


Abstract
Initiation of bacterial translation requires that the ribosome-binding site in mRNAs adopts single-stranded conformations. In Gram-negative bacteria the ribosomal protein S1 (rS1) is a key player in resolving of structured elements in mRNAs. However, the exact mechanism of how rS1 unfolds persistent secondary structures in the translation initiation region (TIR) is still unknown. Here, we show by NMR spectroscopy that Vibrio vulnificus rS1 displays a unique architecture of its mRNA-binding domains, where domains D3 and D4 provide the mRNA-binding platform and cover the nucleotide binding length of the full-length rS1. D5 significantly increases rS1’s chaperone activity, although it displays structural heterogeneity both in isolation and in presence of the other domains, albeit to varying degrees. The heterogeneity is induced by the switch between the two equilibrium conformations and is triggered by an order-to-order transition of two mutually exclusive secondary structures (β-strand-to-α-helix) of the ‘AERERI’ sequence. The conformational switching is exploited for melting of structured 5′-UTR’s, as the conformational heterogeneity of D5 can compensate the entropic penalty of complex formation. Our data thus provides a detailed understanding of the intricate coupling of protein and RNA folding dynamics enabling translation initiation of structured mRNAs.
INTRODUCTION
RNA chaperones form a sequentially and structurally diverse class of proteins. They are important for promoting many RNA-regulated cellular processes (1). By destabilizing secondary structures of RNA (2,3) they accelerate RNA refolding and thus resolve RNAs trapped in misfolded, non-functional conformations. In contrast to RNA helicases, RNA chaperones do not require an external energy source. Early on, it was suggested that RNA chaperones decrease energy barriers between non-native RNA structures and thus assist the conformational search of RNAs for their native fold (4,5). The driving force for the unfolding reaction, however, has remained elusive. An ‘entropy transfer’ model was proposed, in which disordered regions—as frequently found in RNA and in protein chaperones – serve as energy reservoir. According to this model, binding induces a disorder-to-order transition within the chaperone to compensate the order-to-disorder transition of the substrate (6).
RNA chaperone activity is abundant in all organisms and necessary for many RNA-binding proteins required for RNA function, transcription and decay. One prime example is the ribosomal protein S1 (rS1) that exhibits RNA chaperone activity. It is associated to the small ribosomal subunit (30S), where it locates within the cleft between the head and platform on the solvent accessible side of the ribosome (7–9). Particularly, it is close to the 3′-end of the 16S rRNA and thus spatially close to the anti-Shine-Dalgarno (anti-SD) sequence (10). Here, 5′-untranslated regions (5′-UTRs) with their complementary SD-sequence are positioned during translation initiation (11). During translation initiation, proper positioning of the start codon AUG is crucial for setting the correct reading frame. Typically, the positioning for the correct reading frame is guided by ribosomal RNA provided that a single-stranded and strong SD-sequence is present in the mRNA transcript (12–14). However, productive binding to the 16S rRNA is often hampered, as either the SD-sequence found in the mRNA displays poor complementarity or is sequestered in intra-molecular base pairing interactions (15). In these cases, the rS1 is pivotal for translation initiation (16,17).
rS1 has a modular structure. In Gram-negative bacteria it generally consists of altogether six imperfect oligonucleotide binding fold (OB fold) motifs (16,18). These OB fold motifs constitute a five-stranded antiparallel β-barrel composed of ∼75 amino acids and preferentially bind to single-stranded RNAs (19,20). They are connected via linkers of 10–15 amino acids that provide the rS1 with the flexibility and the adaptability needed for recognition of miscellaneous mRNAs. Furthermore, the motifs vary in sequence and thereby tune the biological function of each domain. The two N-terminal domains (D1-D2) function as platform for ribosome-binding through protein-protein interactions with the ribosomal protein S2 (8,21), while the four C-terminal domains (D3-D6) harbor the mRNA-binding site (16). Thus, rS1 contains a protein- and an RNA-binding site in a single protein, mediating interactions between proteins and RNAs. In line with this ability, rS1 is known to activate the RegB endonuclease of the T4 bacteriophage (22,23) and also acts as a host factor during infection with Qβ phage as an integral part of the replication machinery (24,25). Moreover, as the majority of initiation in prokaryotes requires accessibility of both, the SD sequence and the AUG start codon to the ribosome (26,27), the central role of rS1 is to recruit single-stranded regions within the 5′-UTR of mRNA transcripts. Recently, it was shown that rS1 is also critically needed in the establishment of translation by polysomes (28).
Generally, mRNA-based regulation of translation in bacteria can proceed via three different mechanisms: masking of the ribosomal binding site (RBS), preventing accommodation of the mRNA into the decoding channel or indirect competition due to steric clashes (29). These three mechanisms have in common that the initiation region of the mRNA is involved in molecular interaction either in trans with other RNAs and proteins or in cis with sequences upstream from the initiation codon (30–32). To drive translation of highly structured mRNAs that either mask the RBS or prevent accommodation, rS1’s ability to melt local secondary structures is exploited (33). This function is essential as rS1 deletion is lethal to prokaryotic cells (34).
Here, we characterize the interaction of the rS1 protein from the human pathogenic bacterium Vibrio vulnificus with the structured 5′-UTR of the adenine-sensing riboswitch (Asw) from the same organism (35). The riboswitch controls the translation of the adenosine deaminase from the add gene and modulates the expression levels by a novel three-state-mechanism enabling a temperature-compensated regulation of translation by the ligand adenine (36–41). The regulation involves the switch between sequestration and liberation of the ribosome-binding site.
Here, we show for rS1 from V. vulnificus by activity assays and solution NMR that the core region for the mRNA interaction is composed of domains D3, D4 and D5 and that these domains contribute differently in the interplay with structured mRNAs. Domains D3 and D4 adopt stable OB folds and provide a platform for mRNA-binding. In sharp contrast, domain D5 adopts an intrinsically bistable fold that exhibits a conformational equilibrium between a predominantly structured (D5OB) and a predominantly unstructured (D5res-α) state. We find that conformational switching is triggered by a β-strand-to-α-helix transition, thus involves an order-to-order transition on the secondary structure level, leading to a domain-wide order-to-disorder transition. Our study provides insights into how the rS1 exploits its modular domain architecture to enable translation of structured RNAs, by locally melting RNA secondary structure.
MATERIALS AND METHODS
Expression and purification of rS1 constructs
All proteins were cloned with NcoI and BamHI as restriction sites into an in-house modified pKM260 vector (pKM-TX). They were expressed as fusion proteins in BL21(DE3) cells carrying a N-terminal polyhistidine (His6) and thioredoxin (trx) tag (His6-trx-TEV-rS1construct, see also Supplementary Table S1). For isotope labeling cells were grown in M9 medium supplemented with 1 g/l 15NH4Cl and either 2 g/l 13C-glucose or 4–10 g/l 12C-glucose. Expression was induced at OD600 = 0.6–0.8 with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). Proteins were expressed over night at 16–20°C and were purified via HisTrap HP (GE Healthcare) columns using 50 mM Tris/HCl (pH 8), 300 mM NaCl, 10 mM imidazol and 10 mM β-mercapto-ethanol as lysis buffer. The tag was cleaved using TEV (tabacco etch virus) protease and removed via HisTrap HP column. All proteins were further purified via size exclusion chromatography (Superdex 75 or Superdex 200 columns, 26/60, GE Healthcare) with the NMR buffer 25 mM potassium phosphate (pH 7.2) 150 mM KCl, 5 mM DTT. In case of rS1-D4 low yields were obtained in the soluble fraction, hence the size exclusion chromatography was omitted and instead the rS1-D4 was transferred into NMR buffer using PD-10 columns (GE Healthcare).
RNA preparation
The short RNA fragments Asw-6 to Asw-14 were purchased from Dharmacon carrying 2′-ACE protecting groups. They were deprotected and desalted using the manufacturers’ protocol. Lyophilized RNA was reconstituted with NMR buffer (25 mM potassium phosphate (pH 7.2) 150 mM KCl, 5 mM DTT).
Full-length Asw (112 nt) was transcribed with a 5′-hammerhead ribozyme from PCR template via in vitro transcription as described in (39). Asw-42 was transcribed without ribozyme and with an additional G at the 5′-end to enhance transcription yield. The sequences are listed in Supplementary Table S2. For both RNAs 3′-homogeneity was achieved following the instructions in (42). The RNAs were purified either by HPLC or PAGE and were refolded by thermal denaturation at 95°C, subsequent tenfold dilution with ice cold water and incubation on ice for 1 h. They were exchanged into NMR buffer (25 mM potassium phosphate (pH 7.2), 150 mM KCl, 5 mM DTT) using centrifugal concentrators (Sartorius AG).
Electrophoretic mobility shift assay (EMSA)
The EMSAs were performed at constant RNA concentration (4 μM) and increasing amounts of protein (0–25 equivalents). All samples were prepared in EMSA buffer (25 mM potassium phosphate (pH 7.2), 75 mM KCl, 5 mM DTT) and were incubated for 30 minutes on ice. Bands were separated on discontinuous gels (6%/12% PAGE) at low powers (<500 mW, 3.5–5.0 V/cm) in a Tris-acetate buffer (50 mM Tris (pH 8.3), 100 mM sodium acetate). The band separation was optimized for each rS1-construct in order to allow migration of the RNP and the free RNA into the gel (rS1: 500 mW, 14 h; rS1-D3456: 250 mW, 14 h; rS1-D345: 250 mW, 14 h; rS1-D34: 500 mW, 7 h; rS1-D45: 500 mW, 7 h). The RNA was visualized upon staining with GelRed® (Biotium) and subsequent excitation of the bands at 254 nm.
CD RNA chaperone assay
RNA chaperone assays were performed on a JASCO J-810 spectrophotometer using a 1 mm path length cuvette screening a range from 220 to 320 nm. All CD experiments were acquired in NMR buffer (25 mM potassium phosphate (pH 7.2), 150 mM KCl, 5 mM DTT) at a Asw-42 concentration of 10 μM. The assays were performed at 10°C. 0–4 equivalents of the respective protein were stepwise added to the RNA. Each titration point was measured with ten scans and the data was smoothed. In case of rS1-D345 the chaperone assay was additionally performed at 25°C and 35°C. CD melting curve of Asw-42 was measured at a wavelength of 264 nm in a range from 10 to 95°C. Temperature dependent CD spectra of free Asw-42 were acquired in a range from 220 to 320 nm using three scans per temperature interval. The temperature ranged from 10°C to 65°C using 5°C intervals. The CD spectra were smoothed and evaluated using Microsoft Excel 2010 and SigmaPlot 11.0.
In vitro transcription-translation assay
The coupled transcription-translation assays were performed as described previously (43) using the shifted green fluorescent protein to monitor expression levels. The protein was synthesized from plasmid (pIVEX 2.3d) containing T7 regulatory elements, a strong RBS and the sequence of sGFP. To study the influence of rS1-constructs on translation efficiency, the assays were performed as duplicates with rS1-depleted ribosomes reconstituted in S100 extract. The respective rS1-proteins were added in equimolar ratios to the ribosome concentration. 25 μl reactions were performed in 96-well V-shape microplate (Greiner Bio-One, Frickenhausen, Germany) at 30°C for 1 h. 20 μl of the reaction was transferred in μClear® 384-well flat bottom microplates (Greiner Bio-One, Frickenhausen, Germany). The fluorescence measurements were performed with an Inifinite® 200 PRO plate reader (Tecan, Männedorf, Switzerland) using an excitation wavelength of 484 nm and emission wavelength of 510 nm.
NMR spectroscopy
All samples were prepared in NMR buffer (25 mM potassium phosphate (pH 7.2), 150 mM KCl, 5 mM DTT and 5–7% D2O). Trimethylsilylpropanoic acid (TSP) was used as chemical shift standard.
NMR experiments were performed on Bruker spectrometers (either 600, 800, 900 or 950 MHz; Rheinstetten, Germany) equipped with cryoprobes. Standard Bruker pulse sequences as distributed with Topspin 3.5 were used (44–51). Data was processed using Topspin 3.5 (Bruker, Rheinstetten, Germany) and analyzed with Sparky 3.114 (52). Assigned chemical shifts for rS1-D5 and rS1-D345 are deposited under BMRB accession codes 27489 and 27490, respectively.
All NMR titrations were performed at 35°C with protein concentrations of 100 μM. For each titration point 1H–15N-BEST-TROSY was acquired keeping all acquisition parameters constant. For rS1-D34 the RNAs were added stepwise in molar ratios of [RNA]:[protein]: 0, 0.1, 0.2, 0.3, 0.6, 1, 2, 4, 5.5 and the titration experiments were performed at 600 MHz. For rS1-D345 the titration was performed at 950 MHz with stepwise adding Asw-42 in molar ratios of [RNA]:[protein]: 0, 0.1, 0.2, 0.3, 0.6, 1, 2.
Chemical shift perturbations were calculated using 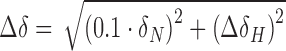 . Dissociation constants were determined as described in (53) by plotting largest CSP against the RNA concentration and fitting with
. Dissociation constants were determined as described in (53) by plotting largest CSP against the RNA concentration and fitting with 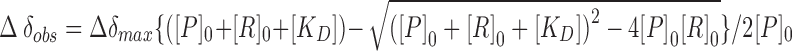 .
.
RESULTS
Delineation of mRNA-interaction-core of rS1 from V. vulnificus
In order to dissect the function of the individual mRNA-binding domains, we investigated several fragments of the binding region (D3–D6, Figure 1A) and studied their interaction with the adenine-sensing riboswitch. We used the 112 nucleotide long full-length add Asw and in addition a 42 nucleotide long fragment of this riboswitch ranging from G85 to U125 (from here on referred as Asw-42, see also Figure 1C). Asw-42 contains the translation initiation region (TIR) of the full-length Asw including the helix P5 followed by the SD sequence (GAA) and the start codon (AUG). This RNA provides a single-stranded 3′-end and an AU-rich sequence within the structured TIR of the riboswitch as constitutive binding platform for rS1 (54).
Figure 1.

RNA-binding and –chaperone activity of rS1 and its truncated versions. (A) Schematic overview of the rS1 constructs used in this study. Fragments are aligned according to their amino acid sequence. Sequence identities compared to domain D3 are displayed on the top. (B) Electrophoretic mobility shift assays (EMSAs) of selected rS1 constructs, using full-length add Asw (4 μM) as RNA substrate. Increasing protein concentrations are depicted on the top of the gels. The EMSAs were performed on discontinuous gels and only the resolving phases are displayed. Bands reporting on complex formation or free RNA are labeled with RNP or Asw, respectively. Two bands are observed for the free RNA, as the ligand-free riboswitch adopts two conformations (apoA and apoB) (38). In case of the full-length rS1 these states are also resolved for the RNP, since the EMSA was performed at higher powers in order to ensure migration of the complex into the resolving phase of the gel. In addition for the full-length rS1 a high-molecular species was observed (<5%) that did not migrate into the resolving phase of the discontinuous gel (not displayed). (C) Schematic overview of add Asw constructs. Asw-42 is highlighted in blue and the location of the P5 helix is indicated. The add gene from V. vulnificus including the transcription start site (TSS) and the open reading frame (ORF) is displayed on the top. (D) Thermal unfolding of Asw-42 (10 μM) to visualize the red-shift of CD maxima upon RNA melting. Temperature-dependent CD spectra of Asw-42 are shown. They were used as a benchmark for CD titration experiments reporting on RNA melting. (E) rS1-induced unfolding of Asw-42 (10 μM) as measured by CD spectra. All CD titration experiments were carried out at 10°C. Molar ratios of [protein]:[RNA] are depicted in the plots. The maximal shifts are displayed.
We probed RNA-binding ability for eight rS1 constructs (Figure 1A and B) with the full-length add Asw as RNA substrate in electrophoretic mobility shift assays (EMSAs). In line with previously reported results (16), the full-length rS1 binds the RNA with an apparent KDrS1 of 3 ± 1 μM. Deletion of the first two domains D1 and D2, responsible for ribosome-binding, results in proteins that retain the full RNA-binding ability. Removal of domain D6 does not significantly reduce the affinity towards RNA. However, deletion of D5 considerably decreases the RNA-binding affinity of rS1 (Figure 1B). Nevertheless, as RNA-binding to the two-domain protein rS1-D34 is detected this construct represents the minimal unit for mRNA-binding. In sharp contrast, no RNA-binding for rS1-D45 (Supplementary Figure S1) or for the single-domain fragments (D3, D4, D5) could be detected within the tested concentration range.
In order to evaluate the RNA-melting properties of the rS1 constructs, we performed CD titration experiments with Asw-42. Thermal unfolding was used as a benchmark for the single-stranded RNA state (Figure 1D and E). The observed red-shift and ellipticity decrease of the CD spectrum of Asw-42 in the presence of four equivalents of rS1 reports on considerable distortions of base pairing within the RNA und thus a protein-induced unfolding. This indicates purely single-stranded conformation of the RNA in the complex. The red-shift is of the same magnitude as observed during thermal unfolding of the RNA. The deletion of the ribosome-binding domains reduces the chaperone activity by 26%. Further deletion of domain D6 leads to no additional reduction in unwinding capacity. In line with the binding affinity, the two-domain construct rS1-D34 is still able to unwind the RNA but with significantly decreased capability. Therefore, the three-domain protein rS1-D345 contains the full chaperone activity of the mRNA-binding region. For this construct, the chaperone activity was also studied as a function of temperature (Supplementary Figure S2). We find that the stability of the RNA is decreased in the presence of four equivalents of rS1-D345 at all temperatures, where the largest destabilization is observed at 10°C (ΔΔG ≈ −3.7 kcal/mol). This further corroborates that the rS1 protein actively melts the RNA.
Consistent with reported results, our data show that even upon deletion of domain D6 and the ribosome-binding domains D1 and D2 mRNA-binding of rS1 is essentially retained (23). This observation emphasizes the exceptional importance of domains D3-D5 for the interplay with mRNAs; they constitute the mRNA-interaction core of rS1. Additional deletions within this core region lead to a severe loss of RNA-binding and chaperone activity. We find that deletion from the N-terminal end has a more dramatic effect, since removal of D3 completely diminishes the RNA-binding ability (Supplementary Figure S1). Furthermore, deletion of domain D5 from the core region significantly reduces the RNA-binding and chaperone activity, suggesting that domain D5 is also crucial in establishing the full magnitude of rS1–mRNA interaction. The binding behavior for V. vulnificus rS1 is therefore in stark contrast to that of E. coli rS1, where activity assays show that domain D4 and D5 are sufficient for mRNA-binding (23).
Delineation of the functional core of rS1
We further investigated the functional importance of rS1-domains for translation by use of in vitro transcription-translation assays (Supplementary Figure S3). Therefore, we studied the influence of the full-length rS1 and also its truncated mutants increasingly lacking the mRNA-binding domains on the expression of sGFP. In line with the EMSAs, the in vitro assays show that deletion of domain D6 does not significantly reduce the expression levels of sGFP, whereas additional deletion of domain D5 reduces the expression level to ∼77%. Further deletion of D4 reduces the sGFP expression to a basal level. Complete lack of mRNA-binding domains leads to inhibition of translation. These findings suggest that the mRNA-binding domains D3–D5 are necessary for efficient translation. Again a significant effect of D5 deletion on rS1 function is observed. Moreover, in case of Vibrio rS1 domains D3 and D4 need to act in tandem, since deletion of D4 diminishes the capability of rS1 to promote translation. In contrast, rS1-D12 inhibits translation probably by preventing ribosome-binding of residual E. coli rS1 that is present in the cell-free reaction mix.
NMR spectroscopic investigation of the mRNA-interaction core
The different rS1 constructs were structurally characterized by solution NMR spectroscopy (Figure 2A, see also Supplementary Figure S4), where the overall protein fold was probed by 1H–15N-BEST-TROSY experiments. Such 1H–15N correlation spectra serve as a fingerprint of the protein fold. A large dispersion of amide signals reports on well-folded proteins. Particularly, for proteins with high beta sheet contents the resonances are generally distributed over a wide ppm range
Figure 2.

Comparison of the mRNA-binding core domains by NMR and sequence. (A) 1H–15N-BEST-TROSY spectra of rS1 constructs as indicated. The spectrum of rS1-D345 was acquired at 35°C and 950 MHz. All other spectra were acquired at 30°C and 600 MHz. (B) Domain sequences were aligned using the Multiple Sequence Alignment tool from Clustal Omega (68–70). Sequence identities in comparison to domain D3 are displayed on the right. Secondary structure elements were computed using SWISS-MODEL (71–74) and are displayed on the top. Identical residues are highlighted.
Here, we find that the multi-domain constructs (rS1-D345, rS1-D34 and rS1-D45) dominantly exhibit well-dispersed resonances and the stepwise removal of domains does not substantially perturb the overall fold. The distribution of the amide signals indicates OB fold motifs. However, the D5 containing multi-domain proteins display severe resonance overlap around 8.5 ppm, pointing towards structural disorder within the respective constructs (Supplementary Figure S5). This crowding of signals can be addressed to domain D5. In isolation, this domain exhibits poorly dispersed resonances that are in addition broadened, indicative of a rather dynamic unstructured state. In stark contrast, the spectra of the isolated domains D3 and D4 display the characteristics of OB-fold motifs. Given the high homology of domains D3, D4 and D5 and further the fact that these are forming together the mRNA-binding core (Figures 1 and 2B), this finding was unexpected.
Further, the resonances of D3 and D4 can be unambiguously assigned and the chemical shifts can subsequently be interpreted as structural reporters of the respective protein structure. The superposition of the individual spectra results overall in the spectrum of the two-domain protein rS1-D34.
The mRNA-interaction-core of rS1 is thus organized in two regions. D3 and D4 together are capable to bind and unwind RNA, whereas domain D5 lacks a single well-defined structure. Nevertheless, its presence significantly increases the activity of the preceding domains. We thus decided to study both regions (rS1-D34 and rS1-D5) in separation, in order to gain insight into each functional contribution to the rS1–mRNA-interaction.
rS1-D34 harbors the mRNA-binding platform
First, we elucidated the RNA-binding behavior of rS1-D34 by solution NMR. We acquired 1H–15N-BEST-TROSY experiments of 15N-labeled rS1-D34 in the presence of increasing amounts of RNA. For our interaction studies we used six different RNAs of increasing length originating from the expression platform of the riboswitch (Table 1).
Table 1.
RNA-binding affinities of rS1-D34 for 5′-truncated Asw constructs obtained by NMR
| Sequence (5′-3′) | RNA length (nt) | K D a (μM) | ΔGfree-complex b (kcal/mol) | |
|---|---|---|---|---|
| Asw-42 | see Figure 1C | 42 | 13 ± 1 | −6.9 ± 0.1 |
| Asw-14 | 112-GAA GA CUC AUG AAU | 14 | 12 ± 1 | −6.9 ± 0.1 |
| Asw-12 | 114-A GA CUC AUG AAU | 12 | 27 ± 3 | −6.4 ± 0.1 |
| Asw-10 | 116-A CUC AUG AAU | 10 | 57 ± 6 | −5.9 ± 0.1 |
| Asw-8 | 118-UC AUG AAU | 8 | 197± 50 | −5.2 ± 0.2 |
| Asw-6 | 120-AUG AAU | 6 | >1000 | >−4.2 |
aThe KD values for the corresponding RNA lengths are listed and they were obtained by fitting largest CSPs with  (see also, Supplementary Figure S6). Errors were obtained from fitting procedure (53).
(see also, Supplementary Figure S6). Errors were obtained from fitting procedure (53).
bFree energy of complex formation was calculated as 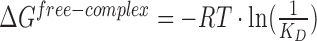 . Errors were calculated using Gaussian error propagation.
. Errors were calculated using Gaussian error propagation.
In addition to the structured Asw-42, we also included RNAs containing only the single-stranded 3′-end of the riboswitch (Asw-6 to Asw-14). They were used for probing the minimal nucleotide length within the RNA-binding site of rS1-D34.
We find that rS1-D34 binds the Asw-42 and Asw-14 with an apparent KD of 12.5 ± 1.5 μM, comparable to the affinities of full-length rS1. Furthermore, from our NMR titrations a minimal length for productive binding of 14 nucleotides can be inferred, as we do not observe an increase in binding affinity for the 42 nt long Asw-42 (see also, Supplementary Figure S6). Further, we find that rS1 preferentially binds to the single-stranded TIR of the riboswitch (Supplementary Figure S7).
Upon addition of increasing amounts of Asw-14 to rS1-D34 we observe large chemical shift perturbations (CSPs) affecting residues that are conserved within both domains. Most of the amide resonances display population-averaged resonances upon addition of the RNA (e.g. H230, R341), where the observed peak position shifts on a line between the free and the complexed protein state. Here, the exchange rate is larger than the frequency difference between the two states (kex > Δν), reporting on a predominantly fast exchanging system. However, the largest shifts (CSP > 0.2 ppm) are accompanied by severe line-broadening of the respective resonances (e.g. Y205, I220, R252, Y290, L303, V306, see also Figure 3A). For those amino acids, the amide signal is not detectable in the early course of titration, as here the larger frequency difference ranges in the same order of magnitude as the exchange rate (kex ∼ Δν). This is indicative of an intermediate exchange process between the free and bound state of the protein. Most of the resonances can be recovered at higher amounts of RNA, as increasing ligand concentrations shift the overall exchange rates towards ‘fast exchange’ conditions ( 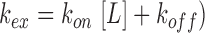 (55). From this, we can estimate a lower limit for the exchange rate between the free and Asw-14 bound rS1-D34 of kex ∼ 200 s−1. In case of L217, T221, V228, F248, D251, V255, S307 and N352 the resonances remain in intermediate exchange (Figure 3C).
(55). From this, we can estimate a lower limit for the exchange rate between the free and Asw-14 bound rS1-D34 of kex ∼ 200 s−1. In case of L217, T221, V228, F248, D251, V255, S307 and N352 the resonances remain in intermediate exchange (Figure 3C).
Figure 3.

Interaction of rS1-D34 with Asw-14. (A) Superposition of 1H-15N-BEST-TROSY acquired at 35°C and 600 MHz. NMR titration was performed with 100 μM 15N-labeled rS1-D34. Unlabeled Asw-14 was added stepwise with ratios ranging from 0 to 5.5 equivalents. The molar ratio ([RNA]:[protein]) is color coded within the superimposed spectra. (B) Cartoon representations of D3 and D4 model structures that were generated in SWISS-MODEL (71–74) using the PDB entry 2KHI (18) as template for homology modeling. Observed chemical shift perturbations of binding site reporters are plotted and color coded according to CSP values. The surface is displayed and solvent exposed residues are additionally shown as sticks. Surface binding site reporters are annotated and basic and aromatic residues are highlighted. (C) Chemical shift perturbations, calculated as described in the method section, between free and bound rS1-D34 are plotted as function of rS1-D34 sequence. Horizontal line indicates threshold value that was used to identify binding site reporters (53). Asterisks mark residues that are undetectable due to RNA-induced exchange broadening. Missing values represent either prolines or undetectable residues (G213, R250, W311, N313, N315 are exchange broadened at 35°C. D249, T253, K314, L346, A269 and C349 could not be assigned as the amide resonances were absent from 1H–15N-correlation spectra).
In line with reported results, our data show that the RNA-binding site involves strands β2, β3 and β5 and contains several key residues characterized by aromaticity or positive charges (1,20,56). In particular, Y205, R252, R254 in D3 and Y290, R341 in D4 display large perturbations in the presence of RNA (Figure 3A). The basic side chains enable charge compensation of the sugar-phosphate backbone, facilitating RNP formation directed by electrostatic interactions. The aromatic side chains of H230 and of Y205 and Y290 can engage in interactions with RNA nucleobases. Presumably, they interfere with base pairing interactions in the RNA and hence maintain residual chaperone activity within rS1-D34, as observed in the CD chaperoning assay. Furthermore, many amide resonances (e.g. I220, V306) distributed within the primary contact site experience large CSPs, due to RNA-induced structural rearrangements of the binding interface.
To investigate structural rearrangements and to probe for changes in the dynamical properties within rS1-D34 further, we acquired and analyzed heteronuclear NOEs, longitudinal and transverse 15N relaxation rates for rS1-D34 in its RNA free form and at saturating RNA concentrations (Supplementary Figure S8). In the absence of RNA both domains tumble together, as they exhibit similar global dynamics. The increase in R2 as well as the decrease in R1 rates in the presence of Asw-14 are indicative of a slower tumbling system and report on complex formation.
Taken together, in rS1-D34 both domains are structurally linked. They present a continuous surface for RNAs, where both domains equally contribute to RNA-binding, accommodating a stretch of 14 nucleotides. This finding is very conspicuous, as an RNA-binding length of 10–15 nucleotides has been reported for the full-length rS1 from E. coli (16,57). In other words, rS1-D34 already covers the size of RNA that the full-length rS1 is known to bind. This strongly supports the idea that domain D5 plays a different role in mRNA-interaction in V. vulnificus rS1.
rS1-D5 from V. vulnificus populates two distinct states
Domains D5 from V. vulnificus and E. coli rS1 display a sequence identity of 90% (Supplementary Figure S9). Both domains contain highly conserved amino acids, where altogether 64 out of 71 amino acids are identical and additional five are homologous. Aliprandi et al. state that even in isolation, the E. coli D5 homolog is well-folded, where the large dispersion of the resonances is in line with the predicted OB-fold motif (56). However, their NMR data evidence for the isolated domain a certain degree of heterogeneity. Further we now show, that the D5 from V. vulnificus exhibits structural disorder, not only present in the isolated domain but also observable in the respective multi-domain rS1 constructs. In order to evaluate the origins of the structural heterogeneity of domain D5, we analyzed rS1-D5 from V. vulnificus in depth by NMR.
We first acquired temperature-dependent 1H–15N-HSQC spectra of rS1-D5, screening a range from 8°C to 35°C (Figure 4, see also Supplementary Figure S10). Intriguingly, low temperatures revealed that for rS1-D5 two distinct conformational states are detectable. The major state (D5res-α) is populated to 92% (Supplementary Table S3) and displays the poor signal dispersion of a predominantly unstructured state. In contrast, the minor state, D5OB, exhibits well-dispersed resonances (Figure 4A) and is in overall agreement with the reported E. coli D5 backbone amide chemical shifts (56). Both D5 states interconvert on the millisecond timescale, causing the observed peak broadening at elevated temperatures.
Figure 4.

The two conformational states of rS1-D5. (A) 1H–15N-HSQC spectra were acquired at the displayed temperatures and at 900 MHz. (B) Annotated resonance assignment for D5res-α. The spectrum (acquired at 12°C and 900 MHz) is identical to the spectrum in panel (A) but to exclusively display the predominantly unstructured D5 state, the 1H–15N-HSQC is plotted at higher contour levels.
Conformational switch of sequence element induces domain heterogeneity
We analyzed the D5res-α state of rS1-D5 by multidimensional NMR. For rS1-D5 altogether 92 resonances in the 1H-15N-HSQC are expected, but at 12°C 154 backbone signals were visible. Using triple resonance experiments, we were able to assign 91 residues to the D5res-α state (Figure 4B). Under native conditions S373 to L383, D388 to S397 and additionally G385 and R407 were not assignable due to the lack of cross peaks in the 3D experiments. Nevertheless, we were able to transfer the respective assignments from non-native conditions using 8 M urea as denaturing agent. The assignment of the D5OB state could not be assured, since this state is at maximum populated to 8%. The signal pattern, however, indicates OB-fold formation.
The temperature series and urea titration experiments reveal residual secondary structure in the D5res-α state of rS1-D5 within residues D423 to G432 (Supplementary Figure S10). In order to identify the fold of proteins, chemical shifts of backbone atoms are utilized, as their values strongly correlate with local structure. Particularly, the deviations of the observed chemical shifts from random coil values are a powerful tool for identification of secondary structure from experimental data. We determined the residual structure in the predominantly unstructured D5res-α state by use of 1H, 15N and 13C secondary chemical shifts (58) and TALOS-N (59) (Figure 5). Our data identifies the existence of an α-helix spanning from A424 to I429. Within the predominantly folded state D5OB the fifth β-strand (E427 to L431) is located exactly in this region. This finding implies that the particular amino acid sequence ‘AERERI’ is capable of switching between two secondary structure elements, a β-strand and an α-helix. Notably, within the OB-fold motif the β5-strand is the closing strand that binds to both, β3 and β4 (Supplementary Figure S11). It potentially locks the anti-parallel β-sheet, following the pattern β3–β2–β1–β4–β5, into a β-barrel structure by forming a parallel β-sheet between strand β3 and β5. The β-strand-to-α-helix transition of this closing strand will inevitably have an impact on the structural integrity of the whole OB-fold motif.
Figure 5.

Structural characteristics of the D5res-α state. The chemical shifts of backbone atoms are sensitive for local structure. Their deviations from random coil values can be used for determination of secondary structure based on experimental data. In the upper plot the fractional secondary structure as calculated from TALOS-N is plotted against the protein sequence (59), where an α-helix is found between A424-I429. The lower plots display 1H, 15N and 13C secondary chemical shifts as a function of residue number. They were calculated as Δ = δobs − δrc (58), where δobs are the observed chemical shifts and δrc are the random coil chemical shifts. δrc were generated using the Javascript provided by Alex Maltsev, on the website of the University of Copenhagen (http://www1.bio.ku.dk/english/research/bms/research/sbinlab/groups/mak/randomcoil/script; 18 April 2018). The expected secondary structure element is indicated within the plot with arrows. All secondary chemical shift values indicate α-helical structure of the sequence stretch A421-G432. The largest values coincide with the AERERI sequence, strongly pointing towards α-helical structure of this particular sequence. In case of ΔCB and ΔN the secondary chemical shifts for N448 are truncated and have a value of 1.9 and 5.7, respectively. Asterisks mark not observed resonances. For clarity primary sequence and secondary structure elements of both rS1-D5 states are displayed on the very top of the figure.
We wondered whether the switching sequence exhibits an intrinsic preference for α-helical structures and thereby promotes the observed order-to-order transition from β-strand to α-helix. Therefore, we analyzed the secondary structure propensities of the switching sequence ‘AERERI’ that is highly conserved amongst gammaproteobacteria (Supplementary Figure S12). For comparison, the corresponding regions of D3 (‘RDRTRV’) and of D4 (‘EERRRI’) were also included into the analysis. We performed sequence-based secondary structure predictions and modeled peptide structures for all core domains (Supplementary Figures S13 and S14). Both prediction methods indicate a preference for α-helical structures for the switching regions of domains D3 to D5. Further, we compared the switching sequences of all core domains with protein structures deposited in the PDB. We were able to identify the respective sequences also in other proteins (right hand side of Supplementary Figure S13), where the peptide sequences dominantly adopt α-helical structures, supporting a general preference for this secondary structure element.
Taken together, the switching regions of all core domains display a tendency towards α-helical structures and this propensity seems to be more pronounced within D4 and D5. The transition to such an α-helix would inevitably destroy the integrity of the OB-fold, yet the bistability is only observed for D5.
Conformational heterogeneity of D5 is preserved in multi-domain constructs
We determined the populations of the two D5 states within the multi-domain rS1 constructs using different reporter signals, well resolved in both states. We chose G378, G389, G412 and G432 as reporters for structural order, as these glycines are located either directly within β-strands or in the direct vicinity. Their assignments within D5OB were achieved by homology, as these glycines are conserved also in D3 and D4. Furthermore, W354 and W398 indole resonances were used as indicators for domain-domain interactions. The unstructured states of both indole resonances could be unambiguously assigned, as W398 is the sole tryptophan within D5 (Figure 4) and W354 is located close to the C-terminus of rS1-D34 and hence is unstructured. Both resonances could be additionally correlated with those stemming from the folded state via an Nzz-exchange experiment using rS1-D345 (Supplementary Figure S15). Particularly, W354 and W398 side chain signals are sensitive reporters of D5 heterogeneity as they exhibit large signal intensities allowing identification of low populations of the D5res-α state even in the context of multi-domain constructs.
We find that the presence of preceding domains stepwise increases the population of the folded D5OB state. While in isolation domain D5 populates the D5OB state only by 8%, the presence of D4 in rS1-D45 leads to a population ratio close to equality (Figure 6E and Supplementary Table S3). Furthermore, the presence of both, D3 and D4 drives domain D5 almost exclusively into the D5OB state (82%). The stabilization of the D5OB state within the respective multi-domain constructs is probably induced by the order promoting properties of tryptophan side chain (60) as the indole moiety can participate in hydrophobic interactions and in hydrogen bonding (61).
Figure 6.

Structural heterogeneity of D5 in presence of preceding domains and RNA. (A) The 1H–15N-HSQC of rS1-D5 was acquired at 35°C and 950 MHz and an excerpt of tryptophan indole region is displayed to illustrate the res-α state. (B) and (C) displays excerpts of tryptophan indole region of rS1-D345 in the absence and presence of two equivalents Asw-42, respectively. For the NMR titration experiments, 1H–15N-BEST-TROSY (NS = 96) spectra were acquired at 35°C and 950 MHz. Unlabeled Asw-42 was added stepwise with ratios ranging from 0 to 2 equivalents. In the presence of RNA a huge intensity decrease of rS1-D345 resonances was observed. Hence, the titration end point was additionally acquired with more number of scans (NS = 256). This spectrum was used for determination of D5-populations and is displayed in panel (C). Positive 1D projections are displayed within the spectra. W-Hϵ1 resonances of D5res-αare annotated in red and of D5OB in blue. (D) Schematic overview of tryptophan-mediated domain stabilization. (E) The populations of the two D5 states are plotted against rS1 constructs. Populations of each reporter signal were determined from their intensities as 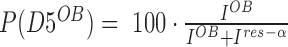 and vice versa. The populations were averaged over all reporter peaks for each rS1-construct. Error bars represent standard deviation of the average. (F) D5 population of rS1-D345 is plotted in the absence and presence (2 equivalents) of Asw-42. The rS1-D345-induced melting of Asw-42 was also monitored by NMR and is displayed in Supplementary Figure S7.
and vice versa. The populations were averaged over all reporter peaks for each rS1-construct. Error bars represent standard deviation of the average. (F) D5 population of rS1-D345 is plotted in the absence and presence (2 equivalents) of Asw-42. The rS1-D345-induced melting of Asw-42 was also monitored by NMR and is displayed in Supplementary Figure S7.
We classify the five tryptophans of rS1-D345 into two groups. Three ‘domain-tryptophans’ are conserved within domains D3 (W225), D4 (W311) and D5 (W398) and they are located in the internal loop between the β3-strand and the β4-strand. Two ‘linker-tryptophans’ (W267 and W354) are conserved within inter-domain linkers between D3–D4 or D4–D5, respectively (Figure 6D). The linker-tryptophans are part of a PWX-motif. This motif is also found in other nucleic acid-binding proteins, in which the PWX-motif packs against the side chain of a phenylalanine that is conserved in the protein core (62). In line, we find evidence that the linker-tryptophans as found here, interact with the preceding domain-tryptophans. In case of the W225–W267 pair we find direct cross peaks in the 1H-15N-NOESY-HSQC of rS1-D34 between the two tryptophan side chains and also between mutual neighboring amino acids (Supplementary Figure S16). Due to signal overlap, such direct NOE evidence for tryptophan-tryptophan interaction could not easily be detected for the larger constructs. However, the interaction can also be detected from chemical shift analysis. Comparison of rS1 constructs of different length reveals that succeeding domains (D4 in rS1-D34 and D5 in rS1-D345) lead to chemical shift perturbations within the internal loop between β3- and β4-strands of the preceding domain (D3 in rS1-D34 and D4 in rS1-D345). Largest CSPs cluster around the indole resonances of domain-tryptophans (Supplementary Figures S17 and S18).
Taken together, domain-tryptophans are engaged in interactions with linker-tryptophans forming hydrophobic clusters and thus guiding and positioning adjacent domains in close proximity. The formation of these two-tryptophan centered hydrophobic clusters are important for the protein-architecture, as they mediate the stabilization of rS1’s fold. The domain-domain contacts induce stabilization of the succeeding domain. Domain D3 is the most stable domain within rS1-D345 and we observe that it increases the stability of domain D4. Both domains together stabilize the OB-fold of domain D5. We do not observe any cooperative influence between domains D5 and D6. In line, domain D6 lacks the stability promoting tryptophan residues both in the linker connecting D5 and D6 as well as within the domain D6.
RNA-binding reintroduces conformational heterogeneity of D5
We investigated the properties of the extended RNA-binding core rS1-D345 upon interaction with Asw-42 by NMR titration experiments (Figure 6). We acquired a series of 1H-15N-BEST-TROSY spectra of rS1-D345 by stepwise increasing the RNA molar ratio from 0 to 2 equivalents. Already after the first titration point the average signal intensities decline at least by a factor of two and remain below 50% during the course of titration. We conclude that the interaction between rS1-D345 and Asw-42 is within the intermediate exchange regime. Therefore, the RNA-binding reporters (Y205, H230, Y290, R341) as found in rS1-D34 cannot be recovered even at higher RNA concentrations. Nevertheless, the overall chemical shift perturbations in domains D3 and D4 (Supplementary Figure S19) agree between the extended three-domain (rS1-D345) and the essential two-domain (rS1-D34) RNA-binding core.
The intensity ratios between the free and bound rS1-D345 state show that particularly for D3 and D4 the signal loss is most prominent (Supplementary Figure S19, left panel). With two equivalents of Asw-42 the average signal intensities of these two domains decrease to 37%. D5 resonances display average intensity ratios between the free and bound state >80%. Notably, the remaining D5 resonances do not display significant CSPs (Supplementary Figure S19, right panel). We observe that the presence of RNA unfolds the OB-fold of D5, as increased population of the res-α state is detected at saturating RNA concentrations (Figure 6C). This shift of the population ratio is most likely driven by the intrinsic property of the ‘AERERI’ sequence to adopt α-helical structures. It is presumably a direct consequence of RNA-binding that induces dissociation of domain D5 by competing out the domain-domain contacts between D4 and D5 leading to the β5-to-α transition as observed for the isolated D5 domain (Figure 6F).
DISCUSSION
We have investigated the structural basis of the RNA-binding and chaperone behavior of the rS1 protein from Vibrio vulnificus. The full-length protein composed of six homologues domains is able to melt RNA secondary structures upon binding. The minimal functional core is composed of domains D3 and D4, covering a stretch of 14 single-stranded nucleotides. We dissected the contribution of each of the four C-terminal domains to binding affinity and chaperone activity. While domain D6 is nearly dispensable, deletion of D5 renders both affinity and activity to a minimum level. Domain D3 and domain D4 form the protein's functional core and operate in tandem. In contrast, for the rS1 from E. coli it was proposed that individual domains consecutively bind the RNA and mediate melting by capturing single-stranded conformations in multiple small substeps (33).
Although, the rS1 from V. vulnificus displays high degree of sequence identity (∼86%) to its homolog from E. coli we find that the V. vulnificus rS1 protein displays remarkable structural differences. For all E. coli rS1 RNA-binding domains it is reported that they adopt structured OB-fold motifs. For the single-domains D4, D5 and D6 NMR structures were reported (18). Whereas the structures of D4 and D6 exhibit a tight bundle of conformations, the structure of D5 is rather loosely defined. Furthermore, it was found that within a fragment containing domains D3 to D5, D4 and D5 are in close contact forming a continuous interaction surface, while D3 is mainly dissociated from the other domains (18,56). Further, the minimal fragment for RegB activation is formed by D4 and D5 (23).
In contrast for the V. vulnificus rS1 only domains D3 and D4 exhibit a single stable and well-ordered conformation. For domain D5 an unusual structural behavior is found that is key to modulate RNA-binding. This domain can populate two conformational states. In one of the two equilibrium conformations D5 exhibits a well-structured OB-fold. In the second conformation, the local refolding of the sequence ‘AERERI’ from the OB-fold closing β5-strand that is essentially required to stabilize the OB-fold to α-helix leads to an overall unstructured state with local residual α-helical secondary structure. Therefore, D5 contains a switching sequence that by forming either the mutual exclusive α-helix or β-strand conformation triggers a full change of the domain's structure. This α-helix to β-strand conformational switch is reminiscent to the changes in transformer proteins, but with the exception that those are bistable systems of two fully folded conformations (63). As the switching sequence is highly conserved in γ-proteobacteria it might be a functional feature of rS1 proteins allowing domain 5 to act as dynamic joint between RNA binding domains 3 and 4 and regulatory domain 6. As the population of the two states will be affected by further interactions throughout the domain, the second conformation could be easily overlooked in proteins from organism other than V. vulnificus where the equilibrium is shifted more to the D5OB state.
The RNA chaperone activity of V. vulnificus rS1 is manifested in its ability to destabilize the secondary structure of RNAs. Whereas in the absence of the protein the expression platform module ASW-42 adopts predominantly its hairpin conformation (14% at 35°C), presence of rS1 induces a significant shift in population towards the single-stranded conformation (86% at 35°C) (Supplementary Figure S2). Delineated from the above described CD RNA chaperone assay, this can be expressed in terms of free energy between the hairpin and single-strand conformation that is changed by interaction with rS1 from ΔGhp-ss(35°C, 0 eq rS1-D345) = 1.1 kcal/mol to ΔGhp-ss(35°C, 4 eq rS1-D345) = –1.0 kcal/mol. Notably, the change towards the single-stranded conformation is strongly dependent on the stoichiometry (Figure S7) and the temperature (Figure S2). Whereas the supra-stoichiometric amounts of rS1 can lead to shifts in the equilibrium of ΔΔG(4 eq) = −2 kcal/mol, sub-stoichiometric amounts only contribute to ΔΔG(0.5 eq) = −0.7 kcal/mol. However, both changes significantly indicate the RNA melting capacity of rS1.
For the rS1 from V. vulnificus the RNA chaperone activity is intimately linked to the structural dynamics of the three domains of the mRNA interaction core. Upon RNA-binding domain D5 is released from its associated state, reflected in a twofold increase of the D5 res-α population from 18% to 36% (Figure 7). The displacement of D5 upon interaction is presumably of allosteric nature as the binding sites for the RNA and D5 onto the mRNA-binding platform D3–D4 only minimally overlap. As the release of D5 increases the structural heterogeneity in the protein, it can compensate the loss of entropy upon complex formation and promote the exothermic (ΔGfree-complex < –7 kcal/mol) binding reaction. Due to the conformational heterogeneity of D5, the resulting RNA-protein complex is a ‘fuzzy’ complex. The positive influence on affinity towards mRNA and the increased ability to stabilize single-stranded conformations (Supplementary Figure S7) in presence of D5 can be attributed to its dynamic disordered behavior in the res-α state. Such a remote effect of complex stabilization has already been documented for several protein-protein complexes. In these cases, the direct interaction is mediated by structured domains but dynamic disordered parts that do not contribute directly increase the affinity of complex formation manifold. A comparable example is the formation of a dynamic ‘fuzzy’ complex of the two protein domains KID and KIX, containing flanking disordered regions close to the interaction site (64). The kinase-inducible domain (KID) binds its interaction domain (KIX) by a segment of 29 amino-acids, while the rest of the protein stays disordered and does not take part in the direct interaction. However, deletion of the flanking disordered region decreases the affinity 5-fold (65).
Figure 7.

Intrinsic structural heterogeneity of domain D5. Schematic representation of the D5OB and D5res-α conformational states within the rS1 constructs (A) rS1-D5, (B) rS1-D45 and (C) rS1-D345. The populations are displayed in the scheme. (D) D5 populations in the absence and presence of Asw-42.
From an overall functional point of view, the protein rS1, as an RNA chaperone, shall be responsible to mediate the translation initiation by melting structured TIRs. Following Sabatier's principle for catalysts (66) it therefore needs to bind, unwind and subsequently release the mRNA chain without falling into an over stabilized conformational state (67). The intricate linkage of nearly equally stable protein and RNA conformational states is exactly what is needed for this function to be fulfilled.
DATA AVAILABILITY
Assigned chemical shifts for rS1-D5 and rS1-D345 are deposited under BMRB accession codes 27489 and 27490, respectively.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Hendrik R.A. Jonker and Christian Richter for help and insightful discussions. We further like to thank Elke Stirnal for excellent technical assistance and Jens Wöhnert for critical reading of the manuscript. Further, we would like to thank the research group of Volker Dötsch for kindly providing the pIVEX-sGFP plasmid.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German funding agency (DFG) in Collaborative Research Center 902: Molecular principles of RNA-based regulation. Work at BMRZ was supported by the state of Hesse and by iNEXT (to S. Sreeramulu). Funding for open access charge: DFG [SFB902].
Conflict of interest statement. None declared.
REFERENCES
- 1. Cristofari G., Darlix J.-L.. The ubiquitous nature of RNA chaperone proteins. Prog. Nucleic Acid Res. Mol. Biol. 2002; 72:223–268. [DOI] [PubMed] [Google Scholar]
- 2. Rajkowitsch L., Chen D., Stampfl S., Semrad K., Waldsich C., Mayer O., Jantsch M.F., Konrat R., Bläsi U., Schroeder R.. RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 2007; 4:118–130. [DOI] [PubMed] [Google Scholar]
- 3. Doetsch M., Fürtig B., Gstrein T., Stampfl S., Schroeder R.. The RNA annealing mechanism of the HIV-1 Tat peptide: Conversion of the RNA into an annealing-competent conformation. Nucleic Acids Res. 2011; 39:4405–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Herschlag D. RNA chaperones and the RNA folding problem. J. Biol. Chem. 1995; 270:20871–20874. [DOI] [PubMed] [Google Scholar]
- 5. Woodson S.A. Taming free energy landscapes with RNA chaperones. RNA Biol. 2010; 7:677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tompa P., Csermely P.. The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 2004; 18:1169–1175. [DOI] [PubMed] [Google Scholar]
- 7. Sengupta J., Agrawal R.K., Frank J.. Visualization of protein S1 within the 30S ribosomal subunit and its interaction with messenger RNA. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:11991–11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Byrgazov K., Grishkovskaya I., Arenz S., Coudevylle N., Temmel H., Wilson D.N., Djinovic-Carugo K., Moll I.. Structural basis for the interaction of protein S1 with the Escherichia coli ribosome. Nucleic Acids Res. 2015; 43:661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilson D.N., Nierhaus K.H.. Ribosomal proteins in the spotlight. Crit. Rev. Biochem. Mol. Biol. 2005; 40:243–267. [DOI] [PubMed] [Google Scholar]
- 10. Shine J., Dalgarno L.. The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc. Natl. Acad. Sci. U.S.A. 1974; 71:1342–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yusupova G.Z., Yusupov M.M., Cate J.H.D., Noller H.F.. The path of messenger RNA through the ribosome. Cell. 2001; 106:233–241. [DOI] [PubMed] [Google Scholar]
- 12. Kaminishi T., Wilson D.N., Takemoto C., Harms J.M., Kawazoe M., Schluenzen F., Hanawa-Suetsugu K., Shirouzu M., Fucini P., Yokoyama S.. A Snapshot of the 30S ribosomal subunit capturing mRNA via the Shine-Dalgarno interaction. Structure. 2007; 15:289–297. [DOI] [PubMed] [Google Scholar]
- 13. Yusupova G., Jenner L., Rees B., Moras D., Yusupov M.. Structural basis for messenger RNA movement on the ribosome. Nature. 2006; 444:391–394. [DOI] [PubMed] [Google Scholar]
- 14. Steitz J.A., Jakes K.. How ribosomes select initiator regions in mRNA: base pair formation between the 3′ terminus of 16S rRNA and the mRNA during initiation of protein synthesis in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1975; 72:4734–4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Studer S.M., Joseph S.. Unfolding of mRNA secondary structure by the bacterial translation initiation complex. Mol. Cell. 2006; 22:105–115. [DOI] [PubMed] [Google Scholar]
- 16. Subramanian A.R. Structure and functions of ribosomal protein S1. Prog. Nucleic Acid Res. Mol. Biol. 1983; 28:101–142. [DOI] [PubMed] [Google Scholar]
- 17. Kolb A., Hermoso J.M., Thomas J.O., Szer W.. Nucleic acid helix-unwinding properties of ribosomal protein S1 and the role of S1 in mRNA binding to ribosomes. Proc. Natl. Acad. Sci. U.S.A. 1977; 74:2379–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Salah P., Bisaglia M., Aliprandi P., Uzan M., Sizun C., Bontems F.. Probing the relationship between Gram-negative and Gram-positive S1 proteins by sequence analysis. Nucleic Acids Res. 2009; 37:5578–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murzin A.G. OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J. 1993; 12:861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Draper D.E., Reynaldo L.P.. RNA binding strategies of ribosomal proteins. Nucleic Acids Res. 1999; 27:381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Loveland A.B., Korostelev A.A.. Structural dynamics of protein S1 on the 70S ribosome visualized by ensemble cryo-EM. Methods. 2017; 2:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ruckman J., Ringquist S., Brody E., Gold L.. The bacteriophage T4 regB ribonuclease. Stimulation of the purified enzyme by ribosomal protein S1. J. Biol. Chem. 1994; 269:26655–26662. [PubMed] [Google Scholar]
- 23. Bisaglia M., Laalami S., Uzan M., Bontems F.. Activation of the RegB endoribonuclease by the S1 ribosomal protein is due to cooperation between the S1 four C-terminal modules in a substrate-dependant manner. J. Biol. Chem. 2003; 278:15261–15271. [DOI] [PubMed] [Google Scholar]
- 24. Wahba A.J., Miller M.J., Niveleau A., Landers T.A., Carmichael G.G., Weber K., Hawley D.A., Slobin L.I.. Subunit I of G beta replicase and 30 S ribosomal protein S1 of Escherichia coli. Evidence for the identity of the two proteins. J. Biol. Chem. 1974; 249:3314–3316. [PubMed] [Google Scholar]
- 25. Takeshita D., Yamashita S., Tomita K.. Molecular insights into replication initiation by Qβ replicase using ribosomal protein S1. Nucleic Acids Res. 2014; 42:10809–10822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Smit M.H., van Duin J.. Control of translation by mRNA secondary structure in Escherichia coli. A quantitative analysis of literature data. J. Mol. Biol. 1994; 244:144–150. [DOI] [PubMed] [Google Scholar]
- 27. de Smit M.H., van Duin J.. Translational initiation on structured messengers. Another role for the Shine-Dalgarno interaction. J. Mol. Biol. 1994; 235:173–184. [DOI] [PubMed] [Google Scholar]
- 28. Andreeva I., Belardinelli R., Rodnina M. V.. Translation initiation in bacterial polysomes through ribosome loading on a standby site on a highly translated mRNA. Proc. Natl. Acad. Sci. U.S.A. 2018; 0:201718029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Duval M., Simonetti A., Caldelari I., Marzi S.. Multiple ways to regulate translation initiation in bacteria: mechanisms, regulatory circuits, dynamics. Biochimie. 2015; 114:18–29. [DOI] [PubMed] [Google Scholar]
- 30. Serganov A., Nudler E.. A decade of riboswitches. Cell. 2013; 152:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kortmann J., Narberhaus F.. Bacterial RNA thermometers: molecular zippers and switches. Nat. Rev. Microbiol. 2012; 10:255–265. [DOI] [PubMed] [Google Scholar]
- 32. Nahvi A., Sudarsan N., Ebert M.S., Zou X., Brown K.L., Breaker R.R.. Genetic control by a metabolite binding mRNA. Chem. Biol. 2002; 9:1043–1049. [DOI] [PubMed] [Google Scholar]
- 33. Qu X., Lancaster L., Noller H.F., Bustamante C., Tinoco I.. Ribosomal protein S1 unwinds double-stranded RNA in multiple steps. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:14458–14463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Duval M., Korepanov A., Fuchsbauer O., Fechter P., Haller A., Fabbretti A., Choulier L., Micura R., Klaholz B.P., Romby P. et al. Escherichia coli ribosomal protein S1 unfolds structured mRNAs onto the ribosome for active translation initiation. PLoS Biol. 2013; 11:12–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mandal M., Breaker R.R.. Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat. Struct. Mol. Biol. 2004; 11:29–35. [DOI] [PubMed] [Google Scholar]
- 36. Rieder R., Lang K., Graber D., Micura R.. Ligand-induced folding of the adenosine deaminase A-riboswitch and implications on riboswitch translational control. ChemBioChem. 2007; 8:896–902. [DOI] [PubMed] [Google Scholar]
- 37. Neupane K., Yu H., Foster D.A.N., Wang F., Woodside M.T.. Single-molecule force spectroscopy of the add adenine riboswitch relates folding to regulatory mechanism. Nucleic Acids Res. 2011; 39:7677–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reining A., Nozinovic S., Schlepckow K., Buhr F., Fürtig B., Schwalbe H.. Three-state mechanism couples ligand and temperature sensing in riboswitches. Nature. 2013; 499:355–359. [DOI] [PubMed] [Google Scholar]
- 39. Warhaut S., Mertinkus K.R., Hollthaler P., Furtig B., Heilemann M., Hengesbach M., Schwalbe H.. Ligand-modulated folding of the full-length adenine riboswitch probed by NMR and single-molecule FRET spectroscopy. Nucleic Acids Res. 2017; 45:5512–5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lemay J.F., Desnoyers G., Blouin S., Heppell B., Bastet L., St-Pierre P., Massé E., Lafontaine D.A.. Comparative study between transcriptionally- and translationally-acting adenine riboswitches reveals key differences in riboswitch regulatory mechanisms. PLoS Genet. 2011; 7:e1001278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tian S., Kladwang W., Das R.. Allosteric mechanism of the V. vulnificus adenine riboswitch resolved by four-dimensional chemical mapping. Elife. 2018; 7:1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Helmling C., Keyhani S., Sochor F., Fürtig B., Hengesbach M., Schwalbe H.. Rapid NMR screening of RNA secondary structure and binding. J. Biomol. NMR. 2015; 63:67–76. [DOI] [PubMed] [Google Scholar]
- 43. Kai L., Dötsch V., Kaldenhoff R., Bernhard F.. Artificial environments for the Co-translational stabilization of cell-free expressed proteins. PLoS One. 2013; 8:e56637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mori S., Abeygunawardana C., Johnson M.O., van Zijl P.C.. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B. 1995; 108:94–98. [DOI] [PubMed] [Google Scholar]
- 45. Favier A., Brutscher B.. Recovering lost magnetization: polarization enhancement in biomolecular NMR. J. Biomol. NMR. 2011; 49:9–15. [DOI] [PubMed] [Google Scholar]
- 46. Schulte-Herbrüggen T., Sørensen O.W.. Clean TROSY: compensation for Relaxation-Induced Artifacts. J. Magn. Reson. 2000; 144:123–128. [DOI] [PubMed] [Google Scholar]
- 47. Solyom Z., Schwarten M., Geist L., Konrat R., Willbold D., Brutscher B.. BEST-TROSY experiments for time-efficient sequential resonance assignment of large disordered proteins. J. Biomol. NMR. 2013; 55:311–321. [DOI] [PubMed] [Google Scholar]
- 48. Bodenhausen G., Ruben D.J.. Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 1980; 69:185–189. [Google Scholar]
- 49. Piotto M., Saudek V., Sklenár V.. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR. 1992; 2:661–665. [DOI] [PubMed] [Google Scholar]
- 50. Sklenar V., Piotto M., Leppik R., Saudek V.. Gradient-tailored water suppression for 1H-15N HSQC experiments optimized to retain full sensitivity. J. Magn. Reson. Ser. A. 1993; 102:241–245. [Google Scholar]
- 51. Lescop E., Schanda P., Brutscher B.. A set of BEST triple-resonance experiments for time-optimized protein resonance assignment. J. Magn. Reson. 2007; 187:163–169. [DOI] [PubMed] [Google Scholar]
- 52. Lee W., Tonelli M., Markley J.L.. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics. 2015; 31:1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Williamson M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013; 73:1–16. [DOI] [PubMed] [Google Scholar]
- 54. Hajnsdorf E., Boni I.V.. Multiple activities of RNA-binding proteins S1 and Hfq. Biochimie. 2012; 94:1544–1553. [DOI] [PubMed] [Google Scholar]
- 55. Kovrigin E.L. NMR line shapes and multi-state binding equilibria. J. Biomol. NMR. 2012; 53:257–270. [DOI] [PubMed] [Google Scholar]
- 56. Aliprandi P., Sizun C., Perez J., Mareuil F., Caputo S., Leroy J.L., Odaert B., Laalami S., Uzan M., Bontems F.. S1 ribosomal protein functions in translation initiation and ribonuclease RegB activation are mediated by similar RNA-protein interactions: an NMR and SAXS analysis. J. Biol. Chem. 2008; 283:13289–13301. [DOI] [PubMed] [Google Scholar]
- 57. Lipecky R., Kohlschein J., Gassen H.G.. Complex formation between ribosomal protein S1, oligo-and polynucleotides: chain length dependence and base specificity. Nucleic Acids Res. 1977; 4:3627–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mielke S.P., Krishnan V.V.. Characterization of protein secondary structure from NMR chemical shifts. Prog. Nucl. Magn. Reson. Spectrosc. 2009; 54:141–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shen Y., Bax A.. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR. 2013; 56:227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Uversky V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta - Proteins Proteomics. 2013; 1834:932–951. [DOI] [PubMed] [Google Scholar]
- 61. Samanta U., Pal D., Chakrabarti P.. Environment of tryptophan side chains in proteins. Proteins Struct. Funct. Genet. 2000; 38:288–300. [PubMed] [Google Scholar]
- 62. Szymczyna B.R., Bowman J., McCracken S., Pineda-Lucena A., Lu Y., Cox B., Lambermon M., Graveley B.R., Arrowsmith C.H., Blencowe B.J.. Structure and function of the PWI motif: a novel nucleic acid-binding domain that facilitates pre-MRNA processing. Genes Dev. 2003; 17:461–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Burmann B.M., Knauer S.H., Sevostyanova A., Schweimer K., Mooney R.A., Landick R., Artsimovitch I., Rösch P.. An α helix to β barrel domain switch transforms the transcription factor RfaH into a translation factor. Cell. 2012; 150:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tompa P., Fuxreiter M.. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008; 33:2–8. [DOI] [PubMed] [Google Scholar]
- 65. Zor T., Mayr B.M., Jane Dyson H., Montminy M.R., Wright P.E.. Roles of phosphorylation and helix propensity in the binding of the KIX domain of CREB-binding protein by constitutive (c-Myb) and inducible (CREB) activators. J. Biol. Chem. 2002; 277:42241–42248. [DOI] [PubMed] [Google Scholar]
- 66. Roduner E. Understanding catalysis. Chem. Soc. Rev. 2014; 43:8226–8239. [DOI] [PubMed] [Google Scholar]
- 67. Espah Borujeni A., Channarasappa A.S., Salis H.M.. Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res. 2014; 42:2646–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011; 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. McWilliam H., Li W., Uludag M., Squizzato S., Park Y.M., Buso N., Cowley A.P., Lopez R.. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 2013; 41:W597–W600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li W., Cowley A., Uludag M., Gur T., McWilliam H., Squizzato S., Park Y.M., Buso N., Lopez R.. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015; 43:W580–W584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Cassarino T.G., Bertoni M., Bordoli L. et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014; 42:W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Benkert P., Biasini M., Schwede T.. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011; 27:343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Guex N., Peitsch M.C., Schwede T.. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009; 30:S162–S173. [DOI] [PubMed] [Google Scholar]
- 74. Bienert S., Waterhouse A., de Beer T.A.P., Tauriello G., Studer G., Bordoli L., Schwede T.. The SWISS-MODEL Repository—new features and functionality. Nucleic Acids Res. 2017; 45:D313–D319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Assigned chemical shifts for rS1-D5 and rS1-D345 are deposited under BMRB accession codes 27489 and 27490, respectively.