

Abstract
We report a new class of artifacts in DNA methylation measurements from Illumina HumanMethylation450 and MethylationEPIC arrays. These artifacts reflect failed hybridization to target DNA, often due to germline or somatic deletions and manifest as incorrectly reported intermediate methylation. The artifacts often survive existing preprocessing pipelines, masquerade as epigenetic alterations and can confound discoveries in epigenome-wide association studies and studies of methylation-quantitative trait loci. We implement a solution, P-value with out-of-band (OOB) array hybridization (pOOBAH), in the R package SeSAMe. Our method effectively masks deleted and hyperpolymorphic regions, reducing or eliminating spurious reports of epigenetic silencing at oft-deleted tumor suppressor genes such as CDKN2A and RB1 in cases with somatic deletions. Furthermore, our method substantially decreases technical variation whilst retaining biological variation, both within and across HM450 and EPIC platform measurements. SeSAMe provides a light-weight, modular DNA methylation data analysis suite, with a performant implementation suitable for efficient analysis of thousands of samples.
INTRODUCTION
The Illumina Infinium HumanMethylation BeadChips are powerful tools for quantifying DNA cytosine modifications in the human genome. The current generation, the HumanMethylationEPIC (EPIC, or HM850) array, shares similar design principles with its predecessor, the HumanMethylation450 (HM450) array. These beadchips have been the most popular platforms for genome-scale DNA methylation studies (1). The Cancer Genome Atlas (TCGA) (2) alone has generated high-quality DNA methylation data on more than 10,000 human samples with the HM450 platform, in addition to more than 100,000 samples jointly contributed by the research community on Gene Expression Omnibus (GEO). Quality control, preprocessing and analytical tools for this beadchip have therefore attracted much attention, and the success in this realm with contributions of many research teams in turn further facilitated the use of this popular technology. The past few years have seen extensive application of this platform to epidemiological association studies (epigenome-wide association studies, or EWAS) of human epigenetic alterations (3), focusing on traits such as body mass index (4), obesity (5) and the impact of periconceptional environment on genomic imprinting (6). In addition, there has been a recent surge in interest of identifying methylation-quantitative trait loci (meQTL/mQTL) in various tissues with this platform (7–9).
DNA methylation readouts from these arrays are usually expressed in 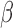 values (10), defined as
values (10), defined as 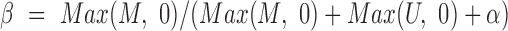 , in which
, in which 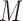 represents the hybridization signal from a methylated version and
represents the hybridization signal from a methylated version and 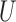 represents the hybridization signal from an unmethylated version of a cytosine nucleotide and
represents the hybridization signal from an unmethylated version of a cytosine nucleotide and 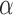 is a offset recommended by Illumina (default to 100), but this offset is unnecessary and later abandoned by common preprocessing pipelines, and this equation is effectively reduced to
is a offset recommended by Illumina (default to 100), but this offset is unnecessary and later abandoned by common preprocessing pipelines, and this equation is effectively reduced to 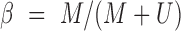 . It has been recognized that this
. It has been recognized that this  value should only be reported when a true signal from DNA hybridization (instead of background fluorescence) is detected. Whether a signal is considered real is determined by a detection P-value, representing the probability of a detected signal being background fluorescence.
value should only be reported when a true signal from DNA hybridization (instead of background fluorescence) is detected. Whether a signal is considered real is determined by a detection P-value, representing the probability of a detected signal being background fluorescence.
In GenomeStudio, the detection P-value is calculated with the background fluorescence level modeled as a Gaussian distribution parameterized by the negative control probes included in the array, and it is recommended that data points associated with P > 0.05 be excluded (GenomeStudio Methylation Module v1.8 User Guide). minfi (11), a widely used pipeline for Infinium array processing (1), calculates the P-values in a similar parametric way, but with the M and U probes combined into one foreground signal and one background distribution to calculate the Z-score. The background distribution is combined based on the color channel of the corresponding probe (i.e. 2*green or 2*red for Type-I and green+red for Type II). A P-value cut-off of 0.01 is recommended per the user manual (version 1.27.1).
TCGA used a non-parametric approach to perform this quality masking (2), fitting the empirical signal intensities from the negative control probes. TCGA required only one of M and U signals to be significantly above background, to accommodate fully methylated or fully unmethylated situations, in which the other signal should be completely absent. This approach has been implemented in methylumi (12). In addition, the TCGA data (referred to as ‘TCGA Legacy data’, as available on the NCI Genomic Data Commons portal) employed the methylumi preprocessing pipeline, but also implemented an experiment-independent masking at the probe level, against probes overlapping single nucleotide polymorphism (SNP) and repeats. We have recently described criteria for experiment-independent masking based on probe design (13). Two other important preprocessing steps for this type of beadchips are dye-bias correction and noob-based background correction. For the comparisons done in this study, it is of note that minfi has adopted the methylumi/TCGA approach in these two steps (14), except for the slight difference in detection P-value calculation described above.
Here we report that the common practices for detection P-value calculation as described above incompletely flag artifacts associated with detection failure. We show that such detection failures are usually attributable to insufficient quantities of the target DNA, due to germline and/or somatic deletions or hyperpolymorphism, and that this problem can be further aggravated by cross-hybridization of the probes to other sites in the genome in the absence of competitive hybridization to the true template sequence, allowing such probes to survive non-detection masking. We show that these artifacts are present in existing datasets, including those from TCGA. We implement a new method, called pOOBAH (P-value with out-of-band (OOB) array hybridization) in the software SeSAMe, that better addresses detection failure as part of a single-sample-based pipeline for DNA methylation data processing. We also show that by sufficiently masking measurements subject to these non-detection failure artifacts, one can greatly reduce technical variations (the most well-known examples being ‘batch effects’) (15), an analytical challenge for DNA methylation. In addition, SeSAMe also substantially improves the consistency between HM450, EPIC and whole genome bisulfite sequencing (WGBS) platforms, and can facilitate combined analyses of HM450 and EPIC data. The latter addresses a rising analytical need as a rich body of data have been generated on the discontinued HM450 as public resources, but new data will be generated with the EPIC array.
MATERIALS AND METHODS
Data
DNA Methylation HM450 IDAT files for 749 normal samples assayed in the TCGA project, together with self-reported sex information, were downloaded from NCI Genomic Data Commons (GDC; https://gdc.cancer.gov/). Infinium HumanMethylationEPIC IDATs were downloaded from GEO with accession GSE86833 (16). Whole-genome bisulfite sequencing data for 47 samples were also obtained from GDC, with the experimental procedure described in (17). DNA copy number segmentation from the same 749 TCGA normal samples (18) using an Affymetrix Human SNP Array 6.0 were also downloaded from GDC. Somatic mutation MAF files for 10,290 TCGA tumor samples were downloaded from a previous study (19). We included mutations independently called by more than two of the seven variant callers (19).
A total of 281 HM450 technical replicates were included in TCGA. These cell line samples were from the same lymphoblastoid cell line (Coriell’s GMO6990), but were independently expanded at two separate institutes (Nationwide Children’s Hospital [NCH], with the code TCGA-07-0227 and International Genomics Consortium [IGC] with the code TCGA-AV-A03D), prior to being sent to a single data production facility (USC Epigenome Center). One replicate aliquot (either TCGA-07-0227 or TCGA-AV-A03D) was assayed together with a batch of TCGA samples, over the course of 5 years, yielding a total of 281 profiles. The same replicate may be packaged into different cancer types due to samples from multiple cancer types being assayed in the same batch. We included only unique IDAT files in this study.
Preprocessing infinium DNA methylation data using methylumi
Signal intensities were extracted from IDAT files using R package illuminaio (20). For each TCGA sample, we first performed background subtraction and dye bias correction across the entire array with methylumi (21), which was the method referenced in TCGA projects (2). We confirmed that for most TCGA packages downloaded from GDC, the two datasets matched perfectly, proving that we reproduced the TCGA preprocessing. However, for LUAD (LUng ADenocarcinoma), the TCGA GDC Legacy data reflected omission of the background correction step, an inconsistency with other TCGA packages. Therefore, we used our methylumi-processed data to represent TCGA GDC Legacy data for comparison, to eliminate this difference. (Therefore, note that the original, inconsistently processed, GDC Legacy data would have exhibited even larger batch effects in our comparison.).
Detection P-value calculation using out-of-band signals of type-I probes
To better identify hybridization failure, we adopted a new method of calculating P-value named pOOBAH (P-value with OOB probes for Array Hybridization), where detection P-value is calculated using empirical cumulative distribution function (implemented in the ecdf function in R stats package) derived from the OOB signal from all Type-I probes. The method is implemented in our SeSAMe pipeline. To compare with other methods, we reprocessed all the TCGA samples and HumanMethylationEPIC array data (described above) with our SeSAMe pipeline.
Benchmarking SeSAMe detection calling against other tools
We used probes from chromosome Y and the GSTT1 germline deletion to evaluate detection calling from different pipelines. We inferred the biological sex of these samples using both chromosome X methylation and chromosome X/Y signal intensity as implemented in SeSAMe and used 745 out of the 749 samples where inferred sex agreed with self-reported gender. Detection from chromosome Y probes (filtered by probe design quality first as described in (13)) in male samples was considered true positive and lack of detection in female samples true negative. Lack of detection from GSTT1 probes in homozygous deletion samples was considered true negative, and detection from either intact diploid or heterozygous deletion cases true positives. Deletion status for each Infinium HM450 probe was determined by overlapping the probe coordinate with copy number segments established using Affymetrix Genome-Wide Human SNP Array 6.0 (SNP6). Probes that did not fall in any segment were discarded. A Log R ratio cut-off of −0.8 from SNP6 array was used to determine the GSTT1 genotype, with <−0.8 considered homozygous deletion.
methylumi (version 2.26) and minfi (version 1.26) was used for the comparison. No documentation of the exact way to calculate the detection P-value in GenomeStudio was available. A prior study (22) characterized it as such: the detection P-value for CpG locus j is given by 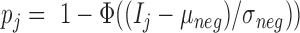 , where
, where 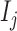 is the sum of foreground intensities, whereas
is the sum of foreground intensities, whereas 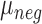 and
and 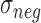 are the mean and standard deviation of signals of internal negative controls and
are the mean and standard deviation of signals of internal negative controls and 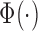 is the normal cumulative probability distribution function. We followed this description in simulating GenomeStudio results. However, this seemed problematic as the background is double-counted in the foreground intensities. Illumina’s manual also described variable detection P-value calculation based on whether background subtraction is performed. Therefore, actual GenomeStudio outputs can have multiple versions and our comparison only represents one scenario.
is the normal cumulative probability distribution function. We followed this description in simulating GenomeStudio results. However, this seemed problematic as the background is double-counted in the foreground intensities. Illumina’s manual also described variable detection P-value calculation based on whether background subtraction is performed. Therefore, actual GenomeStudio outputs can have multiple versions and our comparison only represents one scenario.
Total signal intensity normalization and detection of germline deletion
We summed the methylated and unmethylated signals for each probe (referred to as total signal intensities from here on). The total signal intensity readout from a particular Infinium probe may be affected by the input DNA quality and quantity, as well as differences in the probe design. To control for such covariates, we calculated the Z-score of the log total intensity of a given probe with respect to other probes of the same design type and same color channel in the same sample. This within-sample Z-score was used to compute the prior signal intensity probability for each probe.
To enhance the detection sensitivity of focal deletions, we adopted a bottom-up approach using variations in total signal intensity from 749 normal samples to identify Infinium probes subject to potential germline deletions in the human population with relatively high minor allele frequencies. For each probe, the within-sample Z-scores calculated (above) in the normal samples constitute an empirical prior distribution for the total intensity of that probe. We tested this Z-score distribution for unimodality using the Hartigan’s dip test statistics (23) and identified 14,160 significantly multimodal probes (P ≤ 0.05) covering 94% of the chromosome Y probes and 76% of the chromosome X probes (equivalent to heterozygous deletion in males), but only 1% of the autosomal probes.
In some cases, probes interrogating intermediately-methylated CpGs tend to have higher total intensity than when the targeted CpGs are completely methylated or completely unmethylated (data not shown). This is due to the saturation in signal intensity when the two alleles are either both methylated or both unmethylated. In order to eliminate multimodal probes due to true differential methylation level, we computed the Pearson’s correlation between signal intensity and the deviation of 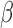 value from intermediate methylation (measured by
value from intermediate methylation (measured by 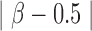 ). We only retained probes with a positive correlation (>0.2). Further filtering probes having small variation in
). We only retained probes with a positive correlation (>0.2). Further filtering probes having small variation in 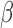 value (SD < 0.1) and probes previously identified to contain SNPs in the neighborhood yields 441 autosomal multimodal probes (Supplementary Table S1 and Figure S1 for
value (SD < 0.1) and probes previously identified to contain SNPs in the neighborhood yields 441 autosomal multimodal probes (Supplementary Table S1 and Figure S1 for 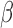 values plotted against total signal intensities).
values plotted against total signal intensities).
Detection of somatic deletion
Using an approach similar to the identification of germline copy number changes, we derived, for each probe, an empirical within-sample Z-score distribution from the TCGA adjacent normal samples. We then evaluated the significance (P-value) of the probe’s Z-score in the tumor sample with respect to this empirical Z-score distribution. We studied adjacent probes that showed co-reduction in total signal intensities and consider them as potential candidates of somatic deletion.
RESULTS
Germline deletion causes artifactual DNA methylation readout
To explore how genomic DNA deletion impinges on DNA methylation readouts, we investigated DNA methylation 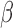 values together with the total signal intensities at the glutathione S-transferase theta (GSTT1) locus (Figure 1A). This locus is known to exhibit deletion polymorphism, with roughly 20% of the human population carrying a homozygous deletion (24). A cluster of samples at the top of the heatmap displayed seemingly distinct DNA methylation patterns within the deleted region. Many of the probes that fell within this region, exemplified by P2, P4 and P5 (Figure 1A), displayed apparent intermediate levels of DNA methylation. These samples also had substantially lower total intensities in this region, and therefore likely represented the homozygous deletion carriers. Comparison of the total intensities of HM450 probes with copy number segmentation using matched Affymetrix SNP 6.0 microarray data from TCGA validated this deletion (Supplementary Figure S2).
values together with the total signal intensities at the glutathione S-transferase theta (GSTT1) locus (Figure 1A). This locus is known to exhibit deletion polymorphism, with roughly 20% of the human population carrying a homozygous deletion (24). A cluster of samples at the top of the heatmap displayed seemingly distinct DNA methylation patterns within the deleted region. Many of the probes that fell within this region, exemplified by P2, P4 and P5 (Figure 1A), displayed apparent intermediate levels of DNA methylation. These samples also had substantially lower total intensities in this region, and therefore likely represented the homozygous deletion carriers. Comparison of the total intensities of HM450 probes with copy number segmentation using matched Affymetrix SNP 6.0 microarray data from TCGA validated this deletion (Supplementary Figure S2).
Figure 1.

Germline deletion causes low total intensity measurements and creates spurious epigenetic silencing patterns at the GSTT1 locus. (A) Heatmaps showing 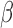 values (top) and total intensities (bottom) of HM450 probes at the GSTT1 locus (columns) in TCGA normal samples (rows). Lines connect probes with actual genomic locations. Yellow box indicates the deleted region. Probe and sample orders are matched in the two heatmaps. Two clusters of samples can be seen on these heatmaps (left sidebar), with black representing samples that carry homozygous GSTT1 deletions. Probes P1-P6 designate example probes shown in Panel C. (B) Mapping quality of probes plotted in Panel A. Arrows indicate probes in the deleted region that did not exhibit the signature intermediate methylation. Color of arrow indicates different combinations of methylation patterns at on-target and off-target sites (M - methylated; U - unmethylated); (C) Expression (y-axis) plotted against
values (top) and total intensities (bottom) of HM450 probes at the GSTT1 locus (columns) in TCGA normal samples (rows). Lines connect probes with actual genomic locations. Yellow box indicates the deleted region. Probe and sample orders are matched in the two heatmaps. Two clusters of samples can be seen on these heatmaps (left sidebar), with black representing samples that carry homozygous GSTT1 deletions. Probes P1-P6 designate example probes shown in Panel C. (B) Mapping quality of probes plotted in Panel A. Arrows indicate probes in the deleted region that did not exhibit the signature intermediate methylation. Color of arrow indicates different combinations of methylation patterns at on-target and off-target sites (M - methylated; U - unmethylated); (C) Expression (y-axis) plotted against 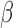 value (x-axis) for the six example probes as indicated in Panel A, showing various spurious correlation patterns, including patterns strongly emblematic of epigenetic silencing. (D) Formular representation of how low signal intensities lead to intermediate DNA methylation readout.
value (x-axis) for the six example probes as indicated in Panel A, showing various spurious correlation patterns, including patterns strongly emblematic of epigenetic silencing. (D) Formular representation of how low signal intensities lead to intermediate DNA methylation readout.
Intriguingly, not all probes in the deleted region showed this general intermediate methylation in samples carrying homozygous deletions. A few exceptions included Probes P1, P3 and P6 (Figure 1A). A low mapping quality score observed for these probe sequences suggested that they might be subject to cross-hybridization with other loci in the genome. In that case, the DNA methylation level in the samples with homozygous deletion of the target locus would reflect the DNA methylation state at off-target loci. We verified this with TCGA WGBS data. For example, the off-target locus of Probe P3 is methylated, giving its higher observed 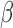 value in samples carrying homozygous deletions of this region (data not shown). The presence of cross-hybridization is also supported by (i) the existence of a slightly higher DNA methylation level in the remaining samples with undeleted GSTT1, relative to nearby probes, and (ii) the existence of a slightly higher total signal intensity of these probes relative to neighboring probes in the cluster of samples with homozygous deletion. Other probes that displayed non-intermediate DNA methylation in the putative deletion region were attributable to different combinations of on-target and off-target DNA methylation patterns as well (Figure 1B). DNA methylation readouts from Infinium DNA Methylation BeadChips at these loci were jointly determined by the presence or absence of target templates and by the DNA methylation status of the off-target loci upon cross-hybridization.
value in samples carrying homozygous deletions of this region (data not shown). The presence of cross-hybridization is also supported by (i) the existence of a slightly higher DNA methylation level in the remaining samples with undeleted GSTT1, relative to nearby probes, and (ii) the existence of a slightly higher total signal intensity of these probes relative to neighboring probes in the cluster of samples with homozygous deletion. Other probes that displayed non-intermediate DNA methylation in the putative deletion region were attributable to different combinations of on-target and off-target DNA methylation patterns as well (Figure 1B). DNA methylation readouts from Infinium DNA Methylation BeadChips at these loci were jointly determined by the presence or absence of target templates and by the DNA methylation status of the off-target loci upon cross-hybridization.
Downregulation of mRNA expression concomitant with promoter DNA hypermethylation is usually deemed as evidence of epigenetic silencing. However, deletion of a gene will also register with a decrease in mRNA expression. Expression of GSTT1 was also reduced in the case of heterozygous DNA deletions. We plotted RNA expression of GSTT1 against the DNA methylation readouts for the six example probes shown in Figure 1A. Probes P2, P5 (representing most probes in this region), and P3 and P6 (representing interactions with off-target sites) all displayed strong negative correlation, a signature emblematic of epigenetic silencing (Figure 1C). Of particular note, probe P6 has been misinterpreted by several prior EWAS studies as the best example of epigenetic silencing (25–28), which caused GSTT1 (or GSTM1 under a similar rationale) to be picked up as an epigenetically silenced gene and meQTL (29,30). Figure 1D illustrates the principle of the observed intermediate methylation pattern associated with detection failure. When true signals in both M and U channels approach 0, foreground signals in both channels approaches background and 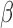 value approaches 0.5.
value approaches 0.5.
Detection calling using out-of-band probes
Prior studies largely rely on negative control probes included in the array to compute detection P-values. The limited number of negative control probes (N = 614 for HM450 array, N = 411 for EPIC array) constrains the precision of the P-value and loses discriminating power when extreme P-values are required, as discussed in the following analysis. The default detection P-value cut-off is set to 0.05 in methylumi. One recent study also suggested using an arbitrarily chosen, stringent P-value cut-off at 1E-16 on the minfi P-values for more accurate detection calling (31).
We developed a new method named pOOBAH (implemented in the R package SeSAMe, see ‘Materials and Methods’ section) using the empirical distribution of OOB signals from Type-I probes. These OOB signals were shown to be better representatives of the background fluorescence in practice, and utilized to perform background subtraction (21). This was used for TCGA data preprocessing (2) and incorporated into methylumi (12), and later also adopted by minfi (14). We compared SeSAMe with minfi and methylumi (used to generate TCGA Legacy packages) for their performance on chromosome Y (chrY) probes (which should not yield signals in female samples) as well as GSTT1.
We also evaluated the performance of SeSAMe masking using probes mapped uniquely to chrY based on our prior characterization of the EPIC probes (13). In female samples, as expected, most chrY probes exhibited low total intensities, associated with the signature intermediate 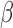 values observed in the case of GSTT1 deletion. In male samples these probes were partitioned into methylated and unmethylated groups, both supported by high total intensities (Figure 2A, top). Using methylumi leaves a substantial number of low total intensity chrY probes not masked in female samples (Figure 2A). These lowest intensity chrY probes continued to display an intermediate level of DNA methylation and almost completely disappeared under SeSAMe masking. Masking with minfi P-value using an extremely low threshold (1E-16) was the next most effective in removing spurious chrY intermediate measurements in TCGA female samples (Figure 2A, see Supplementary Figure S3 for the full ROC curve) but with far higher false positive rate than what the chosen P-value would indicate (Figure 2B).
values observed in the case of GSTT1 deletion. In male samples these probes were partitioned into methylated and unmethylated groups, both supported by high total intensities (Figure 2A, top). Using methylumi leaves a substantial number of low total intensity chrY probes not masked in female samples (Figure 2A). These lowest intensity chrY probes continued to display an intermediate level of DNA methylation and almost completely disappeared under SeSAMe masking. Masking with minfi P-value using an extremely low threshold (1E-16) was the next most effective in removing spurious chrY intermediate measurements in TCGA female samples (Figure 2A, see Supplementary Figure S3 for the full ROC curve) but with far higher false positive rate than what the chosen P-value would indicate (Figure 2B).
Figure 2.

SeSAMe effectively removes non-detection artifacts that survive existing pipelines. (A) 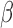 values (y-axis) against total signal intensities of Y-chromosome probes (x-axis) in TCGA normal primary tissue samples (376 males and 369 females), with raw data (top), TCGA Legacy
values (y-axis) against total signal intensities of Y-chromosome probes (x-axis) in TCGA normal primary tissue samples (376 males and 369 females), with raw data (top), TCGA Legacy 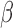 values (middle) and SeSAMe processed data (bottom). (B) Evaluation of false positive and true positive rates of detection associated with different pipelines, using chromosome Y and GSTT1 deletion for benchmarking.
values (middle) and SeSAMe processed data (bottom). (B) Evaluation of false positive and true positive rates of detection associated with different pipelines, using chromosome Y and GSTT1 deletion for benchmarking.
Identification of germline deletion and hyperpolymorphism from Infinium DNA methylation data
The impact of SNPs on the Infinium DNA methylation array measurements has been extensively investigated (13,32), and the exclusion of probes affected by SNPs from analysis is becoming increasingly recognized as an important step in DNA methylation analysis (32). However, impact from structural variations such as insertions and deletions has only been sporadically studied. We set out to discover such regions with germline polymorphism affecting array-based DNA methylation measurement. To approach this, one option would be to use SNP6 array copy number segmentation to flag these regions, when such data are available. However, the boundary resolution using this approach is limited by the distribution of probes on the SNP6 array. For example, in TCGA samples with matched DNA methylation and SNP6 data, the signal intensity at the GSTM1 locus followed a clear bimodal distribution, with samples having lower intensities presumably harboring deletions of the interrogated regions. However, a substantial fraction of these deletions was missed in copy number segmentations because of the moderate SNP6 probe density (Supplementary Figure S4A). Previous studies have shown that copy number alterations could be directly profiled from Infinium data (33–35), but, again, the segmentation algorithm relied on dense probe coverage. Only two Infinium probes were uniquely mapped to the GSTM1 region (Supplementary Figure S4B).
We normalized the total signal intensity for each probe within each sample, stratified by design categories (‘Materials and Method’ section). After this transformation, overall Z-score distributions were stable among all normal samples (Supplementary Figure S5). In addition, the Z-scores were generally comparable between samples for each probe, while different probes exhibited varying level of total intensities, as expected (Supplementary Figure S6). This difference in probe intensity was likely associated with hybridization efficiency, which could be measured by the melting temperature of the hybridized oligos. We observed that GC content, a major determinant of the melting temperature, was also strongly correlated with the Z-score of total intensity of the probe in a normal genome (Supplementary Figure S7, Spearman’s 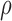 = 0.50, P < 2.2E-16). Germline copy number alterations with high minor allele frequency caused probes inside the alteration to have a multi-modal distribution, with the lowest mode representing hybridization failure (Supplementary Figure S8 for GSTT1). We took an approach of detecting probes multi-modal in total signal intensity to identify germline deletion probes (‘Materials and Method’ section). Visualization of the multi-modality P-value in the GSTT1 and GSTM1 loci confirmed the effectiveness of this method in identifying germline deletions (Supplementary Figure S9). We compared total intensities of these probes with segmental Log R intensities based on the SNP6 array, and many could be validated with this approach, with examples from GSTT1, HLA-DRB6, LOC391322, SLC25A24, ADAM3A, GSTM1, LCE1D, BTNL3, FAM66A, FLJ34503, RHD, ALG1L2 (Supplementary Figure S10).
= 0.50, P < 2.2E-16). Germline copy number alterations with high minor allele frequency caused probes inside the alteration to have a multi-modal distribution, with the lowest mode representing hybridization failure (Supplementary Figure S8 for GSTT1). We took an approach of detecting probes multi-modal in total signal intensity to identify germline deletion probes (‘Materials and Method’ section). Visualization of the multi-modality P-value in the GSTT1 and GSTM1 loci confirmed the effectiveness of this method in identifying germline deletions (Supplementary Figure S9). We compared total intensities of these probes with segmental Log R intensities based on the SNP6 array, and many could be validated with this approach, with examples from GSTT1, HLA-DRB6, LOC391322, SLC25A24, ADAM3A, GSTM1, LCE1D, BTNL3, FAM66A, FLJ34503, RHD, ALG1L2 (Supplementary Figure S10).
We merged probes into segments if two neighboring multimodal probes (<20 kb) showed high correlation (Spearman’s correlation rho > 0.5) in total signal intensity. Most of these probes were mapped to the sex chromosomes as expected. We also identified 40 autosomal segments that were supported by multiple (n ≥ 2) highly correlated probes in signal intensity Z-score (Figure 3 and Supplementary Table S1). These segments include known germline deletions such as GSTT1 (36), BTNL8-BTNL3 (37) together with germline hyper-polymorphic regions such as the Human Leukocyte Antigen (HLA) loci (38). Based on signal intensities, TCGA normal samples were not clustered by tissue type at these loci but strongly impacted by ethnicity (Figure 3), suggesting a genetic, instead of epigenetic, source of the variations observed. Our list provides probes/genes which should be interpreted with caution in microarray-based DNA methylation studies. We had already filtered out probes subject to known SNP polymorphism based on our previous study (13) hence the list here only reflects effects from deletion and hyperpolymorphic region (discussed below).
Figure 3.

Probe total signal intensities in 40 structurally variable and hyperpolymorphic regions supported by more than one probe. Each row corresponds to one of the 749 tumor-adjacent normal samples included in TCGA with the cancer type and patient ethnicity shown on the right. Columns correspond to probes ordered by chromosomes and then genomic locations. Probes are organized by segments (see text) separated with grey vertical bars. An alternating color is assigned on top of the heatmap to distinguish different chromosomes. The design type and color channel of measurement for each probe is shown at the bottom of the heatmap.
It is of note that the HLA regions were recurrently picked up in our screening as well (Figure 4), suggesting that in addition to germline deletions, hyperpolymorphic sites were also susceptible to this artifact. Many of the probes in the HLA/MHC regions have been flagged in our previous annotation for overlapping a SNP that interfered with DNA methylation measurement or for non-unique mapping (‘masking’ track below the heatmaps) for systematic masking. This was expected for hyperpolymorphic regions like HLA. However, there were probes not flagged for this masking and still showed the artifactual intermediate methylation, likely not attributable to a single CpG. With SeSAMe masking, the intermediate methylation readouts due to low signal intensities were more effectively dealt with compared to TCGA legacy processing.
Figure 4.

Examples for HLA loci including MHC class I (top row) and class II (bottom row) demonstrating germline hyperpolymorphism affecting DNA methylation readouts. From left to right: (A) HLA-A, (B) HLA-B, (C) HLA-C, HLA-DQA2 (D), HLA-DRB1 (E) and HLA-DRB6 (F). For each locus, three heatmaps are plotted, with rows representing samples and columns DNA methylation probes. Top panel shows 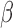 values from TCGA Legacy data. Middle panel shows normalized total signal intensity Z-score. Bottom panel shows
values from TCGA Legacy data. Middle panel shows normalized total signal intensity Z-score. Bottom panel shows 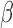 values as masked by SeSAMe based on new detection value. An additional ‘masked’ track below each heatmap indicates probes that SeSAMe masks in general due to overlap with SNPs or non-unique mapping (black). Gray triangles below each probe indicates probes that escape this general masking but effectively masked by SeSAMe.
values as masked by SeSAMe based on new detection value. An additional ‘masked’ track below each heatmap indicates probes that SeSAMe masks in general due to overlap with SNPs or non-unique mapping (black). Gray triangles below each probe indicates probes that escape this general masking but effectively masked by SeSAMe.
Identifying somatic deletion in cancer from infinium DNA methylation data
We explored the extent to which somatic deletions in TCGA cancers could be captured in the Infinium array data, along with the effectiveness of SeSAMe in masking low intensity probes in the case of somatic deletions. Following the rationale used in studying germline deletions, we used a probe-specific empirical prior distribution in evaluating aberrant fluorescent signal as a consequence of variation in the targeted DNA (‘Materials and Methods’ section). We identified copy number alterations across almost all cancer types affecting varying percentages of the genome (Supplementary Figure S11). We were able to pick up recurrent deletions such as RB1 deletion in sarcoma (SARC, Figure 5) and uterine carcinosarcoma (UCS), consistent with previous reports from TCGA (39,40). To investigate recurrent arm-level aneuploidy in the tumor genome, we averaged the log R ratio of all the copy number segments on each chromosome arm in each tumor sample, weighted by the number of supporting probes. We use the order of the matched samples from a previous study based on SNP array platform (41). The gain and loss of the chromosomal arms mirrored what was captured from the SNP array data (Figure 6). Note that the detection sensitivity of somatic copy number alterations was also largely tempered by the proportion of the non-tumor components in the assayed sample. We segmented each tumor genome based on the P-value calculated above. As expected, the number of amplified or deleted segments shrank with a lowering tumor purity (Supplementary Figure S12). The same association was seen in the number of probes with low signal intensity (Supplementary Figure S13), which also included effect from somatic point mutations (‘Discussion’ section).
Figure 5.

SeSAMe masks probes with low total probe intensities caused by somatic deletions of the RB1 locus in cancer. 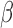 values (top and middle) and total intensities (bottom) of 55 probes at RB1 locus plotted in 265 sarcoma tumor samples from TCGA. Four normal adjacent tissue samples were also included on the top of each heatmap. SeSAMe preprocessing (middle) and contrasted against TCGA level 3 data (top). Tumor samples were clustered based on TCGA preprocessing.
values (top and middle) and total intensities (bottom) of 55 probes at RB1 locus plotted in 265 sarcoma tumor samples from TCGA. Four normal adjacent tissue samples were also included on the top of each heatmap. SeSAMe preprocessing (middle) and contrasted against TCGA level 3 data (top). Tumor samples were clustered based on TCGA preprocessing.
Figure 6.

Arm-level amplifications and deletions inferred for about 10,000 tumors from 33 cancer types using SNP6 array (n = 10,522) (A) and Infinium DNA methylation microarrays (n = 9821) (B). SNP6 array result is adapted from an earlier study (41). Samples are matched between the two panels, with samples present on the SNP6 platform but not the DNA methylation platform replaced with a gray line. For Infinium microarrays, the arm-level average copy number aberration probabilities are plotted in the heatmap from blue to red, with blue indicating arm-level deletion and red indicating amplification following the SNP6 array plot. Rows correspond to mean Log R ratio averaged from probes mapped to the given chromosome arm. Each column corresponds to a primary cancer with color in the top bar showing the cancer type. The color legends for the cancer types are shown on the top.
As in the case for homozygous germline deletions, homozygous somatic deletions could lead to similar spurious DNA methylation measurements. We investigated whether SeSAMe could mask these somatic homozygous deletions in cancer samples and help reduce technical variation caused by low intensities. We studied the retinoblastoma (RB) gene, RB1, mutation of which is known to be associated with osteosarcoma (42) and uterine and ovarian carcinosarcomas (43). We studied 55 probes mapped to the RB1 gene and its flanking region in 151 sarcomas (SARC, ABSOLUTE purity >0.7) included in TCGA. Within TCGA legacy level 3 data, samples with somatic RB1 deletion indeed exhibited spurious DNA methylation measurement caused by the deletion, indicated by low total signal intensity and low mRNA transcription of the RB1 gene (Figure 5, TCGA). SeSAMe was able to correctly mask these data points (Figure 5, SeSAMe). Similar performance of SeSAMe was observed at the CDKN2A locus in bladder cancer where the deletions were more complicated (Supplementary Figure S14A). CDKN2A encodes two famous tumor suppressor genes (TSGs) p16INK4a and p14ARF and therefore attracts a lot of research interests. In particular, p16INK4a was one of the canonical examples for epigenetic silencing of TSGs in cancer. Due to the complexity of this locus, the vast majority of HM450 probes actually do not interrogate the p16INK4a promoter. A single probe (cg13601799) located in the p16INK4a promoter CpG island was routinely used to determine the epigenetic silencing status of this gene (44), and a cutoff of 0.2 was recommended (45). With the TCGA Legacy bladder carcinoma (BLCA) data, 52 out of 178 tumors with relatively high purity had deep deletion of CDKN2A (segment/gene level; without distinguishing p16INK4a and p14ARF exons) based on the copy number array (GISTIC (18) score = −2), 10 of which would have been identified as epigenetically silenced with this existing standard for CDKN2A/p16INK4a. In contrast, all ten data points were masked by the SeSAMe pipeline (Supplementary Figure S14B). Measurement at exon 1β unique to p14ARF appeared to be more affected by the deletion (Supplementary Figure S14A) and again SeSAMe masked the intermediate methylation data points in samples for which the supporting total intensity was low.
In addition, we plotted the 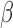 values of chromosome 18 in COAD and READ samples (Supplementary Figure S15) which were shown to be recurrently deleted (46,47). We selected probes with the lowest total intensities and found the same characteristic intermediate
values of chromosome 18 in COAD and READ samples (Supplementary Figure S15) which were shown to be recurrently deleted (46,47). We selected probes with the lowest total intensities and found the same characteristic intermediate 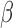 value found in germline deletions. These spurious DNA methylation readouts were mostly masked by SeSAMe. The same was observed for the recurrent chromosome 10 deletion in glioblastoma/low-grade glioma (48) (Supplementary Figure S16). These results again highlight the importance of being aware of copy number alterations when interpreting DNA methylation measurements from Infinium microarrays.
value found in germline deletions. These spurious DNA methylation readouts were mostly masked by SeSAMe. The same was observed for the recurrent chromosome 10 deletion in glioblastoma/low-grade glioma (48) (Supplementary Figure S16). These results again highlight the importance of being aware of copy number alterations when interpreting DNA methylation measurements from Infinium microarrays.
SeSAMe reduces inter- and intra-array technical variations
We hypothesized that variations in background fluorescence levels contribute substantially to between-batch differences. To test whether our improved masking of non-detection would reduce batch effects, we studied 281 HM450 technical replicates of a single lymphoblastoid cell line, expanded in two different institutes prior to being profiled in a single facility for DNA methylation profiles (‘Materials and Methods’ section). We reasoned that independent cultures grown at NCH and IGC could contain minor but real biological differences due to slightly different culturing conditions and/or different population doubling levels. Each replicate also carried microarray-based technical variations associated with each TCGA batch but uncoupled from where the samples were expanded (since samples from both institutes were assayed in the same facility). Some of these biological differences could be smaller than the technical variances, in which case they might only be revealed if unwanted technical variances (49) were removed or reduced.
We studied the TCGA Legacy Level 3 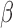 value (with detection failure masked with methylumi; see ‘Materials and Methods’ section) for the lymphoblastoid cell line dataset mentioned above. We selected the top 5% most variable probes based on cross-sample variations among all the technical replicates (Figure 7A). Many Type-II probes displayed intermediate DNA methylation traced by low total signal intensities (Supplementary Figure S17). SeSAMe masking covered most of these intermediate methylation readouts for Type-II probes, while retaining DNA methylation readouts consistent across samples (Figure 7B). After new preprocessing by SeSAMe, we again performed variance-based feature selection and re-clustered the samples. As we had expected, these samples now regrouped according to the institute where the cell line was expanded (Figure 7C), indicating the variation was now being dominated by real biological difference, rather than by technical noise. Indeed, principal component analysis (PCA, Figure 7D) showed that with SeSAMe preprocessing the very first PC was associated with the small biological variation (whether the cell line was expanded at NCH or IGC), while in the TCGA Legacy dataset reproduced by methylumi, only PC2 was associated with this variation. We also observed a reduction in the within-institute variation of the DNA methylation M value (50) with SeSAMe preprocessing compared to the TCGA Legacy dataset, particularly for the Type II probes (Supplementary Figure S18).
value (with detection failure masked with methylumi; see ‘Materials and Methods’ section) for the lymphoblastoid cell line dataset mentioned above. We selected the top 5% most variable probes based on cross-sample variations among all the technical replicates (Figure 7A). Many Type-II probes displayed intermediate DNA methylation traced by low total signal intensities (Supplementary Figure S17). SeSAMe masking covered most of these intermediate methylation readouts for Type-II probes, while retaining DNA methylation readouts consistent across samples (Figure 7B). After new preprocessing by SeSAMe, we again performed variance-based feature selection and re-clustered the samples. As we had expected, these samples now regrouped according to the institute where the cell line was expanded (Figure 7C), indicating the variation was now being dominated by real biological difference, rather than by technical noise. Indeed, principal component analysis (PCA, Figure 7D) showed that with SeSAMe preprocessing the very first PC was associated with the small biological variation (whether the cell line was expanded at NCH or IGC), while in the TCGA Legacy dataset reproduced by methylumi, only PC2 was associated with this variation. We also observed a reduction in the within-institute variation of the DNA methylation M value (50) with SeSAMe preprocessing compared to the TCGA Legacy dataset, particularly for the Type II probes (Supplementary Figure S18).
Figure 7.

SeSAMe preprocessing improves clustering of TCGA cell line replicates driven by small biological differences associated with two different institutes (IGC and NCH with barcodes 0227 and A03D, respectively) performing initial independent expansion of the same cell line (See text). (A) Clustering heatmap showing incomplete separation of 0227 and A03D replicates (columns) in existing TCGA Legacy DNA methylation 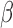 values, based on top variable probes (rows); (B) SeSAMe effectively masks spurious intermediate methylation visible in A; (C) Clustering after SeSAMe masking, with top variable probes (rows) re-selected based on SeSAMe’s masking and re-clustered. Samples are now better clustered by whether they are 0227 or A03D; (D) PCA analysis showing the first two principal components (PC1 and PC2) in TCGA Legacy data and SeSAMe remasked data.
values, based on top variable probes (rows); (B) SeSAMe effectively masks spurious intermediate methylation visible in A; (C) Clustering after SeSAMe masking, with top variable probes (rows) re-selected based on SeSAMe’s masking and re-clustered. Samples are now better clustered by whether they are 0227 or A03D; (D) PCA analysis showing the first two principal components (PC1 and PC2) in TCGA Legacy data and SeSAMe remasked data.
Non-linear dye bias correction
We implemented other functionalities in the SeSAMe package, including improvement on dye bias correction. Dye bias correction is needed for Type II probe preprocessing (21), as Cy3 and Cy5 are directly linked to the methylation status. Both methylumi and minfi adopt a linear scaling based on normalization control probes on both color channels, with a single ratio fit from the median fluorescence for such probes for the two channels. However, we noticed that (i) there was a nonlinear dependence of dye bias on the signal intensities (exemplified by Supplementary Figure S19A) and (ii) the range of intensities for the normalization control probes only captured the lower end while failed to reflect the dye bias in the higher range (blue dots in Supplementary Figure S19A). A substantial portion of Type II probes had higher fluorescence far higher than those of the normalization control probes (Supplementary Figure S19B and C). As a result, after applying the current linear scaling method for dye-bias equalization, Type I probes exhibited worse dye bias in the higher range, while the color channel is not linked to methylation states for this group of probes (Supplementary Figure S19D). We implemented a quantile interpolation-based method that takes advantage of the in-band signal from the Type-I probes. Without prior knowledge of which channel bears the correct signal distribution, we regressed both the Type-I green and red quantile distribution to the mid-point (Supplementary Figure S19E). We applied this regressor to all probes on this platform. Type I probes showed equal distribution as expected (Supplementary Figure S19F). For Type II design since the color was linked to methylation status and therefore not expected to show the same distribution even without dye bias, we chose to evaluate the performance on SNP probes, which are of Type II design and should have a ‘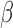 value’ of 0.5 for heterozygous SNPs in the absence of dye bias. Indeed, our nonlinear dye bias correction method, when compared with linear equalization of mean intensities of the two channels, left the
value’ of 0.5 for heterozygous SNPs in the absence of dye bias. Indeed, our nonlinear dye bias correction method, when compared with linear equalization of mean intensities of the two channels, left the 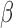 value measurement of heterozygous SNP probes (which are of Type II design) closer to 0.5 in each of the five largest TCGA cancer types investigated (Supplementary Figure S19G–K).
value measurement of heterozygous SNP probes (which are of Type II design) closer to 0.5 in each of the five largest TCGA cancer types investigated (Supplementary Figure S19G–K).
SeSAMe improves between platform consistency
We next investigated whether the SeSAMe pipeline can help improve consistency with WGBS data. On 19 samples with matching HM450 and WGBS data, we first studied the deviation of measured 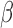 values between the two platforms (minimum depth on cytosine of 10 reads for WGBS). We found that probes with signal masked by SeSAMe indeed had greater deviations in
values between the two platforms (minimum depth on cytosine of 10 reads for WGBS). We found that probes with signal masked by SeSAMe indeed had greater deviations in 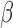 values from WGBS measurement (Figure 8A), suggesting that SeSAMe helps eliminate technical discrepancies between WGBS and HM450 platform. Compared to probes not masked by either approach (methylumi or SeSAMe), probes only masked by SeSAMe are more likely (∼3.5×) to have zero read coverage (suggesting likely deep deletions) in the matched WGBS experiment (Supplementary Figure S20). This comparison suggests the improvement is achieved partly by masking deleted probes and low-intensity probes that in general yielded unreliable results.
values from WGBS measurement (Figure 8A), suggesting that SeSAMe helps eliminate technical discrepancies between WGBS and HM450 platform. Compared to probes not masked by either approach (methylumi or SeSAMe), probes only masked by SeSAMe are more likely (∼3.5×) to have zero read coverage (suggesting likely deep deletions) in the matched WGBS experiment (Supplementary Figure S20). This comparison suggests the improvement is achieved partly by masking deleted probes and low-intensity probes that in general yielded unreliable results.
Figure 8.

SeSAMe reduces inter-platform discrepancies. (A) Distribution of the absolute difference between HM450 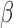 value and methylation level measurement from matched WGBS, with data processed through three different pipelines. (B–F) Distribution of the absolute difference in
value and methylation level measurement from matched WGBS, with data processed through three different pipelines. (B–F) Distribution of the absolute difference in 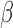 values measured on the HM450 and EPIC platforms on overlapping probes. Each panel shows a different sample assayed on both platforms.
values measured on the HM450 and EPIC platforms on overlapping probes. Each panel shows a different sample assayed on both platforms.
With the recent switch from HM450 to the EPIC platform, a need will rise to combine data generated from both. We investigated whether SeSAMe improves between platform correlations. On all four colorectal samples profiled on both platforms (one sample profiled with two technical replicates on the EPIC platform), SeSAMe improved the consistency between the two platforms, compared to methylumi/TCGA legacy (default P = 0.05 cut-off) and minfi (with P = 0.01 cut-off as suggested in the package vignette) (Figure 8B–F). As previously discussed, minfi and methylumi/ TCGA legacy employ the same preprocessing pipeline (noob + dye-bias correction) except for the detection P-value-based masking step. Therefore, the two methods had similar performance with the difference boiling down to this one single aspect. Between SeSAMe and the other two pipelines, the distributions of abs(EPIC-HM450) were significantly different (P < 2.2e-16 in all cases, Kolmogorov–Smirnov test).
DISCUSSION
Modern ‘omics’ studies employ integration of different molecular data types (51). The interplay between the epigenome and genome in human diseases such as cancer (52) has garnered much attention in recent years, in the hope of finding novel pathways and therapeutic targets. DNA methylation was included as a core measurement in large consortium-based cancer studies such as TCGA and ICGC, while EWAS for population-based studies for phenotypic traits has become increasingly popular. However, non-detection artifacts have not received enough attention in the literature, partly due to the existence of detection P-value designed to safeguard against such effects (but fail to sufficiently do so), and a lack of understanding of the consequences of non-detection as a cause to false-positive methylation and epigenetic silencing calls. In addition, these artifacts could confound integrative analysis combining DNA methylation data and other data types, as the genetic alterations often confounds observed correlations between certain SNPs and DNA methylation, or between DNA methylation and RNA expression.
Most importantly, in the case of EWAS or mQTL/meQTL studies, where (i) mixed tissues are used (implication for this was discussed by Jaffe and Irizarry (53)) and (ii) there often lacks a clonal expansion as in the case of cancer, real ‘signal's are often of a lower scale than the effect size of these artifacts and these artefacts often surface as top hits. As discussed in the results section, GSTT1 was often picked up as a top hit in such studies, likely due to a combination of the germline deletion and off-target hybridization, leading to a strong ‘epigenetic silencing’ pattern (Figure 1C, P6). Most of these studies did not provide data from the original IDATs, and we could not verify that the observed effects were indeed completely caused by non-detection artifacts, but the probes highlighted and patterns of correlation fit what we report in this manuscript. Our analysis also highlights the importance of access to signal intensities in addition to 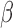 values. In light of this, SeSAMe implements easy access to signal intensities based on the ‘SigSet’ class. This also facilitates copy number inference with DNA methylation microarray data. In TCGA, the IDATs are provided as Level 1 data, but for other studies, more often than not only summarized
values. In light of this, SeSAMe implements easy access to signal intensities based on the ‘SigSet’ class. This also facilitates copy number inference with DNA methylation microarray data. In TCGA, the IDATs are provided as Level 1 data, but for other studies, more often than not only summarized 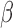 values are made available. Accessibility to the IDATs may be necessary to assess a study and also to facilitate use of the data.
values are made available. Accessibility to the IDATs may be necessary to assess a study and also to facilitate use of the data.
Aside from probe design issues (addressed in (13)), the most common cause of these artifacts is germline and somatic genomic deletion. Cancer samples often display aneuploidy (54) with accompanying broad and focal copy number changes (55). On average, 16% of a typical cancer genome is deleted (56). In the TCGA data, we estimate ∼2–3% of the DNA methylation data points are affected by a deletion deep enough to cause detection failure (Supplementary Figure S21). This number is higher for cancer types with more prevalent copy number variations, such as OV and UCS. Within the same cancer type, the subtypes with more copy number alterations also have a higher number of detection failures. For example, within UCEC, serous-like tumors had a higher failure rate than non-serous tumors using SeSAMe preprocessing (Supplementary Figure S22).
A previous study (57) investigated the relationship between DNA methylation readouts and copy number alterations with the GoldenGate platform, the predecessor to the HM450 and MethylationEPIC arrays, and noted that deletion could cause DNA methylation measurement bias in cancer. However, it was concluded that copy number had little impact on DNA methylation measurements in general. We observe that deletions often cause quite substantial biases when not dealt with properly. In addition, when combined with off-target cross-hybridization, the methylation readout will be from irrelevant sites, as the original target is missing. The effect of DNA amplification is more obscure due to the complexity of dissecting the observed 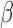 values into contributions from different added copies. Copy number increase is also less detectable with the DNA methylation array because of potential template saturation on the probe. How somatic deletions and amplifications impact DNA methylation readouts are also governed by the fraction of cell populations bearing these abnormal genotypes. For example, when the tumor purity is low, the measurement at sites deleted in cancer cells comes from normal cells present in the tumor. The observation also holds for somatic point mutations, as suggested by comparing known somatic SNVs identified in TCGA with the signal intensities of probes neighboring them. Studying 47,476 mutations close to the 3′-end of an Infinium HM450 probe (including the extension base) revealed that both the total signal intensities and the probe-wise Z-scores were lower in presence of somatic mutations in the targeted tumor sample, most significantly when mutations were of high variant allele frequency (Supplementary Figure S23).
values into contributions from different added copies. Copy number increase is also less detectable with the DNA methylation array because of potential template saturation on the probe. How somatic deletions and amplifications impact DNA methylation readouts are also governed by the fraction of cell populations bearing these abnormal genotypes. For example, when the tumor purity is low, the measurement at sites deleted in cancer cells comes from normal cells present in the tumor. The observation also holds for somatic point mutations, as suggested by comparing known somatic SNVs identified in TCGA with the signal intensities of probes neighboring them. Studying 47,476 mutations close to the 3′-end of an Infinium HM450 probe (including the extension base) revealed that both the total signal intensities and the probe-wise Z-scores were lower in presence of somatic mutations in the targeted tumor sample, most significantly when mutations were of high variant allele frequency (Supplementary Figure S23).
Another source of this type of artifact is germline hyperpolymorphism, which has attracted even less attention from the DNA methylation analysts using hybridization-based arrays. Examples identified in our study included MHC/HLA and olfactory receptor loci. Previous studies show that HLA loci, such as HLA-A (58) and HLA-DQ (59), were candidates for DNA methylation-mediated epigenetic control. Our observations suggest that the Infinium platform might not be an appropriate tool for studying DNA methylation in these regions due to hyperpolymorphism. Even polymerase chain reaction-based methods should be aware of the genetic variations and alternative contigs present in this region before drawing any conclusion about the methylation patterns in this region.
Aside from the interaction between non-detection artifacts and probe design and tumor cellularity discussed above, these artifacts can also interact with technical factors often associated with analytical batches (such as reagent, scanner setting etc; often referred to as ‘batch effects’) which influence background fluorescence levels. The best practice for normalizing Infinium microarray data has been a topic of debate (60). Many of the earlier normalization methods were inherited from the gene expression community and thus often have assumptions that do not hold for DNA methylation (61). It has been noted that between array normalization of DNA methylation data must be handled with care. Most methods bring no or little benefit and actually decrease data quality (62). In fact, correcting for known sources of technical variance (including background and dye bias) yields the safest and best implicit between-array normalization. Indeed, in a recent comparison for EPIC normalization methods, the minfi authors showed that preprocessing with noob (single-sample implementation, dubbed ssNoob) and dye-bias correction outperformed explicit normalization methods exemplified by funnorm (63). Here we showed that after addressing false detection artifacts with SeSAMe, residual technical variations (‘batch effects’) in the TCGA dataset (using noob combined with dye-bias correction) were greatly reduced (Figure 7) revealing minor biological differences. In addition, we demonstrated that the SeSAMe outperformed the existing best standards in improving cross-platform consistency.
Previous studies have shown that Infinium microarrays can also be used to profile copy number alterations (33). Although we focused here on eliminating false positive discoveries primarily due to deletion, our method can also be applied to infer copy number variations, adding to previous methods by deriving a probe-specific prior distribution from normal data. The rediscovery of the same recurrent aneuploidies using the Infinium microarray (Figure 7) confirmed that this platform can cross-validate or partially replace the functionality of SNP arrays in profiling copy number alterations. Because probes with aberrant signal intensities were identified with higher accuracy and merged from bottom up in our approach, focal deletions or copy number of regions with sparse probe coverage may be better captured. The Infinium DNA methylation array could serve as a complementary platform to SNP arrays in that it more densely covers regulatory elements and gene promoter regions and may have greater power of detecting local copy number changes in these regions (33).
DATA AVAILABILITY
The detection calling methods we presented have been implemented in the R package SeSAMe freely available on Bioconductor and at https://github.com/zwdzwd/sesame.
Supplementary Material
ACKNOWLEDGEMENTS
We thank David E. Nadziejka for technical editing and Jay Bowen from Nationwide Children’s hospital for help with information on the TCGA technical replicates.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health/National Cancer Institute [U24 CA143882 to P.W.L., R01 CA170550 to P.W.L., GDAN U24 CA210969 to P.W.L. and H.S.]; Ovarian Cancer Research Fund Grant [373933 to H.S.]; Michelle Lunn Hope Foundation [to T.T.]. Funding for open access charge: Van Andel Research Institute (VARI) New Investigator Funding (Shen).
Conflict of interest statement. None declared.
REFERENCES
- 1. Teschendorff A.E., Relton C.L.. Statistical and integrative system-level analysis of DNA methylation data. Nat. Rev. Genet. 2018; 19:129–147. [DOI] [PubMed] [Google Scholar]
- 2. Cancer Genome Atlas Research Network Ley T.J., Miller C., Ding L., Raphael B.J., Mungall A.J., Robertson A.G., Hoadley K., Triche T.J., Laird P.W. et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013; 368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Flanagan J.M. Epigenome-wide association studies (EWAS): past, present, and future. Methods Mol. Biol. 2015; 1238:51–63. [DOI] [PubMed] [Google Scholar]
- 4. Wahl S., Drong A., Lehne B., Loh M., Scott W.R., Kunze S., Tsai P.-C., Ried J.S., Zhang W., Yang Y. et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017; 541:81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Dijk S.J., Peters T.J., Buckley M., Zhou J., Jones P.A., Gibson R.A., Makrides M., Muhlhausler B.S., Molloy P.L.. DNA methylation in blood from neonatal screening cards and the association with BMI and insulin sensitivity in early childhood. Int. J. Obes. 2018; 42:28–35. [DOI] [PubMed] [Google Scholar]
- 6. Silver M.J., Kessler N.J., Hennig B.J., Dominguez-Salas P., Laritsky E., Baker M.S., Coarfa C., Hernandez-Vargas H., Castelino J.M., Routledge M.N. et al. Independent genomewide screens identify the tumor suppressor VTRNA2-1 as a human epiallele responsive to periconceptional environment. Genome Biol. 2015; 16:118–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Banovich N.E., Lan X., McVicker G., van de Geijn B., Degner J.F., Blischak J.D., Roux J., Pritchard J.K., Gilad Y.. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet. 2014; 10:e1004663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang D., Cheng L., Badner J.A., Chen C., Chen Q., Luo W., Craig D.W., Redman M., Gershon E.S., Liu C.. Genetic control of individual differences in gene-specific methylation in human brain. Am. J. Hum. Genet. 2010; 86:411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen L., Ge B., Casale F.P., Vasquez L., Kwan T., Garrido-Martín D., Watt S., Yan Y., Kundu K., Ecker S. et al. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell. 2016; 167:1398–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bibikova M., Lin Z., Zhou L., Chudin E., Garcia E.W., Wu B., Doucet D., Thomas N.J., Wang Y., Vollmer E. et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006; 16:383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aryee M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., Irizarry R.A.. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014; 30:1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Davis S., Du P., Bilke S., Triche T.J., Bootwalla M.. Methylumi: handle Illumina methylation data. R package version 2.26.0. 2017; doi:10.18129/B9.bioc.methylumi. [Google Scholar]
- 13. Zhou W., Laird P.W., Shen H.. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2017; 45:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fortin J.-P., Triche T.J., Hansen K.D.. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017; 33:558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leek J.T., Scharpf R.B., Bravo H.C., Simcha D., Langmead B., Johnson W.E., Geman D., Baggerly K., Irizarry R.A.. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010; 11:733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pidsley R., Zotenko E., Peters T.J., Lawrence M.G., Risbridger G.P., Molloy P., Van Djik S., Muhlhausler B., Stirzaker C., Clark S.J.. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016; 17:208–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou W., Dinh H.Q., Ramjan Z., Weisenberger D.J., Nicolet C.M., Shen H., Laird P.W., Berman B.P.. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 2018; 50:591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taylor A.M., Shih J., Ha G., Gao G.F., Zhang X., Berger A.C., Schumacher S.E., Wang C., Hu H., Liu J. et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 2018; 33:676–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ellrott K., Bailey M.H., Saksena G., Covington K.R., Kandoth C., Stewart C., Hess J., Ma S., Chiotti K.E., McLellan M. et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 2018; 6:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smith M.L., Baggerly K.A., Bengtsson H., Ritchie M.E., Hansen K.D.. illuminaio: an open source IDAT parsing tool for Illumina microarrays. [version 1; referees: 2 approved]. F1000Res. 2013; 2:264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Triche T.J., Weisenberger D.J., Van Den Berg D., Laird P.W., Siegmund K.D.. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013; 41:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuan P.F., Wang S., Zhou X., Chu H.. A statistical framework for Illumina DNA methylation arrays. Bioinformatics. 2010; 26:2849–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hartigan J.A., Hartigan P.M.. The dip test of unimodality. Ann. Statist. 1985; 13:70–84. [Google Scholar]
- 24. Bruhn C., Brockmöller J., Kerb R., Roots I., Borchert H.H.. Concordance between enzyme activity and genotype of glutathione S-transferase theta (GSTT1). Biochem. Pharmacol. 1998; 56:1189–1193. [DOI] [PubMed] [Google Scholar]
- 25. Liu Y., Ding J., Reynolds L.M., Lohman K., Register T.C., De La Fuente A., Howard T.D., Hawkins G.A., Cui W., Morris J. et al. Methylomics of gene expression in human monocytes. Hum. Mol. Genet. 2013; 22:5065–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reynolds L.M., Taylor J.R., Ding J., Lohman K., Johnson C., Siscovick D., Burke G., Post W., Shea S., Jacobs D.R. et al. Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat. Commun. 2014; 5:5366–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Olsson A.H., Volkov P., Bacos K., Dayeh T., Hall E., Nilsson E.A., Ladenvall C., Rönn T., Ling C.. Genome-wide associations between genetic and epigenetic variation influence mRNA expression and insulin secretion in human pancreatic islets. PLoS Genet. 2014; 10:e1004735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vucic E.A., Chari R., Thu K.L., Wilson I.M., Cotton A.M., Kennett J.Y., Zhang M., Lonergan K.M., Steiling K., Brown C.J. et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am. J. Respir. Cell Mol. Biol. 2014; 50:912–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wagner J.R., Busche S., Ge B., Kwan T., Pastinen T., Blanchette M.. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014; 15:R37–R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heyn H., Moran S., Hernando-Herraez I., Sayols S., Gomez A., Sandoval J., Monk D., Hata K., Marques-Bonet T., Wang L. et al. DNA methylation contributes to natural human variation. Genome Res. 2013; 23:1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lehne B., Drong A.W., Loh M., Zhang W., Scott W.R., Tan S.-T., Afzal U., Scott J., Jarvelin M.-R., Elliott P. et al. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol. 2015; 16:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Y., Lemire M., Choufani S., Butcher D.T., Grafodatskaya D., Zanke B.W., Gallinger S., Hudson T.J., Weksberg R.. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013; 8:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Feber A., Guilhamon P., Lechner M., Fenton T., Wilson G.A., Thirlwell C., Morris T.J., Flanagan A.M., Teschendorff A.E., Kelly J.D. et al. Using high-density DNA methylation arrays to profile copy number alterations. Genome Biol. 2014; 15:R30–R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morris T.J., Butcher L.M., Feber A., Teschendorff A.E., Chakravarthy A.R., Wojdacz T.K., Beck S.. Champ: 450k chip analysis methylation pipeline. Bioinformatics. 2014; 30:428–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morris T.J., Beck S.. Analysis pipelines and packages for Infinium HumanMethylation450 BeadChip (450k) data. Methods. 2015; 72:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bedoyan J.K., Lesperance M.M., Ackley T., Iyer R.K., Innis J.W., Misra V.K.. A complex 6p25 rearrangement in a child with multiple epiphyseal dysplasia. Am. J. Med. Genet. A. 2011; 155A:154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aigner J., Villatoro S., Rabionet R., Roquer J., Jiménez-Conde J., Martí E., Estivill X.. A common 56-kilobase deletion in a primate-specific segmental duplication creates a novel butyrophilin-like protein. BMC Genet. 2013; 14:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shukla S.A., Rooney M.S., Rajasagi M., Tiao G., Dixon P.M., Lawrence M.S., Stevens J., Lane W.J., Dellagatta J.L., Steelman S. et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol. 2015; 33:1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cancer Genome Atlas Research Network Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell. 2017; 171:950–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cherniack A.D., Shen H., Walter V., Stewart C., Murray B.A., Bowlby R., Hu X., Ling S., Soslow R.A., Broaddus R.R. et al. Integrated molecular characterization of uterine carcinosarcoma. Cancer Cell. 2017; 31:411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hoadley K.A., Yau C., Hinoue T., Wolf D.M., Lazar A.J., Drill E., Shen R., Taylor A.M., Cherniack A.D., Thorsson V. et al. Cell-of-origin patterns dominate the molecular classification of 10, 000 tumors from 33 types of cancer. Cell. 2018; 173:291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scholz R.B., Kabisch H., Weber B., Röser K., Delling G., Winkler K.. Studies of the RB1 gene and the p53 gene in human osteosarcomas. Pediatr. Hematol. Oncol. 1992; 9:125–137. [DOI] [PubMed] [Google Scholar]
- 43. Zhao S., Bellone S., Lopez S., Thakral D., Schwab C., English D.P., Black J., Cocco E., Choi J., Zammataro L. et al. Mutational landscape of uterine and ovarian carcinosarcomas implicates histone genes in epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:12238–12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cancer Genome Atlas Research Network Integrated genomic characterization of oesophageal carcinoma. Nature. 2017; 541:169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ma X., Wang Y.-W., Zhang M.Q., Gazdar A.F.. DNA methylation data analysis and its application to cancer research. Epigenomics. 2013; 5:301–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ogunbiyi O.A., Goodfellow P.J., Herfarth K., Gagliardi G., Swanson P.E., Birnbaum E.H., Read T.E., Fleshman J.W., Kodner I.J., Moley J.F.. Confirmation that chromosome 18q allelic loss in colon cancer is a prognostic indicator. J. Clin. Oncol. 1998; 16:427–433. [DOI] [PubMed] [Google Scholar]
- 47. Fearon E.R., Cho K.R., Nigro J.M., Kern S.E., Simons J.W., Ruppert J.M., Hamilton S.R., Preisinger A.C., Thomas G., Kinzler K.W.. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science. 1990; 247:49–56. [DOI] [PubMed] [Google Scholar]
- 48. Fults D., Pedone C.. Deletion mapping of the long arm of chromosome 10 in glioblastoma multiforme. Genes Chromosomes Cancer. 1993; 7:173–177. [DOI] [PubMed] [Google Scholar]
- 49. Maksimovic J., Gagnon-Bartsch J.A., Speed T.P., Oshlack A.. Removing unwanted variation in a differential methylation analysis of Illumina HumanMethylation450 array data. Nucleic Acids Res. 2015; 43:e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Du P., Zhang X., Huang C.-C., Jafari N., Kibbe W.A., Hou L., Lin S.M.. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010; 11:587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hasin Y., Seldin M., Lusis A.. Multi-omics approaches to disease. Genome Biol. 2017; 18:83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shen H., Laird P.W.. Interplay between the cancer genome and epigenome. Cell. 2013; 153:38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jaffe A.E., Irizarry R.A.. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014; 15:R31–R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gordon D.J., Resio B., Pellman D.. Causes and consequences of aneuploidy in cancer. Nat. Rev. Genet. 2012; 13:189–203. [DOI] [PubMed] [Google Scholar]
- 55. Zack T.I., Schumacher S.E., Carter S.L., Cherniack A.D., Saksena G., Tabak B., Lawrence M.S., Zhsng C.-Z., Wala J., Mermel C.H. et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013; 45:1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Beroukhim R., Mermel C.H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J.S., Dobson J., Urashima M. et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010; 463:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Houseman E.A., Christensen B.C., Karagas M.R., Wrensch M.R., Nelson H.H., Wiemels J.L., Zheng S., Wiencke J.K., Kelsey K.T., Marsit C.J.. Copy number variation has little impact on bead-array-based measures of DNA methylation. Bioinformatics. 2009; 25:1999–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ramsuran V., Kulkarni S., O’huigin C., Yuki Y., Augusto D.G., Gao X., Carrington M.. Epigenetic regulation of differential HLA-A allelic expression levels. Hum. Mol. Genet. 2015; 24:4268–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Majumder P., Boss J.M.. DNA methylation dysregulates and silences the HLA-DQ locus by altering chromatin architecture. Genes Immun. 2011; 12:291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liu J., Siegmund K.D.. An evaluation of processing methods for HumanMethylation450 BeadChip data. BMC Genomics. 2016; 17:469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Laird P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010; 11:191–203. [DOI] [PubMed] [Google Scholar]
- 62. Dedeurwaerder S., Defrance M., Bizet M., Calonne E., Bontempi G., Fuks F.. A comprehensive overview of Infinium HumanMethylation450 data processing. Brief. Bioinform. 2014; 15:929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fortin J.-P., Labbe A., Lemire M., Zanke B.W., Hudson T.J., Fertig E.J., Greenwood C.M., Hansen K.D.. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014; 15:503–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The detection calling methods we presented have been implemented in the R package SeSAMe freely available on Bioconductor and at https://github.com/zwdzwd/sesame.