


Abstract
Retroviruses package two complete RNA genomes into a viral particle but generate only one provirus after each infection. This pseudodiploid replication strategy facilitates frequent recombination, which occurs during DNA synthesis when reverse transcriptase switches templates between two copackaged RNA genomes, generating chimeric DNA. Recombination has played an important role in shaping the current HIV-1 pandemic; however, whether recombination is required for HIV-1 replication is currently unknown. In this report, we examined viral replication when recombination was blocked in defined regions of the HIV-1 genome. We found that blocking recombination reduced viral titers. Furthermore, a significant proportion of the resulting proviruses contained large deletions. Analyses of the deletion junctions indicated that these deletions were the direct consequence of blocking recombination. Thus, our findings illustrate that recombination is a major mechanism to maintain HIV-1 genome integrity. Our study also shows that both obligatory and nonobligatory crossovers occur during reverse transcription, thereby supporting both the forced and dynamic copy-choice models of retroviral recombination. Taken together, our results demonstrate that, in most viruses, both packaged RNA genomes contribute to the genetic information in the DNA form. Furthermore, recombination allows generation of the intact HIV-1 DNA genome and is required for efficient viral replication.
INTRODUCTION
Retroviruses are unique among all known virus families in which they package two complete copies of their genome into one viral particle although each infection event yields only a single DNA provirus (1). Thus, retroviruses are pseudodiploid (2). This distinct feature raises a key question that has not been answered yet: why do retroviruses contain two copies of the full-length RNA genome when only one DNA provirus is generated? Two hypotheses that are not mutually exclusive have been proposed. First, packaging two RNA genomes promotes homologous recombination, allowing the reassortment of mutations to increase diversity in the viral population (3). Alternatively, recombination acts as a repair mechanism by enabling the replication of viruses with damaged genomes, such as those containing RNA breaks (4).
Retroviral recombination occurs frequently during reverse transcription of the viral genome (2,5–13). The viral enzyme reverse transcriptase (RT) switches between copackaged RNA templates during minus-strand DNA synthesis, resulting in a hybrid DNA copy that contains a portion of information from each RNA genome (2,14). During infection by homozygous viruses containing two copies of identical RNA, recombination can occur but cannot be genetically detected. In contrast, recombination can be identified during infection by heterozygous particles that contain genetically distinct RNAs such as those from different proviruses (2,15). Currently, there are two proposed mechanisms to describe how recombination occurs during minus-strand DNA synthesis. The forced copy-choice model proposes that recombination occurs when RT encounters a break in the RNA genome, which forces RT to switch to the copackaged RNA to continue DNA synthesis (4). This model is based primarily on the observation that RNA genomes isolated from retroviral particles often contain breaks. On the other hand, the dynamic copy-choice model posits that recombination is not necessarily caused by RNA breaks but depends on the dynamic process of DNA synthesis (16). As RT synthesizes DNA, RNase H degrades the RNA template, leaving the nascent DNA free to anneal to the copackaged RNA. If DNA synthesis slows down or RNase H degrades the RNA template more rapidly, the RT complex becomes less stable, dissociates from the template, and can switch to using the copackaged RNA as a template. Thus, recombination depends on the dynamics of DNA synthesis, is not necessarily an obligatory event, and can occur even in the absence of RNA breaks. This model is based on the finding that recombination is promoted by decreased polymerase or increased RNase H activity (16).
HIV-1 RT is estimated to switch templates 8 to 10 times during the synthesis of a single genome (11,17–21). Thus, HIV-1 recombines frequently during replication. Recombination has led to the emergence of more than 96 circulating recombinant forms (CRFs) of different HIV-1 group M subtypes, and these CRFs cause a substantial proportion of worldwide infections (22). Additionally, recombination has been shown to produce variants that confer multidrug resistance or evade the host immune response (23–25). Thus, frequent recombination acts as a barrier to the development of effective vaccines and antiviral therapies. Taken together, these findings demonstrate that recombination plays an important role in generating viral diversity and shaping the current worldwide HIV-1 pandemic.
Despite the importance of HIV-1 recombination in generating viral diversity, whether it is required for HIV-1 replication has not yet been determined. If all recombination events are required to salvage genetic information, then recombination must be critical to HIV-1 replication, considering the high rate of crossovers. On the other hand, if only a small proportion of recombination events are obligatory, recombination may be dispensable for viral replication. In this report, we sought to answer this long-standing question by blocking homologous recombination in defined regions of the genome and monitoring the survival of progeny with intact genomes. Our results show that blocking recombination in a 2.5-kb region of the HIV-1 genome resulted in the loss of half of the proviruses with intact genomes. These findings indicate that HIV-1 recombination is required for efficient reverse transcription and maintenance of the intact genome structure.
MATERIALS AND METHODS
Construction of HIV-1 plasmids with heterologous sequences
HIV-1 control plasmids pT6S and pH0 were described previously as pWTNL43 and pWTHXB2, respectively (26). Both plasmids express functional Tat and Rev but not Vif, Vpu, Vpr, Env or Nef. Additionally, pH0 expresses functional Gag/Gag-Pol, whereas pT6S does not express functional Gag/Gag-Pol due to an inactivating 4-bp insertion in the capsid-coding region of the gag gene. The pol genes of pT6S and pH0 were derived from NL4-3 and HXB2, respectively. Additionally, pT6S encodes a mouse thy (CD90.2) gene and a green fluorescence protein (gfp) gene that has an inactivating mutation at the 3′ end, whereas pH0 encodes a mouse heat stable antigen (hsa; CD24) gene and a gfp gene that has an inactivating mutation at the 5′ end. To suppress recombination, the pol genes of pH0 and pT6S were modified to generate pH0-SynRT and pT6S-IN2, respectively. For simplicity, the RT- and integrase (IN)-coding region of the pol gene are referred to as rt and in region or rt and in sequence, respectively. HIV-1 plasmids with heterologous sequences were created from these control plasmids as follows. First, a synthetic HXB2 RT-coding sequence (SynRT) was engineered using a custom Python program based on Biopython (27) and developed in Enthought Canopy (v1.6.2). This program silently recoded HXB2 RT based on a human codon usage table but additionally forced codons to switch whenever possible to minimize sequence homology with the NL4-3 rt region. The sequence was further edited manually to remove several restriction enzyme sites and long runs of sequence homology. Although the NL4-3 and HXB2 rt regions share 97% homology at the nucleotide level, SynRT is only 63% homologous to the NL4-3 rt region. A DNA fragment containing the SynRT sequence was synthesized (GenScript) and cloned into pH0 using SpeI and SalI sites, generating pH0-SynRT. To create pT6S-IN2, a DNA fragment containing HIV-2 ROD in region with a silent XbaI site at the 5′ end was synthesized (IDT) and cloned into pT6S using XbaI and AscI sites, generating pT6S-IN2. Single-virion analysis constructs for examining RNA copackaging were derived from the previously described vectors Gag-BglSL, GagCeFP-BglSL, Gag-MSSL and GagCeFP-MSSL (28). To insert the pol sequence into these vectors, small (∼200 bp) NL4-3 or HXB2 linkers containing pro sequence were inserted into the AgeI site of these plasmids. Next, fragments containing wild-type or modified rt and in regions were inserted using SgrAI and AscI sites. All new constructs were verified by restriction digest mapping and sequencing of the cloned region. All helper plasmids have been described previously. Briefly, pHCMV-G expresses the G glycoprotein of vesicular stomatitis virus (VSV-G) (29), pSYNGP encodes human codon-optimized HIV-1 gag and gag-pol (30) and pIIINL(AD8)env expresses the HIV-1 AD8 CCR5-tropic envelope (Env) (31).
Cell culture and flow cytometry
Human 293T cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin. Hut/CCR5 cells are human T cells derived from the Hut78 cell line that stably express the chemokine receptor CCR5 and were maintained in RPMI 1640 medium supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 μg/ml puromycin and 500 μg/ml G418. Cells were analyzed by flow cytometry to determine viral titers as follows. Cells were collected, washed twice with Dulbecco’s phosphate-buffered saline (DPBS) containing 2.5% FBS, stained with 0.4 μg/ml phycoerythrin (PE)-conjugated anti-HSA and/or 2.0 μg/ml allophycocyanin (APC)-conjugated anti-Thy antibodies (BioLegend) for 30 min at 4°C, and washed two additional times with DPBS containing 2.5% FBS. Flow cytometry for PE-HSA, APC-Thy, and GFP was performed 72 h post-infection on an LSR II system (BD Biosciences), while cell sorting was performed on a FACSAria II system (BD Biosciences). Flow cytometry data were analyzed using FlowJo software (TreeStar, LLC).
GFP recombination assay
Dually infected 293T producer cell lines were generated by sequential infection at low multiplicity of infection (MOI; <0.1). Virus stocks were produced by cotransfecting 293T cells with an HIV-1 construct, pHCMV-G, and, in the case of constructs lacking functional Gag/Gag-Pol, pSYNGP, using TransIT-LT1 transfection reagent (Mirus). Virus stocks were harvested 48 h post-transfection, clarified through 0.45-μm pore size filters and used to infect cells. Dually infected cells were enriched by multiple rounds of cell sorting until >93% of the cells were dual positive (HSA+ and Thy+). Each producer cell line consisted of >62 000 independent infection events. As the serial infections were carried out at low MOI (<0.1), most of the cells in the final pool contained one copy of each provirus. Next, to determine GFP reconstitution frequencies, producer cell lines were transfected with pIIINL(AD8)env. Viruses were collected 48 h post-transfection, clarified through 0.45-μm-pore-size filters, and used to infect Hut/CCR5 cells. Infected cells were collected 72 h post-infection, stained and analyzed by flow cytometry. GFP reconstitution frequencies were calculated by dividing the percentage of GFP+ cells by the percentage of all infected (i.e. HSA, Thy and/or GFP+) cells.
Single-virion analysis
As previously described (28), to determine RNA copackaging frequencies, vector pairs were cotransfected into 293T cells, along with the RNA-labeling plasmids MS2-YFP and Bgl-mCherry, using TransIT-LT1 (Mirus). Although only Gag-CeFP constructs are depicted in the Supplementary Figure 2, plasmids encoding Gag-CeFP and untagged Gag were transfected at a 1:1 weight ratio in all experiments. Viral particles were collected 20 h post-transfection, clarified through 0.45-μm-pore-size filters, and added to glass slides (μ-Slide 8 Well ibiTreat; Ibidi) with polybrene (50 μg/ml final concentration). The slides were centrifuged at 1200 × g for 1 h at room temperature and imaged using fluorescence microscopy as described previously. Images were analyzed using custom MATLAB (MathWorks) programs to determine packaging efficiencies (i.e. the percentages of CeFP+ particles that were YFP+, mCherry+, or YFP+ and mCherry+). At least 1000 Gag-CeFP+ particles were analyzed per sample for each biological replicate, with at least three replicates performed for each experimental group.
Single Genome Sequencing (SGS) to determine provirus structures
Genomic DNA from GFP+ cell pools was first purified using the QIAamp DNA blood kit (Qiagen). To achieve single-genome amplification, genomic DNA was serially diluted in 96-well plates to identify a dilution in which ∼20% of wells were positive by polymerase chain reaction (PCR). At this dilution, most amplicons are derived from single templates. PCR was performed in 20-μl reactions containing 1× High Fidelity Buffer, 2 mM MgSO4, 0.2 mM of each deoxyribonucleotide triphosphate (dNTP), 0.2 μM of each primer and 0.025 U/μl Platinum Taq High Fidelity DNA Polymerase (Invitrogen). For the first round of PCR, sense primer HIV.BK5.F1 5′ CCT TGA GTG CTT CAA GTA GTG TGT GCC CGT CTGT and antisense primer pTS-R1: 5′ GTC GAC ACC CAA TTC TGA AAT GG were used. PCR was performed with the following parameters: 1 cycle of 94°C for 2 min; 35 cycles of 94°C for 15 s, 55°C for 30 s and 68°C for 4 min; and a final extension of 68°C for 10 min. For the second round of PCR, 1 μl of first-round PCR product was used with sense primer HIV.BK5.F2: 5′ GTA GTG TGT GCC CGT CTG TTG TGT GAC and antisense primer pTS-R2: 5′ GGC TTC CAA TCC CAT ATG GTG. Second-round PCRs were performed with the same conditions as the first-round PCR, but with a total of 45 cycles. The final amplicon was ∼4.5 kb in size and included the 5′ U5, 5′ UTR, gag and pol regions. All procedures were performed under PCR clean-room conditions with additional safeguards against sample contamination, such as prealiquoting of reagents, use of dedicated equipment and physical separation of pre- and post-PCR sample processing. Amplicons were directly sequenced using BigDye terminator chemistry according to the manufacturer’s recommendations (Applied Biosystems). Individual sequences for each amplicon were assembled and edited using Sequencher (Gene Codes). Chromatograms were inspected for the absence of mixed bases at each nucleotide position throughout the entire amplicon; this quality-control measure confirmed that the amplicons analyzed were derived from single-genome amplification and allowed amplicons that contained PCR-generated recombination events or errors to be excluded from the analysis.
Sequence and statistical analyses
Assembled single-genome sequences were aligned to appropriate parental reference sequences using MAFFT v7 (32). Sequence alignments were manually edited and trimmed to remove primer regions in AliView v1.18 (33). Final Alignments were visualized using Highlighter (34). After trimming, sequences included the last 61 bp of the 5′ UTR, all of gag, and all of pol except the last 64 bp of the in sequence. Crossovers were quantified using a custom Python program based on Biopython (27) and developed in Enthought Canopy (v1.6.2). This program was used to quantify the number of crossovers and reference bases within entire sequences as well as defined subregions such as the pro, rt and in sequences. To distinguish crossovers from mutations or sequencing errors, crossovers were required to switch at least two successive markers. Crossovers that occurred at gene junctions were assigned to the more likely gene. As an example, for the H0/T6S vector pair, the marker segment at the rt/in junction is 31 bp in length, with 28 bp in rt and 3 bp in the in region; crossovers in this segment were thus assigned to rt. Deletions were quantified and characterized manually using AliView. Sequence duplicates containing deletions were collapsed into a single sequence. All statistical analyses were performed in GraphPad Prism v7.01 (GraphPad Software, Inc.).
Calculation of recombination events
We performed the following calculation to estimate the proportion of viruses that would be lost when recombination is blocked in the entire genome. The control virus pair (H0/T6S) and the test virus pair (H0-SynRT/T6S-IN2) generated 5.6 and 2.8% of infected cells with GFP+ intact proviruses, respectively. Therefore, blocking recombination in 28% of the viral genome (2.5 out of 9.2 kb) resulted in the loss of 51% of GFP+ intact proviruses. These results indicate that the rate of forced crossovers is at least 0.51/sequence in the rt and in region. However, some viruses likely would have undergone multiple force crossovers in this region. Recombination events have been shown to be distributed throughout the HIV-1 genome (11,35) and do not exhibit high negative interference (18). Assuming that forced crossovers are randomly distributed, the rate of forced crossovers is 0.71/sequence in the rt and in region and 2.55/sequence in the ∼9.2 kb HIV-1 genome. Under these conditions, ∼92% of viruses would undergo at least one forced crossover during reverse transcription that, when blocked, would lead to a noninfectious virus.
The proportion of obligatory (forced) crossover events was calculated as followed. The observed rate of crossovers in the rt and in region was 1.78/sequence for the control vector pair (H0/T6S) and the estimated rate of forced crossover is 0.71/sequence (see above). Thus, ∼40% (0.71/1.78) of crossover events are forced whereas ∼60% of crossover events are not forced.
RESULTS
Experimental strategy to suppress homologous recombination
During reverse transcription, two template-switching events (minus- and plus-strand DNA transfers) are required to complete viral DNA synthesis (1,36). Therefore, the role of recombination in HIV-1 replication cannot be addressed by blocking all template-switching events. Instead, we adopted a strategy to suppress recombination in part of the viral genome, based on previous observations that frequent HIV-1 recombination depends on high sequence homology (37–39). To suppress recombination, we introduced sequences with reduced homology to replace the wild-type genes in two previously described HIV-1 constructs, H0 and T6S (26) (Figure 1A). These near-full-length constructs contain all the cis-acting elements required for viral replication and encode gag-pol, tat and rev. However, T6S-based constructs do not express Gag or Gag-Pol due to a 4-nt frameshift insertion in the capsid region of gag. Both H0 and T6S are derived from the NL4-3 molecular clone; however, H0 contains the pol gene from HXB2, whereas T6S retains the pol gene from NL4-3. The pol genes from HXB2 and NL4-3 share 97% sequence homology, which is sufficient to mediate frequent recombination while still allowing for the detection of crossovers using polymorphic sites (26). We selected the rt region for sequence alteration because it lacks known cis-acting elements that are essential for viral replication. To suppress homologous recombination, we replaced the rt region in H0 with a synthetic sequence (SynRT; shown as a blue box in Figure 1B) that encodes the same protein but contains multiple silent mutations so that it has ∼63% homology to the rt region in T6S. Additionally, we replaced the in region, which contains the central polypurine tract (cPPT). Previous studies have shown that disruption of the cPPT does not affect reverse transcription kinetics or viral titers (40). Nonetheless, to preserve the cPPT, we replaced the in region in T6S with the in sequence from HIV-2 ROD (IN2; shown as a red box in Figure 1B), which has 67% homology to the in region in H0. Previous studies indicate that this strategy should block >95% of the homologous recombination events in this region (37–39). Similar to T6S, T6S-IN2 contains a frameshift mutation in gag and does not express Gag/Gag-Pol, thereby avoiding potential incompatibility of proteins from different viruses (41).
Figure 1.

Strategy to suppress homologous recombination in HIV-1. (A) Control vector pair. The asterisk denotes an inactivating frameshift mutation, whereas ‘I’ indicates an internal ribosome entry site. (B) Vector pair designed to suppress recombination. Recombination was suppressed in 2.5 kb of the viral genome by replacing the wild-type HIV-1 rt and in regions with synthetic HIV-1 rt (blue) or HIV-2 ROD in (red) regions. The effects of suppressing recombination could be determined by analyzing GFP+ cells, which could only result from heterozygous particles. (C) Experimental protocol for determining the effects of suppressing recombination on GFP titers and GFP+ provirus structures.
In this strategy, heterozygous viruses with one copy of each RNA (H0-SynRT and T6S-IN2) contain two RNAs with different rt and in sequences, thus suppressing recombination in this region. However, recombination in the rt and in regions is not blocked in homozygous viruses containing two copies of the same RNA. To examine the effect of suppressing recombination on viral replication, we identified progeny that were generated from heterozygous particles using a previously described marker system (18). These HIV-1 constructs contain either a mouse hsa or a mouse thy gene; additionally, both constructs contain an internal ribosomal entry site (I) and a defective green fluorescence protein (gfp) gene (Figure 1A and B). HSA and Thy are cell surface markers that can be detected by flow cytometry and used to identify infected cells. The gfp genes in the two vectors contain inactivating mutations at the 5′ (H0) or 3′ (T6S) end (asterisks in Figure 1A and B). In cells infected by heterozygous particles, a functional gfp gene can be restored by recombination during reverse transcription.
To study the replication of heterozygous particles, we first generated producer cell lines that contain one copy of each parental provirus (Figure 1C). For this purpose, we generated H0- or T6S-derived viruses that were pseudotyped with VSV-G and serially infected human 293T cells with these viruses at low MOI (<0.1). Dually infected cells were enriched by multiple rounds of cell sorting until >93% of the cell populations were HSA+ and Thy+. To examine viral replication, we transfected 293T producer cells with a plasmid that expresses HIV-1 Env and used the harvested virus to infect Hut/CCR5 T cells. Infected cells were analyzed by flow cytometry to determine the proportions of cells expressing HSA, Thy and/or GFP. Infected cells express HSA or Thy markers, whereas only cells infected by heterozygous particles can express GFP. Therefore, we could monitor the replication of heterozygous particles in the entire viral population by determining the proportion of GFP+ cells in the infected cells (HSA+ or Thy+). If recombination was not required for replication, RT would be able to complete DNA synthesis regardless of whether recombination was blocked and GFP titers would not be significantly affected. In contrast, if recombination was required to complete DNA synthesis, blocking recombination would inhibit the replication of heterozygous particles, thereby reducing GFP titers. Based on our previous study, most viruses should have at least one crossover in the rt and in regions (26), and we should be able to distinguish between these two possible outcomes. Additionally, it has been shown that HIV-1 recombination events do not exhibit high negative interference; that is, the occurrence of one crossover event does not significantly increase the chance of a second crossover event (18). Therefore, results obtained from studying the GFP+ subpopulation should accurately reflect the entire viral population.
Suppressing recombination reduces the proportion of GFP+ cells in the infected population
To determine whether recombination is required for HIV-1 replication, we generated producer cells containing pairs of proviruses that suppress recombination in 0- (control), 0.9-, 1.7- or 2.5-kb regions (Figure 2). We then produced viruses, infected Hut/CCR5 T cells, and performed flow cytometry to determine HSA, Thy and GFP titers (Figure 1C). To determine whether replacing HIV-1 sequences affects viral replication, we compared HSA and Thy viral titers in Hut/CCR5 cells. We found that relative HSA and Thy titers were similar for viruses from all four producer cell lines (Supplementary Figure S1). These results show that replacing the rt or in sequence in H0-SynRT or T6S-IN2, respectively, did not affect the expression or replication of these vectors. Furthermore, as viruses from these cell lines generated similar relative HSA and Thy titers, we could directly compare their GFP titers. When using virus produced from cells containing the control vector pair, ∼5.7% of infected cells were GFP+ (Figure 2). However, when using viruses generated from vector pairs containing 0.9-, 1.7- or 2.5-kb heterologous sequences, ∼4.1%, ∼3.9% or ∼3.1% of the infected cells were GFP+, respectively; these titers were significantly lower than that of the control (P < 0.05, P < 0.01 and P < 0.001, respectively; one-way ANOVA with Dunnett’s post-test). Furthermore, the decrease in GFP titer correlated with the lengths of heterologous sequences that suppressed recombination (Pearson’s r = −0.96, P = 0.04). These results indicate that GFP titers progressively declined as the region with suppressed recombination was extended.
Figure 2.

Suppressing recombination significantly reduces GFP reconstitution frequencies. Virus pairs were designed to suppress recombination in the rt, in, or both rt and in regions. GFP reconstitution frequencies were determined by measuring GFP, HSA and Thy titers and calculating the proportions of GFP+ cells in all infected cells. The data represent the mean ± standard deviation (SD) of at least three independent experiments; *, P < 0.05; **, P < 0.01; ***, P < 0.001 (one-way ANOVA with Dunnett’s post-test).
Proviruses expressing GFP arise from heterozygous particles (Figure 1A and B). Thus, we hypothesized that decreased GFP titers were caused by inhibiting recombination in heterozygous particles. Alternatively, it is also possible that viral RNAs with different rt and in sequences did not copackage efficiently. We consider this possibility unlikely because the pol sequence does not contribute to RNA genome packaging (28,42), a finding that is supported by the viral titers in this study (Supplementary Figure S1). However, to rule out this possibility, we performed single-virion analysis to detect viral RNAs in individual particles as previously described (28). In this assay, some of the Gag molecules were tagged with cerulean fluorescent protein (CeFP) to identify viral particles, whereas viral RNAs were detected using RNA-binding proteins fused to fluorescent proteins (Bgl-mCherry and MS2-YFP). These RNA-binding proteins specifically recognize stem–loops engineered into the constructs. Assuming there would be equal expression and random copackaging of RNAs from two viral constructs, ∼50% of the particles should be heterozygous, whereas each type of homozygous particles should be ∼25% of the viral population. For the control, we found that Gag-CeFP+ particles were ∼25% MS2-YFP+, ∼26% Bgl-mCherry+ and ∼45% MS2-YFP+/Bgl-mCherry+ (Supplementary Figure S2), consistent with near-random copackaging of viral RNA (28). For the vector pair containing altered rt and in sequences, the Gag-CeFP+ particles were ∼27% MS2-YFP+, ∼25% Bgl-mCherry+ and ∼43% MS2-YFP+/Bgl-mCherry+ (Supplementary Figure S2), which was not significantly different from the control (P = 0.27, unpaired t-test). These results indicate that the RNAs from viral constructs containing SynRT and HIV-2 IN copackage efficiently; thus, decreased GFP titers cannot be explained by reduced copackaging of viral RNAs into particles.
Blocking recombination results in progeny with large deletions in the viral genome
During DNA synthesis of the region where homologous recombination is blocked, RT is unable to switch to the corresponding region in the copackaged RNA when encountering a break in the template (4) or when the RT complex becomes unstable and dissociates from template (16). In this case, RT may not be able to continue DNA synthesis, resulting in premature termination. Alternatively, RT may switch templates in a nonhomologous manner, leading to deletions. To characterize progeny generated from heterozygous particles, we performed single genome sequencing (SGS) of proviruses from GFP+ cells (43). Briefly, we infected Hut/CCR5 T cells as described (Figure 1C) and sorted out GFP+ cell pools that each contained >5000 infection events. We then purified cellular DNA and performed nested, limiting-dilution PCR of a 4.5-kb amplicon that included 5′ U5, 5′ UTR, gag and pol. DNA recombination occurs frequently during PCR (44–46). To ensure that our study is not complicated by PCR-mediated recombination, we used experimental conditions in which only ∼20% of amplification reactions were positive. Furthermore, PCR products were directly sequenced, and sequences with mixed bases were discarded. Thus, most, if not all, of the resulting sequences were derived from single proviruses.
To validate our experimental strategy, we first examined whether introducing heterologous sequences indeed suppressed recombination during viral replication. In our control constructs (H0 and T6S), the region including the pro, rt and in sequences shared 97% homology. In the 86 sequences that we analyzed, 181 homologous recombination events were identified, resulting in an average of 2.1 crossovers per sequence (Table 1), similar to the previously observed rate with this vector pair (26). Within the analyzed sequences, there were 28, 113 and 40 crossovers in the pro, rt and in regions, respectively, indicating that recombination occurred frequently throughout these regions. Furthermore, 78% of the sequences had at least one crossover in the rt or in region.
Table 1.
Decreased nucleotide sequence homology blocks homologous recombination
| pro | rt | in | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Vector pair | Seq | Cross-overs | Cross-overs/seqa | Cross-overs | Cross-overs/seqa | Cross-overs | Cross-overs/seqa | Cross-overs | Cross-overs/seqa |
| H0/T6S | 86 | 28 | 0.33 | 113 | 1.31 | 40 | 0.47 | 181 | 2.10 |
| H0-SynRT/T6S | 130 | 18 | 0.14 | 0 | < 0.01b | 56 | 0.43 | 74 | 0.57 |
| H0-SynRT/T6S-IN2 | 120 | 14 | 0.12 | 0 | < 0.01b | 2 | 0.02 | 16 | 0.13 |
aThe total number of crossovers within each region was divided by the total number of sequences.
bNo crossovers were observed between wild-type and synthetic rt for either vector pair, so the rate of crossovers in rt is less than 1/130 or 1/120 (<0.01).
We next analyzed 130 sequences from GFP+ cell pools generated by the H0-SynRT/T6S virus pair, in which the rt sequences shared 63% homology. Of the 74 homologous recombination events that we identified, we observed 18, 0 and 56 crossovers in pro, rt and in, respectively. Additionally, we analyzed 120 sequences from GFP+ cell pools for the H0-SynRT/T6S-IN2 virus pair, in which both rt and in regions had decreased homology. Of the 16 homologous recombination events that we identified, we observed 14, 0 and 2 recombination events in pro, rt and in, respectively. Therefore, we did not observe homologous crossovers between the wild-type rt and synthetic rt regions for either pair of constructs, indicating that more than 99% of the recombination events were blocked. However, we did detect two homologous crossovers between the HIV-1 and HIV-2 in regions; both events occurred in a conserved stretch of 13 nucleotides. We estimate that ∼96% of crossovers were blocked in the in region. Taken together, these results demonstrate that our strategy of recoding the genome to block recombination was successful.
To determine the consequences of blocking recombination, we next analyzed proviral genome structures. We first examined whether there were deletions outside the region that we altered (rt and in), as these would likely not be due to blocked recombination. Of the 336 sequences that we obtained, there were only two deletions located entirely outside the rt and in regions: one deletion was observed among the progeny of the control virus pair H0/T6S and one deletion was observed among the progeny of the virus pair H0-SynRT/T6S-IN2 (Supplementary Figure S3). Therefore, deletions outside the rt and in regions occurred at similar rates among the progeny from different virus pairs. However, we observed strikingly higher levels of deletions involving rt and in sequences in the progeny of the noncontrol virus pairs. Among progeny from the control, we observed one deletion in 86 sequences, resulting in a deletion frequency of ∼1.2% (Figure 3). In contrast, we observed 9 deletions in 130 progeny sequences from H0-SynRT/T6S (∼6.9%; P = 0.05; Fisher’s exact test; Figure 3) and 16 deletions in 120 progeny sequences from H0-SynRT/T6S-IN2 (∼13.3%; P < 0.01; Fisher’s exact test; Figure 3). Thus, blocking recombination significantly increased deletion frequencies in the targeted regions, demonstrating that recombination is critical to maintenance of the intact genome structure.
Figure 3.

Blocking recombination leads to large deletions. SGS was performed to generate and analyze a 4.5-kb amplicon including the gag, pro, rt and in regions. The total numbers of sequences analyzed are indicated, but only sequences with deletions are shown. Deletions are depicted as dark gray boxes; deletion frequencies, calculated as deletion events per sequence, are also indicated.
Deletions are mediated by nonhomologous recombination
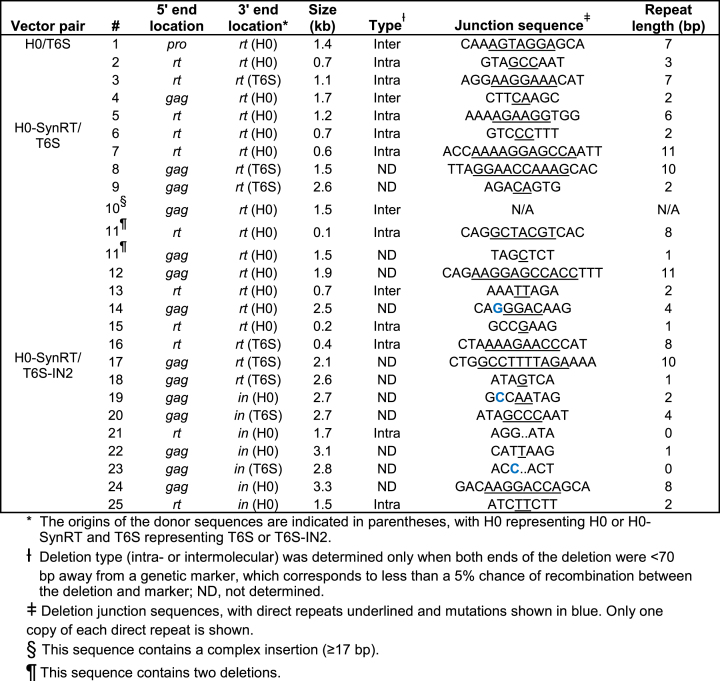
To examine the mechanism(s) of deletions, we analyzed their locations, sizes and junction sequences (Table 2). Among progeny sequences from the control virus pair (H0/T6S), only one deletion was observed in the region of interest (rt and in). Sequence analysis revealed that RT used a 7-bp direct-repeat sequence to perform an intermolecular template switch, resulting in the removal of a 1.4-kb fragment from pro to rt region. For progeny sequences from H0-SynRT/T6S and from H0-SynRT/T6S-IN2, the sizes of the deletions ranged from 0.1 to 3.3 kb, with an average of 1.7 kb. Importantly, all the 3′ ends of the deletion junctions were located in the region where recombination was blocked due to low sequence homology. For H0-SynRT/T6S, all 9 deletions had 3′ ends in the rt sequence; for H0-SynRT/T6S-IN2, 9 deletions had 3′ ends in the rt sequence and 7 deletions had 3′ ends in the in sequence. These results demonstrate that these deletions were the direct consequence of blocking homologous recombination. Furthermore, direct repeats were observed at most deletion junctions, and ranged in size from 1 to 11 bp (Table 2). The presence of short direct repeats at deletion junctions is a known hallmark of RT-mediated nonhomologous recombination (47,48). Taken together, these results show that, when homologous recombination is blocked, HIV-1 undergoes nonhomologous recombination and loses genome integrity.
Table 2.
Features of deletions observed upon blocking homologous recombination
|
Recombination is required for efficient HIV-1 replication
To assess the importance of recombination to HIV-1 replication, we determined the proportion of GFP+ intact proviruses remaining after blocking recombination in 2.5 kb of the viral genome. For the control virus pair (H0/T6S), 5.7% of infected cells expressed GFP and 99% of GFP+ proviruses had intact gag-pol sequences. Therefore, 5.6% of infected cells contained GFP+ intact proviruses. For the virus pair with altered rt and in regions (H0-SynRT/T6S-IN2), 3.1% of infected cells expressed GFP and only 88% of GFP+ proviruses had intact gag-pol sequences. Overall, 2.8% of infected cells contained GFP+ intact proviruses. Thus, blocking recombination in the rt and in regions resulted in the loss of 51% of GFP+ intact proviruses. In these experiments, we blocked recombination in only 28% of the viral genome (2.5 out of 9.2 kb). Recombination events have been shown to occur throughout the HIV-1 genome (11,35). Based on our results and the assumption that the events that eliminated GFP+ intact proviruses are randomly distributed, we predict that ∼92% of GFP+ intact proviruses would be lost if homologous recombination was blocked across the entire 9.2-kb genome (‘Materials and Methods’ section). Thus, recombination is a major mechanism to maintain full-length genome structure and promote efficient HIV-1 replication.
DISCUSSION
A critical step in viral replication is the transfer of genetic information to progeny. Retroviral particles package RNA genomes and must convert the genetic information into a DNA form before generating a provirus in the new target cell. To successfully complete DNA synthesis, RT performs two obligatory strand-transfer events: minus- and plus-strand transfers. In this report, we demonstrate that, most of the time, HIV-1 RT must undergo additional template-switching events via homologous recombination to complete synthesis of full-length DNA. This conclusion comes from our observation that blocking homologous recombination in less than one-third of the HIV-1 genome resulted in the loss of half of proviruses with intact genomes. Although frequent retroviral recombination has been observed for four decades, our study provides a clear demonstration of its role in maintaining genome integrity. Thus, in addition to generating viral diversity, recombination is critical for viral replication. This study answers a long-standing question in the field of retrovirology and reveals previously unknown aspects of HIV-1 replication.
Our results also shed light on the mechanistic basis of HIV-1 recombination. We found that when homologous recombination was blocked in the rt and in regions, only 49% of intact proviruses survived. In regions of the genome where recombination was blocked, deletion frequencies increased significantly (Figure 3), and deletion junctions often contained short stretches of homology between donor and acceptor templates (Table 2). Thus, when homologous recombination is not possible and RT cannot continue synthesis of the same template, RT uses small regions of homology to switch elsewhere in the genome. These findings are consistent with the central thesis of the forced copy-choice model, which proposes that recombination serves as a repair mechanism to salvage damaged genomes (4). However, in our experiments, when recombination was not blocked, 78% of proviruses had at least one crossover in the rt or in region. If all crossovers were obligatory or ‘forced’ — that is, caused by RNA breaks (4) or irreversible kinetic events during DNA synthesis (16) — then only ∼22% of intact proviruses would survive. We observed that 49% of intact proviruses survived, far more than the predicted 22%, indicating that some, but not all, crossovers are forced. These findings are consistent with the central thesis of the dynamic copy-choice model, which proposes that recombination depends on the kinetic stability of the RT complex and that crossovers can be forced or not forced (16). If we assume that the events that force recombination are randomly distributed, our data are consistent with ∼40% of crossovers being forced (See ‘Materials and Methods’ section for calculation). Therefore, we conclude that HIV-1 recombination occurs through both forced and dynamic copy-choice models.
Whether both copackaged retroviral genomes are required for DNA synthesis has been studied previously with varied conclusions. Using spleen necrosis virus, one study concluded that only one RNA genome is required for DNA synthesis, because minus-strand DNA transfer was found to be mostly intramolecular in nature (49). However, spleen necrosis virus, like murine leukemia virus, may not form heterozygous particles efficiently (50–54). Thus, most of the analyzed progeny may have been from homozygous particles, in which case intra- and intermolecular minus-strand transfers cannot be distinguished. Using HIV-1 vectors, another study concluded that one infection event can generate two proviruses, because plaque assays gave rise to rare plaques containing more than one provirus (55). However, these experiments only measured rare recombinants resulting from DNA recombination during cotransfection. This limitation would result in severe underestimation of parental virus titers, such that the observed events were likely due to coinfection. Lastly, using HIV-1 vectors, it was reported that a marker gene was inactivated in target cells more frequently when cotransfected with an empty vector lacking the marker gene (56). However, further analyses revealed that the internal promoter sequence in the vector contained cryptic splice sites, with most inactivation coming from the packaging of aberrantly spliced RNAs lacking the complete marker gene. Therefore, these previous studies did not address whether both packaged RNA genomes are required for DNA synthesis in retroviral replication. In contrast, in our study, we specifically blocked recombination in only part of the viral population—the heterozygous particles—and we observed a significant decline in the progeny of heterozygous particles (Figure 2). Proviral structure analyses showed that deletions were significantly increased specifically in the regions of the genome in which recombination was blocked (Figure 3). Wild-type and altered sequences were used as deletion donors at similar rates (Table 2), suggesting that deletions were not caused by abnormal features of the engineered sequences. Furthermore, the deletions were marked by small regions of homology (Table 2), a known hallmark of nonhomologous recombination. Taken together, these results demonstrate that both copies of the RNA genome must be used during reverse transcription of most HIV-1 particles, although only a single provirus is generated.
Retroviruses transfer their genetic information between RNA and DNA forms, and it is intriguing to contemplate how this transfer evolved to use a pseudodiploid replication strategy. Retroviruses need to perform two strand-transfer steps during DNA synthesis to reconstitute the long terminal repeats. Therefore, RT must be able to switch templates to some extent, and this ability likely enabled template switching elsewhere in the genome through the process of homologous recombination. It is possible that retroviruses evolved to package two copies of the RNA genome to compensate for the tendency of RT to switch templates, thereby preserving genome integrity. On the other hand, it is also possible that because retroviruses package two copies of the RNA genome, they have less selective pressure to evolve a more processive RT or a mechanism to better protect the RNA genome. Regardless of the evolutionary path, the pseudodiploid replication strategy has obviously been highly successful, resulting in hundreds of different retroviral species, including HIV-1.
Our study reveals that efficient HIV-1 replication requires both copackaged RNAs and recombination to maintain genome integrity. These findings unveil the possibility that novel antiviral strategies could be developed to control HIV-1 replication by targeting the essential recombination process or the packaging of two RNA genomes. Such novel strategies could be used in combination with other antivirals such as approved RT inhibitors. Although reverse transcription is a major target for current antiretrovirals, these compounds do not specifically target the essential recombination process during DNA synthesis. Approaches that block different aspects of reverse transcription, such as DNA synthesis and homologous recombination, could potentially achieve synergistic antiviral effects.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Anne Arthur for expert editorial help and John Coffin, Steve Hughes and Eric Freed for discussion.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the National Institutes of Health (NIH); Center for Cancer Research, National Cancer Institute (NCI); NIH Intramural AIDS Targeted Antiviral Program (to W.-S. H.,V.K.P.); Innovation Fund, Office of AIDS Research, NIH; NIH Intramural AIDS Research Fellowship (to J.M.O.R); NCI, NIH [HHSN261200800001E]. Funding for open access charge: Intramural Research Program, NIH, U.S.A.
Conflict of interest statement. None declared.
REFERENCES
- 1. Goff S.P. Knipe DM, Howley PM. Retroviridae: the retroviruses and their replication. Fields Virology. 2007; 2:5th ednPhiladelphia: Lippincott Williams and Wilkins; 1999–2069. [Google Scholar]
- 2. Hu W.S., Temin H.M.. Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:1556–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Temin H.M. Sex and recombination in retroviruses. Trends Genet. 1991; 7:71–74. [DOI] [PubMed] [Google Scholar]
- 4. Coffin J.M. Structure, replication, and recombination of retrovirus genomes: some unifying hypotheses. J. Gen. Virol. 1979; 42:1–26. [DOI] [PubMed] [Google Scholar]
- 5. Hu W.S., Temin H.M.. Retroviral recombination and reverse transcription. Science. 1990; 250:1227–1233. [DOI] [PubMed] [Google Scholar]
- 6. Clavel F., Hoggan M.D., Willey R.L., Strebel K., Martin M.A., Repaske R.. Genetic recombination of human immunodeficiency virus. J. Virol. 1989; 63:1455–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mikkelsen J.G., Lund A.H., Kristensen K.D., Duch M., Sorensen M.S., Jorgensen P., Pedersen F.S.. A preferred region for recombinational patch repair in the 5′ untranslated region of primer binding site-impaired murine leukemia virus vectors. J. Virol. 1996; 70:1439–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhuang J., Jetzt A.E., Sun G., Yu H., Klarmann G., Ron Y., Preston B.D., Dougherty J.P.. Human immunodeficiency virus type 1 recombination: rate, fidelity, and putative hot spots. J. Virol. 2002; 76:11273–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Onafuwa A., An W., Robson N.D., Telesnitsky A.. Human immunodeficiency virus type 1 genetic recombination is more frequent than that of Moloney murine leukemia virus despite similar template switching rates. J. Virol. 2003; 77:4577–4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Galetto R., Moumen A., Giacomoni V., Veron M., Charneau P., Negroni M.. The structure of HIV-1 genomic RNA in the gp120 gene determines a recombination hot spot in vivo. J. Biol. Chem. 2004; 279:36625–36632. [DOI] [PubMed] [Google Scholar]
- 11. Levy D.N., Aldrovandi G.M., Kutsch O., Shaw G.M.. Dynamics of HIV-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:4204–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baird H.A., Galetto R., Gao Y., Simon-Loriere E., Abreha M., Archer J., Fan J., Robertson D.L., Arts E.J., Negroni M.. Sequence determinants of breakpoint location during HIV-1 intersubtype recombination. Nucleic Acids Res. 2006; 34:5203–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen J., Powell D., Hu W.S.. High frequency of genetic recombination is a common feature of primate lentivirus replication. J. Virol. 2006; 80:9651–9658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anderson J.A., Teufel R.J. 2nd, Yin P.D., Hu W.S.. Correlated template-switching events during minus-strand DNA synthesis: a mechanism for high negative interference during retroviral recombination. J. Virol. 1998; 72:1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chin M.P., Rhodes T.D., Chen J., Fu W., Hu W.S.. Identification of a major restriction in HIV-1 intersubtype recombination. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:9002–9007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hwang C.K., Svarovskaia E.S., Pathak V.K.. Dynamic copy choice: steady state between murine leukemia virus polymerase and polymerase-dependent RNase H activity determines frequency of in vivo template switching. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:12209–12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rhodes T., Wargo H., Hu W.S.. High rates of human immunodeficiency virus type 1 recombination: near-random segregation of markers one kilobase apart in one round of viral replication. J. Virol. 2003; 77:11193–11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rhodes T.D., Nikolaitchik O., Chen J., Powell D., Hu W.S.. Genetic recombination of human immunodeficiency virus type 1 in one round of viral replication: effects of genetic distance, target cells, accessory genes, and lack of high negative interference in crossover events. J. Virol. 2005; 79:1666–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Delviks-Frankenberry K., Galli A., Nikolaitchik O., Mens H., Pathak V.K., Hu W.S.. Mechanisms and factors that influence high frequency retroviral recombination. Viruses. 2011; 3:1650–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Onafuwa-Nuga A., Telesnitsky A.. The remarkable frequency of human immunodeficiency virus type 1 genetic recombination. Microbiol. Mol. Biol. Rev. 2009; 73:451–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ramirez B.C., Simon-Loriere E., Galetto R., Negroni M.. Implications of recombination for HIV diversity. Virus Res. 2008; 134:64–73. [DOI] [PubMed] [Google Scholar]
- 22. Hemelaar J., Gouws E., Ghys P.D., Osmanov S.. Global trends in molecular epidemiology of HIV-1 during 2000–2007. AIDS. 2011; 25:679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moutouh L., Corbeil J., Richman D.D.. Recombination leads to the rapid emergence of HIV-1 dually resistant mutants under selective drug pressure. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:6106–6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kellam P., Larder B.A.. Retroviral recombination can lead to linkage of reverse transcriptase mutations that confer increased zidovudine resistance. J. Virol. 1995; 69:669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Streeck H., Li B., Poon A.F., Schneidewind A., Gladden A.D., Power K.A., Daskalakis D., Bazner S., Zuniga R., Brander C. et al. . Immune-driven recombination and loss of control after HIV superinfection. J. Exp. Med. 2008; 205:1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Delviks-Frankenberry K.A., Nikolaitchik O.A., Burdick R.C., Gorelick R.J., Keele B.F., Hu W.S., Pathak V.K.. Minimal contribution of APOBEC3-Induced G-to-A hypermutation to HIV-1 recombination and genetic variation. PLoS Pathog. 2016; 12:e1005646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cock P.J., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B. et al. . Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009; 25:1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen J., Nikolaitchik O., Singh J., Wright A., Bencsics C.E., Coffin J.M., Ni N., Lockett S., Pathak V.K., Hu W.S.. High efficiency of HIV-1 genomic RNA packaging and heterozygote formation revealed by single virion analysis. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:13535–13540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yee J.K., Miyanohara A., LaPorte P., Bouic K., Burns J.C., Friedmann T.. A general method for the generation of high-titer, pantropic retroviral vectors: highly efficient infection of primary hepatocytes. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:9564–9568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kotsopoulou E., Kim V.N., Kingsman A.J., Kingsman S.M., Mitrophanous K.A.. A Rev-independent human immunodeficiency virus type 1 (HIV-1)-based vector that exploits a codon-optimized HIV-1 gag-pol gene. J. Virol. 2000; 74:4839–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Freed E.O., Englund G., Martin M.A.. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J. Virol. 1995; 69:3949–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Katoh K., Standley D.M.. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013; 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Larsson A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics. 2014; 30:3276–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Keele B.F., Giorgi E.E., Salazar-Gonzalez J.F., Decker J.M., Pham K.T., Salazar M.G., Sun C., Grayson T., Wang S., Li H. et al. . Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:7552–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jetzt A.E., Yu H., Klarmann G.J., Ron Y., Preston B.D., Dougherty J.P.. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J. Virol. 2000; 74:1234–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gilboa E., Mitra S.W., Goff S., Baltimore D.. A detailed model of reverse transcription and tests of crucial aspects. Cell. 1979; 18:93–100. [DOI] [PubMed] [Google Scholar]
- 37. An W., Telesnitsky A.. Effects of varying sequence similarity on the frequency of repeat deletion during reverse transcription of a human immunodeficiency virus type 1 vector. J. Virol. 2002; 76:7897–7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Motomura K., Chen J., Hu W.S.. Genetic recombination between human immunodeficiency virus type 1 (HIV-1) and HIV-2, two distinct human lentiviruses. J. Virol. 2008; 82:1923–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nikolaitchik O.A., Galli A., Moore M.D., Pathak V.K., Hu W.S.. Multiple barriers to recombination between divergent HIV-1 variants revealed by a dual-marker recombination assay. J. Mol. Biol. 2011; 407:521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thomas D.C., Voronin Y.A., Nikolenko G.N., Chen J., Hu W.S., Pathak V.K.. Determination of the ex vivo rates of human immunodeficiency virus type 1 reverse transcription by using novel strand-specific amplification analysis. J. Virol. 2007; 81:4798–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Padow M., Lai L., Deivanayagam C., DeLucas L.J., Weiss R.B., Dunn D.M., Wu X., Kappes J.C.. Replication of chimeric human immunodeficiency virus type 1 (HIV-1) containing HIV-2 integrase (IN): naturally selected mutations in IN augment DNA synthesis. J. Virol. 2003; 77:11050–11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu Y., Nikolaitchik O.A., Rahman S.A., Chen J., Pathak V.K., Hu W.S.. HIV-1 sequence necessary and sufficient to package Non-viral RNAs into HIV-1 particles. J. Mol. Biol. 2017; 429:2542–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Palmer S., Kearney M., Maldarelli F., Halvas E.K., Bixby C.J., Bazmi H., Rock D., Falloon J., Davey R.T. Jr, Dewar R.L. et al. . Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 2005; 43:406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Meyerhans A., Vartanian J.P., Wain-Hobson S.. DNA recombination during PCR. Nucleic Acids Res. 1990; 18:1687–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Klug J., Wolf M., Beato M.. Creating chimeric molecules by PCR directed homologous DNA recombination. Nucleic Acids Res. 1991; 19:2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Suspene R., Thiers V., Vartanian J.P., Wain-Hobson S.. PCR mediated recombination impacts the analysis of hepatitis B Virus covalently closed circular DNA. Retrovirology. 2016; 13:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pathak V.K., Temin H.M.. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: deletions and deletions with insertions. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:6024–6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang J., Temin H.M.. Rate and mechanism of nonhomologous recombination during a single cycle of retroviral replication. Science. 1993; 259:234–238. [DOI] [PubMed] [Google Scholar]
- 49. Jones J.S., Allan R.W., Temin H.M.. One retroviral RNA is sufficient for synthesis of viral DNA. J. Virol. 1994; 68:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hu W.S., Bowman E.H., Delviks K.A., Pathak V.K.. Homologous recombination occurs in a distinct retroviral subpopulation and exhibits high negative interference. J. Virol. 1997; 71:6028–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kharytonchyk S.A., Kireyeva A.I., Osipovich A.B., Fomin I.K.. Evidence for preferential copackaging of Moloney murine leukemia virus genomic RNAs transcribed in the same chromosomal site. Retrovirology. 2005; 2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Flynn J.A., An W., King S.R., Telesnitsky A.. Nonrandom dimerization of murine leukemia virus genomic RNAs. J. Virol. 2004; 78:12129–12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rasmussen S.V., Pedersen F.S.. Co-localization of gammaretroviral RNAs at their transcription site favours co-packaging. J. Gen. Virol. 2006; 87:2279–2289. [DOI] [PubMed] [Google Scholar]
- 54. Flynn J.A., Telesnitsky A.. Two distinct Moloney murine leukemia virus RNAs produced from a single locus dimerize at random. Virology. 2006; 344:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Iglesias-Sanchez M.J., Lopez-Galindez C.. Each genomic RNA in HIV-1 heterozygous virus generate new virions. Virology. 2005; 333:316–323. [DOI] [PubMed] [Google Scholar]
- 56. King S.R., Duggal N.K., Ndongmo C.B., Pacut C., Telesnitsky A.. Pseudodiploid genome organization AIDS full-length human immunodeficiency virus type 1 DNA synthesis. J. Virol. 2008; 82:2376–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.