades with at least 0.7 posterior probability support, which meant that a few (3) clades were assessed as older than they may actually be. In each of these cases the transfer involved a genotype found in a human, meaning our hypothesis testing is conservative. Difference between zoonotic transfers and other transfers was assessed using one-tailed t-tests assuming unequal variance on the inferred node age and lower bound of the 95% highest probability density to account for uncertainty in dating of the nodes.
All alignments, metadata and R code are available in the supporting information.
Results
Starting with 2,564 gltA sequences, we analyzed 2,515 277 bp sequences, 1,060 of which were unique and used to construct a phylogenetic hypothesis. Information on identical sequences used to infer host and geography transfers is included in S3 File. In our final dataset, the most commonly sampled taxa were rodents (N = 1,143 of which 94 were rats) and bats (N = 374). We were also able to align a 540bp gltA fragment for 677 genotypes, a subset of the larger dataset.
Evolutionary history
Phylogenetic hypotheses generated from a 277 bp fragment of gltA and a 259 bp fragment of the 16s rRNA gene both support an origin for Bartonella in the environment and in the guts of insects (both ectoparasitic and non-ectoparasitic species; Figs 1 and 2). Twice Bartonella has infected mammals from these environmental samples, which are basal to the main clade of mammal-associated Bartonella (Fig 2), which likely invaded mammals approximately 79 million years ago based on our time calibrated phylogeny, though it is unclear which mammalian host is ancestral (Figs 2 and S1).
Fig 1. Bayesian phylogenetic hypothesis of Bartonella genotypes based on a 259bp fragment of 16s rRNA gene.
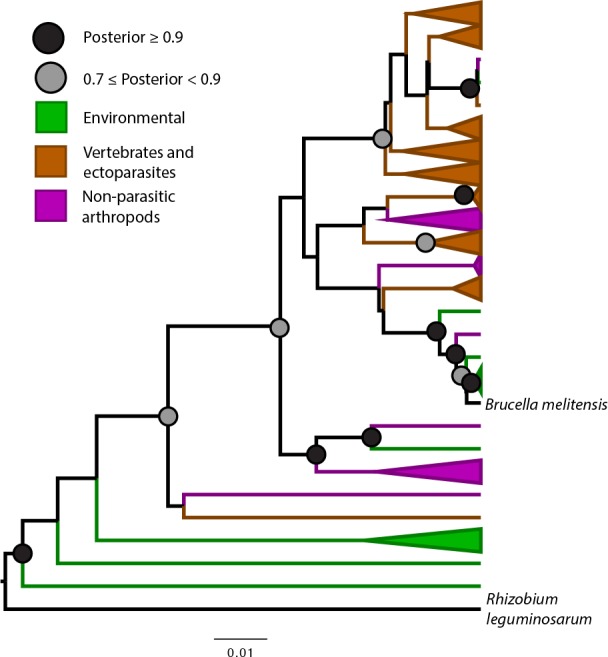
Ectoparasites and their vertebrate hosts are colored brown; environmental sequences are green; non-ectoparasitic arthropods are colored purple. Scale bar indicates substitutions per site. “Posterior” refers to the Bayesian posterior probability of the likelihood.
Fig 2. Bayesian phylogenetic hypothesis of Bartonella genotypes based on a 277 bp fragment gltA.

Tip labels and branches have been colored according to the taxa in which they were identified with ectoparasites colored according to their host and collapsed to highlight specific patterns. For readability, basal clades are shown in A, a subset of the more derived clades is shown in B, and the rest of the clades are shown in C. Branches with less than 0.7 Bayesian posterior probability of the likelihood support (indicating greater uncertainty) are dashed. Accession numbers separated by slashes indicate identical genotypes that were isolated from different hosts. Asterisks indicate strains found in humans, whether zoonotic or human-hosted strains.
Host and geographic conservation
Bartonella are generally highly host-specific with closely related genotypes found in the same order of host; the best model of evolution for Bartonella host order in the 1,039 analyzed 277bp fragments (excluding basal mammalian ectoparasites, rare host orders and taxa for which host order was unknown) was a lambda model in which lambda was 0.96, indicating near Brownian motion evolution along the phylogeny (AICc weight = 1.00; S1 Table). Similarly, in an analysis of 1,027 fragments, closely related genotypes of Bartonella were generally in the same geographic regions, whether analyzed by the continent from which the genotype was isolated or Old World versus New World (best model for both was a lambda transformation; continent: λ = 0.98, AICc weight = 1.00; OW-NW: λ = 0.97; AICc weight = 1.00; S2 and S3 Tables). Basal strains of Bartonella from arthropods were excluded from these analyses as they appeared to be widely distributed, environmental isolates that could skew the analyses.
Exceptions to host specificity and limited geographic range: Zoonosis and the human-domestic-wildlife interface
Despite the overall high host specificity of Bartonella, we observed a number of host shifts in our large phylogenetic hypothesis (S4 File and S2 Fig). Of 19 spillovers into humans (Figs 2 and S2 and S3 and S4 Files; S4 Table), seven were from rodent clades (GQ200856, Z70009, GQ225707, GQ225706, KC633099, AF050108, GQ200861). In some cases, multiple strains of the same Bartonella species have infected humans representing separate spillovers, such as in the case of B. washoensis or the B. vinsonii complex which is found in both rodents and canids [32]. Two human infections appear to stem from bats (B. mayotimonensis [33] and HM116785, found in a Polish forest worker [34] and most closely related to European bat ectoparasite-associated genotypes), one from rabbits (B. alsatica), one from roe deer (HG977196) and four from cats and dogs (B. koehlerae, B. clarridgeiae, B. rochalimae, B. henselae [32]). Four genotypes were of uncertain origin–B. tamiae, a basal infectious strain that causes febrile illness in humans in Asia and has been found in rodent ectoparasites [35,36]; B. bacilliformis, a causative agent of Carrion’s disease and verruga peruana [37]; B. ancashensis, a causative agent of verruga peruana [38] and B. quintana, the causative agent of trench fever. Bartonella quintana has also been found in fleas on gerbils [39] and grouped with Old World rodent and bat-associated genotypes, as well as B. koehlerae and B. henselae. Both B. bacilliformis and B. ancashensis are known primarily from humans [40]; B. quintana is found in both humans and macaques but the genotypes found in macaque are diverged from human B. quintana (95% HPD: 277 ka-5.7 mya; S1 Fig and S5 File).
Rodent-hosted Bartonella has infected domestic carnivores seven times (dogs and cats; representative genotypes: GU056194, FJ946849, GU056198, AF148486, FJ946842, FJ946856, FJ946846) and artiodactyls (a cow ectoparasite; AY517723) once; in five instances rodents and domestic carnivores shared Bartonella but the direction of transmission could not be inferred (representative genotypes: CP019782, EF616739, HM636448, AF214557, AF489539) and in two instances Bartonella from domestic animals has infected rodents (GU354264, GU573930). Bats and rodents have exchanged Bartonella at least five times (representative sequences: EU167549, KP010160, MH234340, AB779518, AF148493). Bats and domestic animals have exchanged Bartonella six times; in four cases bat-associated Bartonella has infected dogs (representative sequences: KP100359, KX300195, HM545140, KT51155); in one case B. bovis infected a bat ectoparasite (JN172054) and in another case Bartonella from a dog may have infected a bat (FJ946847/ HM545137).
In eight cases, there was a transfer of Bartonella between domestic and wild animals (S4 File). In three cases Bartonella associated with domestic carnivores infected wild carnivores. Domestic cat-associated Bartonella has been found in lions (KX499327), mongooses (MF959421) and a cheetah (KX499331); domestic dog-associated Bartonella (B. vinsonii berkhoffii) has infected a fox (KU292568). Additionally, B. rochalimae has been found in a number of wild canids such as coyotes and foxes and has infected domestic dogs. And Bartonella has been shared between (1) a skunk, raccoon, cats and dogs (e.g. CP019782/ CP019786) and (2) domestic cats, lynx, mountain lions, cheetahs and lions (e.g. KX499327/ KX024503). Additionally, in at least two cases Bartonella transferred between domestic artiodactyls and wild artiodactyls–in one case a clade included roe deer, elk, cattle and sheep; in the other B. bovis was found in elk (KB915625).
We also inferred nine transfers of Bartonella between rodents and shrews, which are phylogenetically quite distant but presumably share the same terrestrial habitats and some of the same ectoparasite vectors (S4 File). Similarly, we inferred four transfers of Bartonella between rodents and lagomorphs. Other transfers included B. bovis infecting a cat (Bartonella weissi, AF071190); a Bartonella strain found in a Japanese marten (AB611852) and one in a raccoon (KU292571) in clades of rodent-associated Bartonella; the evolutionary divergence of Bartonella in Antechinus and kangaroos; the divergence of human and non-human primate B. quintana and the grouping of artiodactyl-associated Bartonella with strains found in bats.
We inferred 64 instances where Bartonella was transferred between orders or within orders between wild and domestic animals (S4 File); 42 of these instances were associated with domestic animals, rats or humans, while 21 were not and were presumed to have occurred naturally (including the evolutionary divergence between human and non-human primate B. quintana) and 1 instance involved domestic animals, rats and humans as well as other rodents and shrews. Nearly twice as many instances of host shifts were associated with human influence than natural transitions.
Additionally, we noted 118 instances in which monophyletic clades or single genotypes contained genotypes isolated from more than one continent/ geographic region, 56 of which spanned both the Old World and New World, denoted in parentheses (S4 File and S2 Fig). Of the clades, 55 (27) involved genotypes found in non-rat rodents, 27 (16) involved rats, 12 (11) involved humans, 26 (16) involved cats and dogs, 4 (0) involved domestic hoof stock, 8 (5) involved wild carnivores, 8 (7) involved shrews, 4 (2) involved pikas and hares, 2 (1) involved wild artiodactyls and 38 (7) involved bats. When we analyzed only clades found on more than one continent and grouped humans, cats, dogs, rats and domestic artiodactyls together as human-associated strains, the human-associated strains were more likely to be distributed across both the Old World and New World than other strains (Fisher’s exact test, p = 0.0027, Odds ratio = 3.25). This result was robust regardless of our posterior cut off criteria (0.9 support or above: p = 0.0047, Odds ratio = 3.35; 0.95 support or above: p = 0.013, Odds ratio = 2.98).
Transfers of Bartonella into humans were also more recent than other divergences we inferred based on both the estimated age of the node (meanzoonotic = 4.56 my; meannon-zoonotic = 12.52 my; t-test: t = -2.24, df = 20.9, p = 0.018) and the lower bound of the 95% highest posterior density (meanzoonotic = 0.656 my; meannon-zoonotic = 2.485 my; t-test: t = -2.43, df = 21.8, p = 0.012; S5 File).
Discussion
Our phylogenetic hypothesis based on the 277bp fragment of gltA was broadly concordant with that of other studies of Bartonella. We recovered a monophyletic grouping of B. grahamii, B. rattimasiliensis, B. tribocorum, B. elizabethae and B. queenslandensis; a monophyletic grouping of B. quintana, B. henselae and B. koehlerae; and the clustering of B. vinsonii subspecies consistent with studies from multiple Bartonella genes [6,41]. These groupings are further supported by a large 509 gene phylogeny, which also supported our findings of basal positions for B. apis and B. tamiae and monophyletic grouping of artiodactyl-associated Bartonella [42]. In fact, most of our deeper branching relationships were also consistent with this large, multigene phylogeny, though were significantly less well supported as our conclusions were based on a single gene.
Bartonella as an environmental bacterium turned insect gut symbiont turned vertebrate pathogen
The proliferation of studies investigating Bartonella in various wildlife populations allows for greater insights into the origins and evolution of Bartonella and its potential for spillover more than ever before. Bartonellaceae is nested within the Rhizobiales, a lineage of soil bacteria that contains nitrogen-fixing root-associated members [43]. In our study, the most basal strains of Bartonella were found in environmental samples and arthropods (Figs 1 and 2). Additionally, gut microbiome studies from a variety of insects have revealed that Bartonella is actually widespread across arthropods, occurring in carrion beetles, butterflies, bees, various species of ants and a wide variety of ectoparasitic species [9–15]. Other studies have hypothesized that perhaps Bartonella may have a commensal role in the arthropods that vector it [44,45]. This led us to hypothesize that Bartonella originated as an environmental bacterium that was picked up by arthropods in which it diversified.
Because most metagenomic studies of bacteria amplify the 16s rRNA gene, there is a large amount of 16s data available and also Bartonella can be detected in samples that would not a priori be hypothesized to contain Bartonella, such as non-hematophagous insects or environmental samples. We mined GenBank for Bartonella 16s sequences to test our hypothesis that Bartonella is an environmental bacterium that became an insect commensal before becoming a vertebrate pathogen. The 16s rRNA gene is much less powerful for discriminating Bartonella species than gltA [7] and often metagenomic studies amplify only very small fragments of the gene, making it difficult for us to resolve fine scale diversification but we were able to determine that basal strains of Bartonella were largely found in environmental samples and non-hematophagous insects (Fig 1). Additionally, work on Bartonella has shown that the evolution of a type 4 secretion system, along with selection on other invasion mechanisms [43], has been instrumental in allowing Bartonella to diversify and invade host cells [46,47] while other work has shown Bartonella can incorporate a type 4 secretion system via lateral gene transfer when it coinfects an amoeba with Rhizobium radiobacter [48]. Further, examinations of lateral gene transfer of metabolic genes in Bartonella reveals that many of these genes derive from common insect gut commensal bacteria [49]. We strengthen the suggestions of these previous studies by drawing data from insect and environmental metabarcoding studies and demonstrating their basal phylogenetic position within Bartonella.
Bartonella spillover is predominantly from rodent and domestic animals
Using the literature [32] and isolates from published sequences on GenBank, we identified 19 genotypes of Bartonella that have been detected in humans (most of which are also known to cause disease; S4 Table). Of these, eight of the genotypes are most closely related to genotypes found in rodents and four are distributed in dogs and cats but have spilled over into humans. Bartonella vinsonii forms a species complex that is associated with dogs (subsp. berkhoffi) and rodents (subsp. arupensis). We inferred at least three separate transfers of B. vinsonii (two instances of B. v. arupensis and one instance of B. v. vinsonii) from rodents into humans based on phylogenetic relationships (GQ200861, GQ200857, GQ225708), confirming previous conclusions [6]; however, we treated these as a single spillover for the sake of simplicity. Additionally, we identified a genotype of Bartonella detected in a febrile patient in Thailand (GQ200856) as having over 95% identity with B. queenslandensis, a genotype first found in Australian rodents and also found in numerous Asian rodents, suggesting a previously unappreciated possible rodent-human transmission. Interestingly, one Bartonella genotype that was recovered from a Polish forest worker (HM116785) most closely resembled genotypes found in European Myotis, a genus of bat, and their ectoparasites (JQ695834, JQ695839, KR822802). That most strains isolated from humans are related to domestic or peridomestic animals strongly indicates that spillover of Bartonella requires close contact between humans and the natural reservoirs of these infectious strains.
However, when examined at a broader scale, many of these genotypes are related to genotypes found in wild animals. For example, B. henselae, B. koehlerae and B. quintana were closely related to Bartonella detected in African rodent ectoparasites and an Asian bat. Similarly, B. mayotimonensis was closely associated with genotypes of Central American bats detected in this study. This same isolate has also been found in bats in Europe [33,50] and most recently North America [51]. This suggests that bats may be a possible reservoir of potentially zoonotic Bartonella strains but that infrequent contact between bats and people prevents transmission. Rather most of the transmission we infer requires the transmission of Bartonella into a domestic or peridomestic animal, which can then transmit it to humans. Despite the noted host specificity of Bartonella (S1 Table), the diversity of strains that infect humans and their distribution across the phylogenetic tree of Bartonella suggests that this bacterial genus can and will switch hosts when given the opportunity (especially when hosts are immunocompromised [52,53]). The relative evolutionary lability of these genotypes is further underscored by the instances in the global phylogeny of genotypes being exchanged between bats and rodents (at least five times; S4 File).
Overall, we found that rodents were responsible for more transmission of Bartonella into humans than any other group, followed by domestic carnivores. Rodents also transmitted the most Bartonella to domestic animals and bats, though infections likely originating from wildlife such as bats in domestic animals are also relatively common. One potential explanation for the prominence of rodents in host switching may be the generalist tendencies of their ectoparasites. Bartonella is vectored by arthropods but some ectoparasites, such as blood sucking hippoboscid flies, are very highly host specific [54] potentially preventing cross-species transmission. In contrast, many rodents host fleas which can bite other taxa and have been found to host many genotypes of Bartonella that have originated in rodents and infected other species such as humans (e.g. [55,56]). Considerations of the host specificity of the vector species may be very important for determining the risk for disease spillover and indeed public health officials recommend avoidance of potential vectors as the most important measure for prevention of bartonellosis [32].
It is important to note, however, the constraints on our conclusions due to available data. We only have a small fragment of gltA to examine across these 1,060 genotypes, making inferences at deep nodes uncertain and potentially artificially grouping together isolates that are identical at the sites we examined but that may differ dramatically at other important genetic loci or even other loci within the gltA gene. Additionally, we are limited to the animals that have been sampled, which are overwhelmingly bats and rodents, as well as symptomatic humans. It is possible and highly likely that there are animal intermediates between these transmission events that are missing, which obscures our ability to infer the directionality of transfer. Indeed, to our knowledge, direct Bartonella transmission has not been observed between bats and domestic animals; however, we observed multiple instances in which Bartonella detected in domestic animals and their ectoparasites fell evolutionarily within Bartonella detected in bats. This suggests that these strains may have originated in bats before being transmitted to domestic animals and demonstrates the power of a large-scale phylogenetic approach to identify unlikely sources of Bartonella transfer.
Human movements shape Bartonella diversification and infection patterns
Another interesting pattern that emerged when examining the tree as a whole was the impact of humans in spreading Bartonella strains and infections globally. A few particular mammalian species that are associated with humans, such as dogs, cats, cows and rats, have managed to bring their strains of Bartonella globally [21,32,47,57–61]. Rats, in particular the genus Rattus, were very common in the largest clade of globally distributed rodent Bartonella (the clade containing B. queenslandensis, B. elizabethae, B. tribocorum, B. massiliensis, B. rattimassiliensis and B. grahamii), with representatives on nearly every continent. This clade also contains at least five zoonotic genotypes of Bartonella, as well as genotypes found in dogs and ectoparasites on dogs, bats and a cow, underscoring the important role of human commensals in spreading disease to humans, domestic animals and wildlife and across the globe.
There was a lot of uncertainty in the dating of our divergence times (in one instance three identical genotypes were inferred to be over 700,000 years diverged) perhaps due to the small fragment we were able to analyze and the depth of evolutionary history we were exploring. Additionally, there are many genotypes that may have died out or have not been sampled that mean even our minimum divergence date estimates are likely conservative. We cannot therefore state with certainty that humans are responsible for moving other species around, changing disease risk for themselves and wild animals. However, the finding that strains associated with humans or their domestic animals were more likely to be found globally, the finding that transfers between humans and other groups were the most recent ones, and the diverse placement of human infections across the phylogeny strongly support a role for humans changing their disease risk as they insert themselves and their associated animals into new habitats and ecosystems.
Such movements and increased contact between humans, domestic animals and wildlife not only disguise geographic patterns of Bartonella diversification (e.g. B. queenslandensis, first described in Australia [62], in Rattus norvegicus in Louisiana (AF075162; [57])) but have also led to presumably novel sharing of Bartonella between introduced domestic and peridomestic animals and native wildlife. For example, identical genotypes were found in a Chinese Rattus individual (DQ986952) and a white-footed mouse, a North American native (AY064534). If the introduced bacteria have adverse fitness consequences, this could be another human-mediated conservation concern. Domestic animals also present a health risk to wild animals and other domestic animals; B. bovis was found in a cat, an elk and a bat ectoparasite and Bartonella has been transferred between Old World bats and dogs. The pet trade exacerbates this by shipping exotic animals all over the world, changing the pool of available infections for both the introduced and native species [63]. Introduction of domestic species is causing sharing between these species and wild species, changing the disease risks for both.
Overall our findings show that Bartonella is a rich system for examining the impacts of humans on patterns of infectious disease spread within species and between species, across landscapes and across the globe. Phylogenetic inferences about the origin of infections should be interpreted with caution as they are heavily influenced by available data and the taxa that have been sampled. There may be many missing links between those we inferred but the hosts simply have not been sampled. At least some part of the noted host specificity of Bartonella seems to be due to ecological factors regulating exposure rather than immunological incompatibility. Given the diversity of sources of zoonotic strains, including divergent strains with similar clinical presentations, physicians and researchers should consider a broad range of potential animal hosts and screen for a wide range of Bartonella genotypes when investigating the source of a suspected Bartonella infection.
New sequences generated as part of this study have been uploaded to Genbank (accession numbers MH234314 –MH234380).
Supporting information
(DOCX)
(DOCX)
(DOCX)
Reservoir and geographic data are derived from gltA metadata, Breitschwerdt [32] and cited references. Phylogenetic context refers to placement in both the 277bp and 540bp MCC trees.
(DOCX)
The numbers in brackets indicate the lower and upper bounds of the 95% highest posterior density of the height of that branch, an estimate of the age of divergence of the node that the branch leads to. The width of the circle at each node is proportional to the Bayesian posterior probability support at that node. The blue tick mark numbers indicate the well-supported instances of Bartonella transferring between hosts that are used in inferring the timing of host transfers as detailed in S5 File.
(PDF)
The color of the tip label indicates the order of the host from which the genotype was derived (ectoparasites are colored according to their host). Highlighted tips indicate that multiple samples had the same sequence and the duplicate sequences can be found in S3 File by searching for the Genbank accession number. The number on each branch indicates the Bayesian posterior probability support leading to the node on the right. The icon at each tip indicates the geographic region from which the genotype was derived and the colored dot at each tip indicates the type of host it was isolate from, e.g. Rattus sp. rodents, non-Rattus rodents, wild animals, domestic animals. “SM” indicates a shrew or rodent (small mammal) as sometimes the actual host was not given and “Unk” indicates the continent from which the genotype was isolated is unknown. If a Bartonella species name is listed at a tip but not a Genbank accession number, the accession number is that of the sequence used in Judson et al. [16].
(PDF)
(XLSX)
(XLSX)
Only one representative per group is included in the phylogeny and S2 Fig even though identical genotypes may have been found in other hosts or geographic regions. This file lists all Genbank accession numbers for sequences found in more than one individual in the study. A legend for all categories appears on the second sheet of the file.
(XLSX)
This file lists every inferred host transfer or finding of a genotype or monophyletic clade on more than one continent. A legend for interpreting each category appears on the second sheet of the file.
(XLSX)
This file lists every well-supported host switching event captured in our 540bp phylogenetic hypothesis. A legend for interpreting each column appears on the second sheet of the file.
(XLSX)
(NEX)
(NEX)
(NEX)
(TREES)
(TREES)
(TREES)
(R)
Acknowledgments
We thank C. Mendenhall, F. Oviedo Brenes, R. Zahawi, W. Figueroa, R. Figueroa, J. Figueroa, Y. Lloria, S. Judson, H. Mao, dozens of Costa Rican landowners, the Organization for Tropical Studies, the Las Cruces Biological Station, and especially J. O’Marr for help with collection of data on Costa Rican bat-associated Bartonella, A. Ravenscraft for access to Bartonella from butterflies and K. Roskin for help with obtaining data from Genbank. Additional thanks to the Hadly lab, the Boyd lab, J. Flanders and an anonymous reviewer for useful comments.
Data Availability
All data are publicly available. Genetic alignments and metadata are available in the Supporting Information files. All of the new sequences generated as part of this study are available on Genbank (accession numbers MH234314 – MH234380).
Funding Statement
This work was graciously funded by the Stanford Woods Institute for the Environment Environmental Ventures Program (https://woods.stanford.edu) and a Bat Conservation International (www.batcon.org) Student Scholarship to HKF. HKF was supported by a Bing-Mooney Fellowship in Environmental Science and Conservation and a Stanford Center for Computational, Evolutionary and Human Genomics (https://cehg.stanford.edu) Postdoctoral Fellowship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Warnecke L, Turner JM, Bollinger TK, Lorch JM, Misra V, et al. (2012) Inoculation of bats with European Geomyces destructans supports the novel pathogen hypothesis for the origin of white-nose syndrome. Proc Natl Acad Sci 109: 6999–7003. 10.1073/pnas.1200374109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lips KR, Brem F, Brenes R, Reeve JD, Alford RA, et al. (2006) Emerging infectious disease and the loss of biodiversity in a Neotropical amphibian community. Proc Natl Acad Sci U S A 103: 3165–3170. Available: http://www.pnas.org/cgi/content/long/103/9/3165. 10.1073/pnas.0506889103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, et al. (2008) Global trends in emerging infectious diseases. Nature 451: 990–993. Available: http://www.ncbi.nlm.nih.gov/pubmed/18288193%5Cnhttp://www.nature.com/nature/journal/v451/n7181/pdf/nature06536.pdf. 10.1038/nature06536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lo Iacono G, Cunningham AA, Fichet-Calvet E, Garry RF, Grant DS, et al. (2016) A Unified Framework for the Infection Dynamics of Zoonotic Spillover and Spread. PLoS Negl Trop Dis 10: e0004957 10.1371/journal.pntd.0004957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breitschwerdt EB, Maggi RG, Chomel BB, Lappin MR (2010) Bartonellosis: An emerging infectious disease of zoonotic importance to animals and human beings. J Vet Emerg Crit Care 20: 8–30. 10.1111/j.1476-4431.2009.00496.x [DOI] [PubMed] [Google Scholar]
- 6.Kosoy M, Bai Y, Sheff K, Morway C, Baggett H, et al. (2010) Identification of Bartonella infections in febrile human patients from Thailand and their potential animal reservoirs. Am J Trop Med Hyg 82: 1140–1145. 10.4269/ajtmh.2010.09-0778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.La Scola B, Zeaiter Z, Khamis A, Raoult D (2003) Gene-sequence-based criteria for species definition in bacteriology: the Bartonella paradigm. Trends Microbiol 11: 318–321. 10.1016/S0966-842X(03)00143-4 [DOI] [PubMed] [Google Scholar]
- 8.Kosoy M, McKee C, Albayrak L, Fofanov Y (2017) Genotyping of Bartonella bacteria and their animal hosts: current status and perspectives. Parasitology: 1–20. Available: https://www.cambridge.org/core/product/identifier/S0031182017001263/type/journal_article. [DOI] [PubMed] [Google Scholar]
- 9.Kautz S, Rubin BER, Russell JA, Moreaua CS (2013) Surveying the microbiome of ants: Comparing 454 pyrosequencing with traditional methods to uncover bacterial diversity. Appl Environ Microbiol 79: 525–534. 10.1128/AEM.03107-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kopeck J, Nesvorn M, Hubert J (2014) Bartonella-like bacteria carried by domestic mite species. Exp Appl Acarol 64: 21–32. 10.1007/s10493-014-9811-1 [DOI] [PubMed] [Google Scholar]
- 11.Jones RT, McCormick KF, Martin AP (2008) Bacterial communities of Bartonella-positive fleas: Diversity and community assembly patterns. Appl Environ Microbiol 74: 1667–1670. 10.1128/AEM.02090-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laroche M, Berenger J-M, Mediannikov O, Raoult D, Parola P (2017) Detection of a Potential New Bartonella Species “Candidatus Bartonella rondoniensis” in Human Biting Kissing Bugs (Reduviidae; Triatominae). PLoS Negl Trop Dis 11: e0005297 Available: http://dx.plos.org/10.1371/journal.pntd.0005297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kešnerová L, Moritz R, Engel P (2016) Bartonella apis sp. nov., a honey bee gut symbiont of the class Alphaproteobacteria. Int J Syst Evol Microbiol 66: 414–421. 10.1099/ijsem.0.000736 [DOI] [PubMed] [Google Scholar]
- 14.Kaltenpoth M, Steiger S (2014) Unearthing carrion beetles’ microbiome: Characterization of bacterial and fungal hindgut communities across the Silphidae. Mol Ecol 23: 1251–1267. 10.1111/mec.12469 [DOI] [PubMed] [Google Scholar]
- 15.Kakumanu ML, Reeves AM, Anderson TD, Rodrigues RR, Williams MA (2016) Honey bee gut microbiome is altered by in-hive pesticide exposures. Front Microbiol 7: 1255 10.3389/fmicb.2016.01255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Judson SD, Frank HK, Hadly EA (2015) Bartonellae are Prevalent and Diverse in Costa Rican Bats and Bat Flies. Zoonoses Public Health 62: 609–617. Available: http://doi.wiley.com/10.1111/zph.12188 [DOI] [PubMed] [Google Scholar]
- 17.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, et al. (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649. Available: http://bioinformatics.oxfordjournals.org/content/28/12/1647.short. 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, et al. (2014) BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput Biol 10: e1003537 10.1371/journal.pcbi.1003537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B (2017) PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol Biol Evol 34: 772–773. Available: http://mbe.oxfordjournals.org/lookup/doi/10.1093/molbev/msw260. 10.1093/molbev/msw260 [DOI] [PubMed] [Google Scholar]
- 20.McKee CD, Hayman DTS, Kosoy MY, Webb CT (2016) Phylogenetic and geographic patterns of bartonella host shifts among bat species. Infect Genet Evol 44: 382–394. 10.1016/j.meegid.2016.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayman DTS, McDonald KD, Kosoy MY (2013) Evolutionary history of rat-borne Bartonella: The importance of commensal rats in the dissemination of bacterial infections globally. Ecol Evol 3: 3195–3203. 10.1002/ece3.702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rojas D, Warsi O, Dávalos L (2016) Bats (Chiroptera: Noctilionoidea) challenge recent origin of neotropical diversity. Syst Biol 65: 432–448. 10.1093/sysbio/syw011 [DOI] [PubMed] [Google Scholar]
- 23.Shi JJ, Rabosky DL (2015) Speciation dynamics during the global radiation of extant bats. Evolution (N Y) 69: 1528–1545. 10.1111/evo.12681 [DOI] [PubMed] [Google Scholar]
- 24.Dumont ER, Samadevam K, Grosse I, Warsi OM, Baird B, et al. (2014) Selection for mechanical advantage underlies multiple cranial optima in new world leaf-nosed bats. Evolution (N Y) 68: 1436–1449. 10.1111/evo.12358 [DOI] [PubMed] [Google Scholar]
- 25.Larsen PA, Marchan-Rivadeneira MR, Baker RJ (2010) Natural hybridization generates mammalian lineage with species characteristics. Proc Natl Acad Sci 107: 11447–11452. Available: http://www.pnas.org/cgi/doi/10.1073/pnas.1000133107. 10.1073/pnas.1000133107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fritz SA, Bininda-Emonds ORP, Purvis A (2009) Geographical variation in predictors of mammalian extinction risk: Big is bad, but only in the tropics. Ecol Lett 12: 538–549. 10.1111/j.1461-0248.2009.01307.x [DOI] [PubMed] [Google Scholar]
- 27.Redondo RAF, Brina LPS, Silva RF, Ditchfield AD, Santos FR (2008) Molecular systematics of the genus Artibeus (Chiroptera: Phyllostomidae). Mol Phylogenet Evol 49: 44–58. Available: http://www.ncbi.nlm.nih.gov/pubmed/18662791. Accessed 4 August 2011. 10.1016/j.ympev.2008.07.001 [DOI] [PubMed] [Google Scholar]
- 28.Kumar S, Stecher G, Suleski M, Hedges SB (2017) TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol Biol Evol 34: 1812–1819. Available: https://academic.oup.com/mbe/article-lookup/doi/10.1093/molbev/msx116. 10.1093/molbev/msx116 [DOI] [PubMed] [Google Scholar]
- 29.Baele G, Lemey P, Bedford T, Rambaut A, Suchard MA, et al. (2012) Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol Biol Evol 29: 2157–2167. 10.1093/molbev/mss084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harmon LJ, Weir JT, Brock CD, Glor RE, Challenger W (2008) GEIGER: Investigating evolutionary radiations. Bioinformatics 24: 129–131. 10.1093/bioinformatics/btm538 [DOI] [PubMed] [Google Scholar]
- 31.Kamani J, Morick D, Mumcuoglu KY, Harrus S (2013) Prevalence and Diversity of Bartonella Species in Commensal Rodents and Ectoparasites from Nigeria, West Africa. PLoS Negl Trop Dis 7 10.1371/journal.pntd.0002246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Breitschwerdt EB (2017) Bartonellosis, One Health and all creatures great and small. Vet Dermatol 28: 96–e21. Available: http://www.ncbi.nlm.nih.gov/pubmed/28133871%0Ahttp://doi.wiley.com/10.1111/vde.12413. 10.1111/vde.12413 [DOI] [PubMed] [Google Scholar]
- 33.Veikkolainen V, Vesterinen EJ, Lilley TM, Pulliainen AT (2014) Bats as reservoir hosts of human bacterial pathogen, Bartonella mayotimonensis. Emerg Infect Dis 20: 960–967. 10.3201/eid2006.130956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Podsiadly E, Chmielewski T, Karbowiak G, Kedra E, Tylewska-Wierzbanowska S (2010) The occurrence of spotted fever rickettsioses and other tick-borne infections in forest workers in Poland. Vector-Borne Zoonotic Dis 11: 985–989. Available: http://www.ncbi.nlm.nih.gov/pubmed/21083370. 10.1089/vbz.2010.0080 [DOI] [PubMed] [Google Scholar]
- 35.Kosoy M, Morway C, Sheff KW, Bai Y, Colborn J, et al. (2008) Bartonella tamiae sp. nov., a newly recognized pathogen isolated from three human patients from Thailand. J Clin Microbiol 46: 772–775. 10.1128/JCM.02120-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kabeya H, Colborn JM, Bai Y, Lerdthusnee K, Richardson JH, et al. (2010) Detection of Bartonella tamiae DNA in Ectoparasites from Rodents in Thailand and Their Sequence Similarity with Bacterial Cultures from Thai Patients. Vector-Borne Zoonotic Dis 10: 429–434. Available: http://www.liebertonline.com/doi/abs/10.1089/vbz.2009.0124. 10.1089/vbz.2009.0124 [DOI] [PubMed] [Google Scholar]
- 37.Breitschwerdt EB (2014) Bartonellosis: One Health perspectives for an emerging infectious disease. ILAR J 55: 46–58. 10.1093/ilar/ilu015 [DOI] [PubMed] [Google Scholar]
- 38.Mullins KE, Hang J, Jiang J, Leguia M, Kasper MR, et al. (2013) Molecular typing of “Candidatus bartonella ancashi,” A new human pathogen causing Verruga Peruana. J Clin Microbiol 51: 3865–3868. 10.1128/JCM.01226-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marié JL, Fournier PÉ, Rolain JM, Briolant S, Davoust B, et al. (2006) Molecular detection of Bartonella quintana, B. elizabethae, B. koehlerae, B. doshiae, B. taylorii, and Rickettsia felis in rodent fleas collected in Kabul, Afghanistan. Am J Trop Med Hyg 74: 436–439. [PubMed] [Google Scholar]
- 40.Mullins KE, Hang J, Jiang J, Leguia M, Kasper MR, et al. (2015) Description of Bartonella ancashensis sp. nov., isolated from the blood of two patients with verruga peruana. Int J Syst Evol Microbiol 65: 3339–3343. 10.1099/ijsem.0.000416 [DOI] [PubMed] [Google Scholar]
- 41.Houpikian P, Raoult D (2001) Molecular phylogeny of the genus Bartonella: what is the current knowledge? FEMS Microbiol Lett 200: 1–7. 10.1111/j.1574-6968.2001.tb10684.x [DOI] [PubMed] [Google Scholar]
- 42.Harms A, Segers FHID, Quebatte M, Mistl C, Manfredi P, et al. (2017) Evolutionary Dynamics of Pathoadaptation Revealed by Three Independent Acquisitions of the VirB/D4 Type IV Secretion System in Bartonella. Genome Biol Evol 9: 761–776. 10.1093/gbe/evx042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heavner ME, Qiu WG, Cheng HP (2015) Phylogenetic co-occurrence of ExoR, ExoS, and ChvI, components of the RSI bacterial invasion switch, suggests a key adaptive mechanism regulating the transition between free-living and host-invading phases in rhizobiales. PLoS One 10: 1–22. 10.1371/journal.pone.0135655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morse SF, Olival KJ, Kosoy M, Billeter S, Patterson BD, et al. (2012) Global distribution and genetic diversity of Bartonella in bat flies (Hippoboscoidea, Streblidae, Nycteribiidae). Infect Genet Evol 12: 1717–1723. 10.1016/j.meegid.2012.06.009 [DOI] [PubMed] [Google Scholar]
- 45.Chomel BB, Boulouis HJ, Breitschwerdt EB, Kasten RW, Vayssier-Taussat M, et al. (2009) Ecological fitness and strategies of adaptation of Bartonella species to their hosts and vectors. Vet Res 40: 29 10.1051/vetres/2009011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engel P, Salzburger W, Liesch M, Chang CC, Maruyama S, et al. (2011) Parallel evolution of a type IV secretion system in radiating lineages of the host-restricted bacterial pathogen Bartonella. PLoS Genet 7: e1001296 10.1371/journal.pgen.1001296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guy L, Nystedt B, Toft C, Zaremba-Niedzwiedzka K, Berglund EC, et al. (2013) A Gene Transfer Agent and a Dynamic Repertoire of Secretion Systems Hold the Keys to the Explosive Radiation of the Emerging Pathogen Bartonella. PLoS Genet 9: e1003393 10.1371/journal.pgen.1003393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saisongkorh W, Robert C, La Scola B, Raoult D, Rolain JM (2010) Evidence of transfer by conjugation of type IV secretion system genes between Bartonella species and Rhizobium radiobacter in amoeba. PLoS One 5: 1–14. 10.1371/journal.pone.0012666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu Q, Kosoy M, Olival KJ, Dittmar K (2014) Horizontal transfers and gene losses in the phospholipid pathway of Bartonella reveal clues about early ecological niches. Genome Biol Evol 8: 2156–2169. 10.1093/gbe/evu169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stuckey MJ, Boulouis HJ, Cliquet F, Picard-Meyer E, Servat A, et al. (2017) Potentially zoonotic Bartonella in bats from France and Spain. Emerg Infect Dis 23: 539–541. 10.3201/eid2303.160934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lilley TM, Wilson CA, Bernard RF, Willcox EV., Vesterinen EJ, et al. (2017) Molecular Detection of Candidatus Bartonella mayotimonensis in North American Bats. Vector-Borne Zoonotic Dis 17: 243–246. Available: http://online.liebertpub.com/doi/10.1089/vbz.2016.2080. 10.1089/vbz.2016.2080 [DOI] [PubMed] [Google Scholar]
- 52.Clarridge JE, Raich TJ, Pirwani D, Simon B, Tsai L, et al. (1995) Strategy to detect and identify Bartonella species in routine clinical laboratory yields Bartonella henselae from human immunodeficiency virus- positive patient and unique Bartonella strain from his cat. J Clin Microbiol 33: 2107–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oksi J, Rantala S, Kilpinen S, Silvennoinen R, Vornanen M, et al. (2013) Cat scratch disease caused by Bartonella grahamii in an immunocompromised patient. J Clin Microbiol 51: 2781–2784. 10.1128/JCM.00910-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dick C, Patterson B (2006) Bat flies: Obligate ectoparasites of bats In: Morand S, Krasnov B, Poulin R, editors. Micromammals and macroparasites: from Evolutionary Ecology to Management. Springer-Verlag Publishing; pp. 179–194. [Google Scholar]
- 55.Abbot P, Aviles AE, Eller L, Durden LA (2007) Mixed infections, cryptic diversity, and vector-borne pathogens: Evidence from Polygenis fleas and Bartonella species. Appl Environ Microbiol 73: 6045–6052. 10.1128/AEM.00228-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frye MJ, Firth C, Bhat M, Firth MA, Che X, et al. (2015) Preliminary survey of ectoparasites and associated pathogens from Norway rats in New York City. J Med Entomol 52: 253–259. 10.1093/jme/tjv014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ellis BA, Regnery RL, Beati L, Bacellar F, Rood M, et al. (1999) Rats of the genus Rattus are reservoir hosts for pathogenic Bartonella species: an Old World origin for a New World disease? J Infect Dis 180: 220–224. 10.1086/314824 [DOI] [PubMed] [Google Scholar]
- 58.Rudoler N, Rasis M, Sharir B, Novikov A, Shapira G, et al. (2014) First description of Bartonella bovis in cattle herds in Israel. Vet Microbiol 173: 110–117. 10.1016/j.vetmic.2014.07.006 [DOI] [PubMed] [Google Scholar]
- 59.Cherry NA, Maggi RG, Cannedy AL, Breitschwerdt EB (2009) PCR detection of Bartonella bovis and Bartonella henselae in the blood of beef cattle. Vet Microbiol 135: 308–312. 10.1016/j.vetmic.2008.09.063 [DOI] [PubMed] [Google Scholar]
- 60.Kho K-L, Koh F-X, Jaafar T, Nizam QNH, Tay S-T (2015) Prevalence and molecular heterogeneity of Bartonella bovis in cattle and Haemaphysalis bispinosa ticks in Peninsular Malaysia. BMC Vet Res 11: 153 10.1186/s12917-015-0470-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dahmani M, Sambou M, Scandola P, Raoult D, Fenollar F, et al. (2017) Bartonella bovis and Candidatus Bartonella davousti in cattle from Senegal. Comp Immunol Microbiol Infect Dis 50: 63–69. 10.1016/j.cimid.2016.11.010 [DOI] [PubMed] [Google Scholar]
- 62.Gundi VAKB, Taylor C, Raoult D, La Scola B (2009) Bartonella rattaustraliani sp. nov., Bartonella queenslandensis sp. nov. and Bartonella coopersplainsensis sp. nov., identified in Australian rats. Int J Syst Evol Microbiol 59: 2956–2961. 10.1099/ijs.0.002865-0 [DOI] [PubMed] [Google Scholar]
- 63.Inoue K, Maruyama S, Kabeya H, Hagiya K, Izumi Y, et al. (2009) Exotic small mammals as potential reservoirs of zoonotic Bartonella spp. Emerg Infect Dis 15: 526–532. 10.3201/eid1504.081223 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
(DOCX)
Reservoir and geographic data are derived from gltA metadata, Breitschwerdt [32] and cited references. Phylogenetic context refers to placement in both the 277bp and 540bp MCC trees.
(DOCX)
The numbers in brackets indicate the lower and upper bounds of the 95% highest posterior density of the height of that branch, an estimate of the age of divergence of the node that the branch leads to. The width of the circle at each node is proportional to the Bayesian posterior probability support at that node. The blue tick mark numbers indicate the well-supported instances of Bartonella transferring between hosts that are used in inferring the timing of host transfers as detailed in S5 File.
(PDF)
The color of the tip label indicates the order of the host from which the genotype was derived (ectoparasites are colored according to their host). Highlighted tips indicate that multiple samples had the same sequence and the duplicate sequences can be found in S3 File by searching for the Genbank accession number. The number on each branch indicates the Bayesian posterior probability support leading to the node on the right. The icon at each tip indicates the geographic region from which the genotype was derived and the colored dot at each tip indicates the type of host it was isolate from, e.g. Rattus sp. rodents, non-Rattus rodents, wild animals, domestic animals. “SM” indicates a shrew or rodent (small mammal) as sometimes the actual host was not given and “Unk” indicates the continent from which the genotype was isolated is unknown. If a Bartonella species name is listed at a tip but not a Genbank accession number, the accession number is that of the sequence used in Judson et al. [16].
(PDF)
(XLSX)
(XLSX)
Only one representative per group is included in the phylogeny and S2 Fig even though identical genotypes may have been found in other hosts or geographic regions. This file lists all Genbank accession numbers for sequences found in more than one individual in the study. A legend for all categories appears on the second sheet of the file.
(XLSX)
This file lists every inferred host transfer or finding of a genotype or monophyletic clade on more than one continent. A legend for interpreting each category appears on the second sheet of the file.
(XLSX)
This file lists every well-supported host switching event captured in our 540bp phylogenetic hypothesis. A legend for interpreting each column appears on the second sheet of the file.
(XLSX)
(NEX)
(NEX)
(NEX)
(TREES)
(TREES)
(TREES)
(R)
Data Availability Statement
All data are publicly available. Genetic alignments and metadata are available in the Supporting Information files. All of the new sequences generated as part of this study are available on Genbank (accession numbers MH234314 – MH234380).