

Abstract
Selenium (Se) deficiency brings about defects in the biosynthesis of several selenoproteins and has been associated with aberrant chondrogenesis. Selenocysteine (Sec) Insertion Sequence (SECIS) and SECIS binding protein 2 (SBP2) interaction is a very critical node for the metabolic balance between Se and selenoproteins. The Gpx1, Gpx4 and SelS have different binding affinities with SBP2 in cells. According to our results, both miR‐181a‐5p and SBP2 appeared to be selenium‐sensitive and regulated the expression of selenoproteins in C28/I2 cells under Se sufficient environment. However, they showed significantly opposite expression trend in Se deficiency rats cartilage and SeD C28/I2 cells. The SBP2 is a direct target gene of miR‐181a‐5p in C28/I2 cells as determined by reporter gene and off‐target experiments. And the miR‐181a‐5p could regulate SBP2 and the selenoproteins in C28/I2 cells. Depending upon the Se supply levels, C28/I2 cells were divided into three groups, that is normal Se, SeD and SeS, which underwent through a 7‐day Se deprivation process, then SBP2 was knocked‐down and overexpressed in all the groups. Moreover, the selected selenoproteins were down‐regulated in second‐generation low Se diet rat cartilage. The selenoproteins expression was decreased by Se deficiency which depended on the Selenium‐sensitive miR‐181a‐5p to participate and regulate SBP2 at post‐transcriptional level. It involves a series of antioxidant and ECM (extracellular matrix) genes, to overcome the ROS‐related stress for the protection of essential physiological functions and to maintain the balance between anabolism and catabolism of the cartilage.
Keywords: cartilage, miRNA‐181a‐5p, SBP2, selenoprotein
1. INTRODUCTION
Selenium (Se) is one of essential biological trace element in mammals and is incorporated into selenocysteine (Sec) that is 21st proteinogenic amino acid. Sec is an active‐site residue essential for catalytic activity of selenoproteins (Sel), named after the Se.1, 2, 3 Sel are involved in various biological processes, such as antioxidative stress, anti‐tumour and especially the development.3, 4 Sec is encoded into the Sel by the genetic code “UGA” that is commonly a termination codon in cells.5 Sel biosynthesis is regulated by several special cis‐trans elements and trans‐acting factors, such as selenocysteine insertion sequence (SECIS) and SECIS binding protein 2 (SECISBP2 or SBP2).6, 7 SECIS is located at 3′‐untranslated region (3′‐UTR) of Sel mRNA and can bind with SBP2. SBP2 functions to carry Sec‐tRNASec into ribosomal “A site” that recognizes the “UGA” as the codon for Sec during Sel synthesis.6, 7
Se deficiency leads to altered synthesis of several selenoproteins, affecting chondrogenesis.8 This results in epiphyseal plate abnormalities and articular cartilage degradation.9 Representatively, Kashin‐Beck disease (KBD), an endemic osteoarthropathy in Se deficient regions of China, is characterized by pathological chondronecrosis in the deep zones of articular cartilage and growth plates from different peripheral joints of the endemic children.10, 11, 12 Further, Se supplementation is able to protect the Se deficient rats from epiphysial growth plate abnormality and has been used in the prophylactics of KBD by unknown mechanisms.9, 13, 14
Intriguingly, osteo‐chondroprogenitor‐specific deletion of the selenocysteinyl tRNASec gene leads to the phenotypes similar to KBD, particularly showing chondronecrosis and abnormal skeletal development in mice.15 It is revealed that the termination codon “UGA” is subjected to in‐sufficient Sec‐tRNASec, because of which the inactive truncated selenoproteins are produced.16 Parallelly, the short inactive fragment of TrxR1 with a two‐amino acid‐truncated C‐terminal motif could make human lung carcinoma A549 cells die.17 However, it is poorly understood how Se regulates the selenoprotein biosynthesis in cartilage. Particularly, to find the first‐hand factor of Se‐sensitivity is of great importance to understand the mechanism. In addition, there is little known about the regulatory pathway mediated by SBP2 from Se deficiency to selenoprotein biosynthesis in cartilage.
On the other hand, it has been observed that Dicer‐deficient mice suffer from a severe deficiency in bone development with proliferating chondrocytes and expansion of the hypertrophic region in the limb bud,18, 19 suggesting that miRNAs regulate both proliferation and differentiation of growth plate chondrocytes. MiRNAs have been demonstrated to be associated with both cartilage homoeostasis and development, especially the cartilage‐specific miR‐140.20, 21 About 30 miRNAs are differently expressed during the development of rats femoral articular cartilage.22 For instance, miR‐337 could influence cartilage‐specific gene expression in chondrocytes.23 It would be very exhilarating if we could find a “selenium‐sensitive miRNA” to regulate the expression of SBP2 in cartilage.
In this study, miR‐181a‐5p, one of the differently expressed miRNAs during cartilage development,22 was predicted as a target of hSBP2with TargetScanHuman7.1. Coincidentally, it could repress the expression of Cyclin A2(CCAN2) and Aggrecan(ACAN) in chicken chondrocytes that may act as a negative feedback for cartilage homoeostasis.24 However, to understand the physiological roles of miR‐181a‐5p in cartilage, further investigation was required. GPx1, GPX4 and SELS were selected as selenium phenotype markers in cartilage or chondrocytes, as they not only are regulated in cartilage by Se deficiency and Se supply,9, 25 but also are the representatives of different subcellular localizations: cytoplasm, mitochondria and endoplasmic reticulum, respectively. Hence, the detailed regulatory relationship among “low‐selenium, miRNA, SBP2 and selenoproteins in cartilage” was investigated during this study.
2. MATERIALS AND METHODS
2.1. Rats
The inbred Dark Agouti (DA) rats9, 22, 23 were raised in a climate controlled environment, housed in polystyrene cages containing wood shavings and were fed standard rodent chow and water ad libitum, in the SPF animal house of the Department of Biochemistry and Molecular Biology, Xi'an Jiaotong University, Health Science Center. The DA rats originated from the Section of Medical Inflammation Research, Lund University, Sweden. The two generation low‐selenium rat model was established as described previously, including Se deficient diet group (SeD) and Se sufficient diet group (SeS).9 The experimenters were blinded to the Se condition while processing data and making exclusion decisions. All procedures were in accordance with the Animals (Scientific Procedures) Act, 1986 (UK) (amended 2013). All sections of this report adhere to the ARRIVE Guidelines for reporting animal research.26 A completed ARRIVE guidelines checklist is included in Checklist S.
2.2. Cell culture
C28/I2 cell line, human juvenile chondrocytes, were maintained in Dulbecco's modified Eagle's medium/F‐12 medium (DMEM/F12, Hyclone, USA) containing 10% foetal bovine serum (FBS, Gibico, USA) in a humidified incubator with 5% CO2 at 37°C. For Se deficient and sufficient experiments, C28/I2 cells were preconditioned with medium containing 1% FBS at least for 7 days resulting in Se depletion (Se deficient condition, SeD). Then, 50 nmol L−1 sodium selenite (Se, Sigma) supplementation was performed to rescue the Se level (Se sufficient condition, SeS).
2.3. Histological staining
Knee joints fixed with 4% paraformaldehyde (PFA) were tenderly decalcified in 10% EDTA liquid for 4 weeks. Subsequently paraffin‐embedded, dissected into 5‐μm‐thick pieces and stained to observe the morphological changes in the epiphyseal plate. All the sections were then stained with conventional haematoxylin and eosin (H&E), safranin O and fast green dyes.
2.4. Immunohistochemistry staining
After intrinsic peroxidase activity, the articular cartilage sections were blocked with 3% hydrogen peroxide (H2O2) and then incubated with 1.5% BSA for 1 hour. The tissue sections were covered with the antibodies against SBP2 (12798‐1‐AP, 1:250 dilution), GPX1 (3120‐1, 1:250 dilution), GPX4 (14432‐1‐AP, 1:500 dilution) and SELS (15591‐1‐AP, 1:500 dilution), respectively, which were purchased from Proteintech (Wuhan, China). The samples were incubated at 4°C overnight on a wet box. The sections were rinsed with PBS. Sequentially, they were incubated with biotinylated secondary antibody for 1 hour and DAB reagent (Boster, Wuhan, China) for 5 minute at room temperature. Chromogenic reactions were terminated once claybank regions were observed under the microscope. Rabbit IgG was used as a negative control.
2.5. Luciferase reporter assays
Among hundreds of target genes predicted by the TargetScanHuman7.1 (http://www.targetscan.org/) search programs, hSBP2 was of particular interest (Figure 2A). For luciferase reporter experiments, a 350‐bp DNA segment transcribed from the part of hSBP2 mRNA 3′‐UTR was amplified by PCR from human cDNA and inserted into the psi‐CHECK‐2 Vector (Promega, Fitchburg, USA) between the Xho I and Not I (Fermentas, Canada) site. The information of mature miR‐181a‐5p and stem‐loop miR‐181a is depicted in Tables S1 and S2, respectively.
The primer sequences to generate wild type hSBP2 3′‐UTR fragment (WT group) are as follows: forward primer: 5′‐atactcgagCAGGGAAAGGGCCCTTT‐3′ and reverse primer: 5′‐acagcggccgcTTTCACCAGAGTCTGAAA‐3′. The mutant hSBP2 3′‐UTR DNA fragment sized 350 bp was chemically synthesized with binding sequence completely mutated from TGAATGT to ACTCGTA (Figure 2A) and used the same primers with WT group to be amplified. The insert segment was confirmed by sequencing (data not shown).
HEK‐293T cells were seeded in 48‐well plates at 70% density and co‐transfected with 0.3 μg hSBP2 3′‐UTR/hSBP2 3′‐UTR‐mutant plasmid and 50 nmol L−1 mimic‐miR‐181a‐5p or 200 nmol L−1 inhibitor‐miR‐181a‐5p (Genepharma, Shanghai, China) in each well, using Lipofectamine 2000 (Invitrogen, Carlsbad, USA). Luciferase assays were performed 48 hour after transfection by using the Dual‐Luciferase® Reporter 1000 Assay System (Promega, Fitchburg, USA) on a multiskan spectrum Luminoskan ascent 392 (Thermo, Waltham, USA).
2.6. Cell transfection
Full‐length human hSBP2 cDNA was amplified and subcloned into the pHBAd‐MCMV‐GFP expression vector. Null vectors (without exogenously inserted DNA fragment) were used as the controls. The pHBAd‐MCMV‐GFP‐SBP2(Ad‐SBP2), a recombinant adenoviruses containing hSBP2‐CDS segment, and control (Ad‐GFP) were packaged by Hanbio Biotechnology Inc. (Hanbio, Shanghai, China).
The siRNA of hSBP2 (si‐SBP2) and control siRNA sequences was purchased from Genepharma Biotechnology Inc. (Genepharma, Shanghai, China). Cell transfection was performed according to the manufacturer's instructions. To obtain gene overexpression, the recombinant adenoviruses were used as 300 MOI of final viral titre and added to the cells for incubation. For gene knockdown, the best fit siRNA of hSBP2, hSBP2‐siRNA2527: GAGCCACACUACAUUGAAAtt, was transduced into the cells according to the manufacturer's instructions.
2.7. Total RNA extraction and quantitative PCR analysis
Total RNA was isolated from rats articular cartilage or C28/I2 cell samples using the TRIzol® method (Invitrogen, USA). Two microgram of each total RNA from articular cartilage was timely reversed to cDNA according to the manufacturer's instructions (RevertAid™; Fermentas, Canada) and stored at −20°C until used. In addition, miRNA‐cDNA was obtained using One Step PrimeScript® miRNA‐cDNA Synthesis Kit (Takara, Japan). Because the cartilage were limited and contained very less RNA in one sample, it could not satisfy the test of every gene.
The mRNA or miRNA expression was tested by real‐time quantitative PCR (RT‐qPCR), which was performed on iQ5 real‐time PCR detection system (Bio‐Rad, Hercules, CA, USA) with SYBR® Premix Ex Taq™ II (TaKaRa, Japan). The relative gene expression was normalized by GAPDH, while let‐7a was used as the internal reference of miR‐181a‐5p. The procedure of miRNA‐cDNA qPCR was two‐step amplification:pre‐degeneration 95°C 10 second; PCR amplification: 95°C 5 second, 60°C 20 second, 40 cycles. The information of primers is depicted in Tables 1, 2, 3.
Table 1.
Information of miRNA‐181a‐5p for real‐time PCR
| MicroRNAs | Accession No. | Forward primer |
|---|---|---|
| hsa‐miRNA‐181a | MIMAT0000858 | 5′‐CGCAACATTCAACGCTGTC‐3′ |
| hsa‐let‐7a | MIMAT0000774 | 5′‐CGCTGAGGTAGTAGGTTGT‐3′ |
| Reverse primer: 5′‐ GTGCAGGGTCCGAGGT‐3′ | ||
Table 2.
Information of rat primers for real‐time PCR
| Gene | Sequence (5′‐3′) | Product size (bp) | Annealing temperature (°C) |
|---|---|---|---|
| Sbp2 | Forward: AAGGCACCCTAGCTTCCACT | 116 | 60 |
| Reverse: GGTTGATGCCTCCAGTGAGT | |||
| GPx1 | Forward: GGCTCACCCGCTCTTTACCTTC | 168 | 62 |
| Reverse: CGCACTGGAACACCGTCTGG | |||
| GPx4 | Forward: AATTCGCAGCCAAGGACATCG | 168 | 62 |
| Reverse: CCAGGATTCGTAAACCACACTCG | |||
| SelS | Forward: TTCCTGCACGTCACAGTGGG | 115 | 60 |
| Reverse: TTAAAGCCCTCAGTCGCAGG | |||
| GAPDH | Forward: CGGCAAGTTCAACGGCACAG | 148 | 60 |
| Reverse: GAAGACGCCAGTAGACTCCACGAC |
Table 3.
Information of human primers for real‐time PCR
| Gene | Sequence (5′‐3′) | Productsize (bp) | Annealing temperature (°C) |
|---|---|---|---|
| SBP2 | Forward: CCGCAGATTCAGGGATTACT | 92 | 60 |
| Reverse: CTTGGAAACGGACCAGTTCT | |||
| GPx1 | Forward: AAGCTCATCACCTGGTCTCC | 124 | 60 |
| Reverse: CGATGTCAATGGTCTGGAAG | |||
| GPx4 | Forward: GCTGTGGAAGTGGATGAAGA | 105 | 60 |
| Reverse: TGAGGAACTGTGGAGAGACG | |||
| SelS | Forward: CACCTATGGCTGGTACATCG | 130 | 60 |
| Reverse: AACATCAGGTTCCACAGCAG | |||
| GAPDH | Forward: CACCCACTCCTCCACCTTTG | 110 | 64 |
| Reverse: CCACCACCCTGTTGCTGTAG |
2.8. Western blotting
Total protein were isolated and analysed by Western blotting. Protein samples from rats articular cartilage or C28/I2 cell line (10‐20 μg) were separated on 10% SDS‐PAGE and transferred to PVDF membranes (EMD Millipore, Darmstadt, Germany). After blocked with 3% non‐fat milk in TBST buffer, membrane was incubated with primary antibodies followed by the secondary antibodies conjugated with horseradish peroxidase (HRP), and visualized using the ECL detection system (EMD Millipore, Darmstadt, Germany) on a chemiluminescence imaging system. The antibodies against SBP2 (12798‐1‐AP, 1:500 dilution), GPX1 (3120‐1, 1:2000 dilution), GPX4 (14432‐1‐AP, 1:500 dilution) and SELS (15591‐1‐AP, 1:1000 dilution) were from Epitomics (CA, USA). Antibody against β‐actin (1:2000 dilution), the secondary horseradish peroxidase‐coupled anti‐rabbit (1:5000 dilution) and anti‐mouse (1:5000 dilution) antibodies were purchased from Byotime Biotech (Jiangsu, China).
2.9. Statistical analysis
Data were presented as means ± SEM. The statistical significance of pathological data was calculated by Mann‐Whitney U test. Means of two groups were compared using Student's t test, and statistical significance was considered as P < 0.05 in all tests (*P < 0.05, **P < 0.01and **P < 0.001). All analyses were performed using the software GraphPad Prism 6.0 (GraphPad Software, San Diego, CA, USA).
3. RESULTS
3.1. Both miR‐181a‐5p and SBP2 are selenium‐sensitive in cartilage and chondrocytes
Firstly, the expressions of miR‐181a‐5p, Sbp2 and SBP2 were detected in articular cartilage of the low Se diet rat model.9 In SeD rat cartilage, miR‐181a‐5p showed remarkable up‐regulation as compared to the SeS group (P = 0.0006, Figure 1A). Conversely, the expression of SBP2 was significantly reduced in SeD group compared with SeS group, both at mRNA and protein levels (P = 0.0066, Figure 1B and C). It seemed to be in line with our expectation that low Se could result in Sbp2 down‐regulation in cartilage, with miR‐181a‐5p up‐regulated. In other words, SBP2 was significantly down‐regulated in SeD groups while up‐regulated in SeS groups regardless of interferring or overexpressing Sbp2 and SBP2 (Figure 1A‐C).
Figure 1.
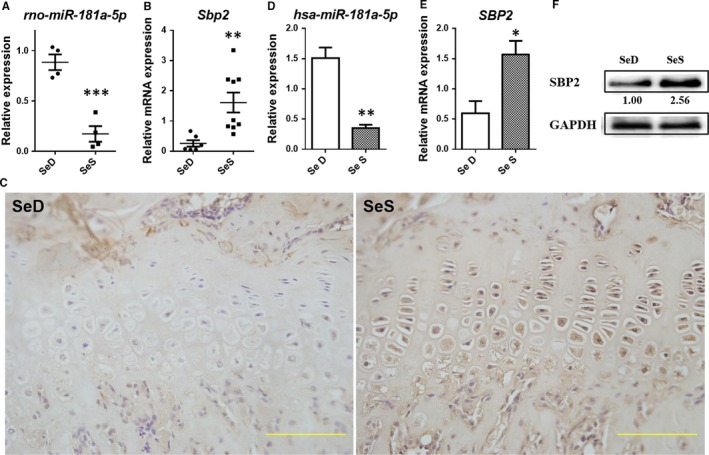
Both miR‐181a‐5pand SBP2 are Se‐sensitive. (A) rno‐miRNA‐181a‐5p expression in low Se rat cartilage (n = 4, 4). (B) Sbp2 expression in low Se rat cartilage (n = 6, 9). (C) SBP2 expression in low Se rat cartilage (bar: 50 μm). (D) hsa‐miRNA‐181a‐5p expression in C28/I2 chondrocytes under Se deficient or Se replenished condition. (E) SBP2 expression in C28/I2 chondrocytes under Se deficient or Se replenished condition. (F) SBP2 expression in C28/I2 chondrocytes under Se deficient or Se replenished condition. Data were presented as means ± SEM. *, ** and *** stand for P < 0.05, 0.01 and 0.001, respectively, between SeS and SeD groups
Further, a seven‐day‐term low medium cell culture was performed as described previously to induce the low Se condition in vitro.25 MiR‐181a‐5p expressed at significantly elevated levels in SeD group compared with Se supplement cultured C28/I2 cells (P = 0.0035, Figure 1D), indicating that miR‐181a‐5p was Se susceptive in chondrocytes, which was in line with the results obtained from the rat model, presented above. Meanwhile, SBP2and SBP2 were significantly down‐regulated, contrarily (P = 0.0335, Figure 1E and F). It is evident from the above results that both miR‐181a‐5p and SBP2 are selenium‐sensitive and are regulated by Se supplement.
3.2. SBP2 is a direct target of miR‐181a‐5p in C28/I2 cells
Subsequently dual‐luciferase reporter gene analysis showed that, the relative activity of Rluc/Fluc of SBP2 WT group was reduced about 37%, compared with the control group, prompting that hsa‐miR‐181a‐5p could bind with SBP2‐3′‐UTR so as to affect Rluc gene expression. Compared with WT group, the relative activity of Rluc/Fluc of SBP2 mutant group Rluc/Fluc was increased about 30%. Furthermore, there was no significant difference between mutant group and control group, suggesting that hsa‐mir‐181a‐5 p could not bind with SBP2‐3′‐UTR because of the mutation in the predicted binding sites (P < 0.0001, Figure 2B).
Figure 2.
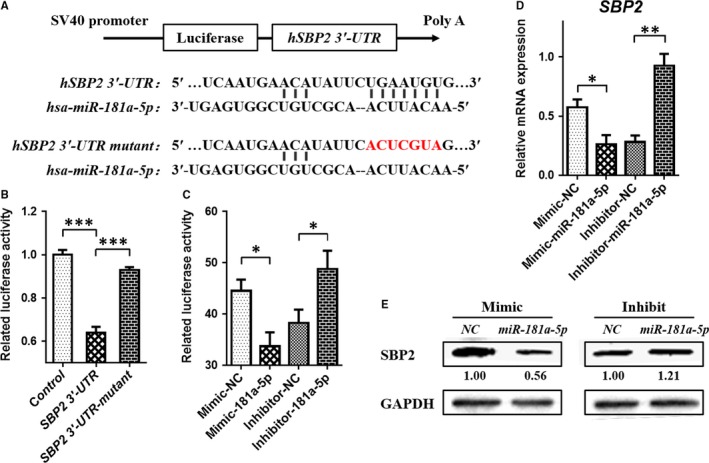
There is a target relationship between miRNA‐181a‐5p and SECISBP2. (A) A Schematic diagram of hsa‐miRNA‐181a‐5p binding site with SBP2 3’‐UTR. (B) Results of dual‐luciferase reporter gene analysis with wild or mutant SBP2 3’‐UTR. (C) Results of dual‐luciferase reporter gene analysis with mimic or inhibitor transfected in C28/I2 chondrocytes. (D) mRNA expression of SBP2 in C28/I2 cell line transfected with mimic or inhibitor of hsa‐miRNA‐181a‐5p. (E) Protein expression of SBP2 in C28/I2 cell line transfected with mimic or inhibitor of hsa‐miRNA‐181a‐5p. Data were presented as means ± SEM. *, ** and *** stand for P < 0.05, 0.01 and 0.001, respectively
Meanwhile, miR‐24‐3p did not target the SBP2‐3′‐UTR in C28/I2 cells although it was predicted. (Figure S1).
Then, mimic‐miR‐181a‐5p and inhibitor‐miR‐181a‐5p were used to alter the level of miR‐181a‐5p, and the relative activity of dual‐luciferase reporter gene and SBP2 expression were detected. Mimic‐miR‐181a‐5p could down‐regulate the dual‐luciferase activity (P = 0.0147), while the inhibitor‐miR‐181a‐5p could up‐regulate it significantly (P = 0.0440) (Figure 2C), in accord with the changes of SBP2 mRNA (P = 0.0364, P = 0.0048, Figure 2D) and protein (Figure 2E) levels. These results suggest that hsa‐miR‐181a‐5p can specifically bind with the SBP2 mRNA 3′‐UTR. In other words, SBP2 is a direct target gene of miR‐181a‐5p.
3.3. Both miR‐181a‐5p and SBP2 could regulate the biosynthesis of three selenoproteins in chondrocytes
The mimic or inhibitor of miR‐181a‐5p was used in normal C28/I2 cells to check the expression of SBP2 and the three selected selenoproteins. After mimic‐miR‐181a‐5p transfection, Western blotting results showed that SBP2, GPx1 and SELS were significantly down‐regulated, while there was no change in GPX4 levels (Figure 3A left). On the other hand, inhibitor‐miR‐181a‐5p resulted in the up‐regulation of SBP2, GPx1 and SELS proteins levels except the GPX4 (Figure 3A right).
Figure 3.
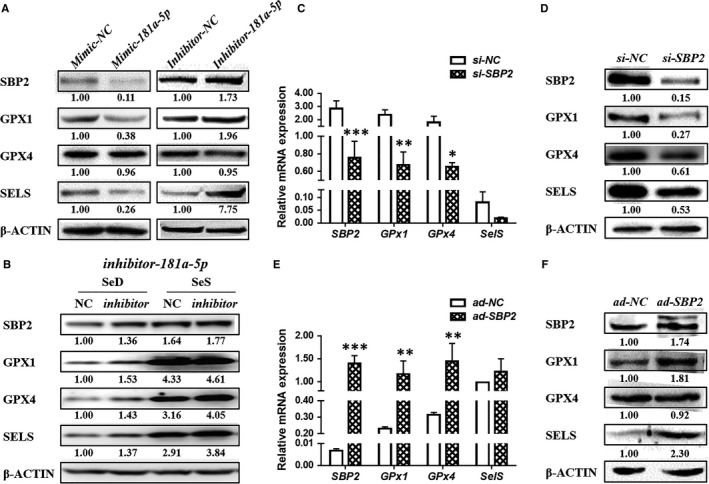
Both miR‐181a‐5pand SBP2 regulate the expression of 3 selenoproteins in chondrocytes. (A) Left: The protein expressions of SBP2 and three selenoproteins in C28/I2 cell line transfected with mimic of hsa‐miRNA‐181a‐5p. Right: The protein expressions of SBP2 and three selenoproteins in C28/I2 cell line transfected with inhibitor of hsa‐miRNA‐181a‐5p.(B) The protein expressions of SBP2 and selenoproteins after inhibitor‐181a‐5p infection in Se deficient or Se replenished C28/I2 chondrocytes.(C) The protein expressions of SBP2 and 3 selenoproteins in C28 cells transfected with siRNA‐SBP2. (D) The mRNA expressions of SBP2 and 3 selenoproteins in C28 cells transfected with siRNA‐SBP2.(E) The protein expressions of SBP2 and 3 selenoproteins in C28 cells infected with Ad‐SBP2 plasmid.(F) The mRNA expressions of SBP2 and 3 selenoproteins in C28 cells infected with Ad‐SBP2 plasmid. Data were presented as means ± SEM. *, ** and *** stand for P < 0.05, 0.01 and 0.001, respectively.
Moreover, after miR‐181a‐5p inhibition in Se deficiency and Se supplemented chondrocytes, both the groups showed an elevation in the expression of SBP2. And, the three selenproteins were also increased in the inhibitor groups compared to the respective control groups.(Figure 3B).
To assess the role of SBP2, regulating the selenoproteins in chondrocyte, we detected the expression of GPx1, GPX4 and SELS in human C28/I2 cells with knockdown or overexpression of SBP2, at RNA and protein levels, respectively. When SBP2 (P = 0.0002) level was reduced by specific siRNA, mRNA levels of Gpx1 (P = 0.0014) andGPX4 (P = 0.0201) were down‐regulated significantly except SELS (P = 0.8900) (Figure 3C); however, at protein level, all the three selenoproteins were significantly down‐regulated (Figure 3D). On the other hand, SBP2 (P = 0.0002) overexpression in C28/I2 cells could elevate the mRNA expression of both Gpx1 (P = 0.0059) and GPX4 (P = 0.0014) but could not change SELS mRNA levels ( 3E). At the protein level, GPX1 and SELS had the obvious response to the SBP2 overexpression, whereas GPX4 showed no evident changes (Figure 3F). These findings suggest that SBP2 can affect the pivotal selenoproteins expression in chondrocyte.
3.4. SBP2 regulates selenoproteins in different Se conditions
In order to further explore the role of SBP2 in cartilage, the knockdown or overexpression of SBP2 was performed in C28/I2 cells by si‐SBP2 or ad‐SBP2, respectively. Under the Se deficient or Se replenished situations, the chondrocytes were treated with si‐SBP2 or Ad‐SBP2, respectively, in order to observe the expression of the above three selected selenoproteins.
In SeD groups of si‐SBP2, the expression of SELS was very low, even GPX1 and GPX4 were difficult to detect, no matter transfected with si‐SBP2 or not. All concerned proteins could be visibly detected with Se supplement, showing a reduced tendency in the si‐SBP2 chondrocytes (Figure 4A).
Figure 4.
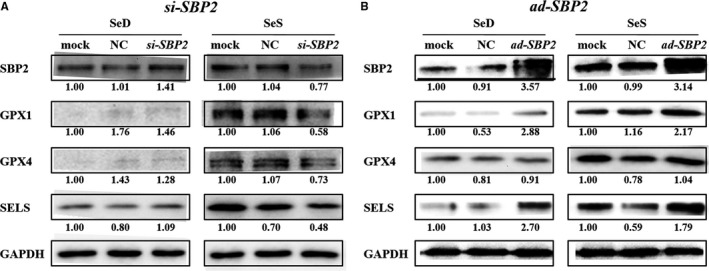
Selenoproteins expression in C28 cells after si‐RNA SBP2 transfection. (A) The protein expressions of SBP2 and selenoproteins after si‐SBP2 transfection in Se deficient or Se replenished C28/I2 chondrocytes. (B) The protein expressions of SBP2 and selenoproteins after Ad‐SBP2 plasmid infection in Se deficient or Se replenished C28/I2 chondrocytes
In the SBP2 overexpression chondrocytes, both SeD and SeS groups showed a remarkable elevation in the expression of SBP2. Interestingly, the GPX1, GPX4 and SELS were robustly up‐regulated compared with control group despite selenium deficient conditions. After the selenium was replenished in to the selenium deficient chondrocytes, the selected selenoproteins expression was rescued significantly. On the other hand, GPX4 showed a little response to the SBP2 overexpression in selenium deficient or supplement chondrcocytes (Figure 4B).
3.5. The selected selenoproteins are down‐regulated in cartilage of low Se diet rat
In the low Se diet rat model, expression of GPx1, Gpx4 and SelS was determined by RT‐qPCR and IHC methods. In the epiphyseal plate of low Se DA rats cartilage (SeD group), mRNA expression of Gpx1 (P = 0.0449, Figure 5A) and Gpx4 (P = 0.0154, Figure 5C) was significantly higher than the control (SeS group) group, while SelS showed no considerable change in the expression at mRNA level (P = 0.9489, Figure 5E). However, at the protein level, GPX1 (Figure 5B), GPX4 (Figure 5D) and SELS (Figure 5F) were all significantly down‐regulated, as determined by IHC. These results suggested that GPx1, GPX4 and SelS participate in the low Se induced cartilage lesions.
Figure 5.
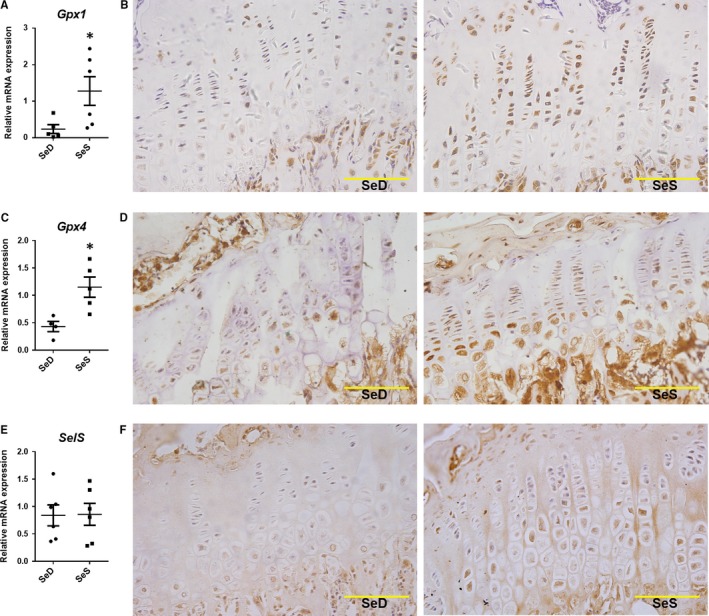
Expression of three selenoproteins in low Se DA rats cartilage model. (A) GPx1 mRNA expression in low Se rats cartilage(n = 6, 5).(B) IHC results of GPx1 protein expression in low Se rats cartilage(bar: 50 μm).(C) GPx4 mRNA expression in low Se rats cartilage (n = 4, 5).(D) IHC results of GPx4 protein expression in low Se rats cartilage(bar: 50 μm).(E) Sel S mRNA expression in low Se rats cartilage (n = 6, 6).(F) IHC results of Sel S protein expression in low Se rats cartilage(bar: 50 μm). Data were presented as means ± SEM. * stand for P < 0.05, respectively, between SeS and SeD groups
4. DISCUSSION
To investigate whether miR‐181a‐5p is involved in cartilage injury process induced by Se deficiency, we established the Se deficiency and Se sufficiency models using C28/I2 human chondrocytes and inbred DA rats.9 The phenotypes of the two generation Se deficiency rats included extracellular matrix (ECM) metabolism, aberration of articular cartilage, epiphyseal dysplasia, delayed skeletal development and the secondary osteoarthritis.9, 27 These phenotypes match exactly with KBD for its Se‐deficient nutritional status.10, 11, 12 Moreover, the main features of epiphyseal dysplasia are the Collagen2α1(COL2α1) and ACAN metabolic imbalances and chondrocytes apoptosis interrelated to abnormal proliferation and differentiation in the Se deficient diet fed rats,9, 27 similar to the symptoms of KBD patients.10, 11, 12 Conformably, miR‐181a‐5p down‐regulated the ACAN in chondrocytes through the unknown mechanism.24 Predictably, miR‐181a‐5p was up‐regulated in Se deficiency both in vivo and in vitro. The expression of miR‐181a‐5p showed a decline with Se deficiency, suggesting that miR‐181a‐5p is susceptive to Se supply in cartilage.
Next, we used mimic and inhibitor sequences of miR‐181a‐5p to up‐ or down‐regulate its expression in C28/I2 cells. The Sbp2/SBP2 showed a significant negative correlation with the miR‐181a‐5p. Further, we proved that SBP2 is a new, direct target of miR‐181a‐5p in chondrocytes. These results establish that Selenium‐sensitive miR‐181a‐5p can regulate the expression of three selected selenoproteins GPx1, GPX4 and SELS to a variable extent through its target gene SBP2 in cartilage.
Meanwhile, the three selenoproteins were out‐of‐step regulated by miR‐181a‐5p mediated by SBP2, because their SECIS had the different affinity and binding efficiency with SBP2,6, 7, 28, 29, 30, 31 and SBP2 affects the expression of selenoproteins at mRNA levels.25, 28, 29, 30 Unexpectedly, GPX4 would not be regulated by miR‐181a‐5p and SBP2 after their knockdown and overexpression in C28/I2 cells. Several mechanisms, such as activated NF‐Y and Sp1, were involved in transcriptional and post‐transcriptional regulation of GPX4.32, 33 Further, its expression is augmented through post‐transcriptional modification by Grsf1 (guanine‐rich sequence‐binding factor 1) which is the Gpx4 mRNA‐binding protein.34 On contrary, the Gpx4expression is decreased after Grsf1 knockdown in mice and leads to developmental retardation of the brain which is similar to the Gpx4+/− mice.34, 35 It is implied that there are multiple modulators and a very complicated regulatory mechanism from Se supplementation to selenoproteins expression.
On the other hand, being the crucial antioxidant enzymes in vivo, both Gpx1 and Gpx4 were up‐regulated in cartilage tissue of the second‐generation Se deficient rats, after they were given the Se sufficient diet, and the two selenoproteins were up‐regulated at post‐transcriptional and translational levels.25 Perceptibly, it might be necessary for GPX1 and GPX4 to simultaneously participate in chondrogenesis, even though we cannot exclude the possibility that their decreased expression, that might influence the other selenoproteins. Hence, Se status may lead to the better utilization of selenoproteins to maintain cartilage homoeostasis, and it may be partly involved in selenoproteins’ function during cartilage formation and degeneration. Our IHC results supported the idea during this study.
Finally, we have discovered a novel pathway of Se‐sensitive miR‐181a‐5p regulated selenoproteins in cartilage. Selenium can mediate miR‐181a‐5p expression, and miR‐181a‐5p then regulates SBP2, resulting in the altered expression of pivotal selenoproteins such as GPx1, GPX4 and SELS, which further play complex roles in the cartilage (Figure 6).
Figure 6.
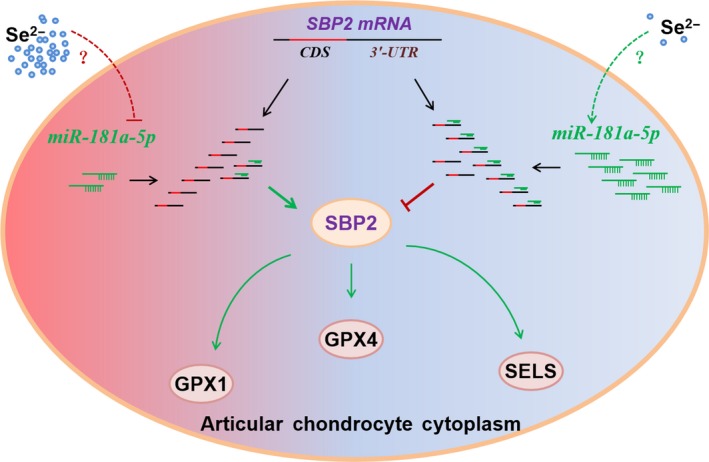
The illustration of possible pathways about selenoproteins regulation by miRNA‐181a‐5p in chondrocytes. Selenium may mediate miRNA‐181a‐5p expression may regulate SBP2, resulting in the altered expression of pivotal selenoproteins GPx1, GPx4 and Sel S, which may further play complex roles in cartilage
In conclusion, the three selenoproteins expression was decreased by Se deficiency which depended on the participation of miR‐181a‐5p to down‐regulate SBP2 at post‐transcriptional level. It involves a series of antioxidant and ECM genes, to overcome the ROS‐related stress for the protection of essential physiological functions and to maintain the balance between anabolism and catabolism of the cartilage. Thus, Se deficiency, the risk factor condition, gives rise to epiphyseal dysplasia in DA rats, similar to KBD patients.
Overall, this study provides the first comprehensive evidence that “Selenium → miR‐181a‐5p → SBP2 → selenoproteins” phenomenon exists in cartilage. Therefore, our data suggest that miR‐181a‐5p, SBP2 and three pivotal selenoproteins can be used to develop novel diagnostic and therapeutic strategies for cartilage diseases.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
Supporting information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
This work was supported by grants from the National Natural Science Foundation of China (Project no. 81371986, 81772410, 81301598), the Shannxi Province Natural Science Foundation (Project no. 2018JM3022, S2016YFJM1171), the Fundamental Research Funds for the Central Universities (xjj2015073).
Jian Sun and Shemin Lu conceived and supervised the study. Zixin Min and Jian Sun finished the most of experiments, analyses and interpretation of the data and wrote the original manuscript. Yuanxu Guo, Mengyao Sun, Safdar Hussain, Yitong Zhao, Dongxian Guo, Huang Huang, Lisong Heng, Fujun Zhang, Qilan Ning, Yan Han, Peng Xu and Nannan Zhong helped the experiments. Jian Sun and Shemin Lu obtained the fundings, critically revised the article for important intellectual content and take responsibility for the integrity of the work as a whole.
The C28/I2 cell line is provided by Prof. Junling Cao, from Institute of endemic diseases, Xi'an Jiaotong University Health Science Center.
Min Z, Guo Y, Sun M, et al. Selenium‐sensitive miRNA‐181a‐5p targeting SBP2 regulates selenoproteins expression in cartilage. J Cell Mol Med. 2018;22:5888–5898. 10.1111/jcmm.13858
Zixin Min and Yuanxu Guo contributed equally to this work.
Contributor Information
Jian Sun, Email: sunjian1@xjtu.edu.cn.
Shemin Lu, Email: lushemin@xjtu.edu.cn.
References
- 1. Kryukov GV, Castellano S, Novoselov SV, et al. Characterization of mammalian selenoproteomes. Science. 2003;300:1439‐1443. [DOI] [PubMed] [Google Scholar]
- 2. Lu J, Holmgren A. Selenoproteins. The Journal of biological chemistry. 2009;284:723‐727. [DOI] [PubMed] [Google Scholar]
- 3. Labunskyy VM, Hatfield DL, Gladyshev VN. Selenoproteins: molecular pathways and physiological roles. Physiol Rev. 2014;94:739‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kohrle J, Jakob F, Contempre B, Dumont JE. Selenium, the thyroid, and the endocrine system. Endocr Rev. 2005;26:944‐984. [DOI] [PubMed] [Google Scholar]
- 5. Driscoll DM, Copeland PR. Mechanism and regulation of selenoprotein synthesis. Annu Rev Nutr. 2003;23:17‐40. [DOI] [PubMed] [Google Scholar]
- 6. Takeuchi A, Schmitt D, Chapple C, et al. A short motif in Drosophila SECIS Binding Protein 2 provides differential binding affinity to SECIS RNA hairpins. Nucleic Acids Res. 2009;37:2126‐2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Donovan J, Caban K, Ranaweera R, Gonzalez‐Flores JN, Copeland PR. A novel protein domain induces high affinity selenocysteine insertion sequence binding and elongation factor recruitment. The Journal of biological chemistry. 2008;283:35129‐35139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zou K, Liu G, Wu T, Du L. Selenium for preventing Kashin‐Beck osteoarthropathy in children: a meta‐analysis. Osteoarthritis Cartilage. 2009;17:144‐151. [DOI] [PubMed] [Google Scholar]
- 9. Min Z, Zhao W, Zhong N, et al. Abnormality of epiphyseal plate induced by selenium deficiency diet in two generation DA rats. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica. 2015;123:697‐705. [DOI] [PubMed] [Google Scholar]
- 10. Diseases Stone R . A medical mystery in middle China. Science. 2009;324:1378‐1381. [DOI] [PubMed] [Google Scholar]
- 11. Cao J, Li S, Shi Z, et al. Articular cartilage metabolism in patients with Kashin‐Beck Disease: an endemic osteoarthropathy in China. Osteoarthritis Cartilage. 2008;16:680‐688. [DOI] [PubMed] [Google Scholar]
- 12. Duan C, Guo X, Zhang XD, et al. Comparative analysis of gene expression profiles between primary knee osteoarthritis and an osteoarthritis endemic to Northwestern China. Kashin‐Beck disease. Arthritis and rheumatism. 2010;62:771‐780. [DOI] [PubMed] [Google Scholar]
- 13. Moreno‐Reyes R, Mathieu F, Boelaert M, et al. Selenium and iodine supplementation of rural Tibetan children affected by Kashin‐Beck osteoarthropathy. The American journal of clinical nutrition. 2003;78:137‐144. [DOI] [PubMed] [Google Scholar]
- 14. Jirong Y, Huiyun P, Zhongzhe Y, et al. Sodium selenite for treatment of Kashin‐Beck disease in children: a systematic review of randomised controlled trials. Osteoarthritis Cartilage. 2012;20:605‐613. [DOI] [PubMed] [Google Scholar]
- 15. Downey CM, Horton CR, Carlson BA, et al. Osteo‐chondroprogenitor‐specific deletion of the selenocysteine tRNA gene, Trsp, leads to chondronecrosis and abnormal skeletal development: a putative model for Kashin‐Beck disease. PLoS Genet. 2009;5:e1000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Papp LV, Wang J, Kennedy D, et al. Functional characterization of alternatively spliced human SECISBP2 transcript variants. Nucleic Acids Res. 2008;36:7192‐7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anestal K, Arner ES. Rapid induction of cell death by selenium‐compromised thioredoxin reductase 1 but not by the fully active enzyme containing selenocysteine. The Journal of biological chemistry. 2003;278:15966‐15972. [DOI] [PubMed] [Google Scholar]
- 18. Harfe BD, McManus MT, Mansfield JH, Hornstein E, Tabin CJ. The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci USA. 2005;102:10898‐10903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kobayashi T, Lu J, Cobb BS, et al. Dicer‐dependent pathways regulate chondrocyte proliferation and differentiation. Proc Natl Acad Sci USA. 2008;105:1949‐1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miyaki S, Asahara H. Macro view of microRNA function in osteoarthritis. Nat Rev Rheumatol. 2012;8:543‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Le LT, Swingler TE, Clark IM. Review: the role of microRNAs in osteoarthritis and chondrogenesis. Arthritis Rheum. 2013;65:1963‐1974. [DOI] [PubMed] [Google Scholar]
- 22. Sun J, Zhong N, Li Q, et al. MicroRNAs of rat articular cartilage at different developmental stages identified by Solexa sequencing. Osteoarthritis Cartilage. 2011;19:1237‐1245. [DOI] [PubMed] [Google Scholar]
- 23. Zhong N, Sun J, Min Z, et al. MicroRNA‐337 is associated with chondrogenesis through regulating TGFBR2 expression. Osteoarthritis Cartilage. 2012;20:593‐602. [DOI] [PubMed] [Google Scholar]
- 24. Sumiyoshi K, Kubota S, Ohgawara T, et al. Novel role of miR‐181a in cartilage metabolism. J Cell Biochem. 2013;114:2094‐2100. [DOI] [PubMed] [Google Scholar]
- 25. Yan J, Zheng Y, Min Z, Ning Q, Lu S. Selenium effect on selenoprotein transcriptome in chondrocytes. Biometals : an international journal on the role of metal ions in biology, biochemistry, and medicine. 2013;26:285‐296. [DOI] [PubMed] [Google Scholar]
- 26. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ren FL, Guo X, Zhang RJ, et al. Effects of selenium and iodine deficiency on bone, cartilage growth plate and chondrocyte differentiation in two generations of rats. Osteoarthritis Cartilage. 2007;15:1171‐1177. [DOI] [PubMed] [Google Scholar]
- 28. Squires JE, Stoytchev I, Forry EP, Berry MJ. SBP2 binding affinity is a major determinant in differential selenoprotein mRNA translation and sensitivity to nonsense‐mediated decay. Mol Cell Biol. 2007;27:7848‐7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bubenik JL, Driscoll DM. Altered RNA binding activity underlies abnormal thyroid hormone metabolism linked to a mutation in selenocysteine insertion sequence‐binding protein 2. The Journal of biological chemistry. 2007;282:34653‐34662. [DOI] [PubMed] [Google Scholar]
- 30. Latreche L, Jean‐Jean O, Driscoll DM, Chavatte L. Novel structural determinants in human SECIS elements modulate the translational recoding of UGA as selenocysteine. Nucleic Acids Res. 2009;37:5868‐5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Touat‐Hamici Z, Legrain Y, Bulteau AL, Chavatte L. Selective up‐regulation of human selenoproteins in response to oxidative stress. The Journal of biological chemistry. 2014;289:14750‐14761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med. 2003;34:145‐169. [DOI] [PubMed] [Google Scholar]
- 33. Kuhn H, Borchert A. Regulation of enzymatic lipid peroxidation: the interplay of peroxidizing and peroxide reducing enzymes. Free Radic Biol Med. 2002;33:154‐172. [DOI] [PubMed] [Google Scholar]
- 34. Ufer C, Wang CC, Fahling M, et al. Translational regulation of glutathione peroxidase 4 expression through guanine‐rich sequence‐binding factor 1 is essential for embryonic brain development. Genes Dev. 2008;22:1838‐1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yant LJ, Ran Q, Rao L, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496‐502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials