

Abstract
We introduce the optimization of the pulping conditions and propose different chemical recovery options for a proven biorefinery concept based on γ-valerolactone (GVL)/water fractionation. The pulping process has been optimized whereby the liquor-to-wood (L:W) ratio could be reduced to 3 L/kg without compromising the pulp properties as raw material for textile fibers production. The recovery of the pulping solvent was performed through combinations of lignin precipitation by water addition, distillation at reduced pressure, and liquid CO2 extraction. With a two-step lignin precipitation coupled with vacuum distillation, more than 90% of lignin and GVL could be recovered from the spent liquor. However, a significant part of GVL remained unrecoverable in the residue, which was a highly viscous liquid with complicated phase behavior. The recovery by lignin precipitation combined with liquid CO2 extraction could recover more than 85% GVL and 90% lignin without forming any problematic residue as in the distillation process. The remaining GVL remained in the raffinate containing a low amount of lignin and other compounds, which can be further processed to isolate the GVL and improve the recovery rate.
1. Introduction
The population growth with increasing demand for energy and materials is depleting the nonrenewable, limited, and unsecured fossil fuel feedstock. Such a crisis has initiated the research for alternative renewable resources, where lignocellulosic biomass has been identified as the prominent candidate to replace crude oil and natural gases for the production of chemicals, materials, and fuels.1,2 The most abundant and important biomass is wood, which is a natural composite mainly composed of cellulose, hemicellulose, and lignin.3 The processing and conversion of wood to products occurs in a biorefinery, which is analogous to an oil refinery, its fossil counterpart. The core of a biorefinery is the pulping process, among which kraft pulping has been predominant for several decades, because of its pulp quality, high chemical recovery rate, and robustness toward raw materials.4 Kraft pulping is currently irreplaceable in the manufacture of paper-grade pulp; however, the production of dissolving pulp, which is the second-most-common pulp grade, via this method encounters several drawbacks, such as the need for an extra prehydrolysis stage, low yield, difficult usability of hemicelluloses, and problematic lignin precipitation.5−9 The traditional source of wood-based dissolving pulp for the production of Rayon fibers by acid sulfite pulping is even more polluting and less attractive, because of the complex and incomplete recovery of the pulping chemicals.10−12 Those technical and environmental issues became the driving force behind the adoption of organic solvents in the alternative pulping methods, among which the major representatives are ALCELL (ethanol–water),13 MILOX (formic acid-peroxyformic acid),14 SEW (SO2–ethanol–water),15 and Clean Fractionation (methyl isobutyl ketone (MEK)–ethanol–water).16 Apart from ALCELL and SEW, which currently operate at pilot scale, most of the existing organosolv fractionation processes are still in early stages with their own disadvantages, particularly with regard to pulp quality and solvent recovery, which prevent them from reaching full-scale commercialization.
As a contribution to the organosolv technologies framework, we previously introduced a novel biorefinery concept where Eucalyptus globulus (E. globulus) could be fractionated in an aqueous solution of γ-valerolactone (GVL) at elevated temperature by a single step into its principal components, namely, cellulose, hemicellulose, and lignin, with adequate quality.17 The main pulping chemical, GVL, is a green solvent that is nontoxic, water-soluble, zeotropic when mixed with water, and nonvolatile (vapor pressure of 0.44 mbar at 25 °C), in addition to having a low melting point (−31 °C) and a high boiling point (207 °C).18,19 The recognizable smell of GVL enables easy detection of leakage or spilling, and more importantly, GVL is a stable chemical that is unsusceptible to degradation and oxidation at room temperature and atmospheric pressure, making it a safe substance for large-scale storage, transportation, and other applications.20 The pulp fraction obtained from the GVL/water fractionation was characterized by high yield, high cellulose purity, and high bleachability. Both the bleached and unbleached pulps were readily spinnable to regenerated cellulosic fibers for textile applications17 or converted to nanofibrillated cellulose.21 The spent liquor contained the fractionation solvent, fragmented lignin, extracted hemicelluloses, and their degradation products, such as furanic compounds (furfural and 5-hydroxymethylfurfural), organic acids (e.g., formic acids, acetic acids, and levulinic acids), and humins. Effective isolation of the extracted wood components and quantitative recovery of the organic solvent are the critical goals that determine the feasibility of the fractionation process.
Distillation is generally the preferred method to recover volatile compounds such as ethanol22,23 or formic acid.24 However, the low volatility of GVL, which offers several advantages in the pulping step, becomes an obstacle for the recovery by distillation, as water must be almost completely evaporated before the GVL removal from the spent liquor, resulting in a surge in energy consumption. Therefore, alternative methods should be considered. Luterbacher et al. suggested in their pioneering works that GVL could be effectively recycled from the biomass fractionation liquor by liquid CO2 extraction.25,26 Our parallel study on the thermodynamics and phase behaviors of the GVL–CO2–water ternary system confirmed the affinity of GVL toward the CO2 phase over the aqueous phase at room temperature and high pressure.27 These two separation methods served as the core techniques of our recovery schemes.
In our previous work, GVL/water fractionation experiments were conducted in an excessive amount of solvent (liquor-to-wood (L:W) ratio of 10 L/kg),17 which is impractical, with regard to the solvent recovery process. Therefore, prior to investigating the recovery of GVL, we optimized the fractionation parameters, with an emphasis on minimizing the L:W ratio while maintaining the viscose-grade quality of the dissolving pulp for textile fibers production. The spent liquor obtained from one optimized pulping experiment was then subjected to solvent recovery using both distillation at reduced pressure and liquid CO2 extraction. In this work, the emphasis was placed on the recovery of the main components with the higher occurrence in the spent liquor, namely GVL, water, and lignin, while the purification and separation of minor substances such as carbohydrates, furanic compounds, and organic acids was not considered.
2. Experimental Section
2.1. Materials
E. globulus wood chips were delivered by ENCE, Spain. The chips were screened according to the SCAN-CM 40:01 method and stored at −20 °C. Some of the chips were air-dried and then ground to sawdust in a Wiley mill (Arthur H. Thomas Co., model No. 2 with a screen opening of 0.5 mm). Only wood particles less than 125 μm in size were collected. The identified chemical composition of the wood was 44.1% glucose, 15.2% xylose, 3.1% other sugars, 28.1% lignin, and 1.3% extractive. Both wood chips and sawdust were used for the GVL/water fractionation experiments. The GVL was supplied by Sigma–Aldrich with ≥98 wt % purity. Pure water was produced on site using a Millipore Synergy ultraviolet (UV) purification system (water resistivity of 18.2 MΩ cm). CO2 was purchased from AGA in the liquid state and was stored in a steel cylinder at ∼56 bar and 20 °C.
2.2. GVL/Water Fractionation
Fractionation of wood chips with different L:W ratios, ranging from 2 L/kg to 10 L/kg, were conducted in the 2.5 L autoclaves heated in an air-bath reactor (Haato-tuote Model 16140-538). The reaction temperature was 180 °C, the residence time was 150 min, and the GVL content in the liquor was 50 wt %. The pulping temperature and GVL concentration was selected based on optimization for the uncatalyzed production of dissolving pulp in our previous work.17 The effect of fractionation time at reduced L:W ratios were investigated at a smaller scale in order to economize on GVL. The experiments were conducted in 225-mL autoclaves heated in a silicon oil-bath reactor (Haato-tuote, Model 43427). The reaction temperature and GVL content in the liquor were 180 °C and 50 wt %, respectively. L:W was either 3 or 4 L/kg, and the residence time ranged from 90 min to 150 min. The penetration of the cooking liquor into the wood cellular structure was facilitated by an impregnation stage at 120 °C for 60 min. The reaction was quenched by submerging the autoclaves in cool water. The temperature profiles of the fractionation in oil-bath and air-bath reactors are presented in the Supporting Information (SI Section 1). The pulp and raw spent liquor were then separated using a nylon filtration bag. The pulp was washed with 50 wt % GVL solution with L:W ratios similar to that of the fractionation. The effective L:W ratio that resulted from the combined amount of the fractionation and washing liquids was 6 or 8 L/kg. The washing GVL solution was combined with the raw spent liquor, and this liquid is referenced as “spent liquor” throughout this paper. The solvent-washed pulp was subjected to a final washing with hot (ca. 80 °C) water until the filtrate appeared clear. The spent liquor and the washing water were collected for subsequent analyses. The fully washed pulp was screened in a table-top screener (G.A. Serlachius A.B., Model 16140-567, with a mesh opening of 0.35 mm) to determine the amount of rejects. The pulp yield was determined gravimetrically.
The recyclability of GVL recovered from the spent liquor by liquid CO2, as described in Section 2.3.3, was tested by the fractionation of E. globulus sawdust in 30 mL vials heated in a microwave reactor (Anton Paar, Model Monowave 300). Three consecutive fractionation cycles were investigated with the protocol illustrated in the Supporting Information (SI Section 2). Fresh GVL was used in the first cycle. For each trial, 1.5 g of oven-dried sawdust were fractionated in 50 wt % GVL solution with L:W = 10 L/kg. The reaction mixture was heated to 180 °C, held at that temperature for 120 min, and then cooled by compressed air to 55 °C. The pulp and the raw spent liquor were separated with a Robu glass crucible (porosity 4). Pulp washing was conducted with the same protocol as in wood chips fractionation trials. The pulp was oven-dried at 105 °C and the yield was determined gravimetrically. The spent liquor and the washing water were collected for subsequent analyses.
2.3. GVL Recovery from the Spent Liquor
2.3.1. Lignin Isolation from Spent Liquor
Lignin was precipitated from the GVL/water fractionation spent liquor by the addition of water (the ratio of water to spent liquor was 0.5:1 or 1:1). The suspension was centrifuged at a relative centrifugal force of 3000g for 30 min. The precipitated lignin was collected and washed three times with water (each time with the same amount as the original spent liquor), followed by ultrasonic treatment for 15 min. The washed lignin was ground to finer particles and dried at 40 °C under a pressure of ∼100 mbar for at least 8 h before the subsequent analyses.
2.3.2. Distillation at Reduced Pressures
2.3.2.1. Equipment
The batch distillation system consisted of a reboiler, a distillation column, a condenser, and a vacuum pump. The reboiler comprised of a Lenz 0.5 L round-bottomed two-necked flask, a Pilz 320W electrical heating mantel, and an IKAMAG REC-G magnetic stirrer. Temperatures of the reboiler and the compartment just below the condenser were measured by Pt100 temperature sensors. The Vigreux column (NORMAG) was coated with silver and vacuum-jacketed with bellows. The column effective length and diameter were 1050 mm and 25 mm, respectively. The condenser was cooled with a small flow of tap water at a temperature of ∼15 °C. The reflux rate was controlled by an electromagnetically operated liquid splitter (NORMAG). The distillation column was evacuated by an Edwards Model RV3 vacuum pump equipped with a Keller LEO2 pressure meter, a liquid nitrogen trap, and an air-bleeding valve for vacuum level adjustment. The Keller pressure meter was calibrated against a Beamex MC2-PE field calibrator.
2.3.2.2. Operation
Prior to distillation, lignin was precipitated from the spent liquor, as described in Section 2.3.1. The diluted lignin-lean spent liquor was fed into the round-bottomed flask of the reboiler and mixed by means of a Teflon-coated magnet bar. After the distillation column was sealed, the heating and mixing started. Once the temperature of the spent liquor was ∼40 °C, the column was evacuated and the vacuum level was gradually increased to prevent the sudden flashing of the liquid upon reaching its boiling point. When the boiling started, the pressure was adjusted to ∼240 mbar in order to evaporate the water fraction in the spent liquor. The system was operated in total reflux mode until the column internal part was heated, the vapor reached the condenser, and the temperature of the column and the reboiler stabilized at ∼60–65 °C. The reflux was then started with a reflux ratio of Rd = 1. The reboiler temperature rose gradually, because of the increasing boiling point corresponding to the increasing GVL mass fraction in the remaining spent liquor upon water removal. When most of the water was removed from the spent liquor, the reboiler temperature increased sharply. However, only 80%–85% of the initial amount of water in the distillation feed was removed from the spent liquor in this step, in order to minimize the GVL collection in the distillate. The distillation was then switched back to total reflux mode and the bleeding valve was gradually closed until the highest vacuum level was reached. The distillation system stabilized at ∼7 mbar and 75–80 °C. After the aqueous distillate was collected, the reflux (Rd = 1) was resumed. The GVL removal increased the lignin concentration in the remaining liquor; therefore, the reboiler temperature gradually increased. The distillation was stopped when the temperature of the reboiler rose sharply. The heating, reflux, and column evacuation was turned off and the organic distillate (GVL) was collected. The column was rinsed with acetone to transfer the distillate retained on the column internal part to the reboiler. The liquid in the reboiler was left until all the washing acetone evaporated. The remaining liquid was referred to as the distillation residue. The distillation of spent liquor can be either a one- or two-staged operation. The GVL recovery schemes by vacuum distillation is illustrated in the Supporting Information (SI Section 3).
2.3.3. Extraction by Liquid CO2
2.3.3.1. Equipment
The semibatch extraction system was constructed based on the JERFI high-pressure phase-behavior sapphire cell by DB Robinson Design & Manufacturing. The extraction column consisted of a sapphire tube with dimensions of 152.5 mm (length), 25.35 mm (internal diameter), and 38.10 mm (external diameter), which was attached between two stainless steel top and bottom caps whose distance was fixed by the tie rods. The gland packing was made from graphite-reinforced polytetrafluoroethylene (PTFE) and tightened between the gland clamps, the top cap, and the bottom caps of the sapphire cell. The temperature of the system was regulated by an air-bath equipped with a blower and an air-to-water heat exchanger tube bank. The water inside the tube bank was thermostated with a LAUDA E200 refrigerating circulator. The air-bath and water circulator temperatures were monitored with Pt100 temperature sensors connected to a NOKEVAL Model RMD680 series 8 channel universal input transmitter. No mixing mechanism was installed.
Spent liquor was fed to the column through a feed line connected to the top cap. CO2 was fed into the extraction column by a Teledyne ISCO 500D syringe pump thermostated at 10 °C. The feed line was equipped with Swagelok valves for flushing the line and feeding CO2 into the sapphire cell. The feeding of CO2 was performed through either the bottom or top cap. Spent liquor and CO2 feeding were controlled by a three-way valve. The CO2 dispersion during the feeding through the bottom cap was facilitated by a capillary distributor that was made of six metal 28-gauge and 75-mm-long capillaries (Hamilton) soldered into the 1/8 in. tubing. For extract collection, the top cap of the extraction cell was equipped with a 1/4 in. outlet line connected to a custom-built Equilibar B6R Series blockage-resistant back-pressure regulator in which the dome pressure was pressurized with nitrogen and controlled by a Series 3000 pressure-reducing regulator. The pressure regulator discharged the extract to a funnel, where a local ventilation evacuated the CO2 out from the laboratory while the liquid extract was collected. The sapphire tube and safety valve were tested up to 15.0 MPa for leaks with pressurized deionized water prior to any experiments. The extraction cell was connected with a Swagelok high-pressure proportional relief valve adjusted to 10.3 MPa for protection against abnormal pressure rise. Schematic of the extraction unit is presented in the Supporting Information (SI Section 4).
2.3.3.2. Operation
CO2 was loaded from AGA gas cylinder at room temperature to the ISCO pump and then compressed to 70 bar. The Equilibar B6R pressure regulator was set to 75 bar. The spent liquor was loaded into the extraction column by injection with a plastic syringe until the liquid level was ∼50%–60% of the sapphire tube (ca. 30–40 g spent liquor). At the beginning of the extraction, CO2 feeding was performed through the top cap of the sapphire tube for safe pressurization of the system, preventing the shooting of spent liquor into the extract exit line by heavy flashing of liquid CO2. When the liquid level inside the column (spent liquor and CO2) was ∼80%, the feeding from the top was terminated and replaced by the feeding from the bottom through the capillaries. During the filling of the extraction column, the CO2 flow was limited to 10 mL/min. When the pressure inside the extraction column equilibrated with that of the ISCO pump, i.e., 70 bar, the ISCO pump was set to a constant flow mode of 2 mL/min. The constant injection of CO2 to the column gradually increased the pressure to 75 bar, at which the Equilibar B6R valve was forced open, to release the extract to the exit line. A spent liquor/CO2 mass ratio of 1:4 was used for all the experiments. An extraction typically lasted for ∼90–120 min. After the extraction, the ISCO pump was stopped, the dome pressure of the pressure regulator was gradually released for safely flashing the remaining liquid CO2 inside the column. After the flashing, the extract was collected from the top and the raffinate was collected from the bottom of the extraction column. If necessary, the raffinate can be subjected to centrifugation at 3000 g for 30 min to sediment the suspending lignin particles. The precipitated lignin deposited on the interior wall of the extraction column was washed with acetone. The lignin solution was left until all the acetone evaporated and the precipitated lignin was collected. The entire system was washed intensively with acetone in order to clean all the tubing of the top exit line from the retained extract liquid. The washing liquid was collected and left until all the acetone evaporated. The remaining liquid was referenced as extraction residue. Prior to the extraction, the spent liquor could be pretreated, for example, by lignin precipitation, as described in Section 2.3.1, or by water removal by vacuum distillation, or by combination of both. The GVL recovery schemes by liquid CO2 extraction are illustrated in the Supporting Information (SI Section 4).
2.4. Analyses of Pulp, Lignin, and Liquid Samples
This section demonstrates an overview of the analysis routine for the solid and liquid samples. Detailed descriptions of the methods are presented in the Supporting Information (SI Section 5).
The carbohydrate content of the pulp and precipitated lignin samples was analyzed by high-performance anion-exchange chromatography (HPAEC), in accordance to the NREL/TP-510-42618 standard. The acid-insoluble lignin in pulp was determined gravimetrically, while the acid-soluble lignin in pulp was quantified by measuring the absorbance at 25 °C at a wavelength of 205 nm (using a Shimadzu Model UV-2550 spectrophotometer). An extinction coefficient of 148 L/(g cm) was used for quantification of ASL.17 The pulps were analyzed for intrinsic viscosity, in accordance to the SCAN-CM 15:88 standard. The molecular weight distributions, the number-average molecular weights (Mn), and weight-average molecular weights (Mw) of the precipitated lignin and lignin retained in the residue of vacuum distillation were determined by gel permeation chromatography (GPC).
The carbohydrate and lignin contents in the liquid samples were analyzed in accordance to the NREL/TP-510-42623 standard. The lignin content in the spent and washing liquors was determined by ultraviolet-visible light (UV-vis) spectrophotometry (Shimadzu, Model UV-2550) at 25 °C by diluting in ethanol 50 wt % and measuring the absorption at a wavelength of 205 nm, with an extinction coefficient of 148 L/(g cm). The content of furanic compounds and organic acids in the liquid samples was determined by high-performance liquid chromatography (HPLC). The GVL/water mass ratio in the liquid samples was determined by gas chromatography (GC).
3. Results and Discussion
3.1. E. globulus Wood Fractionation at Reduced L:W Ratios
The effect of the L:W ratio on the removal of the wood main components, namely, cellulose, hemicelluloses, and lignin, during fractionation is shown in Figure 1. The wood was not extracted before the fractionation trials, and thus a small amount of extractives (∼1.3%) in the starting material might appear as lignin in the ASL analysis via UV spectrometry.
Figure 1.
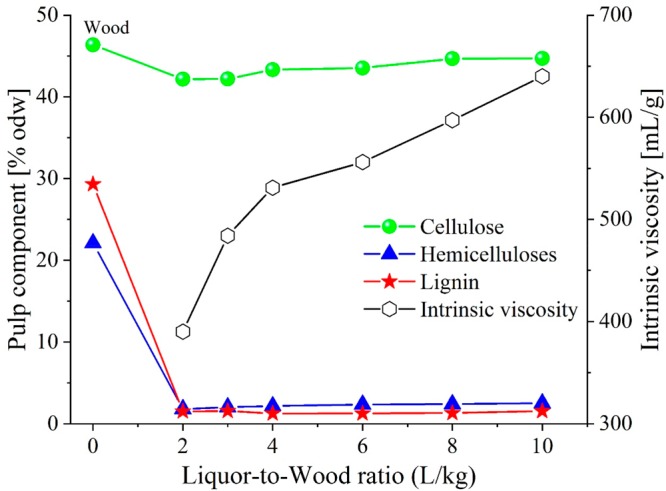
Effect of the L:W ratio on the removal of eucalyptus wood chip components and on the intrinsic viscosity of the pulp. The fractionation trials were conducted in 50 wt % GVL solution, at 180 °C, for 150 min, and with varying L:W ratios, from 2 L/kg to 10 L/kg in the air-bath digester. [Note: the abbreviation odw represents oven-dried wood.]
No reject fraction (uncooked wood) was detected for any of the fractionation trials, which is consistent with our previous work.17 The results in Figure 1 indicate a similar degree of delignification and hemicellulose removal upon reducing the L:W ratio. The cellulose yield was relatively well-preserved, comprising ∼91%–96% of the original cellulose in wood. The small losses can be attributed to a low-crystalline, low-molecular-weight cellulose fraction. The L:W reduction has the most pronounced effect on the degree of polymerization of the pulp cellulose. The pulp intrinsic viscosity gradually decreased from ∼640 mL/g at L:W = 10 L/kg to 531 mL/g at L:W = 4 L/kg; beyond that point, the viscosity plunged further to 390 mL/g at L:W = 2 L/kg. This phenomenon can be explained by the enhanced hydrolytic activity corresponding to the increased acidity of the fractionation medium, when a similar amount of hemicellulose-originated organic acids was formed28,29 in a lesser amount of liquid. This hypothesis was partly confirmed by the pH of the spent liquors, as shown in Table 1. Simultaneously, the increase in ionic strength of the bulk liquor might enhance the proton activity in the cell wall due to the Donnan equilibrium.30 The higher hydrolytic activity of the cooking liquor was also demonstrated by a reduction in the hemicellulose content in the pulp.
Table 1. Pulp Characterization and pH of the Spent Liquor of the E. globulus Wood Chips Fractionation Experiments in 50 wt % GVL Solution at 180 °C, for 150 min with Varying L:W Ratios in the Air-Bath Digester.
| Solid Fraction (Pulp) |
|||||||
|---|---|---|---|---|---|---|---|
| L:W ratio (L/kg) | yielda (% odw) | celluloseb (% odp) | C5c (% odp) | C6d (% odp) | lignin (% odp) | viscosity (mL/g) | spent liquor pH |
| 10 | 48.8 | 90.8 | 5.1 | 1.2 | 2.9 | 640 | 3.68 |
| 8 | 48.4 | 91.2 | 5.0 | 1.3 | 2.5 | 597 | 3.66 |
| 6 | 47.2 | 91.3 | 5.0 | 1.2 | 2.5 | 556 | 3.53 |
| 4 | 47.2 | 91.8 | 4.6 | 1.1 | 2.5 | 531 | 3.45 |
| 3 | 46.3 | 91.4 | 4.4 | 1.0 | 3.2 | 517 | 3.06 |
| 2 | 45.5 | 92.0 | 4.0 | 0.8 | 3.2 | 390 | 2.99 |
Percent on oven-dried wood.
Percent on oven-dried pulp.
C5 hemicelluloses (xylan and arabinan).
C6 hemicelluloses (galactan, mannan, and rhamnan).
As shown in Table 1, L:W could be reduced to 3 L/kg without compromising the pulp properties required for its conversion to regenerated cellulosic fibers by, for example, the viscose process31 or IONCELL-F process,32 which are high cellulosic content (>90%) and suitable intrinsic viscosity (400–600 mL/g) after bleaching. Further decreases of L:W significantly impaired the pulp properties.
L:W ratios of 3 and 4 L/kg are economically reasonable for upscaling the process. Therefore, the effect of fractionation time to the pulp quality at these two L:W ratios was further investigated, and the results are summarized in Table 2. Detailed mass balances of these experiments are shown in the Supporting Information (SI Section 6).
Table 2. Properties of the Pulps Obtained from the Fractionation of E. globulus Wood Chips in 50 wt % GVL Solution at 180 °C, for 90–150 min with L:W = 3 or 4 L/kg in the Oil-Bath Digester.
| Solid Fraction (Pulp) |
||||||
|---|---|---|---|---|---|---|
| samplea | yieldb (% odw) | cellulosec (% odp) | C5d (% odp) | C6e (% odp) | lignin (% odp) | viscosity (mL/g) |
| 4–90 | 50.7 | 87.5 | 6.6 | 1.6 | 4.4 | 846 |
| 4–120 | 48.8 | 88.5 | 6.2 | 1.6 | 3.7 | 769 |
| 4–150 | 47.1 | 90.4 | 5.3 | 1.5 | 2.8 | 562 |
| 3–90 | 48.9 | 88.1 | 5.9 | 1.7 | 4.3 | 773 |
| 3–120 | 47.0 | 89.1 | 5.3 | 1.6 | 4.1 | 597 |
| 3–150 | 45.6 | 89.8 | 5.6 | 1.2 | 3.4 | 493 |
The sample is named as fractionation L:W (in L/kg)-fractionation time (in minutes).
Percent on oven-dried wood.
Percent on oven-dried pulp.
C5 hemicelluloses (xylan and arabinan).
C6 hemicelluloses (galactan, mannan, and rhamnan).
All six of the pulps introduced in Table 2 can be potentially processed to regenerated cellulosic fibers. Pulps 4-150 and 3-150, with high cellulose content and proper intrinsic viscosity, may be directly converted to textile fibers via the IONCELL-F process without further purification. Pulp 3-120 may be processed to fiber after a short elemental-chlorine-free (ECF) bleaching sequence of D0-Ep-P, similar to our previous work,17 which typically bleach the pulp to more than 90% ISO brightness while dropping the intrinsic viscosity ∼100 mL/g. The other three pulps with higher intrinsic viscosity (samples 4-90, 4-120, and 3-90) may be treated with total-chlorine-free (TCF) bleaching, for example, with oxygen delignification, followed by ozone bleaching and alkaline extraction, to full brightness. The better-preserved cellulose chain and slightly higher hemicellulose content in pulp can compensate for the inferior lignin/carbohydrate selectivity of oxygen and ozone, in comparison to chlorine-containing bleaching chemicals. The TCF-bleached pulp should be of viscose-grade.
Among the six aforementioned experiments, no particular pulping condition is the optimum one. L:W = 4 L/kg generally delivers better pulp yield and quality but the recycling of GVL would be more costly. Higher fractionation time offers slightly better delignification and hemicellulose removal at the expense of some cellulose loss (see the Supporting Information (SI Section 6)). The choice of pulping parameters, particularly the cooking time, L:W, and possibly the bleaching technology, should therefore be considered under specific conditions of each pulp mill (e.g., utilities price, environmental legislation, economic environment, product spectrum, price, and quality of the regenerated cellulosic fibers).
In this work, we proceeded with the recovery of GVL from the spent liquor obtained from the fractionation of wood in 50 wt % GVL, 180 °C, 150 min, and L:W = 4 L/kg. These are the conditions of a hypothesized biorefinery, where the main product is unbleached dissolving pulp readily convertible to lignin-containing textile fibers by the IONCELL-F process. The spent liquor from this fractionation consisted of (by mass) 47.07% GVL, 47.27% water, 3.73% lignin, 0.65% carbohydrate (in both monomeric and oligomeric form), 0.57% furanic compounds and 0.71% organic acids. The sum of individual components was normalized to 100%; unidentified substances such as humins were considered minor and therefore not taken into account.
3.2. GVL Recovery by Distillation at Reduced Pressure
3.2.1. One-Stage Distillation (Recovery Scheme 1)
The first half of Figure 2 presents the mass balance of a representative experiment for recovering GVL from the spent liquor by a lignin precipitation followed by distillation at reduced pressure (recovery scheme 1). The mass balance is not perfect; there is ∼5% loss from the total input, which can be accounted for by the handling and operational losses and analytical errors.
Figure 2.

Flow diagram of the two-stage vacuum distillation of spent liquor for GVL recovery (recovery scheme 2), showing the distribution of the main components into the product streams. The mass balance is based on 100 g of spent liquor. The mass balance of single-stage vacuum distillation (recovery scheme 1) is also included in the first half of the diagram, before the second lignin precipitation by the addition of water.
The presence of GVL in the aqueous distillate, originated from the GVL coevaporation with water, was unavoidable with our existing instruments. This phenomenon can be limited by three different approaches. First, the column packing can be modified to enhance the number of separation stages; however, such investment was not possible in our case. Second, the purity of aqueous distillate can be improved by alternating the distillation parameter, such as by increasing the pressure or the reflux ratio, which, in turn, lengthen the distillation, for both cases, leading to a more-energy-intensive process. The third choice is to terminate the water distillation before the selectivity declines. The last one was employed in this work, since the fractionation requires only a 50 wt % GVL solution; therefore, water does not need to be distilled to completion in the first step, and the presence of some water in the organic distillate is tolerable.
This scheme could recover ∼86% of the GVL, of which 4% was collected in the aqueous distillate. In the precipitation before distillation, ∼47% of the lignin in spent liquor was recovered in high purity (>90%). Increasing the water-to-spent-liquor ratio to 1:1 could improve the lignin isolation to ∼67%–75% of the lignin in spent liquor, at the expense of the process energy economy in the subsequent water removal step. More than 7% of the original spent liquor, most of which consisted of GVL and lignin, was unrecoverable in the form of distillation residue. The residue is a highly viscose lignin solution in GVL with a complicated phase behavior, as demonstrated in our earlier study.33 As the residue cannot be reliably analyzed, its composition was calculated based on the mass balance of lignin, carbohydrates, furanic compounds, and organic acids; the remainder was assumed to be GVL. The presence of lignin in high concentration (ca. 25 wt %) further decreased the volatility of GVL. Hence, the reboiler temperature increased sharply and the distillation must thus be stopped before the complete evaporation of the liquid in the reboiler for safety reason. The lignin isolation and GVL recovery could be improved by extending the precipitation and distillation to another cycle (recovery scheme 2).
3.2.2. Two-Stage Distillation (Recovery Scheme 2)
The introduction of a successive distillation cycle slightly improved the GVL recovery (from 86% to 90%), while almost doubling the lignin removal from the spent liquor (from 47% to 93%), as demonstrated in Figure 2. The second precipitation isolated the lignin fraction with lower molecular weight (see the Supporting Information (SI Section 7)), which remained soluble in the first cycle. The high GVL concentration (ca. 80 wt %) in the organic distillate from the first stage allowed the distillate in the second distillation to be collected in just one fraction. The distillate obtained in the last stage could be combined with the organic distillate from the previous stage to a 55 wt % GVL solution, which is sufficient for the pulping process.
However, in our experiment, ∼7% of the GVL remained unrecoverable in the residue, which originated from two technical limitations. First, because of the presence of involatile compounds, such as lignin and carbohydrate in the reboiler, a certain amount of GVL had to remain there for safety issues. Full evaporation of GVL would cause overheating, which might damage the equipment. Second, at the end of a batch distillation process, a certain amount of GVL retained on the column internal wall and packing and, therefore, was washed back to the reboiler and combined with the residue (as described in Section 2.3.2). Such a loss of GVL could have been prevented by modifying the column washing procedure (by acetone), so that the washing liquor was collected as a separate fraction, instead of being combined with the residue. After the acetone evaporation from the washing liquor, the remaining liquid would be almost pure GVL and could be combined with the organic distillate, thus improving the overall GVL recovery rate by ∼2%–4%.
The GVL removal efficiency already declined in the second distillation cycle (see Figure 2). An extension to a third stage, or even a fourth stage, would slightly improve the lignin and GVL recovery, at the expense of energy for distilling the amount of water added in the lignin precipitation step. Even with an optimized distillation process, the presence of the distillation residue is inevitable, which requires a more advanced processing to remove lignin and the nonvolatile carbohydrate to recover GVL. Ultra/nanofiltration34 or absorption by activated carbon.35 Besides, because of the low molecular weight of the lignin remaining in the residue (see the Supporting Information (SI Section 7)), one approach could be the lignin depolymerization by hydrothermolysis to bio-oil,36 in combination with carbohydrate hydrolysis and dehydration to furanic compounds.37 The resulting liquid could be further distilled for its components. The viability and energy requirement for such approach might be an interesting research topic.
3.2.3. Preliminary Design of a Continuous Distillation Process
Based on the experience from the batch distillation, we propose a continuous vacuum distillation process for GVL recovery (see Figure 3). Unlike the batch distillation, where the spent liquor is fed at the reboiler, the feed point is in the middle of the column in a continuous process. Therefore, the presence of lignin in the distillation column must be avoided, which is realized by the inclusion of a spent liquor pre-evaporation stage. A standard black liquor evaporator design is expected to be suitable for such application. The vapor fraction, containing GVL, water, furanic compounds, and organic acids, enters the distillation column for further separation. A fraction of GVL is left in the residue, together with other involatile compounds such as lignin, carbohydrates, and humins. Lignin and humins can be separated by the addition of water. The remaining lignin-lean diluted liquid is recycled to the evaporator. The recycled liquid and product streams might require further purification, e.g., by filtration or absorption. The possible accumulation of dissolved compounds in the recycled liquid should also be investigated. A preliminary energy evaluation of the continuous distillation process is presented in Section 3.4.
Figure 3.

Proposed GVL recovery scheme by continuous vacuum distillation.
3.3. GVL Recovery by Liquid CO2 Extraction
3.3.1. Extraction of the Spent Liquor after Partial Lignin Removal (Recovery Scheme 3)
Before the extraction, ∼65% of the lignin was precipitated from the spent liquor with high purity by dilution with a water-to-spent-liquor mass ratio of 1:1. The remaining diluted lignin-lean spent liquor, with a GVL concentration of ∼25 wt %, was extracted by liquid CO2 (see Figure 4). Most of the GVL and furanic compounds in the original spent liquor were collected in the extract, while the others remained in the raffinate. The rate of GVL removal from the spent liquor decreased as the extraction proceeded, because the partition coefficient approached unity at low GVL concentration.27 Moreover, the lack of a mixing mechanism in our system severely limited the mass transfer of GVL to the CO2 phase. Both phenomena resulted in a high leftover GVL rate (∼10%) in the raffinate, even at excessive CO2 dosage. Suspended lignin particles were separated from the raffinate by sedimentation and centrifugation. This lignin fraction was more fragmented than the one precipitated by the addition of water (see the Supporting Information (SI Section 7)). About 14% of the original lignin, with even lower molecular mass, remained dissolved in the raffinate. More advanced lignin removal methods, such as ultrafiltration or adsorption, are necessary before further processing of the raffinate to valorize the extracted carbohydrates and their degradation products. It was noteworthy that, in contrast to the distillation residue, which was a highly concentrated lignin solution in GVL with complicated behavior, the residue liquid from the extraction was only the liquid entrapped in the dead volume of the piping system and, thus, consisted only of GVL and water that can be combined with the extract. Therefore, extraction is more promising than distillation for a complete recovery of GVL. However, there is a ca. 7% gap of GVL in the mass balance, which can be assigned to handling losses, CO2 leakage, and analysis errors. The most severe disadvantage of this strategy is the particularly high extractant consumption, because of the dilution of the original spent liquor.
Figure 4.

Flow diagram of the spent liquor extraction after partial lignin precipitation for GVL recovery (recovery scheme 3), showing the distribution of the main components into the product streams. The mass balance is based on 100 g of spent liquor.
3.3.2. Extraction of the Original Spent Liquor (Recovery Scheme 4)
In this scheme, the original spent liquor, with a GVL concentration of ∼50 wt %, was extracted without any pretreatment. About 87% of the GVL, 60% of the furanic compounds, and 20% of the organic acids were recovered in the extract and residue, while ∼90% of the lignin was collected as a single fraction with decent purity (see Figure 5). The extraction of GVL was also not complete in this scheme, with ∼12% of the GVL remaining in the raffinate. Similar to the previous scheme, the lignin-containing raffinate must be purified before further separation and valorization steps. A better GVL balance (ca. 98%) was obtained for this recovery scheme, because the leaking problem that was experienced in Section 3.3.1 was fixed. The elimination of a lignin precipitation step via the addition of water significantly reduced the amount of extraction feed, which led to an extractant savings of 50%, with comparable separation efficiency. We then investigated the possibility of further CO2 savings by partial water removal before the extraction.
Figure 5.

Flow diagram of the spent liquor extraction without any pretreatment for GVL recovery (recovery scheme 4), showing the distribution of the main components into the product streams. The mass balance is based on 100 g of spent liquor.
3.3.3. Extraction of the Spent Liquor after Partial Water Removal (Recovery Scheme 5)
Water was evaporated at reduced pressure, resulting in a liquid with ∼75% GVL (by weight) for extraction. Unfortunately, the higher lignin content in the concentrated spent liquor complicated the extraction procedure. The GVL removal induced a rapid and uncontrollable lignin precipitation, creating a sticky lignin agglomerate. In addition, a strong interaction with lignin trapped the GVL in the raffinate and formed a highly viscous mixture with an undesirable solid–liquid–liquid equilibrium.33 The presence of such a mixture complicated the handling process and subsequent analyses; therefore, a reliable mass balance could not be provided.
This recovery approach is definitely not suitable for industrial scale, especially not for continuous operation, where the sticky lignin precipitate would lead to scaling and clogging of the piping system. However, the reduction of the amount of water is economically essential. To achieve this goal, a substantial amount of lignin must be separated prior to the extraction step.
3.3.4. Extraction of the Spent Liquor after Partial Lignin and Water Removal (Recovery Scheme 6)
This recovery strategy enabled further reduction of CO2 consumption, as illustrated in Figure 6. Approximately 83% of the GVL could be collected in the extract and residue, while the amount of unextracted GVL in the raffinate was reduced to 5%. Lignin was recovered at a high rate (ca. 91%) and high purity in two fractions with different molecular weights (see the Supporting Information (SI Section 7)). In the distillation pretreatment, ∼10% GVL and ∼42% furanic compounds were removed, together with water, but this could be prevented by optimizing the distillation parameters. Otherwise, the distillate must be used in the lignin precipitation of the next batch of spent liquor, whereby the trapped GVL would be transferred into the next cycle, which could improve the GVL recovery rate. This possibility should be confirmed with a process simulation model.
Figure 6.

Flow diagram of the spent liquor extraction after partial lignin precipitation and water evaporation for GVL recovery (recovery scheme 6), showing the distribution of the main components into the product streams. The mass balance is based on 100 g of spent liquor.
Note that furanic compounds were prone to be extracted together with GVL (see Figures 4 and 5). The zeotropic behavior,27 together with the significant difference in boiling points of the components (207 °C for GVL, 161 °C for furfural, and 115 °C for 5-hydroxymethylfurfural), would facilitate the subsequent purification by vacuum distillation. This would be an advantage in comparison to the furfural recovery from an aqueous solution, which would require advanced approaches such as extractive distillation38 or reactive distillation.39
3.4. Assessment of the Proposed Recovery Schemes
From the six recovery schemes introduced in this paper, only recovery schemes 2, 4, and 6 demonstrated the potential to be further developed and integrated to the GVL pulping process. Generally, liquid CO2 extraction exhibited a distinct advantage over the vacuum distillation with regard to the formation of a sticky residue, which rendered a significant amount of GVL difficult to separate (comparing Figure 2 and Figure 6). Distillation is not a feasible method unless this specific limitation is resolved. The treatment of extraction raffinate containing dissolved lignin at low concentration theoretically demands less effort.
The energy intensity is a vital criterion for the selection of a recovery process. However, the instrumental limitations and lack of optimization, as previously discussed, prevented the adoption of the experimental data for assessing the energy consumption of the recovery scheme. To provide certain insights to the magnitude of energy consumption for the GVL recovery process, simulation models were constructed in the ASPEN PLUS v.10 environment, as presented in the Supporting Information (SI Section 8). The distillation process was modeled in accordance to the proposal in Section 3.2.3. The extraction was modeled as a three-stage extraction unit, followed by a four-stage decompression–recompression of CO2. For simplification, only the two main components—namely, GVL and water—were included in the simulation. The energy consumption presented in the Supporting Information (SI Section 8) was based on a 1 kg/s flow rate of liquid containing 50 wt % GVL and 50 wt % water. With the assumption of a pulping process similar to the one selected for the GVL recovery study in this paper (50 wt % GVL, L:W = 4 L/kg, 180 °C, 150 min), Table 3 summarizes the basic energy requirement for the solvent recovery processes, based on a unit mass of wood.
Table 3. Evaluation of the Energy Consumption of the GVL Recovery Processes Based on Vacuum Distillation and Liquid CO2 Extraction, Using the Simplified Simulation Modelsa.
| vacuum distillation | liquid CO2 extraction | |
|---|---|---|
| heating duty | 19.55 GJ/ton of wood | 0.68 GJ/ton of wood |
| cooling duty | –19.47 GJ/ton of wood | –0.49 GJ/ton of wood |
| CO2 compression | 0.00 GJ/ton of wood | 0.41 GJ/ton of wood |
See the Supporting Information (SI Section 8). The energy of mixing and vacuum pumping and efficiency were not considered.
From one metric ton of wood, ∼235 kg of lignin could be recovered (26.2% extracted to the spent liquor, from which 90% could be precipitated). With the calorific value of 21.8 MJ/kg,40 ∼5.1 GJ/ton of wood could be generated by lignin combustion.
The energy intensity of the vacuum distillation was 1 order of magnitude higher than the liquid CO2 extraction, as the recovered GVL had to be evaporated from the spent liquor. The heat released from the hot stream (the vapor entering the distillation column condenser, the water, and the GVL products) was of low quality and could only be utilized in preheating the spent liquor. Therefore, the process still consumed an immense amount of energy for the evaporation and distillation (∼17.5 GJ/ton of wood) and the lignin-derived energy was not sufficient for this recovery scheme. On the other hand, in the liquid CO2 extraction, the heat generated from the operation of the compressor can be directed to thermostatting the extractor and flash tanks. Part of the lignin could be combusted to cover the rest of the heating duty and to produce electricity for the compressor. The surplus lignin could be used as fuel for the pulping process. The preliminary assessment indicated that the GVL recovery process by liquid CO2 extraction could be energetically self-sustained, while the distillation required an external fuel source. Hence, the liquid extraction is a more sustainable alternative for the solvent recovery of a GVL biorefinery.
Similar to the determination of pulping parameters, the selection of a recovery scheme is not one-dimensional. Several factors must be taken into consideration, such as the biorefinery product portfolio, energy balance, solvent purity requirement, and pulping conditions. Generally, the precipitated lignin is preferably used as fuel to cover the process energy consumption, only the excess lignin is further processed into value-added products such as resins, composites, or aromatic compounds. For example, recovery scheme 6 might be preferred over recovery scheme 4 only if there was a surplus in lignin production, because the division into two lignin fractions with a distinct degree of polymerization would improve the selectivity of the subsequent chemical conversion processes.
3.5. Recyclability of GVL in a Study of Three Consecutive Fractionation Cycles
The performance of recycled GVL in successive wood fractionation steps was investigated (see the Supporting Information (SI Section 2)). The experiments were conducted on a small scale, whereby the sawdust was fractionated in a monowave reactor to maintain the controllability of reaction conditions and thereby achieve better reproducibility. The results summarized in Table 4 exhibit similar properties of the pulps obtained from the fractionation using either fresh (cycle 1) or recycled GVL (cycle 2 and 3). The nuclear magnetic resonance (NMR) spectra of recycled GVL (see the Supporting Information (SI Section 9)) exhibited no visible alteration of the chemical structure of the solvent. The extent of GVL extraction was consistent after three cycles with a GVL purity of >98.5%, which was confirmed by GC. There was a slow accumulation of furanic compounds after each cycle, because of their high affinity to GVL as previously discussed; however, such impurity did not yield any visible effect on the pulp quality. The separation of furanic compounds from the recycled GVL at such low concentration would be too costly. Therefore, the separation of the resulting furanic compounds by distillation would only be economically reasonable after a sufficient enrichment.
Table 4. Pulp Properties and Recycled GVL Purity after Three Fractionation Cyclesa.
| Pulp |
Recycled GVL |
|||||||
|---|---|---|---|---|---|---|---|---|
| cycle | yieldb (% odw) | cellulosec (% odp) | hemicellulosec (% odp) | ligninc (% odp) | GVL (wt %) | H2O (wt %) | furanics (wt %) | acidsd (wt %) |
| 1 | 50.37 | 86.44 | 8.00 | 5.57 | 98.62 | 1.21 | 0.10 | 0.06 |
| 2 | 51.03 | 86.19 | 7.99 | 5.82 | 98.51 | 1.26 | 0.16 | 0.07 |
| 3 | 50.67 | 86.84 | 7.73 | 5.43 | 98.72 | 1.08 | 0.20 | 0.00 |
Pulping conditions: E. globulus sawdust, 50 wt % GVL, 180°C, 120 min, L:W = 10 L/kg in a monowave reactor.
Percent on oven-dried wood.
Percent on oven-dried pulp.
Organic acids, including formic acid, acetic acid, and levulinic acid.
Analysis of the liquors obtained from the fractionation experiments (spent liquor and washing water) indicated a virtually quantitative GVL mass balance of ∼97%–98.5% (see the Supporting Information (SI Section 2)). The minor gap can be attributed to the handling losses or analysis error. However, the chemistry of GVL pulping has not been investigated; therefore, the possibility of GVL undergoing degradation reactions has not been excluded yet. In such case, the solvent loss could be compensated by the synthesis of GVL from furanic compounds via the levulinic acid intermediate in a GVL medium.37,41
4. Conclusion
This paper demonstrated various possibilities for optimizing the GVL/water fractionation process and the subsequent recovery processes based on lignin precipitation by water addition, distillation at reduced pressure, and liquid CO2 extraction. In the pulping phase, the ratio of liquor-to-wood was reduced to 3–4 L/kg, while the pulp quality was maintained, confirming the suitability of the process for further scaleup. A further reduction of the L:W ratio to 2.5 L/kg, comparable to acid sulfite pulping, might also be possible and should be investigated.
An overview of the proposed recovery schemes is given in Table 5. The combination of lignin precipitation and vacuum distillation was limited to 90% GVL recovery by the formation of a sticky residue, which was the GVL being trapped together with the remaining lignin. Advanced treatment of the resulting residue is necessary to make distillation a viable recovery process. The distillation of GVL was particularly energy-intensive, which shifted the favor toward a more sustainable technique: liquid CO2 extraction. In extraction, up to 87% of the GVL could be recovered in the extract, while ∼10% of the GVL remained in the raffinate, as the mass exchange was limited, because of the lack of an effective mixing mechanism. The extraction raffinate contained dissolved lignin at low concentration, which must be purified before further processing. Preliminary evaluation indicated that liquid CO2 extraction was a mild and energetically self-sufficient treatment. Moreover, the extractant CO2 could be quantitatively recycled by the multistage flashing–compressing cycle, which ensure the carbon neutrality of the process. With a green recovery process and the previously proven environmental advantages, the GVL-based fractionation process would potentially comply with the sustainability standards for a modern biorefinery.
Table 5. Comparison of the Six Proposed Recovery Schemes.
| description of proposed scheme | pros | cons |
|---|---|---|
| Recovery Scheme 1 | ||
| single-stage (lignin precipitation + vacuum distillation) | • simple | • low lignin recovery rate |
| • low GVL recovery rate | ||
| • energy- and time-consuming | ||
| • GVL trapped in sticky residue | ||
| • collection of GVL in the aqueous distillate | ||
| Recovery Scheme 2 | ||
| two-stage (lignin precipitation + vacuum distillation) | • high lignin recovery rate | • more time- and energy-consuming |
| • two recovered lignin fractions with distinctive molecular weight | • GVL trapped in sticky residue | |
| • more reasonable GVL recovery rate | • collection of GVL in the first-stage aqueous distillate | |
| Recovery Scheme 3 | ||
| lignin precipitation + liquid CO2 extraction | • less energy and time-consuming than distillation | • low extraction selectivity due to diluted feed |
| • high lignin recovery rate | • significant GVL remaining in raffinate due to limited mass transfer | |
| • two recovered lignin fractions with distinctive molecular weight | ||
| • furanics recovery in the extract (GVL) stream | ||
| Recovery Scheme 4 | ||
| liquid CO2 extraction | • simple | • significant GVL remaining in raffinate due to limited mass transfer |
| • better energy and time economy | • risk of clogging by lignin precipitation | |
| • higher extraction selectivity | ||
| • high lignin recovery rate | ||
| • furanics recovery in the extract (GVL) stream | ||
| Recovery Scheme 5 | ||
| vacuum distillation + liquid CO2 extraction | • clogging of extraction equipment due to uncontrollable lignin precipitation | |
| Recovery Scheme 6 | ||
| lignin precipitation + vacuum distillation + liquid CO2 extraction | • best extraction selectivity | • more time- and energy-consuming than recovery scheme 4 |
| • high lignin recovery rate | • collection of GVL in the aqueous distillate | |
| • two recovered lignin fractions with distinctive molecular weight | ||
The recovery schemes were presented in this paper rather as concepts, where the liquid CO2 extraction of the original spent liquor (recovery scheme 4) or the reconcentrated lignin-lean spent liquor (recovery scheme 6) was highlighted as a feasible method. Therefore, further optimization and techno-economic analysis on these two recovery schemes will be the subjects of future research. The introduction of a mixing mechanism in extraction could completely change the energy balance and motivate a revision regarding the introduced recovery schemes. The isolation and valorization of other minor substances such as furanic compounds, carbohydrates, and carboxylic acids, which have not been treated in this paper, is another topic for future studies. The recycling of the washing water must also be investigated.
Acknowledgments
Funding from Aalto University, School of Chemical Technology and Finnish Bioeconomy Cluster Oy (FIBIC) via the Advanced Cellulose to Novel Products (ACel) research program is gratefully acknowledged. The authors would like to thank Ms. Rita Hataka, Ms. Johanna Hakonen, and Ms. Heidi Meriö-Talvio for their support with the chromatographic analyses. The authors acknowledge the COST Association (Action No. FP1306) for supporting the dissemination of this work. This work was a part of the Academy of Finland's Flagship Programme under Project Nos. 318890 and 318891 (Competence Center for Materials Bioeconomy, FinnCERES).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.iecr.8b03723.
Temperature profiles of the GVL fractionation in oil-bath and air-bath reactors; experimental protocol for investigating the recyclability of GVL recovered from spent liquor by liquid CO2 extraction; experimental protocols for GVL recovery from spent liquor by vacuum distillation; experimental protocols for GVL recovery from spent liquor by liquid CO2 extraction; analyses of pulp, lignin, and liquids samples; mass balance (with Janson calculation): effect of time on fractionation of eucalyptus wood chips in 50 wt % GVL solution at 180 °C, with L:W = 3 or 4 L/kg in the oil-bath digester; molecular weight of lignin fractions isolated during GVL recovery processes; preliminary energy assessment for GVL recovery by vacuum distillation and liquid CO2 extraction; NMR analysis of GVL recycled by liquid CO2 extraction in the three-fractionation-cycle investigation (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Balan V.; Chiaramonti D.; Kumar S. Review of US and EU initiatives toward development, demonstration, and commercialization of lignocellulosic biofuels. Biofuels, Bioprod. Biorefin. 2013, 7, 732–759. 10.1002/bbb.1436. [DOI] [Google Scholar]
- Huber G. W.; Iborra S.; Corma A. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering. Chem. Rev. 2006, 106, 4044–4098. 10.1021/cr068360d. [DOI] [PubMed] [Google Scholar]
- Sjöström E.Wood Chemistry: Fundamentals and Applications, 2nd Edition; Academic Press: San Diego, CA, 1993. [Google Scholar]
- Iakovlev M.; Pääkkönen T.; van Heiningen A. Kinetics of SO2-Ethanol-Water pulping of spruce. Holzforschung 2009, 63, 779–784. 10.1515/HF.2009.109. [DOI] [Google Scholar]
- Håkansson H.; Germgård U.; Sens D. Influence of xylan on the degradability of laboratory kraft pulps from hardwood and reed canary grass in acid hydrolysis. Cellulose 2005, 12, 621–628. 10.1007/s10570-005-9011-6. [DOI] [Google Scholar]
- van Heiningen A. Converting a kraft pulp mill into an integrated forest biorefinery. Pulp Pap. Can. 2006, 107, 38–43. [Google Scholar]
- Mendes C. V. T.; Carvalho M. G. V. S.; Baptista C. M. S. G.; Rocha J. M. S.; Soares B. I. G.; Sousa G. D. A. Valorisation of hardwood hemicelluloses in the kraft pulping process by using an integrated biorefinery concept. Food Bioprod. Process. 2009, 87, 197–207. 10.1016/j.fbp.2009.06.004. [DOI] [Google Scholar]
- Leschinsky M.; Weber H. K.; Patt R.; Sixta H. Formation of insoluble components during autohydrolysis of Eucalyptus globulus. Lenzinger Ber. 2009, 87, 16–25. [Google Scholar]
- Sixta H.; Schild G. A new generation kraft process. Lenzinger Ber. 2009, 87, 26–37. [Google Scholar]
- Rydholm S. A.Pulping Chemistry; R. E. Krieger Publishing Company: Malabar, FL, 1985. [Google Scholar]
- Sixta H.Handbook of Pulp, Vol. 1; Wiley–VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; p 608. [Google Scholar]
- Sixta H.; Iakovlev M.; Testova L.; Roselli A.; Hummel M.; Borrega M.; van Heiningen A.; Froschauer C.; Schottenberger H. Novel concepts of dissolving pulp production. Cellulose 2013, 20, 1547–1561. 10.1007/s10570-013-9943-1. [DOI] [Google Scholar]
- Pye E. K.; Lora J. H. The Alcell process: A proven alternative to kraft pulping. Tappi J. 1991, 74, 113–118. [Google Scholar]
- Dapia S.; Santos V.; Parajo J. C. Formic Acid-Peroxyformic Acid Pulping of Fagus sylvatica. J. Wood Chem. Technol. 2000, 20, 395–413. 10.1080/02773810009351891. [DOI] [Google Scholar]
- Iakovlev M.; van Heiningen A.. SO2–Ethanol–Water (SEW) Fractionation of Lignocellulosics. In Proceedings of the 11th European Workshop on Lignocellulosics and Pulp, Aug. 16–19, 2010, Hamburg, Germany; p 70. [Google Scholar]
- Bozell J. J.; Black S. K.; Myers M.; Cahill D.; Miller W. P.; Park S. Solvent fractionation of renewable woody feedstocks: Organosolv generation of biorefinery process streams for the production of biobased chemicals. Biomass Bioenergy 2011, 35, 4197–4208. 10.1016/j.biombioe.2011.07.006. [DOI] [Google Scholar]
- Lê H. Q.; Ma Y.; Borrega M.; Sixta H. Wood biorefinery based on gamma-valerolactone/water fractionation. Green Chem. 2016, 18, 5466–5476. 10.1039/C6GC01692H. [DOI] [Google Scholar]
- Horvath I. T. Solvents from nature. Green Chem. 2008, 10, 1024–1028. 10.1039/b812804a. [DOI] [Google Scholar]
- Emel’yanenko V. N.; Kozlova S. A.; Verevkin S. P.; Roganov G. N. Vapour pressures and enthalpies of vapourization of a series of the γ-lactones. J. Chem. Thermodyn. 2008, 40, 911–916. 10.1016/j.jct.2008.02.002. [DOI] [Google Scholar]
- Horvath I. T.; Mehdi H.; Fabos V.; Boda L.; Mika L. T. Gamma-Valerolactone-a sustainable liquid for energy and carbon-based chemicals. Green Chem. 2008, 10, 238–242. 10.1039/B712863K. [DOI] [Google Scholar]
- Lê H. Q.; Dimic-Misic K.; Johansson L.; Maloney T.; Sixta H. Effect of lignin on the morphology and rheological properties of nanofibrillated cellulose produced from γ-valerolactone/water fractionation process. Cellulose 2018, 25, 179–194. 10.1007/s10570-017-1602-5. [DOI] [Google Scholar]
- Sklavounos E.; van Heiningen A.. Conditioning of SO2–Ethanol–Water Fractionation Liquor of Spruce for ABE Fermentation. In Proceedings of the 11th European Workshop on Lignocellulosics and Pulp, Aug. 16–19, 2010, Hamburg, Germany; pp 205–208. [Google Scholar]
- Viell J.; Harwardt A.; Seiler J.; Marquardt W. Is biomass fractionation by Organosolv-like processes economically viable? A conceptual design study. Bioresour. Technol. 2013, 150, 89–97. 10.1016/j.biortech.2013.09.078. [DOI] [PubMed] [Google Scholar]
- Muurinen E.; Sohlo J.. Heat Integration of a New Formic Acid Pulping and Recovery Process. In Energy Efficiency in Process Technology; Pilavachi P. A., Ed.; Springer: Dordrecht, The Netherlands, 1993; pp 996–1004. [Google Scholar]
- Luterbacher J. S.; Rand J. M.; Alonso D. M.; Han J.; Youngquist J. T.; Maravelias C. T.; Pfleger B. F.; Dumesic J. A. Nonenzymatic Sugar Production from Biomass Using Biomass-Derived γ-Valerolactone. Science 2014, 343, 277–280. 10.1126/science.1246748. [DOI] [PubMed] [Google Scholar]
- Shuai L.; Questell-Santiago Y.; Luterbacher J. S. A mild biomass pretreatment using γ-valerolactone for concentrated sugar production. Green Chem. 2016, 18, 937–943. 10.1039/C5GC02489G. [DOI] [Google Scholar]
- Pokki J.; Lê H. Q.; Petri U.; Sixta H.; Alopaeus V.. Isobaric vapor–liquid equilibrium of furfural + γ-valerolactone at 30 kPa and isothermal liquid–liquid equilibrium of carbon dioxide + γ-valerolactone + water at 298 K. submitted to J. Chem. Eng. Data, 2018. [Google Scholar]
- Theander O.; Nelson D. A.. Aqueous, high-temperature transformation of carbohydrates relative to utilization of biomass. In Advances in Carbohydrate Chemistry and Biochemistry; Tipson R. S., Horton D., Eds.; Harcourt Brace Jovanovich: San Diego, CA, 1988; pp 273–326. [Google Scholar]
- Girisuta B.; Janssen L. P. B. M.; Heeres H. J. Kinetic Study on the Acid-Catalyzed Hydrolysis of Cellulose to Levulinic Acid. Ind. Eng. Chem. Res. 2007, 46, 1696–1708. 10.1021/ie061186z. [DOI] [Google Scholar]
- Zaranyika M. F.; Madimu M. Heterogeneous dilute acid hydrolysis of cellulose: A kinetic model for the hydrolysis of the difficultly accessible portion of cellulose based on donnan’s theory of membrane equilibria. J. Polym. Sci., Part A: Polym. Chem. 1989, 27, 1863–1872. 10.1002/pola.1989.080270607. [DOI] [Google Scholar]
- Strunk P.; Lindgren Å; Eliasson B.; Agnemo R. Chemical changes of cellulose pulps in the processing to viscose dope. Cellul. Chem. Technol. 2012, 46, 559–569. [Google Scholar]
- Sixta H.; Michud A.; Hauru L.; Asaadi S.; Ma Y.; King A. W. T.; Kilpelainen I.; Hummel M. Ioncell-F: A High-strength regenerated cellulose fibre. Nord. Pulp Pap. Res. J. 2015, 30, 043–057. 10.3183/NPPRJ-2015-30-01-p043-057. [DOI] [Google Scholar]
- Lê H. Q.; Zaitseva A.; Pokki J.; Ståhl M.; Alopaeus V.; Sixta H. Solubility of Organosolv Lignin in Gamma-Valerolactone/Water Binary Mixtures. ChemSusChem 2016, 9, 2939–2947. 10.1002/cssc.201600655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jönsson A.; Nordin A.; Wallberg O. Concentration and purification of lignin in hardwood kraft pulping liquor by ultrafiltration and nanofiltration. Chem. Eng. Res. Des. 2008, 86, 1271–1280. 10.1016/j.cherd.2008.06.003. [DOI] [Google Scholar]
- Gütsch J. S.; Sixta H. Purification of Eucalyptus globulus water prehydrolyzates using the HiTAC process (high-temperature adsorption on activated charcoal). Holzforschung 2011, 65, 511–518. 10.1515/hf.2011.065. [DOI] [Google Scholar]
- Hashmi S. F.; Meriö-Talvio H.; Hakonen K. J.; Ruuttunen K.; Sixta H. Hydrothermolysis of organosolv lignin for the production of bio-oil rich in monoaromatic phenolic compounds. Fuel Process. Technol. 2017, 168, 74–83. 10.1016/j.fuproc.2017.09.005. [DOI] [Google Scholar]
- Alonso D. M.; Wettstein S. G.; Mellmer M. A.; Gurbuz E. I.; Dumesic J. A. Integrated conversion of hemicellulose and cellulose from lignocellulosic biomass. Energy Environ. Sci. 2013, 6, 76–80. 10.1039/C2EE23617F. [DOI] [Google Scholar]
- Buell C. K.; Boatright R. G. Furfural Extractive Distillation. Ind. Eng. Chem. 1947, 39, 695–705. 10.1021/ie50450a003. [DOI] [Google Scholar]
- Metkar P. S.; Till E. J.; Corbin D. R.; Pereira C. J.; Hutchenson K. W.; Sengupta S. K. Reactive distillation process for the production of furfural using solid acid catalysts. Green Chem. 2015, 17, 1453–1466. 10.1039/C4GC01912A. [DOI] [Google Scholar]
- Demirbaş A. Relationships between lignin contents and heating values of biomass. Energy Convers. Manage. 2001, 42, 183–188. 10.1016/S0196-8904(00)00050-9. [DOI] [Google Scholar]
- Qi L.; Horváth I. T. Catalytic Conversion of Fructose to γ-Valerolactone in γ-Valerolactone. ACS Catal. 2012, 2, 2247–2249. 10.1021/cs300428f. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.