

Abstract
Intestinal epithelial barrier repair is vital for remission in inflammatory bowel disease (IBD). Extracellular adenosine signaling has been implicated in promoting restoration of epithelial barrier function. Currently, no clinically approved agents target this pathway. Adenosine signaling is terminated by uptake from the extracellular space via equilibrative nucleoside transporters (ENTs). We hypothesized that ENT inhibition could dampen intestinal inflammation. Initial studies demonstrated transcriptional repression of ENT1 and ENT2 in IBD biopsies or in murine IBD models. Subsequent studies in mice with global Ent1 or Ent2 deletion revealed selective protection of Ent2–/– mice. Elevated intestinal adenosine levels in conjunction with abolished protection following pharmacologic blockade of A2B adenosine receptors implicate adenosine signaling as the mechanism of gut protection in Ent2–/– mice. Additional studies in mice with tissue-specific deletion of Ent2 uncovered epithelial Ent2 as the target. Moreover, intestinal protection provided by a selective Ent2 inhibitor was abolished in mice with epithelium-specific deletion of Ent2 or the A2B adenosine receptor. Taken together, these findings indicate that increased mucosal A2B signaling following repression or deletion of epithelial Ent2 coordinates the resolution of intestinal inflammation. This study suggests the presence of a targetable purinergic network within the intestinal epithelium designed to limit tissue inflammation.
Keywords: Gastroenterology
Keywords: Inflammatory bowel disease
Suppression of adenosine transport at the epithelial surface increases extracellular adenosine to promote barrier protective signaling pathways.
Introduction
Inflammatory bowel disease (IBD), chronic relapsing-remitting conditions of the gastrointestinal tract encompassing Crohn’s disease (CD) and ulcerative colitis (UC), is increasing in incidence and prevalence (1). IBD has a long natural history requiring a range of pharmaceutical interventions and regular admission to the hospital, with significant numbers of patients progressing to surgical intervention (2, 3). Current therapeutic strategies seek to provide symptom control, but more recent evidence suggests that mucosal healing is a key end point to validate disease remission and successful therapeutic response in IBD (4–6). Indeed, achieving mucosal healing in IBD has been shown to improve quality of life and reduce complications, including hospitalizations and the need for surgical intervention (7, 8). When considering mucosal healing, the most critical component of the intestinal mucosal barrier being assessed is the intestinal epithelial cell layer (8). Increased permeability of the epithelial barrier and significant damage to the epithelial layer, including ulceration, are observed in IBD patients (9–13). This is also observed in animal models of IBD, where increased intestinal permeability is an early hallmark of disease (14–17). In addition, several studies suggest that IBD occurs due to disruption of the intestinal epithelial barrier; this leads to increased bacterial contact with the mucosal immune system, resulting in proinflammatory cytokine production and uncontrolled tissue inflammation in genetically predisposed individuals (18). Given the primary role of epithelial barrier dysfunction in disease and the correlation between barrier restitution and therapeutic response, there is great interest in identifying new therapies to restore epithelial barrier function in IBD (reviewed in ref. 19).
It was recently reported that adenosine signaling to the colonic epithelium improves epithelial barrier function in experimental colitis (20). Adenosine is an endogenous nucleoside that is rapidly produced at the extracellular surface through the breakdown of ATP by the membrane-bound enzymes ectonucleoside triphosphate diphosphohydrolase 1 (CD39) and ecto-5′-nucleotidase (CD73), which convert ATP to AMP and on to adenosine (21). The process of enzymatic production of extracellular adenosine is dramatically increased under inflammatory or hypoxic conditions — such as occurs during intestinal inflammation (22–24). Adenosine can signal through 4 different transmembrane G protein–coupled receptors (A1, A2A, A2B, and A3 receptors; gene names, Adora1, Adora2a, Adora2b, Adora3) (25). Indeed, studies in IBD patients highlight that nonfunctional mutations of CD39 are associated with a higher incidence of IBD, suggesting that extracellular adenosine production and signaling play a role in an endogenous protective pathway that dampens intestinal inflammation (26). This is supported by studies demonstrating that signaling through the A2A (27–30), A2B (20, 31, 32), or A3 (33) receptors is protective in experimental colitis. Most of these studies show a broad antiinflammatory effect of adenosine signaling using whole-body knockout of the receptors or receptor agonist and/or antagonist treatments (27–29, 33). Using a tissue-specific approach we recently demonstrated that deletion of the A2B receptor on the intestinal epithelium was particularly deleterious in acute colitis and highlighted its critical function in mucosal healing (20).
Adenosine signaling events are terminated via uptake of adenosine from the extracellular toward the intracellular compartment through equilibrative nucleoside transporters — particularly ENT1 and ENT2 (34–40). These channels allow adenosine to freely cross the extracellular membrane along a concentration gradient. As discussed above, extracellular adenosine production from precursor nucleotides is dramatically increased during intestinal inflammation. Therefore, conditions of hypoxia or inflammation are associated with a transcellular adenosine gradient directed from the extracellular toward the intracellular compartment. Based on these considerations, we examined the possibility that blocking ENT-dependent adenosine uptake — either pharmacologically or via specific genetic deletion — would enhance extracellular adenosine signaling and thereby mediate adenosine-dependent intestinal protection. Indeed, our studies reveal that mucosal adenosine transporters of the “ENT2 type” can be targeted to enhance intestinal epithelial A2B signaling and thereby promote mucosal barrier restitution in IBD.
Results
Ent1 and Ent2 expression is decreased in IBD patients and in experimental colitis.
In order to address a functional role of ENTs in IBD, we first examined expression levels of both adenosine transporters (ENT1 and ENT2) during conditions of intestinal inflammation. Previous studies in the intestine (39) and lung (37, 40) had demonstrated that Ent1 and Ent2 expression is downregulated during tissue hypoxia and inflammation via the actions of hypoxia-inducible factor 1-alpha (HIF-1α) and NF-κB. Both transcriptional pathways are active in the inflamed intestine in IBD (41, 42). Therefore, we hypothesized that Ent1 and Ent2 expression could be downregulated in IBD biopsies and experimental colitis. Our studies revealed a decrease in ENT1 mRNA expression in biopsies from either CD or UC patients that did not reach significance compared with controls (Figure 1A). Interestingly, ENT2 mRNA expression was significantly decreased in both CD and UC compared with controls (Figure 1B). These findings were mirrored at the level of the whole colon in the dextran sulfate sodium (DSS) model of murine colitis (Figure 1, C and D). DSS colitis injury is primarily localized to the mucosal surface of the distal colon (43); therefore we tested whether there were regional differences in proximal or distal mucosal expression of the Ent transporters during DSS. We performed colonic mucosal scrapings following DSS exposure to isolate the mucosal layer, which is enriched for intestinal epithelial cells. We observed that Ent1 mRNA expression was not significantly altered in the proximal colon mucosal scrapings, whereas in the distal colon mucosal layer, Ent1 mRNA expression decreased by approximately 70% by day 6 of DSS (Figure 1E). Similarly, Ent2 mRNA expression was not significantly diminished in the proximal colon mucosal scrapings but was decreased by almost 80% by day 6 of DSS in the distal colonic mucosal layer (Figure 1F). Taken together, these studies demonstrate that both ENT1/Ent1 and ENT2/Ent2 mRNA expression levels are decreased in the inflamed colonic mucosal layer in IBD and in murine colitis. These findings implicate ENT repression as an endogenous response during intestinal inflammation.
Figure 1. Ent 1 and Ent2 expression is repressed in IBD and murine colitis.

(A and B) cDNA from control, active Crohn’s disease, or ulcerative colitis biopsies (Origene) was probed with specific primers (QuantiTect, QIAGEN) for human ENT1, ENT2, and β-actin. ENT1 and ENT2 levels were normalized to β-actin and are expressed as fold change relative to control biopsies. n = 5 control, n = 17–21 Crohn’s disease, and n = 18–20 ulcerative colitis patients. (C–F) Sex-, age-, and weight-matched C57BL/6 mice were exposed to DSS. After 3, 6, or 7 days, whole-colon (C and D) or mucosal scrapings from the proximal and distal colon (E and F) were harvested, and total RNA was extracted. TaqMan RT-PCR for Ent1, Ent2, and 18s was performed. (C–F) mRNA transcript levels were calculated relative to 18s and are expressed as the fold change compared with water-treated (H2O) mice. In all cases data are displayed as mean ± SEM. Results in C and D represent n = 5–10 mice/group from 2 independent experiments. Results in E and F represent 6–8 mice/group from 2 independent experiments. One-way ANOVA with post hoc Dunnett’s multiple-comparisons test was performed to determine statistical differences compared with control or water. *P < 0.05.
Nonspecific pharmacologic ENT inhibition is protective in acute experimental colitis.
Previous studies demonstrate an antiinflammatory role for ENT inhibition (37, 39, 40), and having observed ENT repression during intestinal inflammation above, we hypothesized that ENT blockade could be protective in IBD. To address this hypothesis, we treated mice with dipyridamole (5 mg/kg), an inhibitor of ENT1 and ENT2 transporters during DSS colitis (44). Dipyridamole-treated mice demonstrated significantly less weight loss and colonic shortening compared with vehicle-treated controls (Figure 2, A and B). Treatment with dipyridamole decreased flux of orally gavaged FITC-labeled dextran into the serum by almost 50% compared with vehicle treatment (Figure 2C). Blinded histologic analysis showed a significant reduction in tissue injury and inflammation in mice treated with dipyridamole compared with vehicle (Figure 2D). Taken together, our studies show that dipyridamole treatment is beneficial in acute murine colitis by decreasing mucosal barrier permeability and injury.
Figure 2. Ent1 and Ent2 inhibition is protective in acute experimental colitis.

Sex-, age-, and weight-matched C57BL/6 WT mice were treated with dipyridamole (combined Ent1 and Ent2) inhibitor, 5 mg/kg, i.p., or vehicle 2–3 times daily from 1 day prior to exposure to DSS. (A) Daily weight measurements were obtained for each group of mice and are displayed as percentage of the body weight average from day 0–3. (B) Following sacrifice, colons were harvested and measured. (C) Mice were administered FITC-dextran by oral gavage (0.6 mg/g at 100 mg/ml) 4 hours prior to sacrifice on day 7. Serum was harvested at sacrifice, and fluorescence measurement was used to determine FITC levels. n = 7 mice/group from 1 independent experiment. (D) Blinded histological analysis of whole colon from each group following DSS exposure. Representative histological sections from whole colon harvested on day 7 after DSS (scale bars: 100 μm; images acquired at ×10). Unless otherwise stated, results of 2 independent experiments are presented as mean ± SEM (n = 4–17 mice/group). Two-way ANOVA with post hoc Bonferroni’s t test was used to determine statistical weight change. Two-way ANOVA with post hoc Tukey’s multiple comparison test was used to determine statistical colon length change. In all other cases, unpaired Student’s t test was used. *P < 0.05.
Genetic loss of Ent1 does not alter the outcome of DSS colitis.
Based on the above findings showing that dipyridamole treatment provided robust protection to the colon during DSS colitis, we next pursued studies to address the individual contributions of Ent1 or Ent2 — the main intestinal adenosine transporters (39, 45). Therefore, we next performed studies in mice with genetic deletion of Ent1 (Ent1–/–) (36) or Ent2 ( Ent2–/–) (37) to determine their individual contributions during acute colitis. Studies utilizing real-time PCR (RT-PCR) for individual adenosine transporters demonstrated abolished Ent1 expression in the colon, while the expression of Ent2, Ent3, or Ent4 channels was unaltered during acute colitis in Ent1–/– mice (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.121521DS1). Subsequent functional studies in Ent1–/– mice failed to demonstrate any difference in the degree of weight loss, colonic shortening, or colonic histologic damage when comparing Ent1-deficient mice with WT controls (Ent1+/+) during DSS colitis (Figure 3, A–C). Taken together, these findings suggest that the absence of Ent1 function provides no benefit in acute experimental colitis.
Figure 3. Loss of Ent1 does not protect the colonic mucosa during experimental colitis.

Ent1-deficient mice (Ent1–/–) or C57BL/6 WT controls (Ent1+/+) matched by sex, age, and weight were exposed to water or DSS. (A) Weight measurements were obtained for each group of mice and are displayed as percentage of body weight on day 0. (B) Following sacrifice, colons were harvested and measured. (C) Representative histological sections from whole colon harvested on day 7 after DSS (scale bars: 200 μm; images acquired at ×10). Results of 3 independent experiments are shown as mean ± SEM (n = 6–24 mice/group). Two-way ANOVA with post hoc Bonferroni’s t test was used to determine statistical weight change. Two-way ANOVA with post hoc Tukey’s multiple comparison test was used to determine statistical colon length change. P < 0.05.
Genetic loss of Ent2 provides robust protection during DSS colitis.
Having observed a beneficial effect of global Ent inhibition with dipyridamole (Figure 2) that was not recapitulated by genetic deletion of Ent1 (Figure 3), we proceeded to investigate a potential role for Ent2 channels in murine colitis through studying mice with genetic deletion of Ent2 (Ent2–/–) (37). Ent2–/– mice had abolished expression of Ent2 mRNA in the colon, while there was no compensatory alteration in the expression of the other Ent channels, Ent1, Ent3, or Ent4, when comparing Ent2–/– mice and WT controls during DSS colitis (Supplemental Figure 2). In functional studies, Ent2–/– mice exhibited considerably less weight loss and colonic shortening compared with their WT controls (Ent2+/+) in DSS colitis (Figure 4, A and B). The FITC-dextran concentration in the serum was reduced by greater than 50% in Ent2–/– mice compared with Ent2+/+ mice following DSS exposure (Figure 4C). RT-PCR demonstrated a 3-fold decrease in expression of inflammatory cytokines in colonic tissue of Ent2–/– mice compared with WT mice in DSS colitis (Figure 4D). Blinded histologic analysis showed significantly diminished colonic tissue damage in Ent2–/– mice compared with their WT controls (Figure 4, E and F). These findings demonstrate improved barrier function and reduction in colonic inflammation in mice lacking Ent2 during colitis, suggesting that inhibition of Ent2 function is protective in murine colitis.
Figure 4. Deletion of Ent2 protects from inflammation and injury in DSS colitis.

Matched Ent2-deficient mice (Ent2–/–) or WT controls (Ent2+/+, mice on a B6/129 background) were exposed to DSS. (A) Weights were obtained for each group of mice and are displayed as percentage of body weight on day 0. (B) Following sacrifice, colons were harvested and measured. (C) Mice were administered FITC-dextran by oral gavage (0.6 mg/g at 80 mg/ml) 4 hours prior to sacrifice on day 7. Serum was harvested at sacrifice, and fluorescence measurement was used to determine FITC levels. n = 5–7 mice/group from 1 independent experiment. (D) Following sacrifice on day 7, whole colonic tissue was snap frozen. Total RNA was extracted and RT-PCR. performed. mRNA transcript levels were calculated relative to β-actin and are expressed as fold change compared with DSS-treated WT mice. Data represent 5–8 mice per group from 1 independent experiment. (E) Representative histological sections from whole colon harvested on day 7 after DSS (scale bars: 100 μm; images acquired at ×10). (F) Histological analysis of whole colon harvested on day 7 after DSS provided by a pathologist blinded to the groups and the study. Unless otherwise stated, there were n = 7–9 mice/group. presented are representative of at least 3 independently performed experiments. Graphs show data as the mean ± SEM. Two-way ANOVA with post hoc Bonferroni’s t test was used to determine statistical weight change; in all other cases, unpaired Student’s t test was used. *P < 0.5, **P < 0.001.
Ent2–/– mice experience attenuated disease activity in TNBS colitis.
To assess the importance and reproducibility of our finding that loss of Ent2 was protective in DSS colitis (Figure 4), we subsequently performed TNBS (2,4,6-trinitrobenzenesulfonic acid) colitis studies in mice with genetic deletion of Ent2 (Figure 5). TNBS administration leads to excessive Th1 cell–mediated inflammation characterized by CD4+ T cell infiltration, excessive weight loss, and diarrhea with enhanced mucosal permeability (46). This model closely represents the pathology of Crohn’s colitis (46, 47) and was used to determine whether our findings with DSS are applicable to an alternative IBD model with mucosal damage. Similar to our findings in DSS colitis (Figure 4), we observed that Ent2–/– mice were significantly protected from colitic injury in this model compared with WT controls (Ent2+/+) as measured by weight loss (Figure 5A). TNBS colitis is characterized by infiltration of the colonic lamina propria with pathogenic CD4+ Th1 cells, which we define here as CD4+T-bet+ T cells (48). The frequency of these pathogenic CD4+ T cells in the colonic lamina propria of Ent2–/– mice was significantly reduced (6%± 0.8%) compared with control mice (12% ± 1.8%) following TNBS administration (Figure 5B). Blinded histologic analysis revealed a dramatic reduction in tissue injury in Ent2–/– mice compared with WT controls (Figure 5, C and D). Thus, loss of the Ent2 nucleoside transporter significantly reduces intestinal inflammation in an alternative model of experimental IBD, speaking to a broader significance of our findings. Taken together, our results provide a strong rationale for targeting ENT2 as a novel mechanism for gut protection.
Figure 5. Loss of Ent2 results in a marked reduction in the severity of TNBS colitis.
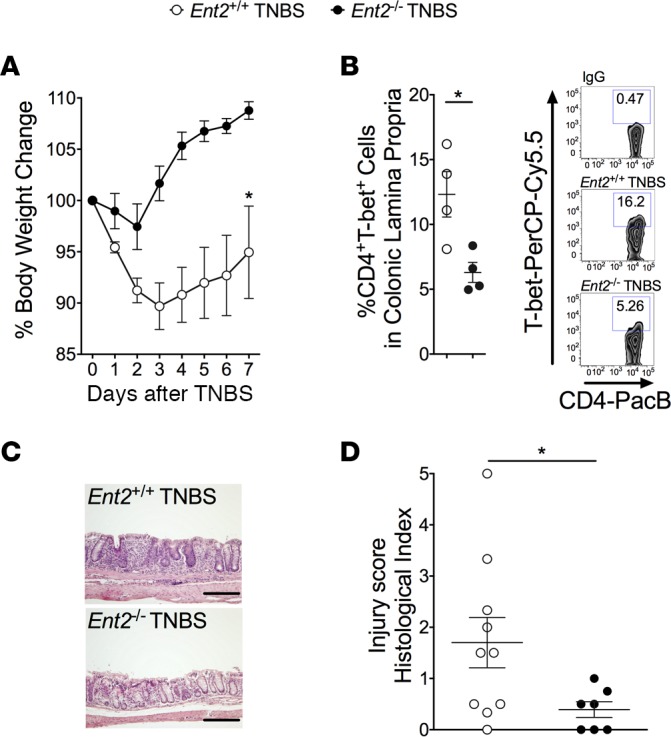
Matched Ent2-deficient mice (Ent2–/–) or WT controls (Ent2+/+, mice on B6/129 background) were given a TNBS (2,4,6-trinitrobenzenesulfonic acid) enema 7 days following skin sensitization. (A) Weights for each group of mice are presented as percentage of body weight on day 0. (B) Lamina propria leukocytes were isolated from mouse colon 7 days after TNBS enema. Flow cytometric analysis of CD4+T-bet+ cells was performed. Left: Percentage of CD4+T-bet+ cells in the colonic lamina propria in each group. Right: Representative zebra plots of the percentage of CD4+T-bet+ cells in the colonic lamina propria. IgG served as an antibody control for antibody staining. Data represent n = 4 mice per group. (C) Representative histological sections from distal colon harvested on day 7 after TNBS (scale bars: 200 μm; images acquired at ×10). (D) Histological analysis of distal colon harvested on day 7 after TNBS enema provided by a pathologist blinded to the groups and the study. Results are displayed as mean ± SEM; n = 7–11 mice/group from 1 independent experiment. Two-way ANOVA with post hoc Bonferroni’s t test was used to determine statistical weight change; in all other cases, unpaired Student’s t test was used. *P < 0.05
Loss of intestinal epithelial Ent2 expression protects the colon during acute colitis.
Previous studies suggest that Ent2 is expressed at a high level on the cell membrane of intestinal epithelial cells (39). In order to study whether Ent2 functions in a tissue-specific fashion at the level of the intestinal epithelium, we generated an Ent2 flox mouse (Supplemental Data and Supplemental Figure 3). Subsequently, transgenic mice with a floxed Ent2 gene (Ent2fl/fl) were crossed with an intestinal epithelium–specific Cre mouse (VillinCre) in order to generate a mouse strain with intestinal epithelium–specific deletion of Ent2 (Ent2fl/flVillinCre+; see Methods). RT-PCR on whole colon was performed to confirm deletion of Ent2 in Cre-positive mice (Supplemental Figure 4A). An almost 90% reduction in Ent2 mRNA expression was noted in Ent2fl/flVillinCre+ mice compared with WT controls (Ent2fl/flVillinCre–) at baseline (Supplemental Figure 4A). Further analysis determined that there was no significant difference in expression of other adenosine transporters (Ent1, Ent3, Ent4) or adenosine receptors (Adora1, Adora2a, Adora2b, Adora3) in the colon of Ent2fl/flVillinCre+ mice compared with WT controls at baseline (Supplemental Figure 4A). Furthermore, during DSS colitis, a significant reduction in colonic Ent2 expression was maintained in Ent2fl/flVillinCre+ mice compared with Ent2fl/flVillinCre– mice, but no significant alterations in Ent1, Ent3, or Ent4 were noted between the 2 strains (Supplemental Figure 4B). Ent2fl/flVillinCre+ mice had greatly reduced weight loss and colon shortening compared with littermate controls (Figure 6, A and B, respectively). Histologic damage was also significantly improved in Ent2fl/flVillinCre+ mice compared with their littermates during colitis (Figure 6, C and D). Intestinal epithelial permeability as measured by FITC-dextran flux was reduced by 50% in Ent2fl/flVillinCre+ compared with littermate controls in DSS (Supplemental Figure 5). Taken together, our findings suggest that inhibition of Ent2 expressed on the intestinal epithelium protects the colonic barrier and reduces tissue injury during acute inflammation.
Figure 6. Ent2 expression on the intestinal epithelium is detrimental during experimental colitis.
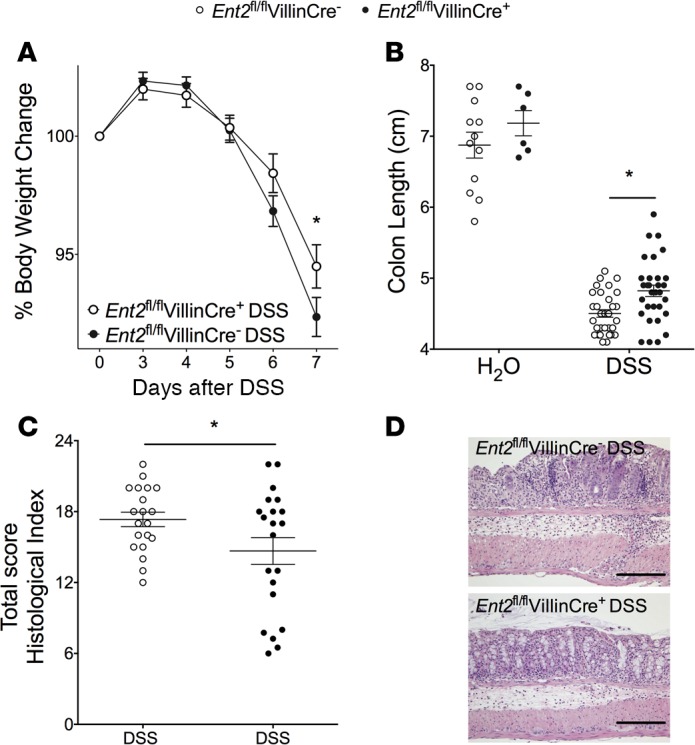
Mice with Ent2 deletion in the intestinal epithelium (Ent2fl/flVillinCre+) or matched WT littermates (Ent2fl/flVillinCre–) were given water or DSS. (A) Weights for each group are presented as percentage of bodyweight on day 0. (B) On day 7 following DSS, mice were sacrificed. Colons were harvested, and length was measured. (C) Histological analysis of distal colon harvested on day 7 after DSS. Scores were provided by a pathologist blinded to the groups and the study. (D) Representative histological sections from distal colon harvested on day 7 after DSS (scale bars: 200 μm; images acquired at ×10). Results are displayed as mean ± SEM. In A and B, results represent n = 6–32 mice/group from 3 independent experiments. In C and D, results represent n = 20–21 mice/group from 2 independent experiments. Two-way ANOVA with post hoc Bonferroni’s t test was used to determine statistical weight change. Two-way ANOVA with post hoc Tukey’s multiple comparison test was used to determine statistical colon length change. In all other cases, unpaired Student’s t test was used. *P < 0.05.
Ent2–/– mice have elevated extracellular adenosine levels in the colonic lumen during acute colitis.
In order to ascertain whether the functional role of Ent2 during acute inflammation involves the transport of adenosine at the colonic luminal surface, colonic lavage was performed on WT (Ent2+/+) and Ent2–/– mice following acute colitis (Figure 7A). HPLC analysis demonstrated an approximately 3-fold increase in adenosine concentration in the colonic lavage of Ent2–/– mice compared with WT controls during DSS colitis (Figure 7A). These data suggest that loss of Ent2 results in enhanced adenosine availability at the colonic epithelial surface.
Figure 7. Adenosine generation and signaling play a central role in the protective effect of Ent2 loss in experimental colitis.

(A) Sex-, age-, and weight-matched Ent2-deficient mice (Ent2–/–) or WT controls (Ent2+/+, mice on a B6/129 background) were exposed to DSS for 6 days, followed by water for 24 hours. Colonic lavage was performed on terminally anesthetized mice with nucleoside preserving cocktail. Adenosine concentration in the lavage fluid was determined by HPLC and normalized to protein content. Results are shown as mean ± SEM, n = 6 mice/group, and are representative of 2 independent experiments. (B–D) Sex-, age-, and weight-matched Ent2-deficient mice (Ent2–/–, mice on a B6/129 background) were treated with an A2B receptor–specific antagonist (PSB 1115, 1 mg/kg/mouse, oral gavage) on days –1, 0, 2, 4, and 6 of DSS colitis. (B) Mice were weighed daily. Results are presented as percentage of bodyweight on day 0. (C) Histological analysis of whole colon harvested on day 7 after DSS. Scores were provided by a pathologist blinded to the groups and the study. (D) Representative histological sections from whole colon harvested on day 7 after DSS (Bar represents 100 μm; images acquired at ×10). Results are presented as mean ± SEM, n = 7–8 mice/treatment group, from 1 independent experiment. Two-way ANOVA with post hoc Bonferroni’s t test was used to determine statistical weight change; in all other cases, paired Student’s t test was used. *P < 0.05.
Adenosine A2B receptor signaling is protective in Ent2–/– mice.
Previous studies have demonstrated a tissue-protective effect of adenosine signaling through its endogenous receptors during acute colitis (20, 27–31, 33). Having observed enhanced mucosal levels of adenosine in Ent2–/– mice during acute colitis (Figure 7A), we tested the hypothesis that adenosine signaling was responsible for the amelioration of acute colitis in Ent2–/– mice. In particular, the A2B receptor has been shown to be a key adenosine receptor involved in protecting colonic barrier function (20). Since we have seen preservation of colonic barrier function in experimental colitis as a result of the loss of Ent2 function (Figures 2 and 4), we pursued the role of A2B receptor signaling in mediating tissue protection in Ent2–/– mice. For the purpose of these studies, Ent2–/– mice were treated with a specific A2B receptor antagonist (PSB 1115, 1 mg/kg) or vehicle during acute colitis (Figure 7, B–D). A2B receptor antagonism resulted in increased weight loss (Figure 7B) and an approximately 50% increase in histological damage in Ent2–/– mice during acute colitis compared with vehicle-treated controls (Figure 7, C and D). These data suggest that the protected phenotype of Ent2–/– mice can be abolished via treatment with an A2B adenosine receptor antagonist. In sum, these findings demonstrate that elevated mucosal adenosine levels and A2B signaling events are mediating the protected phenotype of Ent2–/– mice during intestinal inflammation.
Specific pharmacologic targeting of Ent2 is tissue protective during acute colitis.
Combining our insights from pharmacologic (Figure 2) and genetic (Figures 4–6) studies targeting Ents, we postulated that specific inhibition of Ent2 could be protective in experimental colitis. To address this hypothesis, we next pursued studies utilizing a novel nucleoside transport inhibitor with described selectivity for ENT2 (soluflazine; Figure 8A) (49–51). The ENT2 inhibitor or vehicle was delivered via subcutaneous osmotic pump allowing for steady-state, continuous delivery of treatment during the course of our studies (Figure 8, B–D). Mice receiving soluflazine during DSS colitis experienced significantly decreased weight loss, colon shortening, and histologic injury compared with vehicle-treated controls (Figure 8, B–D). These studies demonstrate that specific inhibition of the Ent2 transporter can protect the colon during acute inflammation.
Figure 8. Specific pharmacological inhibition of Ent2 ameliorates experimental colitis.

Sex-, age-, and weight-matched C57BL/6 WT mice were treated with soluflazine (Ent2 inhibitor, 7.7 μg/kg, Alzet pump) or vehicle 1 day prior to exposure to DSS. (A) Chemical structure of Ent2 inhibitor (soluflazine). (B) Each group of mice was weighed daily. Weights are presented as percentage of body weight on day 0. (C) Following sacrifice, colons were harvested and measured. (D) Histological analysis of whole colon from each group following DSS. Scores were provided by a pathologist blinded to the groups and the study. Representative histological sections from whole colon harvested on day 7 after DSS (scale bars: 200 μm; images acquired at ×10). Results of 2 independent experiments are presented as mean ± SEM (n = 2 mice/water group and n = 10 mice/DSS group). Two-way ANOVA with post hoc Bonferroni’s t test was used to determine statistical weight change; in all other cases, unpaired Student’s t test was used. *P < 0.05.
Increased IL-10 expression and decreased neutrophil accumulation are not responsible for tissue protection following Ent2 deletion or inhibition.
Having observed that blockade of the A2B adenosine receptor reversed protection in Ent2–/– mice (Figure 7, B–D) we performed studies to investigate the role of A2B receptor signaling following genetic or pharmacologic inhibition of Ent2 in colitis. A2B receptor signaling has been suggested to inhibit neutrophil migration to protect the inflamed colon in colitis (31). We measured the level of myeloperoxidase (MPO) in the tissue of mice following DSS exposure as a marker of neutrophil content (Figure 9, A and B). We observed no significant difference in the concentration of MPO in the colon in Ent2–/– mice compared with controls (Figure 9A) or following specific pharmacologic inhibition of Ent2 (Figure 9B). This suggests that suppression of neutrophil migration to the inflamed intestine is not responsible for the tissue protection observed following genetic or pharmacologic Ent2 inhibition. In addition, A2B receptor signaling has been implicated in inducing the pro-resolution cytokine IL-10 to mediate tissue protection during colitis (32). However, we observed a significant decrease in IL-10 protein concentration in the colon of Ent2–/– mice compared with controls in DSS colitis (Figure 9C) that was mirrored in mice treated with the ENT2-specific inhibitor compared with vehicle during DSS (Figure 9D). These findings indicate that enhanced IL-10 expression is not responsible for tissue protection following genetic or pharmacologic Ent2 inhibition.
Figure 9. Genetic or pharmacologic Ent2 inhibition does not affect neutrophil migration or increase IL-10 expression in DSS colitis.
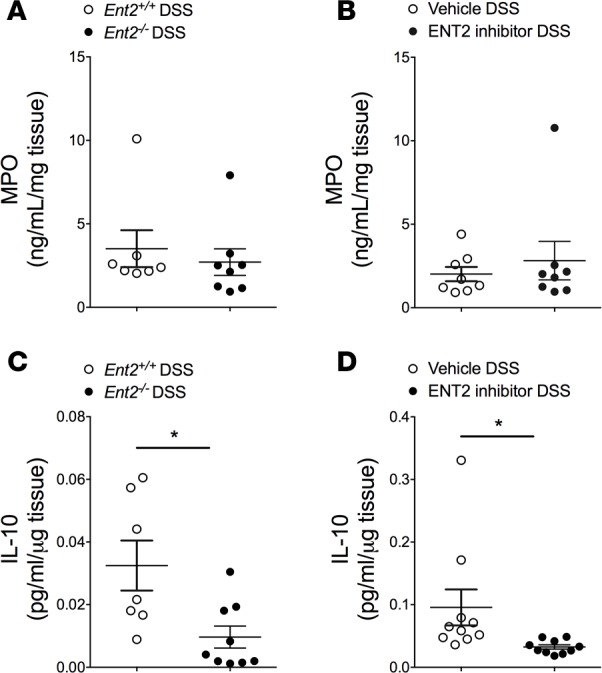
Matched Ent2-deficient mice (Ent2–/–) or WT controls (Ent2+/+, mice on a B6/129 background) were exposed to DSS (A and C), or C57BL/6 WT mice were treated with soluflazine (Ent2 inhibitor, 7.7 μg/kg, Alzet pump) or vehicle 1 day prior to exposure to DSS for 7 days (B and D). Following sacrifice, colons were harvested. (A and B) Whole colon was homogenized and myeloperoxidase (MPO) activity measured by specific ELISA. (C and D) Whole colon was placed in cell culture media for 24 hours (C, ex vivo culture) or whole colon was homogenized (D). Media from ex vivo culture or tissue lysate were loaded onto a Meso Scale assay plate for specific detection of mouse IL-10. Data in A, B, and C are presented relative to tissue weight. Data in D are relative to protein concentration as determined by BCA. Data represent 7–8 mice per group (A), 8 mice per group (B), 7–9 mice per group (C), and 10 mice per group (D), from 1 independent experiment. Results are displayed as mean ± SEM. Unpaired Student’s t test was used to test for statistical changes. *P < 0.05.
Protective effects of specific ENT2 inhibition are abolished following intestinal epithelial Ent2 or Adora2b deletion.
The above studies demonstrate that utilizing an ENT2-specific inhibitor of adenosine transport (soluflazine) is associated with intestinal protection during experimental colitis (Figure 8). To demonstrate in proof-of-principle studies that soluflazine protects during IBD via inhibition of epithelial Ent2 and attenuated mucosal A2B signaling, we next attempted treatment studies in genetic models. First, we treated mice with intestinal epithelial deletion of Ent2 (Ent2fl/flVillinCre+) with soluflazine. Indeed, treatment of Ent2fl/flVillinCre+ mice with an Ent2 inhibitor did not affect the outcome of DSS compared with vehicle treatment, as measured by weight loss (Figure 10A), colon shortening or histologic injury (Supplemental Figure 6, A–C). This suggests that the beneficial effect of ENT2 inhibitor treatment in colitis occurs via inhibition of intestinal epithelial Ent2 function. Next we performed therapeutic studies with soluflazine in mice with deletion of the A2B receptor on the intestinal epithelium (Adora2bfl/flVillinCre+/+) (20). A previous study highlighted adenosine signaling through this receptor as an endogenous mechanism for colonic barrier protection in experimental colitis (20). These mice were treated with the ENT2 inhibitor or vehicle during acute colitis (Figure 10B and Supplemental Figure 7, A–C). No difference in weight loss (Figure 10B), colon shortening, or histologic injury (Supplemental Figure 7, A–C) was observed between ENT2 inhibitor–treated and vehicle-treated animals. Taken together, these findings demonstrate that the therapeutic effect of Ent2 inhibition requires Ent2 on the intestinal epithelium as a target and functions via enhancing A2B receptor signaling on the intestinal epithelium in colitis.
Figure 10. Ent2 and the A2B receptor on the colonic intestinal epithelium provide a targetable signaling network to protect the inflamed colon.
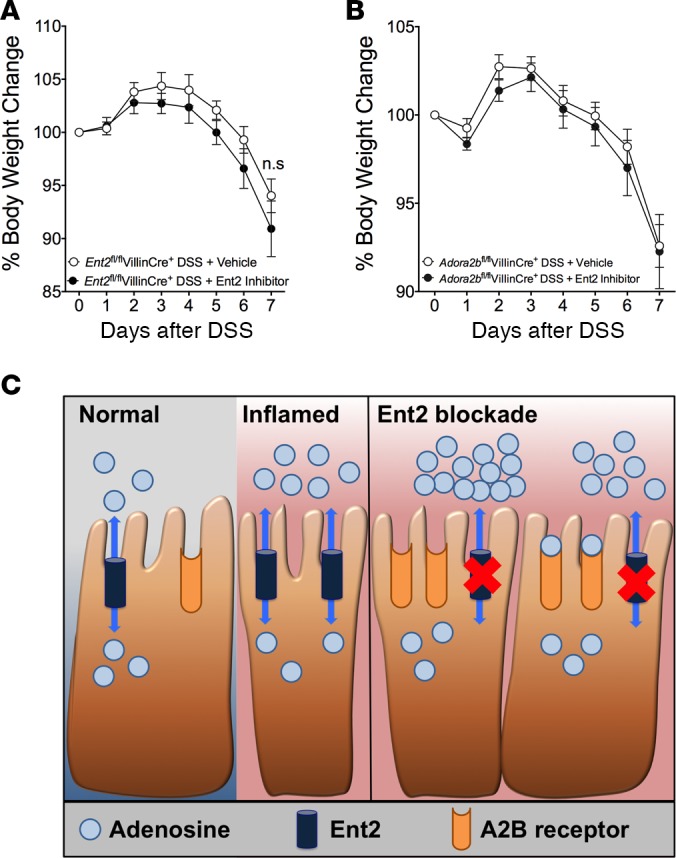
Matched mice with (A) Ent2 deletion on the intestinal epithelium (Ent2fl/flVillinCre+) or (B) A2B receptor deletion on the intestinal epithelium (Adora2bfl/flVillinCre+) were treated with soluflazine (Ent2 inhibitor, 7.7 μg/kg, Alzet pump) or vehicle 1 day prior to exposure to DSS or water. Mice were weighed, and results are presented as percentage of body weight on day 0. Two-way ANOVA with post hoc Bonferroni’s t test was used to determine statistical weight change. n = 6–8 mice/group from 1 independent experiment. (C) Schematic of functional consequence of Ent2 inhibition in colitis. Intestinal inflammation as observed in IBD is associated with decreased intestinal epithelial Ent2 expression, which leads to increased extracellular adenosine that can signal through the epithelial A2B adenosine receptor to protect the mucosal barrier.
Discussion
In the present study, we examined the contribution of adenosine transporters to the resolution of intestinal inflammation, such as occurs during IBD. Initially we examined transcriptional expression of adenosine transporters in intestinal biopsies obtained from IBD patients or in the colon of mice during experimental colitis. These studies demonstrated repression of ENT1/Ent1 and ENT2/Ent2 during ongoing intestinal inflammation. Based on previous studies showing that ENT repression may serve as an endogenous protective pathway that can be enhanced pharmacologically (37, 39, 40), we subsequently performed pharmacologic and genetic studies that implicate ENT2 as a suitable target for IBD treatment. Indeed, selective pharmacologic inhibition of ENT2 was associated with colonic protection. Similarly, mice with global or mucosal Ent2 deletion were protected in models of IBD. Additional pharmacologic and genetic studies of A2B signaling implicated elevated adenosine levels and A2B signaling events in the observed protection. Taken together, these results suggest that pharmacologic inhibition of Ent2 on intestinal epithelia functions to enhance extracellular adenosine, which can engage the intestinal epithelial A2B receptor to protect the colonic barrier during acute colitis (Figure 10C).
Several previous studies have implicated extracellular adenosine signaling in mucosal protection during IBD (21). Adenosine signals via G protein–coupled receptor A1, A2A, A2B, or A3 to mediate its cellular response (25). Limited data suggest a protective role for A1 and A3 receptor signaling in inflammation (33, 52–56). In contrast, the role of A2A and A2B receptor function during tissue inflammation is better understood. In a landmark study using both pharmacologic and genetic approaches, it was demonstrated that A2A adenosine receptors play a nonredundant role in attenuating tissue inflammation and injury in multiple in vivo models (57). Subsequent studies have identified that the A2A receptor is the dominant adenosine receptor expressed on a broad range of immune cells and suppresses proinflammatory responses of these cells (reviewed in ref. 58). In the context of tissue inflammation, pharmacologic and genetic studies reveal that the A2B receptor plays a substantial antiinflammatory role in multiple models in vivo by signaling on both hematopoietic and non-hematopoietic cells (25, 59). Recent studies have sought to define the contribution of adenosine receptor signaling to intestinal inflammation as observed in IBD. Studies suggest that A2A receptor activation is a protective mechanism in IBD (27–30), in part by suppressing T cell cytokine production (29, 30). A2B signaling is particularly relevant to IBD, as the A2B receptor is the predominant receptor expressed on intestinal epithelial cells and A2B expression is upregulated in biopsies from IBD patients as well as in experimental colitis (32, 60). Studies using experimental models suggest a tissue-protective role for the A2B receptor in murine colitis via induction of the pro-resolution cytokine IL-10 (32) or suppression of neutrophil chemotaxis (31). To date, these studies demonstrate a broad antiinflammatory effect of adenosine signaling using whole-body knockout of the receptors or receptor agonist and/or antagonist treatments (27–29, 31–33). In order to study cell-specific effects of A2B signaling in the context of IBD, we recently employed tissue-specific genetic deletion of the A2B receptor in murine colitis (20). These studies demonstrate that A2B receptor signaling on the intestinal epithelium promotes intestinal epithelial barrier repair during colitis (20). Specifically, mice with intestinal epithelial deletion of the A2B receptor experienced earlier loss of epithelial barrier function during experimental colitis compared with control mice (20). Agonist treatment or genetic deletion studies showed that A2B receptor signaling promoted epithelial barrier repair in vitro and in vivo (20). It was demonstrated that the A2B receptor mediated its barrier-protective effects through cAMP -induced phosphorylation of vasodilator-stimulated phosphoprotein (VASP) in intestinal epithelial cells in vitro and in vivo (20). Upon phosphorylation, VASP colocalizes with the tight protein zonula occludens 1 (ZO-1) to facilitate closure and repair of intestinal epithelial junctions (61). Therefore, signaling via the A2B receptor at the intestinal epithelial surface during colitis drives phosphorylation of VASP, which moves to the epithelial cell junctions to promote closure of the damaged epithelium and drive mucosal protection in IBD.
In the present study, we were interested in the expression and function of adenosine transporters in the context of IBD. Adenosine is hydrophilic, and therefore its availability at the extracellular surface is dependent on specific transporters that allow movement of adenosine across cell membranes (62). The equilibrative nucleoside transporters (ENT/Ent) are passive transporters that allow bidirectional movement of adenosine across the cell membrane to normalize adenosine concentrations (reviewed in ref. 62). Currently, this family comprises 4 isoforms, Ent1, Ent2, Ent3, and Ent4 (62). Ent1 and Ent2 were originally defined based on their sensitivity to the nucleoside transport inhibitor S-(4-nitrobenzyl)-6-thioinosine (NBTI/NBMPR) (34, 35). Ent3 and Ent4 were discovered more recently, and less in known about their biological roles (62). There is a range of nucleoside transporter inhibitors that can target Ent1 and/or Ent2 channels, and have been shown to prevent the clearance of adenosine from the extracellular space and enhance adenosine signaling in models of inflammation and alcohol intoxication (36–40). Our studies focused on Ent1 and Ent2 transporters for a number of reasons. ENT1 and ENT2 are expressed in the gastrointestinal tract (45, 63). The presence of functional Ent1 and Ent2 transporters in human intestinal epithelial cell lines has been demonstrated (39, 64–66). As described above, Ent1 and Ent2 expression is decreased during intestinal hypoxia (38, 39) and lung inflammation (37, 40), with inhibition of Ent1 and/or Ent2 activity eliciting a tissue-protective response in these studies. Importantly, an inhibitor targeting both Ent1 and Ent2 (dipyridamole) is used clinically as a vasodilator for stress echocardiography and as an inhibitor of platelet aggregation, suggesting that Ent1 and/or Ent2 inhibition could be safely progressed to the clinic as a therapeutic option in IBD (67, 68). In contrast, little is understood about the biological role of Ent3 or Ent4, and they display low sensitivity to available nucleoside inhibitors, making them unattractive targets for our study (69, 70).
Consistent with other studies in inflamed tissues (37, 40), we observed a decrease in ENT1 and ENT2 mRNA expression in mucosal biopsies from CD or UC patients, as well as in the whole colon of mice during the course of DSS colitis. Interestingly, while the decrease in ENT1 expression was not significant, we observed a profound decrease in ENT2 mRNA expression in CD and UC biopsies as well as in the whole murine colon in DSS colitis. When we studied the injured colonic mucosal layer (distal colon) in isolation in our murine colitis model, we observed a comparable decrease in Ent1 and Ent2 expression during DSS colitis compared with controls. The reason for the nonsignificant decrease in Ent1 expression in whole colonic samples may be the fact that Ent1 is expressed on many cell types, with Ent2 being suggested to have a more restricted expression profile (71). Therefore, we hypothesize that we did not observe a significant loss of Ent1 expression in our biopsy or whole-colon studies because there is a broader expression profile of Ent1 in these tissues compared with our murine mucosal scrapings. In contrast, a previous study indicated that ENT1 and ENT2 expression is upregulated in IBD biopsies compared with control (63). However, in a previous publication, this group failed to detect ENT1 in normal human biopsies (45), calling into question how their findings would relate to IBD. In addition, ENT expression analysis was done relative to Villin-1 expression and thus does not take into account overall expression in the samples, which is likely to account for the differences between our studies (63). In summary, in all intestinal samples studied, Ent1 and Ent2 mRNA expression was decreased compared with controls during inflammation. Our observations correspond to other studies in which Ent1 and Ent2 expression is downregulated during inflammation (37, 39, 40). We have demonstrated a consistent and significant reduction in Ent2 expression in the colonic mucosa in IBD and murine colitis. This may suggest a more prominent or tissue-specific role for Ent2 during intestinal inflammation.
Our current study and evidence from others supports the concept that amplifying endogenous adenosine signaling pathways may be a critical next step in controlling mucosal inflammation in IBD (26, 72–76). Indeed consistent with other studies (37, 39, 40), we observed that blocking the adenosine transporters Ent1 and Ent2 using dipyridamole treatment supported mucosal barrier function and suppressed tissue injury in murine colitis. This provides proof of concept that there is a potential for off-label use of dipyridamole for IBD. Our findings in mice with genetic deletion of Ents demonstrated that Ent1 does not play a role in intestinal inflammation as observed in IBD. However, genetic deletion or pharmacologic inhibition of Ent2 revealed that Ent2 inhibition plays a prominent role in protecting the mucosal barrier in IBD. Given that Ent1 and Ent2 function to clear adenosine from the extracellular space, it may be surprising that Ent1 deletion had no effect in our studies. However, this could be explained by the fact that Ent1 has been shown to be ubiquitously expressed to high levels in many cell types and tissues of the body, with the expression profile of Ent2 being more limited (71). Indeed studies suggest that Ent2 may be expressed to an even greater degree in the colon than Ent1 (39, 45). Therefore, use of dipyridamole may be undesirable due to nonspecific effects on Ents, which could result in inhibition of platelet function or even hypotension. Given the possible off-target effects of a nonspecific Ent inhibitor and having demonstrated a specific role for genetic deletion of Ent2 in supporting mucosal barrier function during murine colitis, we propose that directly targeting ENT2 may be a highly specific therapeutic approach in IBD. We identified a compound known as soluflazine that has been demonstrated to have 20- to 100-fold increased specificity for ENT2 relative to ENT1 in vitro (49–51). In vivo studies demonstrate that soluflazine is efficacious via increasing extracellular adenosine (77). Continuous delivery of soluflazine mimicked the protective effect observed with dipyridamole treatment in murine colitis. However, soluflazine treatment was not as effective as genetic deletion of Ent2. This may be due to issues of concentration or bioavailability of the drug. Future studies in animal models would need to focus on dose and route of administration to determine the potential efficacy of soluflazine as a clinical agent.
Our current findings using genetic and pharmacologic approaches illustrate a specific role for Ent2 inhibition in protecting the colonic barrier in murine colitis. We have demonstrated that enhanced extracellular adenosine signaling via the A2B receptor is responsible for the tissue protection observed in Ent2–/– mice. We investigated pathways known to be mediated by A2B signaling, including IL-10 production and suppression of neutrophil migration, to assess their role in tissue protection mediated by Ent2 genetic or pharmacologic inhibition. We observed a nonsignificant trend toward a decrease in MPO in Ent2–/– mice, suggesting that specific inhibition of neutrophil migration was not responsible for tissue protection in these mice. We observed a significant decrease in IL-10 expression following genetic or pharmacologic Ent2 inhibition, indicating that an A2B-mediated increase in IL-10 was not responsible for the tissue protection observed. Indeed, this significant reduction in IL-10 expression correlates with decreases in other tissue cytokines measured (Figure 4D), suggesting a nonspecific decrease in tissue inflammation in Ent2–/– mice. To further explore the role of A2B receptor signaling, we used mice with intestinal epithelium–specific deletion of the A2B receptor. Recent studies highlight a colonic barrier–protective role for the intestinal epithelial A2B receptor in colitis (20). Selective Ent2 inhibitor treatment of mice with intestinal epithelial deletion of the A2B receptor was ineffective. Therefore, we propose that Ent2 inhibition enhances extracellular adenosine, which signals through the epithelial A2B receptor to protect the colonic barrier.
Our study has addressed the tissue-specific role of Ent2 in colitis using mice with intestinal epithelial Ent2 deletion (Ent2fl/flVillinCre+). We observed marked improvement in the outcome of DSS colitis in Ent2fl/flVillinCre+ mice compared with controls, suggesting a significant protective role for intestinal epithelial Ent2 in the inflamed colon. However, the protective effect of intestinal epithelial deletion of Ent2 was not as pronounced as that observed in mice with whole-body Ent2 knockout during colitis. Furthermore, barrier permeability studies failed to demonstrate a significant difference in epithelial barrier permeability in Ent2fl/flVillinCre+ mice compared with controls during DSS, which had been observed in whole-body knockouts. These data could be explained by the fact that the strains of mice are on different genetic backgrounds, with potentially different susceptibilities to DSS. It may also suggest that there is a role for Ent2 expression on other cell types during colitis that we cannot rule out. Furthermore, Ent2 inhibition may play a role in dampening tissue inflammation through a mechanism that does not involve protecting barrier permeability that has yet to be identified. However, the fact that the ENT2 specific inhibitor was ineffective in Ent2fl/flVillinCre+ mice supports our findings that intestinal epithelial Ent2 plays a key role in protecting the colonic barrier in colitis.
In summary, our studies in mice with intestinal epithelium–specific deletion of Ent2 and the A2B receptor demonstrate a significant effect of intestinal epithelial Ent2 inhibition and A2B receptor signaling in protecting the colonic mucosal barrier. Therefore, we suggest that a coordinated network of adenosine transport and signaling between Ent2 and the A2B receptor exists at the epithelial surface that can be pharmacologically targeted for therapeutic benefit in colitis (Figure 10C).
Methods
Study design.
This study was designed to investigate the role of equilibrative nucleoside transporters in IBD. To do this, we used mice with genetic deletion of Ent1 and Ent2 as well as mice with intestinal epithelial deletion of Ent2 or the A2B receptor (described below in Experimental animals). Mice were age, weight, and sex matched prior to commencement of our studies. Mice between the ages of 8 and 14 weeks were used, and both male and female mice were used. Based on previous experience, a minimum of 5 mice per colitis group were used per study, and studies were repeated up to 3 times, as indicated in individual figure legends (20, 31). Mice were numbered at sample collection to blind researchers to the study and processed in subsequent analysis as numbered samples only. The pathologist was provided with numbered slides and was blinded to the study and the experimental groups.
Experimental animals.
All experimental animals were bred and housed at the University of Colorado Anschutz Medical Campus. C57BL/6 mice were purchased from the Jackson Laboratory and bred in-house. C57BL/6 mice were used in expression and therapeutic studies and also served as WT controls for Ent1–/– mice. Mice with whole-body Ent1 deletion (Ent1–/–) were donated by Doo-Sup Choi (36). Ent2–/– mice and their B6/129 control strain were purchased from Taconic. Ent2 flox mice were generated by Ozgene. The flox strategy involved flanking exon 2 of Ent2 with loxP sites to achieve gene deletion by Cre-mediated recombination (see Supplemental Data and Supplemental Figure 3). These mice were bred with VillinCre mice (B6.Cg-Tg[Vil-cre]997Gum/J, The Jackson Laboratory) to achieve mice with intestinal epithelium–specific deletion of Ent2. At least 3 rounds of backcrossing were performed to generate homozygous flox Cre-positive (Ent2fl/flVillinCre+) mice. Homozygous flox Cre-negative littermates were used as controls for our studies (Ent2fl/flVillinCre–). Genotyping PCR performed on tails was used to determine Cre and flox expression as well as ensure specific deletion of the floxed area of Ent2 by testing for null PCR (GeneTyper; Supplemental Figure 8). Mice with intestinal epithelium-specific deletion of the A2B receptor (Adora2bfl/flVillinCre+/+) were previously described (20). All mice were bred and housed in standard conditions.
DSS colitis and pharmacological treatments.
DSS (36,000–50,000 MW, MP Biomedicals) was added to mouse drinking water (3%–4%), followed by monitoring of mice over 7 days, as previously described (31). The ENT inhibitor dipyridamole (5 mg/kg, Sigma-Aldrich) or vehicle (2% DMSO, 10% ethanol, 88% corn oil) was administered by i.p. injection 2–3 times daily from 1 day prior to DSS exposure to day 6 of DSS. A2B receptor antagonist (PSB 1115: 1 mg/kg, Tocris Bioscience) or vehicle (sterile water) was administered by oral gavage 1 day prior to DSS and once on days 0, 2, 4, and 6 of DSS. Ent2 inhibitor (soluflazine, 7.7 μg/kg; supplied by Janssen Pharmaceuticals) or vehicle (sterile water) was delivered by subcutaneous osmotic pump (Alzet) implanted 1 day prior to DSS. Mice were weighed daily, and changes in weight were calculated relative to initial body weight. On completion of DSS treatment, mice were euthanized and colons harvested for measurement and processing. Whole colons were fixed in 10% buffered formalin, paraffin embedded, and stained with H&E. For expression studies, strips of whole colon or scrapings from the proximal or distal colon were snap frozen in liquid nitrogen. Histological scoring was performed by a board-certified pathologist blinded to the study using the following scoring system, as previously described (20). The inflammation index gives 0 points for no lamina propria inflammation, 1 point for increased lamina propria inflammatory cells, 2 points for confluence of inflammatory cells extending into the submucosa, and 3 points for transmural inflammation. The injury index gives 0 points for no crypt damage, 1 point for partial (up to 50%) crypt drop-out, 2 points for partial to complete (50%–100%) crypt drop-out, and 3 points for complete (100%) crypt drop-out. The score from each category is then multiplied by 1 for 1%–25% of intestinal length involved, 2 for 25%–50% of intestinal length involved, 3 for 50%–75% of intestinal length involved, or 4 for 75%–100% of intestinal length involved. It is also noted whether the section is from the proximal or distal colon. The resulting products for each category are then added for an overall score of 0–24. The total histological index encompasses both injury and inflammation scores as described above. Representative images were selected based on histologic scores. Images were acquired at ×10 using an Olympus BX51 with an Olympus DP72 camera and cellSens imaging software (version 1.6).
TNBS colitis.
TNBS colitis involves skin sensitization of mice with the haptenating agent TNBS 1 week prior to single rectal administration to mice of TNBS in ethanol (46). When TNBS couples with proteins with high molecular weight, it presents them as immunogenic to the host immune system (47). Mice were sensitized by epicutaneous application of TNBS (1% in 100% ethanol, Sigma-Aldrich) or vehicle (100% ethanol) 1 week prior to rectal instillation of TNBS (5% in 40% ethanol) or vehicle (40% ethanol), as previously described (46). Weight changes in mice were monitored for 1 week, and tissue was collected and processed as described for DSS colitis.
Intestinal permeability.
FITC-labeled dextran (4 kDa, Sigma-Aldrich) was administered to mice by oral gavage (0.6 mg/g) as previously described (31). Blood was collected 3–4 hours later and plasma separated. FITC levels were measured at 485 nm using a BioTek Synergy 2 and calculated using a standard curve of the gavaged FITC-dextran solution.
RT-PCR.
A cDNA array plate of human control, CD, and UC biopsies (Origene) was probed with specific human ENT1, ENT2, and β-actin primers (QuantiTect primer assays, QIAGEN) in an RT-PCR protocol using Sybr Green (PowerSybr, Applied Biosystems). For murine studies, RNA isolation, cDNA synthesis, and RT-PCR from flash-frozen whole colonic tissue or mucosal scrapings were performed as previously described (31). Sybr Green–based (PowerSybr, Applied Biosystems) RT-PCR assays were performed for IL-6, IL-1β, IFNγ, Ent1, Ent2, Ent3, Ent4, and the internal control β-actin (QuantiTect primer assays, QIAGEN). In some instances TaqMan RT-PCR assays were also performed for Ent1, Ent2, Ent3, Ent4, Adora1, Adora2a, Adora2b, Adora3, and the internal control 18s (Applied Biosystems, Thermo Fisher Scientific). The 2–ΔΔCt method was used to calculate relative changes in gene expression based on the internal control.
Flow cytometry.
Lamina propria leukocytes were harvested as previously described (31, 78). Briefly, following removal of intraepithelial lymphocytes and epithelial cells, whole colonic tissue was collagenase digested and cell viability assessed prior to staining. Cells were incubated with a fluorescent viability dye (LIVE/DEAD, Invitrogen) and fluorescent antibodies against CD45.2 (clone 30.F11, eBioscience), CD4 (clone RM4-5, eBioscience), and T-bet (clone 4B10, BD Biosciences). Stained cells were analyzed using the FACSCanto II system. Post-analyses were performed using FlowJo software. Percentage of live total cells of each population were calculated.
Adenosine measurement.
Following DSS exposure mice, were terminally anesthetized and colons exposed by blunt dissection. The proximal colon was ligated at the cecal junction and the colonic lumen flushed with a nucleoside preserving cocktail (10 μM dipyridamole, 10 μM adenosine 5′-[α,β- methylene] diphosphate, and 10 μM 5′-deoxycoformycin). Samples were pelleted, and the concentration of adenosine was measured by HPLC. The protein concentration in the sample was determined by Bradford assay (Thermo Fisher Scientific). Data are displayed as adenosine concentration normalized to protein content of the sample.
MPO assay.
Flash-frozen whole colonic tissue collected from DSS-treated mice was weighed and homogenized using a hand-held tissue homogenizer (Tissue Master 125, Omni International) in MPO lysis buffer as per the manufacturer’s instructions (Mouse MPO Kit, Hycult Biotech). MPO activity was determined in the samples according to the kit instructions. Absorbance at 450 nm was determined using BioTek Synergy 2. MPO concentration in each sample was determined from the provided standard curve and was normalized to the weight of tissue.
Meso Scale assay.
Following DSS colitis, flash-frozen whole colon was homogenized in Tris lysis buffer containing protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific), using a hand-held tissue homogenizer. Alternatively, a small piece of fresh colonic tissue (approx 1 cm2) was incubated in 1 ml RPMI (Invitrogen, ex vivo culture). After overnight incubation, tissue and media were frozen at –80°C. IL-10 concentration was determined in tissue lysate or in ex vivo culture media using the V-Plex Mouse IL-10 Kit (Meso Scale Discovery) as per the manufacturer’s instructions. Cytokine concentrations were determined using a Sector Imager 2400 (Meso Scale Discovery). Total protein concentration in the tissue lysate was determined using the BCA Protein Assay Kit (Thermo Fisher Scientific). Cytokine concentrations in tissue homogenates were normalized to total protein. Cytokine concentrations in media from ex vivo cultures were normalized to mass of tissue used in culture.
Statistics.
Data analysis was performed using GraphPad Prism Analysis software (version 6.0). Statistical changes in weight loss between groups was assessed by 2-way ANOVA with post hoc Bonferroni’s t test. For mRNA expression studies, 1-way ANOVA with post hoc Dunnett’s test was performed. For studies comparing more than 1 group, 2-way ANOVA with post hoc Tukey’s multiple-comparisons test was used. Otherwise, statistical significance was determined by unpaired 2-tailed Student’s t test. Data are displayed as mean ± SEM. Statistical significance was set at P < 0.05. Measurements were considered to be outliers if they were greater than 3 SDs from the mean and were identified by the Grubbs’ test (79). These observations were investigated further to ensure correct data collection, processing, and handling of the samples.
Study approval.
All experiments were performed in accordance with protocols approved by the University of Colorado Denver Institutional Animal Care and Use Committee, reference numbers B97511(10)1D, B97513(10)1D, and B97516(10)1E.
Author contributions
CMA designed, performed, and analyzed the experiments and wrote the manuscript; CBC proofed the manuscript, and performed and analyzed the flow cytometric analysis; CRR designed, performed, and analyzed mouse DSS experiments; KEO designed, performed, and analyzed mesoscale and MPO experiments, as well as assisting with writing; LP designed and performed the colonic lavage experiments; PJ is a board-certified pathologist who provided histological analysis of all tissues; JLB reviewed the original data, provided statistical input, and assisted with writing the manuscript; TWM designed and performed the HPLC and protein assays; HKQ analyzed the HPLC and protein assays; MRB provided technical advice on HPLC assays; HKE provided research direction, reviewed the original data, made most of the genetic mouse models available, and assisted with writing the manuscript.
Supplementary Material
Acknowledgments
The authors would like to acknowledge technical assistance from Maria I. Wong, Kristann Magee, Dan Koyanagi, Eóin N. McNamee, Kristen Allison, Dan Kao, Joseph S. Lee, and Melissa Ledezma. Ent1-deficient mice were provided by Doo-Sup Choi (The Mayo Clinic, Rochester, Minnesota, USA). The Ent2 inhibitor soluflazine was provided by Johnson & Johnson Pharmaceutical Research & Development, a division of Janssen Pharmaceutica N.V., Beerse, Belgium. The present study was supported by NIH grants R01 DK097075, P0I-HL114457, R01-HL109233, R01-DK109574, R01-HL119837, and R01-HL133900 to HKE. Support for this study was also provided by NIH grant K01 DK099485 and R03 DK114545, a grant from the American Gastroenterological Association (AGA), and grants from the Crohn’s & Colitis Foundation (CCFA 2865 and 276536) to CMA.
Version 1. 10/18/2018
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
License: Copyright 2018, American Society for Clinical Investigation.
Reference information: JCI Insight. 2018;3(20):e121521. https://doi.org/10.1172/jci.insight.121521.
Contributor Information
Carol M. Aherne, Email: carol.aherne@ucdenver.edu.
Colm B. Collins, Email: colm.collins@ucdenver.edu.
Loni Perrenoud, Email: loni.perrenoud@ucdenver.edu.
Paul Jedlicka, Email: paul.jedlicka@ucdenver.edu.
Jessica L. Bowser, Email: jessica.l.bowser@uth.tmc.edu.
Harry Karmouty-Quintana, Email: hkarquin@gmail.com.
Michael R. Blackburn, Email: michael.r.blackburn@uth.tmc.edu.
Holger K. Eltzschig, Email: holger.eltzschig@uth.tmc.edu.
References
- 1.Shivashankar R, Tremaine WJ, Harmsen WS, Loftus EV. Incidence and prevalence of Crohn’s disease and ulcerative colitis in Olmsted County, Minnesota from 1970 through 2010. Clin Gastroenterol Hepatol. 2017;15(6):857–863. doi: 10.1016/j.cgh.2016.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peyrin-Biroulet L, Loftus EV, Colombel JF, Sandborn WJ. The natural history of adult Crohn’s disease in population-based cohorts. Am J Gastroenterol. 2010;105(2):289–297. doi: 10.1038/ajg.2009.579. [DOI] [PubMed] [Google Scholar]
- 3.Monstad I, Hovde O, Solberg IC, A Moum B. Clinical course and prognosis in ulcerative colitis: results from population-based and observational studies. Ann Gastroenterol. 2014;27(2):95–104. [PMC free article] [PubMed] [Google Scholar]
- 4.Beigel F, et al. Rate and predictors of mucosal healing in patients with inflammatory bowel disease treated with anti-TNF-alpha antibodies. PLoS ONE. 2014;9(6):e99293. doi: 10.1371/journal.pone.0099293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pineton de Chambrun G, Peyrin-Biroulet L, Lémann M, Colombel JF. Clinical implications of mucosal healing for the management of IBD. Nat Rev Gastroenterol Hepatol. 2010;7(1):15–29. doi: 10.1038/nrgastro.2009.203. [DOI] [PubMed] [Google Scholar]
- 6.Sandborn WJ, et al. Treating beyond symptoms with a view to improving patient outcomes in inflammatory bowel diseases. J Crohns Colitis. 2014;8(9):927–935. doi: 10.1016/j.crohns.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 7.Dave M, Loftus EV. Mucosal healing in inflammatory bowel disease-a true paradigm of success? Gastroenterol Hepatol (N Y) 2012;8(1):29–38. [PMC free article] [PubMed] [Google Scholar]
- 8.Neurath MF, Travis SP. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut. 2012;61(11):1619–1635. doi: 10.1136/gutjnl-2012-302830. [DOI] [PubMed] [Google Scholar]
- 9.Söderholm JD, et al. Increased epithelial uptake of protein antigens in the ileum of Crohn’s disease mediated by tumour necrosis factor alpha. Gut. 2004;53(12):1817–1824. doi: 10.1136/gut.2004.041426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Söderholm JD, et al. Epithelial permeability to proteins in the noninflamed ileum of Crohn’s disease? Gastroenterology. 1999;117(1):65–72. doi: 10.1016/S0016-5085(99)70551-2. [DOI] [PubMed] [Google Scholar]
- 11.DeRoche TC, Xiao SY, Liu X. Histological evaluation in ulcerative colitis. Gastroenterol Rep (Oxf) 2014;2(3):178–192. doi: 10.1093/gastro/gou031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerova VA, Stoynov SG, Katsarov DS, Svinarov DA. Increased intestinal permeability in inflammatory bowel diseases assessed by iohexol test. World J Gastroenterol. 2011;17(17):2211–2215. doi: 10.3748/wjg.v17.i17.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivanov AI, Nusrat A, Parkos CA. The epithelium in inflammatory bowel disease: potential role of endocytosis of junctional proteins in barrier disruption. Novartis Found Symp. 2004;263:115–124. [PubMed] [Google Scholar]
- 14.Madsen KL, Malfair D, Gray D, Doyle JS, Jewell LD, Fedorak RN. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm Bowel Dis. 1999;5(4):262–270. doi: 10.1097/00054725-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Olson TS, et al. The primary defect in experimental ileitis originates from a nonhematopoietic source. J Exp Med. 2006;203(3):541–552. doi: 10.1084/jem.20050407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitajima S, Takuma S, Morimoto M. Changes in colonic mucosal permeability in mouse colitis induced with dextran sulfate sodium. Exp Anim. 1999;48(3):137–143. doi: 10.1538/expanim.48.137. [DOI] [PubMed] [Google Scholar]
- 17.Laukoetter MG, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204(13):3067–3076. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474(7351):307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neurath MF. New targets for mucosal healing and therapy in inflammatory bowel diseases. Mucosal Immunol. 2014;7(1):6–19. doi: 10.1038/mi.2013.73. [DOI] [PubMed] [Google Scholar]
- 20.et al. Epithelial-specific A2B adenosine receptor signaling protects the colonic epithelial barrier during acute colitis. Mucosal Immunol. 2015;8(6):1324–1338. doi: 10.1038/mi.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colgan SP, Eltzschig HK. Adenosine and hypoxia-inducible factor signaling in intestinal injury and recovery. Annu Rev Physiol. 2012;74:153–175. doi: 10.1146/annurev-physiol-020911-153230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364(7):656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012;367(24):2322–2333. doi: 10.1056/NEJMra1205750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson EK, Boison D, Schwarzschild MA, Kochanek PM. Purines: forgotten mediators in traumatic brain injury. J Neurochem. 2016;137(2):142–153. doi: 10.1111/jnc.13551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aherne CM, Kewley EM, Eltzschig HK. The resurgence of A2B adenosine receptor signaling. Biochim Biophys Acta. 2011;1808(5):1329–1339. doi: 10.1016/j.bbamem.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedman DJ, et al. From the Cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci USA. 2009;106(39):16788–16793. doi: 10.1073/pnas.0902869106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Odashima M, et al. Activation of A2A adenosine receptor attenuates intestinal inflammation in animal models of inflammatory bowel disease. Gastroenterology. 2005;129(1):26–33. doi: 10.1053/j.gastro.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 28.Pallio G, et al. Adenosine receptor stimulation by polydeoxyribonucleotide improves tissue repair and symptomology in experimental colitis. Front Pharmacol. 2016;7:273. doi: 10.3389/fphar.2016.00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurtz CC, et al. Extracellular adenosine regulates colitis through effects on lymphoid and nonlymphoid cells. Am J Physiol Gastrointest Liver Physiol. 2014;307(3):G338–G346. doi: 10.1152/ajpgi.00404.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naganuma M, Wiznerowicz EB, Lappas CM, Linden J, Worthington MT, Ernst PB. Cutting edge: critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J Immunol. 2006;177(5):2765–2769. doi: 10.4049/jimmunol.177.5.2765. [DOI] [PubMed] [Google Scholar]
- 31.Aherne CM, et al. Neuronal guidance molecule netrin-1 attenuates inflammatory cell trafficking during acute experimental colitis. Gut. 2012;61(5):695–705. doi: 10.1136/gutjnl-2011-300012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frick JS, MacManus CF, Scully M, Glover LE, Eltzschig HK, Colgan SP. Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J Immunol. 2009;182(8):4957–4964. doi: 10.4049/jimmunol.0801324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mabley J, et al. The adenosine A3 receptor agonist, N6-(3-iodobenzyl)-adenosine-5’-N-methyluronamide, is protective in two murine models of colitis. Eur J Pharmacol. 2003;466(3):323–329. doi: 10.1016/S0014-2999(03)01570-X. [DOI] [PubMed] [Google Scholar]
- 34.Griffiths M, et al. Cloning of a human nucleoside transporter implicated in the cellular uptake of adenosine and chemotherapeutic drugs. Nat Med. 1997;3(1):89–93. doi: 10.1038/nm0197-89. [DOI] [PubMed] [Google Scholar]
- 35.et al. Molecular cloning and characterization of a nitrobenzylthioinosine-insensitive (ei) equilibrative nucleoside transporter from human placenta. Biochem. 1997;328(pt 3):739–743. doi: 10.1042/bj3280739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi DS, et al. The type 1 equilibrative nucleoside transporter regulates ethanol intoxication and preference. Nat Neurosci. 2004;7(8):855–861. doi: 10.1038/nn1288. [DOI] [PubMed] [Google Scholar]
- 37.et al. Crosstalk between the equilibrative nucleoside transporter ENT2 and alveolar Adora2b adenosine receptors dampens acute lung injury. FASEB J. 2013;27(8):3078–3089. doi: 10.1096/fj.13-228551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eltzschig HK, et al. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med. 2005;202(11):1493–1505. doi: 10.1084/jem.20050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136(2):607–618. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 40.Morote-Garcia JC, et al. Repression of the equilibrative nucleoside transporters dampens inflammatory lung injury. Am J Respir Cell Mol Biol. 2013;49(2):296–305. doi: 10.1165/rcmb.2012-0457OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114(8):1098–1106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schreiber S, Nikolaus S, Hampe J. Activation of nuclear factor kappa B inflammatory bowel disease. Gut. 1998;42(4):477–484. doi: 10.1136/gut.42.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan Y, et al. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS ONE. 2009;4(6):e6073. doi: 10.1371/journal.pone.0006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Visser F, Vickers MF, Ng AM, Baldwin SA, Young JD, Cass CE. Mutation of residue 33 of human equilibrative nucleoside transporters 1 and 2 alters sensitivity to inhibition of transport by dilazep and dipyridamole. J Biol Chem. 2002;277(1):395–401. doi: 10.1074/jbc.M105324200. [DOI] [PubMed] [Google Scholar]
- 45.Meier Y, et al. Regional distribution of solute carrier mRNA expression along the human intestinal tract. Drug Metab Dispos. 2007;35(4):590–594. doi: 10.1124/dmd.106.013342. [DOI] [PubMed] [Google Scholar]
- 46.Neurath MF, Fuss I, Kelsall BL, Stüber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182(5):1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kiesler P, Fuss IJ, Strober W. Experimental models of inflammatory bowel diseases. Cell Mol Gastroenterol Hepatol. 2015;1(2):154–170. doi: 10.1016/j.jcmgh.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neurath MF, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J Exp Med. 2002;195(9):1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Griffith DA, Conant AR, Jarvis SM. Differential inhibition of nucleoside transport systems in mammalian cells by a new series of compounds related to lidoflazine and mioflazine. Biochem Pharmacol. 1990;40(10):2297–2303. doi: 10.1016/0006-2952(90)90726-2. [DOI] [PubMed] [Google Scholar]
- 50.Hammond JR. Interaction of a series of draflazine analogues with equilibrative nucleoside transporters: species differences and transporter subtype selectivity. Naunyn Schmiedebergs Arch Pharmacol. 2000;361(4):373–382. doi: 10.1007/s002100000214. [DOI] [PubMed] [Google Scholar]
- 51.Hammond JR. Comparative pharmacology of the nitrobenzylthioguanosine-sensitive and -resistant nucleoside transport mechanisms of Ehrlich ascites tumor cells. J Pharmacol Exp Ther. 1991;259(2):799–807. [PubMed] [Google Scholar]
- 52.Cronstein BN, Daguma L, Nichols D, Hutchison AJ, Williams M. The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J Clin Invest. 1990;85(4):1150–1157. doi: 10.1172/JCI114547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salmon JE, Cronstein BN. Fc gamma receptor-mediated functions in neutrophils are modulated by adenosine receptor occupancy. A1 receptors are stimulatory and A2 receptors are inhibitory. J Immunol. 1990;145(7):2235–2240. [PubMed] [Google Scholar]
- 54.Gallos G, Ruyle TD, Emala CW, Lee HT. A1 adenosine receptor knockout mice exhibit increased mortality, renal dysfunction, and hepatic injury in murine septic peritonitis. Am J Physiol Renal Physiol. 2005;289(2):F369–F376. doi: 10.1152/ajprenal.00470.2004. [DOI] [PubMed] [Google Scholar]
- 55.Chen Y, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314(5806):1792–1795. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- 56.Lee HT, Kim M, Joo JD, Gallos G, Chen JF, Emala CW. A3 adenosine receptor activation decreases mortality and renal and hepatic injury in murine septic peritonitis. Am J Physiol Regul Integr Comp Physiol. 2006;291(4):R959–R969. doi: 10.1152/ajpregu.00034.2006. [DOI] [PubMed] [Google Scholar]
- 57.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414(6866):916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 58.Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7(9):759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dubey RK, et al. Adenosine attenuates human coronary artery smooth muscle cell proliferation by inhibiting multiple signaling pathways that converge on cyclin D. Hypertension. 2015;66(6):1207–1219. doi: 10.1161/HYPERTENSIONAHA.115.05912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kolachala V, et al. TNF-alpha upregulates adenosine 2b (A2b) receptor expression and signaling in intestinal epithelial cells: a basis for A2bR overexpression in colitis. Cell Mol Life Sci. 2005;62(22):2647–2657. doi: 10.1007/s00018-005-5328-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lawrence DW, Comerford KM, Colgan SP. Role of VASP in reestablishment of epithelial tight junction assembly after Ca2+ switch. Am J Physiol, Cell Physiol. 2002;282(6):C1235–C1245. doi: 10.1152/ajpcell.00288.2001. [DOI] [PubMed] [Google Scholar]
- 62.Young JD. The SLC28 (CNT) and SLC29 (ENT) nucleoside transporter families: a 30-year collaborative odyssey. Biochem Soc Trans. 2016;44(3):869–876. doi: 10.1042/BST20160038. [DOI] [PubMed] [Google Scholar]
- 63.Wojtal KA, et al. Changes in mRNA expression levels of solute carrier transporters in inflammatory bowel disease patients. Drug Metab Dispos. 2009;37(9):1871–1877. doi: 10.1124/dmd.109.027367. [DOI] [PubMed] [Google Scholar]
- 64.Senyavina NV, Gerasimenko TN, Fomicheva KA, Tonevitskaya SA, Kaprin AD. Localization and expression of nucleoside transporters ENT1 and ENT2 in polar cells of intestinal epithelium. Bull Exp Biol Med. 2016;160(6):771–774. doi: 10.1007/s10517-016-3306-5. [DOI] [PubMed] [Google Scholar]
- 65.Ward JL, Tse CM. Nucleoside transport in human colonic epithelial cell lines: evidence for two Na+-independent transport systems in T84 and Caco-2 cells. Biochim Biophys Acta. 1999;1419(1):15–22. doi: 10.1016/s0005-2736(99)00045-0. [DOI] [PubMed] [Google Scholar]
- 66.Mun EC, Tally KJ, Matthews JB. Characterization and regulation of adenosine transport in T84 intestinal epithelial cells. Am J Physiol. 1998;274(2 Pt 1):G261–G269. doi: 10.1152/ajpgi.1998.274.2.G261. [DOI] [PubMed] [Google Scholar]
- 67.Sacco RL, et al. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med. 2008;359(12):1238–1251. doi: 10.1056/NEJMoa0805002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Picano E, Trivieri MG. Pharmacologic stress echocardiography in the assessment of coronary artery disease. Curr Opin Cardiol. 1999;14(6):464–470. doi: 10.1097/00001573-199911000-00003. [DOI] [PubMed] [Google Scholar]
- 69.Baldwin SA, et al. Functional characterization of novel human and mouse equilibrative nucleoside transporters (hENT3 and mENT3) located in intracellular membranes. J Biol Chem. 2005;280(16):15880–15887. doi: 10.1074/jbc.M414337200. [DOI] [PubMed] [Google Scholar]
- 70.Barnes K, et al. Distribution and functional characterization of equilibrative nucleoside transporter-4, a novel cardiac adenosine transporter activated at acidic pH. Circ Res. 2006;99(5):510–519. doi: 10.1161/01.RES.0000238359.18495.42. [DOI] [PubMed] [Google Scholar]
- 71.Pennycooke M, Chaudary N, Shuralyova I, Zhang Y, Coe IR. Differential expression of human nucleoside transporters in normal and tumor tissue. Biochem Biophys Res Commun. 2001;280(3):951–959. doi: 10.1006/bbrc.2000.4205. [DOI] [PubMed] [Google Scholar]
- 72.Louis NA, Robinson AM, MacManus CF, Karhausen J, Scully M, Colgan SP. Control of IFN-alphaA by CD73: implications for mucosal inflammation. J Immunol. 2008;180(6):4246–4255. doi: 10.4049/jimmunol.180.6.4246. [DOI] [PubMed] [Google Scholar]
- 73.Bynoe MS, Waickman AT, Mahamed DA, Mueller C, Mills JH, Czopik A. CD73 is critical for the resolution of murine colonic inflammation. J Biomed Biotechnol. 2012;2012:260983. doi: 10.1155/2012/260983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Siegmund B, et al. Adenosine kinase inhibitor GP515 improves experimental colitis in mice. J Pharmacol Exp Ther. 2001;296(1):99–105. [PubMed] [Google Scholar]
- 75.Antonioli L, et al. Inhibition of adenosine deaminase attenuates inflammation in experimental colitis. J Pharmacol Exp Ther. 2007;322(2):435–442. doi: 10.1124/jpet.107.122762. [DOI] [PubMed] [Google Scholar]
- 76.Brown JB, Lee G, Grimm GR, Barrett TA. Therapeutic benefit of pentostatin in severe IL-10–/– colitis. Inflamm Bowel Dis. 2008;14(7):880–887. doi: 10.1002/ibd.20410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maes SS, Pype S, Hoffmann VL, Biermans M, Meert TF. Antihyperalgesic activity of nucleoside transport inhibitors in models of inflammatory pain in guinea pigs. J Pain Res. 2012;5:391–400. doi: 10.2147/JPR.S35108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Collins CB, et al. Inhibition of N-terminal ATPase on HSP90 attenuates colitis through enhanced Treg function. Mucosal Immunol. 2013;6(5):960–971. doi: 10.1038/mi.2012.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Analytical Methods Committee, AMCTB No 69. Using the Grubbs and Cochran tests to identify outliers. Anal Methods. 2015;7(19):7948–7950. doi: 10.1039/C5AY90053K. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.