

Abstract
We report a rare etiology of vocal cord paralysis secondary to undiagnosed severe pulmonary hypertension from a de novo ACVRL1 variant identified by whole genomic sequencing. The patient had a partial response to intravenous trepostinil in addition to inhaled nitric oxide, bosentan, and sildenafil.
Keywords: pulmonary arterial hypertension, Hereditary hemorrhagic telangiectasia type 2, VA ECMO, Ortner Syndrome
Background:
Sporadic pediatric idiopathic pulmonary arterial hypertension (PAH) is uncommon, and rarely has been ascribed to pathologic genetic variants in the activin A receptor type II-like Kinase 1(ACVRL1). ACVRL1 variants are implicated in the development of Hereditary Hemorrhagic Telangiectasia (HHT) with late-onset penetrance, such that pediatric PAH may develop significantly prior to any overt symptoms of HHT1,2. HHT manifests as progressive mucocutaneous telangiectasias, recurrent epistaxis, and the development of arteriovenous malformations that may affect multiple organs including the lungs, liver or brain (Figure 1).3 Other genetic variants implicated in HHT include variants in Endoglin, SMAD-4 and GDF-2.4
Figure 1:
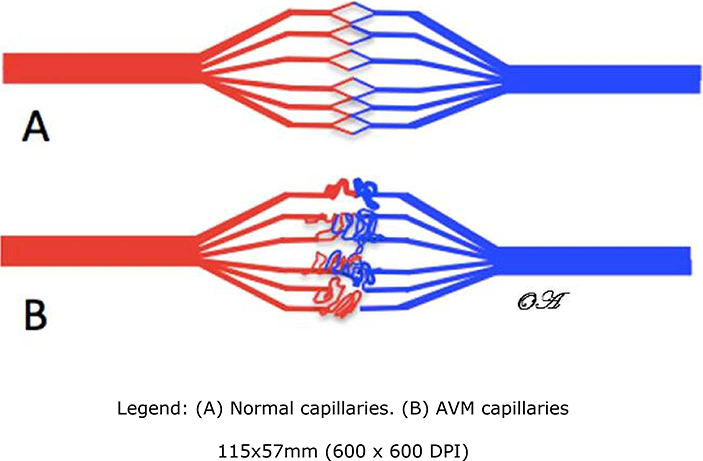
AVM depiction
There are three pulmonary manifestations of HHT: pulmonary arteriovenous malformations, pulmonary hypertension secondary to increased pulmonary blood flow from a high cardiac output state, and rarely pulmonary arterial hypertension.5 Pulmonary hypertensive crisis may be precipitated after minor procedures or due to inter-current respiratory infections. With significant long-standing pulmonary artery dilation from pulmonary hypertension, compression of the left recurrent laryngeal nerve may occur causing vocal changes.6 This was first described in 1897 by Norbert Ortner in an adult female with mitral stenosis and left atrial enlargement but is also reported in cases of aortic aneurysm, chronic obstructive pulmonary disease, and recurrent pulmonary embolism. 7 We report a case of a 14-month-old male with Ortner syndrome secondary to pulmonary arterial hypertension associated with a de novo ACVRL1 variant known to be associated with HHT.
Case Presentation:
A previously healthy 14 m/o male presented with syncope in the setting of four weeks of progressive “noisy” breathing. The patient did not have a prior history of syncopal episodes, dyspnea on exertion, congenital heart disease, telangiectasias or epistaxis. The family history was negative for bleeding disorders, epistaxis or PAH. His physical exam was unrevealing and benign other than stridulous breathing and he was admitted to the hospital for observation and workup of the respiratory symptoms. A bedside laryngoscopy was performed which revealed a left vocal cord paralysis. The procedure provoked a pulmonary hypertensive crisis manifesting as cardiogenic shock and identified by new onset hypotension, tachycardia, poor perfusion, delayed peripheral pulses and a prominent S2 with a gallop. Further evaluation by ECG revealed severe right ventricular strain and an echocardiograph demonstrated supra-systemic PAH with bowing of the ventricular septum in a structurally normal heart. CT images of the chest and neck confirmed right ventricular hypertrophy and a severely dilated main pulmonary artery that likely contributed to left recurrent laryngeal nerve compression. Further workup for idiopathic pulmonary hypertension included negative hepatitis and HIV serologies, normal coagulation studies, normal thyroid function and a negative ANA as a screen for rheumatologic disease.
The patient’s right ventricular failure progressed despite treatment with inhaled nitric oxide, sildenafil, bosentan, and treprostinil. His clinical course was complicated by an inter-current rhinovirus upper respiratory tract infection. He was placed on veno-atrial ECMO for right ventricular failure and exhibited a partial response to escalating dose of treprostinil allowing for ECMO decannulation after seven days. Due to severity of his illness, cardiac catheterization was performed after ECMO decannulation confirming the diagnosis of PAH with a mean pulmonary artery pressure of 59 mmHg and indexed pulmonary vascular resistance of 9.5 wood units/m^2. Cardiac output was 3.05 L/min estimated by Fick equation. The pulmonary artery wedge pressure was 13mmHg and pulmonary vasoreactivity testing was negative. Serial echocardiograms demonstrated improvement in right ventricular function and the patient was discharged after 3 months on subcutaneous trepostinil in addition to the above-mentioned oral therapies.
Rapid trio whole genome sequencing revealed a de novo heterozygous missense variant (c.1450C>T; p.Arg484Trp) in the ACVRL1 gene that has been associated with HHT type 2. This variant was predicted to be pathologic and confirmed by Sanger sequencing8.
Discussion:
When PAH occurs in a familial context, pathologic variants in bone morphogenic protein receptor 2 (BMPR2) are identified in approximately 70% of patients. Furthermore, in patients with sporadic idiopathic PAH 10–40% will have a pathologic variant in BMPR2 detected.9 Heritable pulmonary arterial hypertension can be associated with a variety of additional gene mutations including ACVRL1, GDF-2, SMAD-4, SMAD-8 and Endoglin.10 According to Girerd et al, carriers with pathogenic ACVRL1 variants in comparison to carriers and non-carriers of BMPR2 variants were diagnosed at a younger age and had overall worse prognosis.11 Utilization of rapid trio whole genome sequencing to identify pathologic genetic variants maybe advantageous in several regards. Firstly, since patients with ACVRL1 variants typically do not respond to acute vasoreactivity testing, as was found with our patient, they may not respond to calcium channel blockade12. Secondly, while current recommendations for genetic screening in idiopathic pulmonary hypertension without a positive family history can be controversial, it may also be helpful for family planning13. Lastly, knowledge of the genetic variant may change long term management in our patient resulting in periodic surveillance for development of cerebral arteriovenous malformations. Successful treatment of early pediatric ACVRL1 patients is rarely reported, and in our patient a response was obtained to a combination of a phosphodiasterase-5 inhibitor, an endothelin receptor antagonist, and a prostacyclin. Progressive PAH despite medical management warrants lung transplant evaluation.
Conclusion:
Pediatric PAH may manifest with Ortner’s syndrome. We present a pediatric patient with severe PAH found to have a de novo autosomal dominant ACVRL1 pathologic variant by rapid trio whole genomic sequencing who required advanced medical management and exhibited a short term favorable outcome to multimodal therapy. A multidisciplinary approach and long term follow up is key for patients with HHT as long-term outcomes are not well described.
Acknowledgements:
We would like to thank Justin Yeh, M.D, Rohit Rao M.D, MBA, Raghav Murthy M.D, Shylah Haldeman, MSN, NP-C and the remainder of team at Genomics Institute including: Stephen Kingsmore MB,ChB,BAO, Lauge Farnaes MD PhD, Amber Hildreth DO, Nathaly M. Sweeney MD MPH, Shareef Nahas PhD, Julie A. Cakici BSN, Yan Ding MD, Narayanan Veeraraghavan PhD, and David Dimmock MD
Funding: National Human Genome Research Institute. Award Number: U19HD077693. Recipient: Stephen F. Kingsmore
Footnotes
Competing Interests: none
Ethics approval and consent to participate: Rady Children’s Hospital has approved of the research study in which genetic testing was performed. “Genomic Biorepository: Protocol for the Collection, Storage, Analysis and Distribution of Biological Samples and Clinical Data.”
Consent: Written informed consent for publication of their clinical details and/or clinical images was obtained from parent of the patient. A copy of the consent form is available for review by the Editor of this journal.
Data Generation: All data generated or analyzed during this study are included in this published article.
Reference
- 1.Girerd B, Montani D, Coulet F, et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med. 2010;181(8):851–861. [DOI] [PubMed] [Google Scholar]
- 2.Abman SH, Hansmann G, Archer SL, et al. Pediatric pulmonary hypertension: Guidelines from the american heart association and american thoracic society. Circulation. 2015;132(21):2037–2099. doi: 10.1161/CIR.0000000000000329 [doi]. [DOI] [PubMed] [Google Scholar]
- 3.Vorselaars VM, Velthuis S, Snijder RJ, Vos JA, Mager JJ, Post MC. Pulmonary hypertension in hereditary haemorrhagic telangiectasia. World J Cardiol. 2015;7(5):230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hernandez F, Huether R, Carter L, et al. Mutations in RASA1 and GDF2 identified in patients with clinical features of hereditary hemorrhagic telangiectasia. Hum Genome Var. 2015;2:15040. doi: 10.1038/hgv.2015.40 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vorselaars VM, Velthuis S, Snijder RJ, Vos JA, Mager JJ, Post MC. Pulmonary hypertension in hereditary haemorrhagic telangiectasia. World J Cardiol. 2015;7(5):230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mulpuru SK, Vasavada BC, Punukollu GK, Patel AG. Cardiovocal syndrome: A systematic review. Heart Lung Circ. 2008;17(1):1–4. doi: S1443-9506(07)00204-1 [pii]. [DOI] [PubMed] [Google Scholar]
- 7.Mulpuru SK, Vasavada BC, Punukollu GK, Patel AG. Cardiovocal syndrome: A systematic review. Heart Lung Circ. 2008;17(1):1–4. doi: S1443-9506(07)00204-1 [pii]. [DOI] [PubMed] [Google Scholar]
- 8.Farnaes L, Nahas SA, Chowdhury S, Nelson J, Batalov S, Dimmock DM, Kingsmore SF; RCIGM Investigators. Rapid whole-genome sequencing identifies a novel GABRA1 variant associated with West syndrome. Cold Spring Harb Mol Case Stud. 2017. September 1;3(5). pii: a001776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abman SH, Hansmann G, Archer SL, et al. Pediatric pulmonary hypertension: Guidelines from the american heart association and american thoracic society. Circulation. 2015;132(21):2037–2099. doi: 10.1161/CIR.0000000000000329 [doi]. [DOI] [PubMed] [Google Scholar]
- 10.Chida A, Shintani M, Yagi H, et al. Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am J Cardiol. 2012;110(4):586–593. [DOI] [PubMed] [Google Scholar]
- 11.Girerd B, Montani D, Coulet F, et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med. 2010;181(8):851–861. [DOI] [PubMed] [Google Scholar]
- 12.Girerd B, Montani D, Coulet F, et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med. 2010;181(8):851–861. [DOI] [PubMed] [Google Scholar]
- 13.Abman SH, Hansmann G, Archer SL, et al. Pediatric pulmonary hypertension: Guidelines from the american heart association and american thoracic society. Circulation. 2015;132(21):2037–2099. doi: 10.1161/CIR.0000000000000329 [doi]. [DOI] [PubMed] [Google Scholar]