


Abstract
Bacterial small RNAs (sRNAs) are a heterogeneous group of post-transcriptional regulators that often act at the heart of large networks. Hundreds of sRNAs have been discovered by genome-wide screens and most of these sRNAs exert their functions by base-pairing with target mRNAs. However, studies addressing the molecular roles of sRNAs have been largely confined to gamma-proteobacteria, such as Escherichia coli. Here we identify and characterize a novel sRNA, ChvR, from the alpha-proteobacterium Caulobacter crescentus. Transcription of chvR is controlled by the conserved two-component system ChvI-ChvG and it is expressed in response to DNA damage, low pH, and growth in minimal medium. Transient over-expression of ChvR in combination with genome-wide transcriptome profiling identified the mRNA of the TonB-dependent receptor ChvT as the sole target of ChvR. Genetic and biochemical analyses showed that ChvR represses ChvT at the post-transcriptional level through direct base-pairing. Fine-mapping of the ChvR-chvT interaction revealed the requirement of two distinct base-pairing sites for full target regulation. Finally, we show that ChvR-controlled repression of chvT is independent of the ubiquitous RNA-chaperone Hfq, and therefore distinct from previously reported mechanisms employed by prototypical bacterial sRNAs. These findings have implications for the mechanism and evolution of sRNA function across bacterial species.
INTRODUCTION
The ability of bacteria to survive and grow in constantly changing conditions requires them to continuously adapt to their physical and chemical environments. The primary way in which bacteria adapt to different conditions is by altering their gene expression profiles, which they achieve by a complex interplay of transcriptional, post-transcriptional, translational, and post-synthetic processes. The most common molecular sensors in bacteria are two-component signal transduction systems (TCSs), which often span the bacterial inner membrane to translate external stimuli into intracellular, regulatory responses (1). Typically, bacterial TCSs consist of a membrane-inserted sensor kinase, which relays an external signal to a cognate cytoplasmic response regulator. When activated, most response regulators bind to genomic promoter elements, acting as transcriptional modulators (2). At the post-transcriptional level, bacteria frequently employ small, regulatory RNAs (sRNAs) to fine-tune gene expression. This versatile class of heterogeneously sized and structured RNA molecules predominantly acts by direct base-pairing to cognate target mRNAs, which typically requires the RNA chaperone, Hfq. Upon base-pairing, translation and/or stability of targeted transcripts are affected, leading to either repression or activation of gene expression (3).
Studies on the molecular functions of sRNAs have been strongly focused on enterobacterial organisms including Escherichia coli and Salmonella Typhimurium, and it has been suggested that every major regulon in these species contains at least one regulatory RNA (4). For example, ∼150–200 sRNAs have been discovered in E. coli to date, a small fraction of which has been characterized in more detail (5). In contrast, the roles of regulatory RNAs in the stalked alpha-proteobacterium Caulobacter crescentus, a well-established model of bacterial cell biology, have been barely addressed. Transcriptomic studies suggest that there are ∼140 sRNAs expressed from the Caulobacter genome during growth in rich and minimal medium (6). Most C. crescentus sRNAs remain uncharacterized with the exception of CrfA, an sRNA which is induced in response to carbon starvation and functions in remodeling the profile of outer membrane transport proteins under this condition (7), as well as GsrN, a conserved sRNA which is directly controlled by the general stress sigma factor, σT, and facilitates expression of katG mRNA under hydrogen peroxide stress (8).
In many species, the activity of sRNAs depends on the ubiquitous RNA chaperone Hfq, which protects RNA from decay by ribonucleases and mediates base-pairing between sRNAs and cognate target transcripts (9). Given that hfq is an essential gene under certain growth conditions in Caulobacter (10), and that absence of Hfq is associated with a severe loss of fitness and an elongated cell morphology (11), Hfq-mediated sRNA activities are likely to also play a key role in this organism. Because post-transcriptional regulation represents a rapid mechanism for altering gene expression, harmful stress such as DNA damage, e.g. through UV-radiation, represents a potentially important context for studying sRNAs. Indeed, all bacteria have developed sophisticated stress response systems to ensure maintenance of genome integrity in the face of DNA damage. However, the Caulobacter DNA damage response differs significantly from the well-studied stress programs of enterobacteria such as E. coli (12–15). Consequently, here we set out to determine if and how regulatory RNAs are integrated into the Caulobacter response to DNA damage. Specifically, we performed a transcriptomic analysis of cells treated with the DNA-crosslinking agent mitomycin C (MMC). We identified one sRNA candidate, CCNA_R0100, which was induced by MMC, but also expressed during growth in minimal medium. Expression of CCNA_R0100 is controlled by the activity of the conserved ChvI-ChvG TCS, and we thus renamed the sRNA ChvR (ChvI-ChvG regulated RNA). ChvI-ChvG has been previously implicated in virulence regulation and the response to low pH in related alpha-proteobacteria, but our work represents its first implication in DNA damage and sRNA induction. We further discovered that ChvR acts as a negative regulator of the TonB-dependent receptor CCNA_03108/CC_3013 (hereafter: ChvT), and that ChvT production is repressed by ChvR under different physiological conditions. Mechanistically, ChvR employs two distinct base-pairing sites to interact with the chvT target transcript, and both sites are required for full regulation. Surprisingly, testing regulation in an hfq mutant strain revealed that both ChvR expression and ChvT repression occur independent of Hfq. In summary, our work presents the first in depth characterisation of an sRNA-target mRNA interaction in C. crescentus, and suggests that Hfq-independent processes could be an important aspect of post-transcriptional gene regulation in this organism.
MATERIALS AND METHODS
DNA oligonucleotides
Sequences of all oligonucleotides employed in this study are listed in Supplementary Table S2.
Construction of plasmids
All plasmids used in this study are summarized in Supplementary Table S3.
Gibson assembly (16) was used to fuse flanking fragments of genes vanAB (f1: KFO-0065/KFO-0066, f2: KFO-0067/KFO-0068; pKF323-2), chvR (f1: KFO-0252/KFO-0253, f2: KFO-0254/KFO-0255; pKF379-1), chvIG-hprK (f1: KFO-0345/KFO-0346, f2: KFO-0347/KFO-0348; pKF389-6), and chvT (f1: KFO-0336/KFO-0338, f2: KFO-0337/KFO-0339; pKF436-1), with plasmid pNPTS138 at the multiple cloning site (KFO-0059/KFO-0060). The same approach was chosen to construct the ChvR transcriptional reporter pKF383-7; an E. coli lacZ fragment (amplified from pPR9TT using KFO-0286/KFO-0289) was fused with the chvR promoter region (spanning –109 to +9 relative to the transcriptional start; amplification by KFO-0282/KFO-0283 on C. crescentus gDNA) in backbone pXGFP-5 (PCR amplification by KFO-0277/KFO-0287).
To obtain pKF416-15, 3XFLAG::hfq was amplified from KFS-0297 via KFO-0069/KFO-0072, restricted with MluI/BamHI and ligated to an equally treated pNPTS backbone.
The translational reporter plasmid pKF310-3 was constructed by inserting a XhoI/XbaI restricted fragment (spanning –480 to +30 of the bapE gene relative to the translational start; KFO-0029/KFO-0035 amplification of gDNA) into equally treated pKF308-1. pKF308 is a derivative of pPR9TT in which lacZ has been replaced by sfgfp (amplified from pXG10sf by KFO-0026/KFO-0028) via BamHI/SacI.
Plasmid pVan-ChvR (pKF382-1) was constructed by ligation of the chvR fragment (PCR-amplified from C. crescentus gDNA using KFO-0230/KFO-0231; the sense primer starts at the sRNA +1 site determined by transcriptome analysis and carries a 5′ phosphate modification; XbaI restriction) to the pBVMCS-6 backbone ((17); amplification with KFO-0056/KFO-0144 at the +1 site of the vanillate-inducible promoter; XbaI restriction).
A high-copy plasmid expressing ChvR under the control of its native promoter (pKF370-1; spanning region –96 relative to the TSS of chvR up to the 10th aa of recF) was constructed by restricting pBXMCS-6 ((17); removal of xylose-responsive promoter) with PstI/XbaI, and ligation of an equally treated insert (PCR-amplified from Caulobacter gDNA using KFO-0227/KFO-0230).
For complementation of the chvIG-hprK deletion in the chromosome, the operon was amplified by PCR (KFO-0349/KFO-0350), and ligated to backbone pXGFP-5 ((17); integration into the xyl locus) via NheI/NdeI restriction sites (pKF390-14).
To express gfp reporter fusions under the control of PrsaA, plasmid pGFPC-2 (17) was PCR-amplified (KFO-0278/KFO-0321), restricted with NdeI and BglII, and ligated to an equally digested fragment spanning the rsaA upstream and promoter region to the transcriptional start site (amplified from gDNA using KFO-0323/KFO-0331). The resulting plasmid (pKF384-1) served as backbone to introduce different inserts amplified from gDNA at the at the second codon of gfp via cloning into EcoRI and KpnI restriction sites: pPrsaA-rsaA::gfp (pKF385-1; insert KFO-0483/KFO-0484); pPrsaA-chvT::gfp (pKF386-1; insert KFO-0326/KFO-0327); pPrsaA-chvT-M1::gfp (pKF397-4; insert KFO-0372/KFO-0327), pPrsaA-chvT-del5::gfp (pKF402-1; insert KFO-0382/KFO-0327).
Plasmids expressing single nucleotide mutants were constructed via PCR amplification of the original plasmids, DpnI digestion of template DNA, and self-ligation of purified PCR products. Plasmid pKF382-1 served as a template for PCR amplification with primer pairs KFO-0354/KFO-0355 (pVan-ChvR-M1; pKF395-1), KFO-0417/KFO-0418 (pVan-ChvR-M2; pKF414-1), and pKF395-1 was amplified using KFO-0417/KFO-0418 to obtain pVan-ChvR-M1M2 (pKF418-1). Correspondingly, plasmid pKF386-1 or pKF397-4 were amplified with primer pairs KFO-0426/KFO-0427 to obtain pPrsaA-chvT-M2::gfp (pKF420-1), and pPrsaA-chvT-M1M2::gfp (pKF466-1), respectively.
Bacterial strains and growth conditions
A complete list of bacterial strains employed in this study is provided in Supplementary Table S4. The Caulobacter crescentus strain NA1000 (KFS-0006; lab stock Z. Gitai) is referred to as the wild-type strain and was used for mutant construction. Deletions and insertions in the C. crescentus chromosome were obtained by using a two-step recombination procedure (18). Chromosomal mutations were transferred by transduction with phage Cr30 following standard protocols.
C. crescentus was cultivated aerobically at 30°C in either complex PYE medium, or in minimal M2 salts containing 0.2% glucose (19). Where appropriate, media were supplemented with antibiotics at the following concentrations (liquid/solid): kanamycin (5/25 μg/ml); chloramphenicol (2/1 μg/ml); tetracycline (2/1 μg/ml); nalidixic acid (-/20 μg/ml). A final concentration of 0.5 mM vanillate was added to cultures to induce expression from the vanAB promoter. To induce DNA damage, bacteria were grown to mid-exponential phase (OD660 of 0.4) and treated with mitomycin C (1 μg/ml). The response to low pH was tested by growing cells in PYE (pH 7.0) to mid-exponential phase (OD660 of 0.5) when the cultures were split, collected by centrifugation and resuspended in fresh PYE medium (pH 7.0 or pH 5.5, respectively).
E. coli strains were grown aerobically at 37°C in LB broth. Where appropriate, medium was supplemented with antibiotics at the following concentrations: kanamycin (50 μg/ml); chloramphenicol (20 μg/ml); tetracycline (12 μg/ml); ampicillin (100 μg/ml).
Transposon screen
For transposon mutagenesis, plasmid pRL27 ((20); carrying a hyperactive Tn5 transposase) was transferred to KFS-0172 by conjugation from an E. coli donor. Conjugants were selected on PYE agar supplemented with X-gal (40 μg/ml), kanamycin and nalidixic acid. White or light blue colonies were screened for integrity of the lacZ reporter, and Tn5 insertion sites were mapped as described previously (21).
Protein sample analysis
To prepare whole-cell protein samples, bacteria were collected by centrifugation (3 min; 9 000 rpm; 4°C) and resuspended in 1× protein loading buffer (62.5 mM Tris–HCl, pH 6.8, 100 mM DTT, 10% (v/v) glycerol, 2% (w/v) SDS, 0.01% (w/v) bromophenol blue) to a final concentration of 0.01 OD/μl. To analyze protein levels by Western blotting, 0.1 OD per lane were separated by SDS-PAGE and transferred onto PVDF membranes. 3XFLAG-tagged fusion proteins were detected using anti-FLAG antibody (1:1 000; mouse; Sigma #F1804), respectively. DivJ served as a loading control, and was probed with an antiserum (1:1 000; rabbit; (22)).
Fluorescence intensity measurements
Unless stated otherwise, GFP expression of translational reporter fusions was determined from cells cultivated overnight in PYE supplemented with the appropriate antibiotics and supplements. Samples were collected by centrifugation (3 min; 9 000 rpm; 4°C), washed once in phosphate buffer, and resuspended in phosphate buffer. A control sample not expressing GFP was used to determine background fluorescence. Fluorescence intensity in the presence of the control plasmid was set to 1, and relative expression was calculated from three biological replicates (error bars represent standard deviation).
RNA isolation and Northern blot analysis
Total bacterial samples were collected, mixed with 0.2 volumes of stop-mix (95% ethanol and 5% phenol, v/v) and snap-frozen in liquid nitrogen. Total RNA was isolated using the Hot Phenol method with modifications (23). Pellets were resuspended in 600 μl lysozyme solution (0.5 mg/ml lysozyme in TE buffer, pH 8.0), and 60 μl of 10% (w/v) SDS. The suspension was incubated at 65°C in a water bath for 1–2 min. The pH was equilibrated by addition of 0.1 vol of sodium acetate (pH 5.2), and samples were mixed with 750 μl phenol. Tubes were incubated at 65°C for 5 min and frequently mixed. Samples were centrifuged (10 min; 13 000 rpm; 4°C), and the aqueous layer mixed with 750 μl chloroform and centrifuged again (10 min; 13 000 rpm; 4°C). The RNA was ethanol-precipitated from the aqueous layer, washed with 70% ethanol, dried and dissolved in water.
For Northern blot analysis, 5–10 μg of total RNA were separated on 6% polyacrylamide (7M urea) gels and electroblotted. Membranes were hybridized with gene-specific 5′ end-labelled DNA-oligonucleotides at 42°C in Roti-Hybri-Quick hybridization solution (Roth), and washed in three subsequent steps with SSC wash buffers (5×/1×/0.5× SSC) supplemented with 0.1% SDS.
Hfq coIP
C. crescentus wild-type and cells expressing 3XFLAG-Hfq (KFS-0344) were grown in minimal M2G medium to OD660 of 1. Lysates of cell pellets corresponding to 50 OD660 were subjected to immunoprecipitation as described previously (24).
Transcriptomic analysis using RNA-seq
Libraries for Illumina sequencing of cDNA were constructed by vertis Biotechnology AG, Germany (http://www.vertis-biotech.com/), as described previously for eukaryotic microRNAs (25) but omitting the RNA size-fractionation step prior to cDNA synthesis. For the depletion of processed transcripts, equal amounts of RNA were incubated with Terminator 5′-phosphate-dependent exonuclease (TEX; Epicentre) as previously described (26). The transcripts were not fragmented in order to get mainly sequencing reads of the 5′-end of the transcripts. In a second sample set, biological replicates were depleted from ribosomal RNA (Ribo-Zero rRNA Removal Kit (Bacteria); Epicentre) and fragmented using ultrasound (4 pulses of 30s each). Afterwards, RNAs <20 nt were removed using the Agencourt RNAClean XP kit (Beckman Coulter Genomics). Equal amounts of RNA samples were poly(A)-tailed using poly(A) polymerase. Then, the 5′-triphosphates were removed by applying tobacco acid pyrophosphatase (TAP) resulting in 5′-monophosphates. Afterwards, an RNA adapter was ligated to the 5′-phosphate of the RNA. First-strand cDNA was synthesized by an oligo(dT)-adapter primer and the M-MLV reverse transcriptase. In a PCR-based amplification step using a high fidelity DNA polymerase the cDNA concentration was increased to 20–30 ng/μl. A library-specific barcode for multiplex sequencing was part of a 3′-sequencing adapter. The resulting cDNA libraries were sequenced using an Illumina NextSeq HiSeq 2500 in single-end mode with 100 cycles for the unfragmented TEX treated and untreated sample, or an Illumina NextSeq 500 in single-end mode with 75 cycles for the fragmented libraries, respectively. The raw, de-multiplexed reads files have been deposited in the National Center for Biotechnology Information's Gene Expression Omnibus (GEO) (27), and are accessible via the GEO accession GSE104186 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104186).
The Illumina reads in FASTQ format were trimmed with a cut-off phred score of 20 and cleaned from adapter sequences using cutadapt version 1.13. The following steps were performed using the subcommand ‘create’, ‘align’ and ‘coverage’ of the tool READemption (28) version 0.4.3. The poly(A)-tail sequences were removed and a size filtering step was applied in which sequences shorter than 12 nt were eliminated. The remaining reads were mapped to the reference genome sequences of C. crescentus NA1000 (accession number NC_011916.1, retrieved from NCBI Genbank) using segemehl (29,30). Based on the alignment files in BAM format, coverage files in wiggle format representing the number of aligned reads per base were created and visualized in the Integrated Genome Browser (30). Each graph was normalized to the total number of reads that could be mapped for the respective library. To restore the original data range each graph was then multiplied by the minimum number of mapped reads calculated over all libraries. For the differential gene expression analysis the number of aligned reads per genes were quantified and DESeq2 ((31); version 1.12.4) was applied to compare the two conditions. The data analysis workflow is compiled in a Unix Shell script that can be retrieved from Zenodo (https://doi.org/10.5281/zenodo.1028768).
qRT PCR
To prepare cDNA samples for qRT PCR, RNA was extracted from two biological replicates using the SV Total RNA Isolation System (Promega), and reverse transcribed using SuperScriptIII (Invitrogen) following the manufacturers’ recommendations. Real-time PCR reactions were performed in 384-well optical reaction plates in technical triplicates on an ABI Prism 7900HT Sequence Detection System using with Sybr Green mix (Applied Biosystems). As an internal control, RNA abundances were normalized to rpoD mRNA levels.
Microarray analysis
Total RNA samples were prepared from two independent biological replicates using the Hot Phenol method. Preparation of cDNA libraries, microarray hybridization (to customized Agilent microarrays, 0304061531; Agilent Technologies) and scanning were performed as described previously (32). Data analysis was performed using the Princeton University Microarray Database (PUMAdb), and microarray data were submitted to PUMAdb for archiving (https://puma.princeton.edu/cgi-bin/publication/viewPublication.pl?pub_no=580).
RESULTS
The C. crescentus transcriptome upon DNA damage
To analyze the involvement of sRNAs in the response to DNA damage in Caulobacter, we performed RNA sequencing before and after treatment with mitomycin C (MMC), a DNA-crosslinking drug and potent inducer of the DNA damage response. To confirm the induction of the stress response we used the previously characterized Caulobacter-specific endonuclease BapE as a reporter (12), and monitored expression of a transcriptional PbapE::gfp fusion in response to MMC treatment (Supplementary Figure S1). We isolated RNA from Caulobacter crescentus NA1000 grown to mid-exponential phase (OD660 of 0.4) in rich PYE medium, and after 240 min of growth in the presence of 1 μg/mL MMC (final OD660 of ∼1.0) when expression of the PbapE::gfp reporter had increased ∼5-fold (Supplementary Figure S1).
We used dRNA-seq (26) to quantify changes in transcript abundance under both conditions, and to define transcriptional start sites (TSS). The dRNA-seq protocol enriches primary transcripts by treating input RNA with terminator exonuclease, an enzyme selectively degrading mono-phosphorylated RNAs (as found on processed transcripts) while not affecting tri-phosphorylated RNAs (as found on primary transcripts).
Mapping the cDNA reads to the C. crescentus NA1000 genome revealed differential expression (>3-fold induction) of 185 genes (Supplementary Table S1), including 9 transcripts annotated as sRNAs (Table 1; Figure 1A and Supplementary Figure S2). The master regulator of the SOS-response is LexA which, in the non-induced state, represses its target genes by binding to a regulatory motif, the so-called SOS box (13,33). Within the set of 185 genes induced in the presence of MMC, we identified 20 (out of 45 known) genes which have previously been identified to be controlled by LexA in Caulobacter (13). However, no SOS box (GTTCN7GTTC; (13,34)) was identified in proximity to the transcriptional start sites of MMC-induced sRNAs genes (Table 1).
Table 1.
sRNAs upregulated in response to treatment with MMC
| sRNA | Size (nt) | Own promoter | 5′ flanking gene | 3′ flanking gene | Orientation | Comment | Fold change | Specific induction by MMC |
|---|---|---|---|---|---|---|---|---|
| CCNA_R0158 | 95 | yes | CCNA_02277 | CCNA_02278 | > > > | 36.9 | no | |
| CCNA_R0097 | 84 | no | CCNA_00028 | CCNA_00027 | < < < | 18.3 | no | |
| CCNA_R0180 | 87 | yes | CCNA_03118 | CCNA_03117 | > < < | 4.9 | no | |
| CCNA_R0100 | 84 | yes | CCNA_00157 | CCNA_00158 | > > > | ChvR | 4.7 | yes |
| CCNA_R0051 | 209 | yes | CCNA_02253 | CCNA_02254 | > > < | 4.4 | no | |
| CCNA_R0132 | 116 | no | CCNA_01141 | CCNA_01142 | > > > | 3′ UTR of CCNA_01141 (recA) | 4.1 | yes |
| CCNA_R0004 | 133 | yes | CCNA_00197 | CCNA_00196 | < < < | 3.7 | no | |
| CCNA_R0063 | 112 | yes | CCNA_02725 | CCNA_02726 | < > < | 3.3 | no | |
| CCNA_R0155 | 89 | yes | CCNA_02158 | CCNA_02158 | > > > | internal TSS | 3.1 | no |
Figure 1.

Transcriptome analysis of C. crescentus in response to DNA damage. (A) Diagram summarizing the results of the dRNA-seq experiment. RNA was collected from C. crescentus grown in PYE prior to (OD660 of 0.4) or 240 min after treatment with the DNA-damaging agent MMC. Under these conditions, 169 genes were induced >3-fold, including 9 sRNAs. No conserved SOS box was identified in proximity to transcriptional start sites of sRNAs. (B) Expression of ChvR sRNA in wild-type C. crescentus. Cells were grown in PYE, and RNA samples were collected prior to (OD660 of 0.4) and at indicated time-points after MMC treatment, or at different time-points over growth (OD660 of 0.2, 0.5, 1.0, 3h after cells had reached OD660 of 1.0, and overnight [on]), respectively. ChvR levels were determined by Northern blot analysis; 5S rRNA served as loading control. (C) cDNA reads of +/- MMC libraries mapping to the chvR/recF locus of C. crescentus NA1000. Libraries established from terminator exonuclease (TEX)-treated RNA samples are represented in blue, libraries established from untreated RNA in black, respectively. All libraries were adjusted to the same scale. Annotation and genome position are indicated in the centre. Transcriptional start sites are marked by arrows.
Caulobacter can continue to grow in the presence of MMC such that genes induced by prolonged MMC exposure could represent genes induced by DNA damage or genes induced by changes in growth state such as entry into stationary phase. To distinguish these possibilities, we used Northern blot analysis to compare the RNA levels of our candidate sRNAs in the presence of MMC to the RNA levels in stationary phase (Supplementary Figure S3). Only two sRNAs, ChvR (CCNA_R0100) and CCNA_R0132, showed higher abundance in the presence of MMC compared to stationary phase growth in PYE (Figure 1B and Supplementary Figure S3E). Our dRNA-seq analysis revealed that sRNA CCNA_R0132 was not transcribed from its own promoter, but rather represented a stable RNA fragment derived from the 3′ UTR of the recA transcript (Supplementary Figure S2E). While such RNA species are in principle able to exert regulatory functions, they may also reflect accumulated RNA decay intermediates (24). We thus focused the remainder of our analysis on the sRNA ChvR, which is expressed under the control of its own promoter from the CCNA_00157/CCNA_00158 (recF) intergenic region as an 84 nt long transcript, and a less abundant processed RNA (Figure 1B, C and Supplementary Figure S4).
Transcription of ChvR is controlled by the ChvI-ChvG TCS
While ChvR is robustly induced from its own promoter upon DNA damage, we could not identify an SOS box in proximity to its transcriptional start site, indicating that it is controlled by a regulator other than LexA (Table 1). A tool for studying ChvR regulation emerged from a previous genomic analysis suggesting that ChvR becomes enriched during growth in minimal medium (6). To verify this result, we compared ChvR levels of Caulobacter grown in complex PYE or minimal M2G medium on Northern blots, and detected strong expression of the sRNA that increased during growth in minimal medium (Figure 2A). Gene synteny analysis revealed a high degree of plasticity in the region upstream of the recF gene (i.e. the genomic location of chvR in C. crescentus) in other Caulobacter species (Figure 2B), and BLAST searches (35) did not reveal conservation of the chvR gene beyond C. crescentus.
Figure 2.

ChvR expression is controlled by the ChvI-ChvG TCS. (A) ChvR is induced in minimal medium. RNA was collected from C. crescentus grown in either PYE or M2G to indicated growth phases (OD660 of 0.2, 0.5, 1.0 and overnight [o/n]), and ChvR expression was determined by Northern blot analysis. (B) chvR is specific to C. crescentus. Synteny analysis of the genomic locus upstream the recF/gyrB operon between C. crescentus, C. sp. K31 and C. segnis. Conserved regions are marked by gray boxes. (C) Mapping of the transposon insertion site. A transposon mutant with reduced PchvR::lacZ reporter activity was recovered, and the Tn5 insertion site was mapped to chvI (first gene of the chvIG-hprK operon). Flanking genes on the C. crescentus chromosome are indicated in white. (D) Expression of ChvR in minimal medium. ChvR expression was determined by Northern blot analysis of RNA samples collected from C. crescentus cultures grown in minimal M2G medium to mid-exponential phase. ChvR levels were compared between wild-type cells, the chvR deletion strain, the ΔlacA reporter strain, the Tn5 mutant recovered from the transposon screen, a chromosomal deletion of chvIG-hprK, and a complementation strain in which the chvIG-hprK operon is expressed under control of its own promoter from the xyl locus on the C. crescentus chromosome. (E) Expression of ChvR upon DNA damage requires integrity of the chvIG-hprK operon. RNA was collected from C. crescentus grown in PYE 240 min after treatment with MMC, and ChvR levels were determined by Northern blot analysis. (F) Expression of ChvR in acidified growth medium. C. crescentus was grown in PYE to mid-exponential phase when cultures were split, and growth was continued in PYE at either neutral (N; pH 7) or acidic pH (A; pH 5.5). RNA samples were collected at indicated time-points, and ChvR levels were determined by Northern blot analysis.
To determine the regulator responsible for ChvR induction we constructed a lacZ transcriptional reporter of chvR. In the presence of the chromogenic substrate X-gal, Caulobacter appears blue on solid media, and this native β-galactosidase activity is dependent on lacA (36). We thus chromosomally integrated our reporter into a lacA mutant background, in which the basal activity of X-gal hydrolysis is abolished. We randomly mutagenized this reporter strain using Tn5, and selected white or light blue colonies from PYE plates containing X-Gal. Tn5 insertions in the lacZ gene were excluded using PCR analysis. We isolated a single light-blue clone, and localized the transposon insertion site within chvI, interrupting the first gene of the chvIG-hprK operon (Figure 2C). We confirmed that the chvI::Tn5 mutant disrupts ChvR induction in M2G by Northern blot analysis. When compared to wild-type, ChvR expression was not affected by the deletion of lacA, but dropped significantly in chvI::Tn5 (Figure 2D; lanes 1, 3 and 4). Furthermore, ChvR expression was absent when the entire chvIG-hprK operon was deleted, but restored in a complementation strain in which the chvIG-hprK operon was expressed under control of its own promoter from the the xyl locus (lanes 5 and 6).
We next asked whether the chvIG-hprK operon was likewise required for induction of ChvR in response to DNA damage. To this end, we probed ChvR expression in RNA samples collected from MMC-treated cells and discovered that loss of the chvIG-hprK operon abolished MMC-dependent ChvR induction (Figure 2E). Thus, the chvIG-hprK operon is necessary for ChvR induction upon grown in both minimal media and MMC treatment.
Together, ChvG and ChvI form a TCS that is highly conserved in the alpha-proteobacteria (37–40), and ChvG has previously been shown to become activated in low pH environments (38). To test whether this was also the case in Caulobacter, we used ChvR induction as a proxy for ChvI-ChvG activity at neutral and acidic pH. Specifically, we cultivated wild-type C. crescentus in PYE medium to an OD660 of 0.5, pelleted the cells, and resuspended aliquots in either neutral or acidified PYE medium (pH of 7 or pH of 5.5, respectively). Total RNA was isolated prior to and after reinoculation, and ChvR levels were determined by Northern blot analysis. We discovered that ChvR expression was strongly induced during growth at low pH (Figure 2F, lanes 1–4), and that the chvIG-hprK locus was strictly required for ChvR induction (Figure 2F, lanes 8–10 versus 1–4 / 11–13). Taken together, our results suggest that ChvR sRNA expression is dependent on the ChvI-ChvG TCS, and that this TCS is largely inactive in rich PYE medium but active in minimal medium, at acidic pH, and during the DNA damage response of C. crescentus.
ChvR is a trans-acting small RNA
To test if ChvR functions as a post-transcriptional regulator of gene expression in C. crescentus, we took a transcriptomic approach to screen for its direct targets. To minimize potential secondary effects of sRNA expression, i.e. the altered expression of an unrelated gene in response to deregulation of a direct target, we examined global mRNA changes in response to a brief pulse of ChvR overexpression (41,42). Caulobacter chvR mutant cells carrying either a plasmid expressing chvR under the control of the inducible vanAB promoter (pVan-ChvR), or an empty control vector were grown in M2G to an OD660 of 0.6. Since we had observed increased expression of ChvR in minimal medium, we suspected the sRNA to be active, and potential targets to be expressed under this condition. ChvR expression was induced by the addition of vanillate, and total RNA was collected prior to and at several time-points post induction. Northern blot analysis showed that ChvR was undetectable in the absence of vanillate, and that ChvR rapidly accumulated in the presence of the inducer (Figure 3A).
Figure 3.

ChvR acts as a repressor of chvT mRNA. (A) C. crescentus ΔchvR cells carrying either plasmid pPvan::ChvR (pKF382-1), or a control plasmid (pBVMCS-6) were grown in minimal M2G medium to mid-exponential phase (OD660 of 0.6) when vanillate was added to induce ChvR expression. RNA samples were collected prior to and at indicated time-points after sRNA induction, and ChvR levels were determined by Northern blot analysis. (B) Microarray analysis of C. crescentus genes affected by pulse overexpression of ChvR. Changes in transcript abundances between C. crescentus ΔchvR in response to ChvR overexpression and a control sample were scored on C. crescentus-specific microarrays. Dashed vertical or horizontal lines in the volcano plot indicate cut-off criteria of target selection (>3-fold change; P-value < 0.005), and chvT is marked in blue. (C) Verification of microarray results by qRT-PCR analysis. Abundances of chvT and recF mRNAs were determined in independent RNA samples collected as described in (A). The signal obtained in the control sample was set to 1; error bars represent the standard deviation calculated from two independent biological replicates.
We scored changes in mRNA abundance on microarrays, and identified a single deregulated transcript (>3-fold) in response to ChvR over-expression (Figure 3B). The mRNA of CCNA_3108 (CC_3013; hereafter named chvT), encoding a TonB-dependent receptor, was repressed ∼15-fold in the presence of ChvR. To corroborate the transcriptome data, we also determined chvT levels using qRT-PCR. Consistent with our microarray results, the abundance of chvT mRNA was reduced ∼30-fold in the ChvR overexpression strain (Figure 3C). As a control, we also determined the effect of ChvR expression on recF mRNA, which is transcribed from the genomic locus just downstream of chvR in Caulobacter (Figure 3C, and Supplementary Figure S4). Our data showed that ChvR had no effect on the expression of the recF gene, indicating that ChvR acts in trans to specifically repress the chvT transcript.
ChvR represses ChvT expression under various environmental conditions
To further characterize ChvR-mediated control of chvT, we investigated whether the repression we identified from an overexpression pulse also occurred under physiological conditions when ChvR was expressed from its endogenous promoter. To this end, we added a C-terminal 3XFLAG affinity tag to the chromosomal locus of chvT, and monitored production of ChvT::3XFLAG in wild-type C. crescentus as well as ΔchvR mutant cells carrying either an empty control vector or a high-copy plasmid complementing chvR under control of its native promoter. We first compared the levels of ChvT::3XFLAG from cells grown in M2G and observed increased expression of ChvT in the absence of ChvR in all growth phases (Figure 4A, lanes 1–4 and 5–8). ChvT::3XFLAG became undetectable in cells in which the ChvR sRNA deletion was complemented by a high-copy plasmid expressing chvR (Figure 4A, lanes 9–12). As expected, the ChvT::3XFLAG protein pattern was anti-correlated with the expression of the ChvR sRNA (Figure 4A, lower panel).
Figure 4.

ChvR controls ChvT::3XFLAG production under different environmental conditions. (A–C) Samples were collected from C. crescentus wild-type (carrying a control plasmid (pBXMCS-6)), or a chvR deletion strain (carrying either a control plasmid, or a multi-copy construct expressing chvR from its own promoter (pKF370-1)). In all strains, chvT is marked by a C-terminal 3XFLAG affinity tag. Protein and RNA levels were determined by Western blot and Northern blot analysis, respectively. (A) C. crescentus cells were grown in minimal M2G medium to indicated growth phases (OD660 of 0.2, 0.5, 1.0 and overnight [o/n]). (B) Expression of ChvR and ChvT::3XFLAG was determined in C. crescentus grown in PYE prior to (OD660 of 0.5; [-]), and 240 min after treatment with MMC [+]. (C) C. crescentus was grown in PYE to mid-exponential phase (OD660 of 0.5) when cultures were split, and growth was continued in PYE at either neutral (N; pH 7) or acidic pH (A; pH 5.5).
ChvR sRNA was also induced in Caulobacter cells treated with MMC (Figures 1B and 4B), and expression of ChvT::3XFLAG decreased in response to DNA damage (Figure 4B, lanes 1 and 2). In contrast, ChvT levels remained constantly high in a chvR mutant irrespective of the addition of MMC (lanes 3 and 4). Overexpression of the sRNA from the complementation plasmid reduced ChvT::3XFLAG both prior to and after addition of MMC to the culture (lanes 5 and 6).
In contrast to our findings with minimal media and MMC treatment, monitoring ChvT::3XFLAG expression revealed no detectable changes in protein levels, independent of ChvR expression within 30 min of growth at low pH (Figure 4C). Bacteria are generally able to adapt rapidly to mildly acidic conditions (43). Indeed, monitoring ChvR expression during extended growth at low pH for up to 120 min revealed that the sRNA was only transiently induced with a peak in expression at 30 min after shifting the cultures (Supplementary Figure S5). Given that membrane proteins are usually highly stable (44), and that the growth rate of C. crescentus is reduced under acidic conditions, transient induction of ChvR appears to be insufficient to significantly change ChvT protein levels.
ChvR base-pairs with chvT mRNA via two distinct base-pairing sites
How does ChvR regulate chvT? Bacterial sRNAs frequently function at the post-transcriptional level by base-pairing with the 5′ region of target mRNAs, altering transcript stability and translation (3). To uncouple the expression of chvT mRNA from its endogenous transcriptional control, we constructed a post-transcriptional reporter expressing the 5′ UTR and the first 15 codons of chvT fused to the green fluorescent protein (GFP) and drove transcription of this reporter with the constitutive PrsaA promoter (Figure 5A). As validated by qRT-PCR, overexpression of ChvR did not influence rsaA expression levels (Supplementary Figure S6). We integrated our reporter construct at the native rsaA locus, and co-transformed cells with either plasmid-borne pVan-ChvR or an empty control vector. Analyzing GFP production from this reporter fusion in cells grown for 12 h in the presence of vanillate revealed strong (∼15-fold) repression of GFP (Figure 5B), confirming that ChvR regulates chvT at the post-transcriptional level.
Figure 5.

ChvR interacts with two independent sites of chvT mRNA. (A) Schematic representation of the translational chvT::gfp fusions (under control of the constitutive PrsaA promoter). Reporters comprise the 5′ untranslated region plus the first 45 nucleotides of the chvT CDS. Location of ChvR binding sites 1 and 2 are indicated by blue boxes, positions of single nucleotide exchanges are marked in red. (B) Partial repression of chvT::gfp by ChvR via binding site 1. C. crescentus ΔvanAB carrying the indicated gfp reporter fusion were co-transformed with either a control plasmid (pBVMCS6), or a plasmid overexpressing ChvR or ChvR-M1 under the control of the vanillate-inducible promoter Pvan (pKF382-1 or pKF395-1, respectively) were grown overnight in PYE supplemented with vanillate. GFP fluorescence was determined by plate reader measurements. For each fusion, GFP levels in the presence of the control plasmid were set to 1, and relative changes were determined for cells expressing ChvR. GFP levels were calculated from three biological replicates; error bars indicate the standard deviation. (C, D) Secondary structures of ChvR and the 5′ UTR of chvT mRNA (from the transcriptional start site to the start codon) based on bioinformatic predictions (45). (E) Predicted base-pairing interactions forming between ChvR and chvT mRNA. Both interactions at binding site 1 (base-pairing of the second stem-loop of ChvR (nts 31–42) and chvT mRNA (nts −70 to −59 relative to the translational start site)) and at binding site 2 (base-pairing of the third stem-loop of ChvR (nts 63–73) and chvT mRNA (nts −20 to −10 relative to the translational start site). Positions of single-nucleotide exchanges generating the compensatory mutants M1 and M2 are indicated. Expression of all ChvR variants was confirmed by Northern blot analysis (Supplementary Figure S7B). (F-G) Analysis of GFP fluorescence of C. crescentus ΔvanAB carrying the indicated gfp reporter fusion in combination with either a control construct, or plasmids overexpressing ChvR, ChvR-M1, ChvR-M2 (pKF414-1) or ChvR-M1M2 (pKF418-1). Experimental details as in (B).
Bioinformatic predictions (using RNAfold; (45)) indicated that ChvR is likely highly structured, forming three hairpins with the last hairpin potentially acting as a rho-independent transcription terminator (Figure 5C). To investigate how ChvR regulates the expression of chvT, we used the RNAhybrid algorithm to predict base-pairing between the sRNA and the target transcript (46). This analysis predicted the formation of a continuous 12 bp interaction between the very 5′ end of the chvT mRNA and the second stemloop of ChvR (Figure 5C–E). To validate this prediction, we introduced a point mutation at position 35 in ChvR, replacing a guanosine with a cytosine (ChvR-M1; Figure 5E). While this mutation strongly reduced the repression of the reporter, ChvR-M1 still significantly down-regulated ChvT::GFP levels when compared to the control sample (∼5-fold; Figure 5B). Regulation of the chvT reporter was specific as expression of a control reporter PrsaA::gfp was only mildly affected by the presence of ChvR (Supplementary Figure S7A). We next introduced a compensatory mutation in the chvT::gfp reporter (chvT-M1, replacing a guanosine at position –62 relative to the translational start site with a cytosine residue; Figure 5E) and observed that wild-type ChvR had an intermediate effect (∼3-fold repression) on this reporter, whereas co-expression of ChvR-M1 fully restored regulation (Figure 5B). These results suggest that full repression of chvT::gfp by ChvR depends on the formation of a second base-pairing site between the two RNAs.
To identify candidates for the second interaction site we first shortened the chvT::gfp reporter from the 5′ end to eliminate the primary binding site (chvT-del5::gfp; Figure 5A). Both ChvR and ChvR-M1 repressed chvT-del5::gfp to a similar extent (∼3-fold; Figure 5F), confirming our hypothesis that ChvR uses an additional base-pairing site to repress this reporter. Based on this information, we predicted an alternative interaction between the third stem–loop of ChvR and a region just upstream of the translational start site of chvT mRNA (Figure 5D and E). A single-nucleotide exchange in ChvR (ChvR-M2; replacing cytosine at position –13 with a guanosine; Figure 5E) partially reduced GFP levels of the chvT wild-type reporter and fully abrogated repression of chvT-del5::gfp (Figure 5F).
We confirmed the requirement of both interaction sites by introducing two individual single nucleotide exchanges in ChvR. The sRNA variant ChvR-M1M2 completely failed to repress the wild-type chvT::gfp reporter (Figure 5G). Likewise, a chvT-M1M2::gfp reporter was no longer regulated by wild-type ChvR, but was strongly repressed by ChvR-M1M2. Together, our data shows that ChvR controls chvT at the post-transcriptional level, and that two distinct base-pairing sites are required for full regulation.
ChvR-mediated regulation is independent of the chaperone Hfq
In most well-characterized bacteria the formation of intermolecular base-pairing between cognate sRNA/mRNA partners is aided by the RNA chaperone, Hfq (9). Due to a role in the maintenance of central metabolism, deletion of hfq is associated with severe phenotypes including loss of cell morphology in C. crescentus (11), and is even an essential gene under certain growth conditions (10). To investigate the potential involvement of Hfq in regulating chvT mRNA by ChvR, we tested regulation of the chvT::gfp reporter in a C. crescentus hfq mutant strain. We determined that absence of the chaperone mildly reduced basal expression levels of chvT::gfp, but did not affect repression by ChvR (Figure 6A). It has previously been reported that certain Hfq-dependent sRNAs also exert regulatory activity in the absence of the chaperone when strongly overexpressed (47). To verify our observation that ChvR functions without Hfq, we compared the synthesis of ChvT::3XFLAG in wild-type C. crescentus and ΔchvR mutant cells (carrying either an empty control vector or a high-copy plasmid complementing chvR under its native promoter) to isogenic Δhfq or Δhfq ΔchvR mutant strains grown in minimal M2G medium (Figure 6B). Similar to the expression pattern of wild-type C. crescentus, ChvT::3XFLAG was more abundant in the hfq mutant strain in the absence of ChvR, and barely detectable in cells in which ΔchvR was complemented. We also observed that ChvR expression was increased in the hfq mutant (Figure 6B, lower panel). We next examined the abundance of ChvR in the hfq mutant strain in comparison to wild-type cells at different time-points over growth. In further support of the Hfq-independent phenotype observed before, Northern blot analysis revealed that expression of ChvR was indeed not reduced in the absence of Hfq, but that the sRNA was present at even higher levels when compared to the wild-type strain (Figure 6C).
Figure 6.
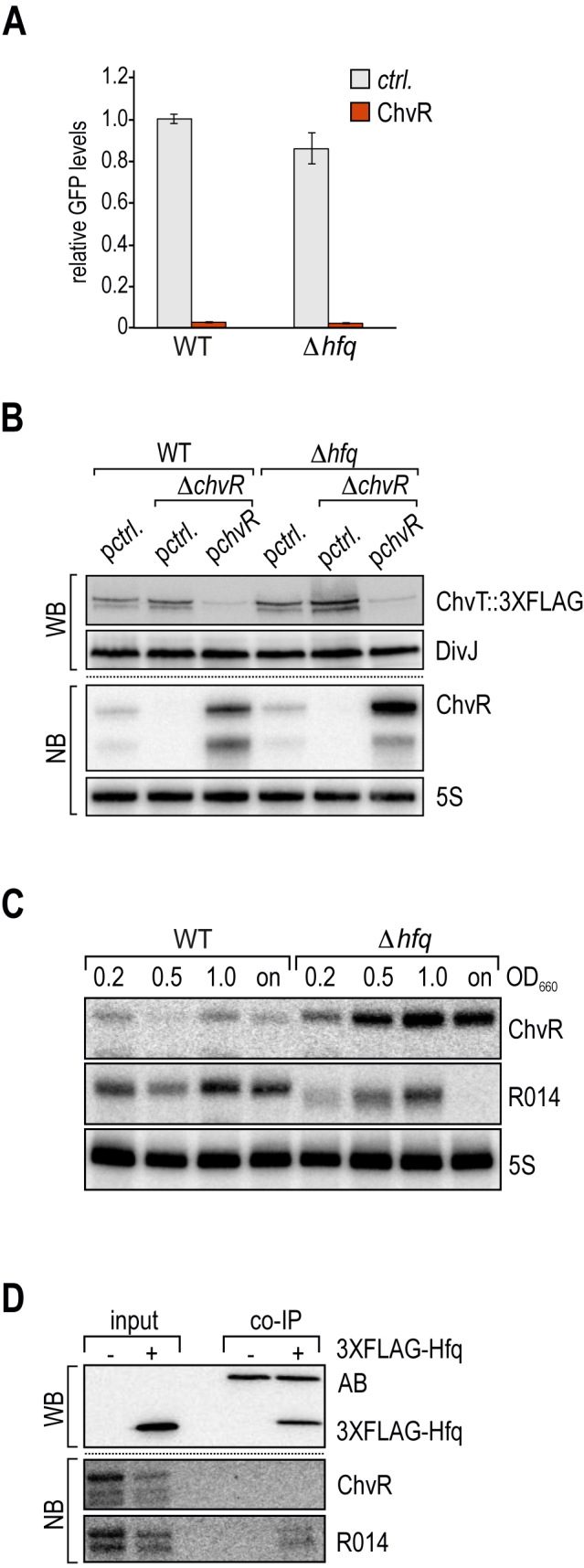
Post-transcriptional regulation by ChvR is independent of Hfq. (A) Analysis of GFP fluorescence of C. crescentus ΔvanAB (WT) or ΔvanAB Δhfq (Δhfq) cells carrying the indicated gfp reporter fusion in combination with either a control construct, or a plasmid overexpressing ChvR. Experimental details as in Figure 5 (B). (B) Samples were collected from C. crescentus wild-type (carrying a control plasmid (pBXMCS-6)), or a chvR deletion strain (carrying either a control plasmid, or a multi-copy construct expressing chvR from its own promoter (pKF370-1)), as well as isogenic hfq mutants of these strains grown in minimal M2G medium to an OD660 of 0.5. In all strains, ChvT is marked by a C-terminal 3XFLAG affinity tag. Protein and RNA levels were determined by Western blot and Northern blot analysis, respectively. (C) Expression of ChvR in an hfq mutant strain. C. crescentus wild-type and Δhfq cells were grown in M2G to indicated time-points over growth (OD660 of 0.2, 0.5, 1.0 and overnight [on]). RNA expression of ChvR and CCNA_R0014 were determined by Northern blot analysis. (D) ChvR does not interact with Hfq in vivo. C. crescentus wild-type or a 3XFLAG::hfq strain were grown in minimal M2G medium to OD660 of 1.0, and cell lysates were subjected to immunoprecipitation using a monoclonal anti-FLAG antibody. Protein and RNA samples of lysate (input) and co-immunoprecipitation fractions (co-IP) were analysed by Western and Northern blot analysis, respectively. AB: recovered anti-FLAG antibody.
Since we did not observe increased levels of ChvR in C. crescentus Δhfq when expressing the sRNA from the vanillate-inducible promoter in our reporter assays (Supplementary Figure S9) we speculate that deletion of hfq results in activation of the chvR promoter.
Since the independence of ChvR function and expression from Hfq was surprising, we sought an additional method to investigate ChvR’s relation to Hfq. We thus asked if ChvR biochemically interacts with Caulobacter Hfq. To this end, we subjected wild-type and 3XFLAG::hfq C. crescentus lysates to immunoprecipitation with a monoclonal anti-FLAG antibody and compared co-purified RNA to total RNA samples of both strains. When probing for ChvR, we found the sRNA to be absent from co-IP fractions but detected a signal in total RNA samples (Figure 6D). We furthermore probed for sRNA CCNA_R0014, which is a homologue of AbcR1, an sRNA associated with Hfq in the distantly related alpha-proteobacterium Sinorhizobium meliloti (48). We detected CCNA_R0014 in both samples of total RNA, but also specifically enriched in the Hfq co-IP fraction. Moreover, expression of R0014 was reduced in the hfq mutant strain compared to wild-type C. crescentus (Figure 6C). This result suggests that only a subset of sRNAs in Caulobacter associate with the RNA-binding protein Hfq, and that Caulobacter has a second, Hfq-independent class of sRNAs that includes the sRNA ChvR. While C. crescentus does not encode homologues of the two other global sRNA binding protein families in bacteria, CsrA/RsmA and ProQ, respectively, we cannot exclude that ChvR requires a different, yet-to-be-identified accessory factor to inhibit chvT expression.
DISCUSSION
Gene regulation by sRNAs plays an important role in the physiology of many microorganisms (49). The impact of bacterial sRNAs on post-transcriptional regulation has been intensively studied in the gamma-proteobacteria E. coli and Salmonella, but the biological function of the majority of sRNAs identified to date is unknown. In this work, we characterized a novel sRNA, ChvR, in C. crescentus which is expressed under the control of the conserved ChvI-ChvG TCS in response to DNA damage, acidic pH and growth in minimal medium. ChvR functions as a dedicated repressor of the TonB-dependent receptor, ChvT. Acting as a post-transcriptional regulator, ChvR recognizes chvT mRNA at two distinct sites, and both base-pairing interactions are required for full repression. Importantly, the expression and regulatory activity of ChvR are independent of the conserved RNA chaperone, Hfq. Our results suggest that while Caulobacter Hfq does associate with some sRNAs, ChvR represents a class of Hfq-independent sRNAs that contribute to the adjustment of gene expression in this species.
ChvR represses the TonB-dependent receptor ChvT
ChvT is one of 65 TonB-dependent receptor proteins in C. crescentus (50) and has a possible function for the survival of C. crescentus under nutrient scarce conditions (51). TonB-dependent receptors can facilitate the transport of siderophores, vitamins or carbohydrates (52), but as for most of the members of this class, the substrate of ChvT is not known. In general, bacterial outer membrane proteins serve as selective barriers controlling the exchange of both harmful and beneficial substances, and thus their expression is usually tightly regulated (53). One important layer of control of outer membrane protein expression is constituted by sRNAs, which have been repeatedly identified to regulate membrane composition and architecture at the post-transcriptional level (54). Indeed, another characterized sRNA from C. crescentus, CrfA, also modulates the expression of a large set of outer membrane proteins (7). It is currently not fully understood why mRNAs translating into outer membrane proteins could be more prone to regulation by sRNAs than other transcripts. One possible explanation is the relatively long half-live paired with the high copy number of some outer membrane protein-coding transcripts, and the potential need to rapidly reduce transcript numbers when environmental conditions change. Under these circumstances, efficient sRNA-mediated regulation could be superior to conventional transcriptional control mechanism of outer membrane protein synthesis.
ChvR expression is controlled by the ChvI-ChvG TCS
Caulobacter thrives in nutrient-poor aquatic habitats (55), a lifestyle which requires the bacterium to cope with a diverse range of physiological stresses. The ability to adapt to ever changing conditions is reflected in the high number of two-component systems (34 sensor kinases, 44 response regulators and 27 sensor kinase/response regulator hybrid genes) encoded in the C. crescentus genome (50), which function in the transmission of signals to regulate response processes.
The ChvI-ChvG TCS is highly conserved among the alpha-proteobacteria, and is required for the association of bacteria with higher organisms. For example, mutations in either chvI or chvG abrogate the ability of Agrobacterium tumefaciens to form tumors on plants (56), and mutants of the homologous BvrR-BvrS TCS in Brucella abortus display reduced virulence in animal and cell culture models (40). The ChvI-ExoS TCS of S. meliloti is crucial for the establishment of symbiosis between the bacterium and its host plant alfalfa, and regulating the production of succinoglycan (37).
In this study, we showed that expression of the ChvR sRNA in C. crescentus is controlled by the ChvI-ChvG TCS (Figure 2D), and that ChvR is produced in response to DNA damage, acidic pH and during growth in minimal medium (Figure 2D–F). Since C. crescentus is a free-living, non-pathogenic bacterium, our results suggest an additional role of the ChvI-ChvG TCS other than sensing a host cell environment. The environmental cues perceived by membrane receptors are oftentimes unknown (40,57), and this is also the case for ChvG. Whether one common or several different cues activate signaling through the ChvI-ChvG TCS in Caulobacter under the different inducing conditions remains to be determined.
Requirement of two individual binding sites
Post-transcriptional regulation of chvT mRNA by ChvR is based on the formation of two distinct RNA-RNA interactions between the sRNA and its target (Figure 5E). While full repression requires ChvR to bind both target sites on the chvT mRNA (Figure 5G), each individual base-pairing confers an intermediate effect on target gene expression (Figure 5B, F). Given that ChvR uses two different sequence stretches to base-pair at the very 5′ end of the chvT transcript or close to the ribosome binding site, respectively, it remains to be established whether one or two sRNA molecules are required to form these interactions. Multi-site pairing of an sRNA on its mRNA target is not common, but has been observed previously. For example, the E. coli sRNA Spot42 employs two different seed regions to repress nanC, galK, sthA and ascF mRNAs. OxyS sRNA, involved in the E. coli response to oxidative damage, forms two kissing loop complexes with the fhlA transcript, and repression of lpxR mRNA by MicF likewise involves two distinct base-pairing interactions between the binding partners. Unlike ChvR, however, these sRNAs require Hfq to function (58–60). Structurally, Hfq can only accommodate one sRNA-mRNA pair at a time (61). Thus, the activity of the E. coli sRNAs may be restricted from targeting both mRNA sites simultaneously. In contrast, the Hfq-independent two-site pairing discovered here for ChvR (Figure 6) could provide a novel mechanism for using two sites simultaneously to enhance both the strength and efficiency of target regulation by the sRNA.
The locations of the two pairing sites within its mRNA target suggest that the two base-pairing regions of ChvR regulate chvT expression via distinct mechanisms. The base-pairing between ChvR and chvT at site 2 is formed by 7 + 2 bp interrupted by a 2 bp bulge (–15.5 kcal/mol (46); Figure 5E). By binding close to the start codon (residues –10 to –20 relative to the AUG of chvT mRNA), ChvR could mask the recognition site for the 30S ribosomal subunit (ranging from residue -20 in the 5′ UTR to +19 in the coding sequence; (62)) and thereby interfere with translation initiation (63). In contrast, base-pairing site 1 is located at the very 5′ end of the chvT mRNA at residues –70 to –59 relative to the AUG (–27.6 kcal/mol (46); Figure 5E), and thus is in considerable distance from translation initiation signals. Bioinformatic predictions of the secondary structure of the chvT leader (using RNAfold; (45)) reveal the formation of a weak stem-loop structure that would be interrupted by interacting with ChvR (see Figure 5D and E). Similar structural elements have previously been shown to confer stability to transcripts by occluding the access of exonucleases with 5′ to 3′ directionality, including the major cellular nuclease RNase E (64,65). The presence of such a protective hairpin that is disrupted by ChvR binding could thus explain how ChvR represses chvT via the upstream binding site. In line with this model, deletion of the 5′ region of the chvT transcript (as in chvT-del5::gfp) reduces basal expression of the reporter in the absence of ChvR by approximately one third (Supplementary Figure S8). While the exact molecular mechanism of ChvR regulation still awaits experimental validation, the use of complementary approaches targeting distinct aspects of RNA biology could serve as a paradigm for effective gene regulation.
Regulation by ChvR is independent of Hfq
Hfq contains three principal sites that interact with RNA: the proximal and distal surfaces of the hexameric ring structure, and the rim (66). By binding to two different RNAs at once, Hfq acts as a matchmaker to bring together cognate RNA interaction partners (67,68). Thereby, Hfq contributes to both the specificity of the pairing, as well as to the efficiency of forming base-pairing interactions. In contrast, ChvR sRNA post-transcriptionally regulates expression of its cognate target, chvT mRNA, independently of Hfq (Figure 6A and B). Given that C. crescentus expresses functional Hfq, is there an advantage for not engaging Hfq in ChvR-mediated regulation? One reason could be an increase in robustness of the regulation. Even though the exact in vivo concentration of Hfq is uncertain (in E. coli, estimates range from 400–10 000 hexamers per cell (69–71)), the total number of all binding-competent RNAs are clearly in molar excess over the protein. To overcome this limitation, RNAs are thought to rapidly cycle on and off of Hfq to maximize the time associated with the RNA binding protein (72). Profiling of Hfq-associated RNAs has shown that individual, highly expressed RNAs are able to influence the pool of bound species (24,73). As a consequence, competition between sRNAs for Hfq can result in displacement of sRNAs from the chaperone, and reduction of their regulatory potential (74,75). In contrast, the functionality of Hfq-independent sRNAs, like ChvR, is not affected by fluctuation in the transcriptomic output of the cell. Thus, regulation via this class of sRNAs controls gene expression robustly independent of other cellular activities.
While Hfq-independent sRNA regulation may have the benefit of being insulated from the expression levels of other sRNAs, there may also be a cost associated with this type of regulation. Specifically, since Hfq stabilizes RNA–RNA interactions, Hfq-independent regulation may require far more stable associations, potentially explaining why the ChvR:chvT interaction involves two distinct sites that include an unusually-long 12 bp continuous homology region. Co-evolving such stable interaction sites may be difficult, which also potentially explains why ChvR has only a single target. Considerably less is known about Hfq-independent sRNAs than their Hfq-dependent counterparts, but our findings with ChvR suggest that sRNA-mediated gene regulation may have initiated through strong base-pairing associations that were later relaxed upon stabilization by Hfq. In extant cases, the trade-off between maintaining strong interactions and insulation from other sRNA levels could dictate which sRNAs use which mechanisms for regulation. In this context, ChvR could represent a useful model for studying sRNA-controlled gene regulation in the absence of Hfq.
DATA AVAILABILITY
Files of the raw, de-multiplexed reads of C. crescentus transcriptomes upon MMC treatment are accessible via the GEO accession GSE104186 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104186). Microarray data are available from PUMAdb (https://puma.princeton.edu/cgi-bin/publication/viewPublication.pl?pub_no=580).
Supplementary Material
ACKNOWLEDGEMENTS
We thank J. Chen (San Francisco State University), C. Jacobs-Wagner (Yale University) and U. Jenal (Basel University) for sharing strains, and A. Siryaporn for help with the microarray experiment. We further thank K. Papenfort for comments on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
K.S. Fröhlich was supported by a Research Fellowship of the German Research Foundation (DFG) [FR3502/1-1]; Center for Integrated Protein Science Munich (CiPSM); LMU Mentoring program of the LMU Faculty of Biology. Z. Gitai acknowledges funding from the National Institutes of Health (NIH) [5R01GM107384-04, DP1 AI124669-01]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Capra E.J., Laub M.T.. Evolution of two-component signal transduction systems. Annu. Rev. Microbiol. 2012; 66:325–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gao R., Mack T.R., Stock A.M.. Bacterial response regulators: versatile regulatory strategies from common domains. Trends Biochem. Sci. 2007; 32:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Waters L.S., Storz G.. Regulatory RNAs in bacteria. Cell. 2009; 136:615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gottesman S. Micros for microbes: non-coding regulatory RNAs in bacteria. Trends Genet. 2005; 21:399–404. [DOI] [PubMed] [Google Scholar]
- 5. Wang J., Liu T., Zhao B., Lu Q., Wang Z., Cao Y., Li W.. sRNATarBase 3.0: an updated database for sRNA-target interactions in bacteria. Nucleic Acids Res. 2016; 44:D248–D253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schrader J.M., Zhou B., Li G.W., Lasker K., Childers W.S., Williams B., Long T., Crosson S., McAdams H.H., Weissman J.S. et al. The coding and noncoding architecture of the Caulobacter crescentus genome. PLoS Genet. 2014; 10:e1004463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Landt S.G., Lesley J.A., Britos L., Shapiro L.. CrfA, a small noncoding RNA regulator of adaptation to carbon starvation in Caulobacter crescentus. J. Bacteriol. 2010; 192:4763–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tien M., Fiebig A., Crosson S.. Gene network analysis identifies a central post-transcriptional regulator of cellular stress survival. Elife. 2018; 7:e33684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vogel J., Luisi B.F.. Hfq and its constellation of RNA. Nat. Rev. Microbiol. 2011; 9:578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Christen B., Abeliuk E., Collier J.M., Kalogeraki V.S., Passarelli B., Coller J.A., Fero M.J., McAdams H.H., Shapiro L.. The essential genome of a bacterium. Mol. Syst. Biol. 2011; 7:528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Irnov I., Wang Z., Jannetty N.D., Bustamante J.A., Rhee K.Y., Jacobs-Wagner C.. Crosstalk between the tricarboxylic acid cycle and peptidoglycan synthesis in Caulobacter crescentus through the homeostatic control of alpha-ketoglutarate. PLoS Genet. 2017; 13:e1006978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bos J., Yakhnina A.A., Gitai Z.. BapE DNA endonuclease induces an apoptotic-like response to DNA damage in Caulobacter. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:18096–18101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. da Rocha R.P., Paquola A.C., Marques Mdo V., Menck C.F., Galhardo R.S.. Characterization of the SOS regulon of Caulobacter crescentus. J. Bacteriol. 2008; 190:1209–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Modell J.W., Hopkins A.C., Laub M.T.. A DNA damage checkpoint in Caulobacter crescentus inhibits cell division through a direct interaction with FtsW. Genes Dev. 2011; 25:1328–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Modell J.W., Kambara T.K., Perchuk B.S., Laub M.T.. A DNA damage-induced, SOS-independent checkpoint regulates cell division in Caulobacter crescentus. PLoS Biol. 2014; 12:e1001977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A. 3rd, Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 17. Thanbichler M., Iniesta A.A., Shapiro L.. A comprehensive set of plasmids for vanillate- and xylose-inducible gene expression in Caulobacter crescentus. Nucleic Acids Res. 2007; 35:e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Skerker J.M., Prasol M.S., Perchuk B.S., Biondi E.G., Laub M.T.. Two-component signal transduction pathways regulating growth and cell cycle progression in a bacterium: a system-level analysis. PLoS Biol. 2005; 3:e334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ely B. Genetics of Caulobacter crescentus. Methods Enzymol. 1991; 204:372–384. [DOI] [PubMed] [Google Scholar]
- 20. Larsen R.A., Wilson M.M., Guss A.M., Metcalf W.W.. Genetic analysis of pigment biosynthesis in Xanthobacter autotrophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch. Microbiol. 2002; 178:193–201. [DOI] [PubMed] [Google Scholar]
- 21. Rutherford S.T., van Kessel J.C., Shao Y., Bassler B.L.. AphA and LuxR/HapR reciprocally control quorum sensing in Vibrios. Genes Dev. 2011; 25:397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wheeler R.T., Shapiro L.. Differential localization of two histidine kinases controlling bacterial cell differentiation. Mol. Cell. 1999; 4:683–694. [DOI] [PubMed] [Google Scholar]
- 23. Blomberg P., Wagner E.G., Nordstrom K.. Control of replication of plasmid R1: the duplex between the antisense RNA, CopA, and its target, CopT, is processed specifically in vivo and in vitro by RNase III. EMBO J. 1990; 9:2331–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chao Y., Papenfort K., Reinhardt R., Sharma C.M., Vogel J.. An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J. 2012; 31:4005–4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berezikov E., Thuemmler F., van Laake L.W., Kondova I., Bontrop R., Cuppen E., Plasterk R.H.. Diversity of microRNAs in human and chimpanzee brain. Nat. Genet. 2006; 38:1375–1377. [DOI] [PubMed] [Google Scholar]
- 26. Sharma C.M., Hoffmann S., Darfeuille F., Reignier J., Findeiss S., Sittka A., Chabas S., Reiche K., Hackermuller J., Reinhardt R. et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010; 464:250–255. [DOI] [PubMed] [Google Scholar]
- 27. Edgar R., Domrachev M., Lash A.E.. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Förstner K.U., Vogel J., Sharma C.M.. READemption-a tool for the computational analysis of deep-sequencing-based transcriptome data. Bioinformatics. 2014; 30:3421–3423. [DOI] [PubMed] [Google Scholar]
- 29. Hoffmann S., Otto C., Kurtz S., Sharma C.M., Khaitovich P., Vogel J., Stadler P.F., Hackermuller J.. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009; 5:e1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Freese N.H., Norris D.C., Loraine A.E.. Integrated genome browser: visual analytics platform for genomics. Bioinformatics. 2016; 32:2089–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Siryaporn A., Kuchma S.L., O’Toole G.A., Gitai Z.. Surface attachment induces Pseudomonas aeruginosa virulence. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:16860–16865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Butala M., Zgur-Bertok D., Busby S.J.. The bacterial LexA transcriptional repressor. Cell. Mol. Life Sci. 2009; 66:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Erill I., Jara M., Salvador N., Escribano M., Campoy S., Barbe J.. Differences in LexA regulon structure among Proteobacteria through in vivo assisted comparative genomics. Nucleic Acids Res. 2004; 32:6617–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 36. Arellano B.H., Ortiz J.D., Manzano J., Chen J.C.. Identification of a dehydrogenase required for lactose metabolism in Caulobacter crescentus. Appl. Environ. Microbiol. 2010; 76:3004–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng H.P., Walker G.C.. Succinoglycan production by Rhizobium meliloti is regulated through the ExoS-ChvI two-component regulatory system. J. Bacteriol. 1998; 180:20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li L., Jia Y., Hou Q., Charles T.C., Nester E.W., Pan S.Q.. A global pH sensor: Agrobacterium sensor protein ChvG regulates acid-inducible genes on its two chromosomes and Ti plasmid. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:12369–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Quebatte M., Dehio M., Tropel D., Basler A., Toller I., Raddatz G., Engel P., Huser S., Schein H., Lindroos H.L. et al. The BatR/BatS two-component regulatory system controls the adaptive response of Bartonella henselae during human endothelial cell infection. J. Bacteriol. 2010; 192:3352–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sola-Landa A., Pizarro-Cerda J., Grillo M.J., Moreno E., Moriyon I., Blasco J.M., Gorvel J.P., Lopez-Goni I.. A two-component regulatory system playing a critical role in plant pathogens and endosymbionts is present in Brucella abortus and controls cell invasion and virulence. Mol. Microbiol. 1998; 29:125–138. [DOI] [PubMed] [Google Scholar]
- 41. Masse E., Vanderpool C.K., Gottesman S.. Effect of RyhB small RNA on global iron use in Escherichia coli. J. Bacteriol. 2005; 187:6962–6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Papenfort K., Pfeiffer V., Mika F., Lucchini S., Hinton J.C., Vogel J.. σE-dependent small RNAs of Salmonella respond to membrane stress by accelerating global omp mRNA decay. Mol. Microbiol. 2006; 62:1674–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kanjee U., Houry W.A.. Mechanisms of acid resistance in Escherichia coli. Annu. Rev. Microbiol. 2013; 67:65–81. [DOI] [PubMed] [Google Scholar]
- 44. Maier T., Schmidt A., Guell M., Kuhner S., Gavin A.C., Aebersold R., Serrano L.. Quantification of mRNA and protein and integration with protein turnover in a bacterium. Mol. Syst. Biol. 2011; 7:511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gruber A.R., Lorenz R., Bernhart S.H., Neubock R., Hofacker I.L.. The Vienna RNA websuite. Nucleic Acids Res. 2008; 36:W70–W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rehmsmeier M., Steffen P., Hochsmann M., Giegerich R.. Fast and effective prediction of microRNA/target duplexes. RNA. 2004; 10:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Soper T., Mandin P., Majdalani N., Gottesman S., Woodson S.A.. Positive regulation by small RNAs and the role of Hfq. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:9602–9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Torres-Quesada O., Reinkensmeier J., Schluter J.P., Robledo M., Peregrina A., Giegerich R., Toro N., Becker A., Jimenez-Zurdo J.I.. Genome-wide profiling of Hfq-binding RNAs uncovers extensive post-transcriptional rewiring of major stress response and symbiotic regulons in Sinorhizobium meliloti. RNA Biol. 2014; 11:563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Storz G., Vogel J., Wassarman K.M.. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell. 2011; 43:880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nierman W.C., Feldblyum T.V., Laub M.T., Paulsen I.T., Nelson K.E., Eisen J.A., Heidelberg J.F., Alley M.R., Ohta N., Maddock J.R. et al. Complete genome sequence of Caulobacter crescentus. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:4136–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Marks M.E., Castro-Rojas C.M., Teiling C., Du L., Kapatral V., Walunas T.L., Crosson S.. The genetic basis of laboratory adaptation in Caulobacter crescentus. J. Bacteriol. 2010; 192:3678–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Noinaj N., Guillier M., Barnard T.J., Buchanan S.K.. TonB-dependent transporters: regulation, structure, and function. Annu. Rev. Microbiol. 2010; 64:43–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fröhlich K.S., Gottesman S.. Small Regulatory RNAs in the enterobacterial response to envelope damage and oxidative stress. Microbiol. Spectr. 2018; 6:doi:10.1128/microbiolspec.RWR-0022-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guillier M., Gottesman S., Storz G.. Modulating the outer membrane with small RNAs. Genes Dev. 2006; 20:2338–2348. [DOI] [PubMed] [Google Scholar]
- 55. Poindexter J.S. Biological properties and classification of the Caulobacter Group. Bacteriol. Rev. 1964; 28:231–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Charles T.C., Nester E.W.. A chromosomally encoded two-component sensory transduction system is required for virulence of Agrobacterium tumefaciens. J Bacteriol. 1993; 175:6614–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mascher T., Helmann J.D., Unden G.. Stimulus perception in bacterial signal-transducing histidine kinases. Microbiol. Mol. Biol. Rev. 2006; 70:910–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Argaman L., Altuvia S.. fhlA repression by OxyS RNA: kissing complex formation at two sites results in a stable antisense-target RNA complex. J. Mol. Biol. 2000; 300:1101–1112. [DOI] [PubMed] [Google Scholar]
- 59. Beisel C.L., Updegrove T.B., Janson B.J., Storz G.. Multiple factors dictate target selection by Hfq-binding small RNAs. EMBO J. 2012; 31:1961–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Corcoran C.P., Podkaminski D., Papenfort K., Urban J.H., Hinton J.C., Vogel J.. Superfolder GFP reporters validate diverse new mRNA targets of the classic porin regulator, MicF RNA. Mol. Microbiol. 2012; 84:428–445. [DOI] [PubMed] [Google Scholar]
- 61. Updegrove T.B., Correia J.J., Chen Y., Terry C., Wartell R.M.. The stoichiometry of the Escherichia coli Hfq protein bound to RNA. RNA. 2011; 17:489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hüttenhofer A., Noller H.F.. Footprinting mRNA-ribosome complexes with chemical probes. EMBO J. 1994; 13:3892–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bouvier M., Sharma C.M., Mika F., Nierhaus K.H., Vogel J.. Small RNA binding to 5′ mRNA coding region inhibits translational initiation. Mol. Cell. 2008; 32:827–837. [DOI] [PubMed] [Google Scholar]
- 64. Emory S.A., Bouvet P., Belasco J.G.. A 5′-terminal stem-loop structure can stabilize mRNA in Escherichia coli. Genes Dev. 1992; 6:135–148. [DOI] [PubMed] [Google Scholar]
- 65. Celesnik H., Deana A., Belasco J.G.. Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol. Cell. 2007; 27:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sauer E. Structure and RNA-binding properties of the bacterial LSm protein Hfq. RNA Biol. 2013; 10:610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kawamoto H., Koide Y., Morita T., Aiba H.. Base-pairing requirement for RNA silencing by a bacterial small RNA and acceleration of duplex formation by Hfq. Mol. Microbiol. 2006; 61:1013–1022. [DOI] [PubMed] [Google Scholar]
- 68. Soper T.J., Woodson S.A.. The rpoS mRNA leader recruits Hfq to facilitate annealing with DsrA sRNA. RNA. 2008; 14:1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ali Azam T., Iwata A., Nishimura A., Ueda S., Ishihama A.. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J. Bacteriol. 1999; 181:6361–6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Short J., Tsukada K., Rudert W.A., Lieberman I.. Cyclic adenosine 3′:5′-monophosphate and the induction of deoxyribonucleic acid synthesis in liver. J. Biol. Chem. 1975; 250:3602–3606. [PubMed] [Google Scholar]
- 71. Carmichael G.G., Weber K., Niveleau A., Wahba A.J.. The host factor required for RNA phage Qβ RNA replication in vitro. Intracellular location, quantitation, and purification by polyadenylate-cellulose chromatography. J. Biol. Chem. 1975; 250:3607–3612. [PubMed] [Google Scholar]
- 72. Fender A., Elf J., Hampel K., Zimmermann B., Wagner E.G.. RNAs actively cycle on the Sm-like protein Hfq. Genes Dev. 2010; 24:2621–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Papenfort K., Said N., Welsink T., Lucchini S., Hinton J.C., Vogel J.. Specific and pleiotropic patterns of mRNA regulation by ArcZ, a conserved, Hfq-dependent small RNA. Mol. Microbiol. 2009; 74:139–158. [DOI] [PubMed] [Google Scholar]
- 74. Hussein R., Lim H.N.. Disruption of small RNA signaling caused by competition for Hfq. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:1110–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Moon K., Gottesman S.. Competition among Hfq-binding small RNAs in Escherichia coli. Mol. Microbiol. 2011; 82:1545–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Files of the raw, de-multiplexed reads of C. crescentus transcriptomes upon MMC treatment are accessible via the GEO accession GSE104186 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104186). Microarray data are available from PUMAdb (https://puma.princeton.edu/cgi-bin/publication/viewPublication.pl?pub_no=580).