

Abstract
Objective:
The aim of this study was to evaluate children with pulmonary arterial hypertension (PAH) regarding epidemiological characteristics, clinical status with respect to the WHO functional class (WHO-FC), prognostic factors, and efficacy of medical treatment.
Methods:
A retrospective evaluation of 41 patients with PAH was made in the Pediatric Cardiology Unit, Gazi University Medical Faculty, between February 2006 and October 2015.
Results:
Of the 41 patients included in this study, 51.2% were female. The median age was 60 months at first evaluation. The median follow-up was 60 months. At the start of the treatment, 43.9% patients were receiving combined drug therapy, and this rate increased to 60.9% by the last evaluation. The median time of adding a new medication to the therapy was 20 months. The 1- and 5-year survival rates were 94% and 86%, respectively. At the time of diagnosis, only pro-brain natriuretic peptide (proBNP) levels were associated with mortality (p=0.004), but at the last evaluation, 6-min walking test, proBNP and uric acid levels, and WHO-FC were also associated with survival (p=0.02, p=0.001, p=0.002, and p=0.05, respectively).
Conclusion:
With current treatment choices in experienced centers, positive results are obtained with respect to the functional status and survival rates of patients with PAH. At the time of diagnosis, only proBNP had a prognostic value, whereas at the last evaluation, WHO-FC, 6-min walking test, proBNP, and uric acid were reported prognostic factors. For preventing rapid progression, determination of factors that have an effect on prognosis, in particular, is extremely important.
Keywords: pulmonary arterial hypertension, children, survival, congenital heart disease
Introduction
Pulmonary arterial hypertension (PAH) is a chronic and progressive disease that may affect all age groups, i.e., from newborns to adults (1). However, PAH in children is different from that in adults with respect to etiology and therapeutic approach (2). Although there has been an increase in pediatric studies related to PAH, knowledge about the diagnosis and treatment of pediatric PAH is still limited.
Specific treatment protocols for adult patients are based on the results of randomized controlled studies, and the adoption of these protocols has distinctly improved the quality of life and survival of these patients (3, 4). In addition, beneficial and hazardous effects of the drugs that have been developed for PAH might considerably vary in children and adults (5). Therefore, in this study, we focus on monitoring and treating children diagnosed with PAH.
It is of great importance to determine prognostic factors that can be used to monitor children with PAH and specify the efficiency of the administered drugs. Although echocardiographic findings and hemodynamic characteristics may indicate prognosis and therapeutic efficacy, noninvasive parameters, such as WHO functional class (WHO-FC), 6-min walk test (6MWT), and pro-brain natriuretic peptide (proBNP) levels, can also be used (2, 6).
This study aims to evaluate pediatric patients with PAH regarding epidemiological characteristics, clinical status with respect to WHO-FC, prognostic factors, and efficacy of medical treatment.
Methods
This is a retrospective review of 41 patients who were diagnosed with PAH in the Department of Pediatric Cardiology at Gazi University Medical School Hospital between February 2006 and October 2015. The inclusion criteria were age <18 years at the time of diagnosis and mean pulmonary artery pressure (mPAP) ≥25 mm Hg, pulmonary capillary wedge pressure (PCWP) ≤15 mm Hg, and pulmonary vascular resistance index (PVRI) ≥3 WU.m2, measured by right heart catheterization (7). The exclusion criteria were (i) age ≤28 days at the time of diagnosis and (ii) withdrawal from clinical follow-up.
Data related to patient age, sex, underlying etiology, clinical symptoms (fatigue, exercise-induced cyanosis, and dyspnea), and medical treatment were obtained from hospital files. Serum uric acid and pro BNP levels were measured; WHO-FC categorization, cardiac catheterization findings, 6MWT results, and vasoreactivity test positivity were recorded at the time of diagnosis.
Cardiac catheterization was performed in 40 patients (97.6%) patients, whereas it could not be performed in one patient because of deterioration in his general status. In this patient, the diagnosis of PAH was made using Doppler echocardiography, according to the European Society of Cardiology guidelines (8). The vasoreactivity test was conducted with inhaled iloprost in 24 patients who underwent cardiac catheterization (60%), and a positive response was obtained in nine patients (37.5%), based on the criteria recommended by the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL) record study (9). Moreover, 6MWT was conducted in all patients aged >7 years (n=25, 61%) according to the American Thoracic Society guidelines, with adjustment of the track distance from 30 to 8 m to prevent children from getting distracted on a long track (10).
All patients diagnosed with PAH were monitored at 3-month intervals. Serum uric acid and proBNP levels were measured, WHO-FC categorization was done, and 6MWT was performed at all follow-up visits.
Statistical analysis
Collected data were analyzed using Statistical Package for Social Sciences version 15.0 (SPSS IBM, Armonk, NY, USA). Continuous variables are expressed as median, interquartile range (IQR), or range (minimum–maximum), whereas categorical variables were denoted as numbers or percentages. Data distribution was tested by the Shapiro–Wilk test. Wilcoxon test and McNemar test were used for comparing continuous and categorical variables, respectively. The univariate Cox regression analysis was performed for determining predictors of survival, and the Kaplan–Meier curve was drawn for illustrating survival of the patients. Two-tailed p values <0.05 were considered to be statistically significant.
Results
Table 1 demonstrates demographic and clinical characteristics of pediatric patients with PAH. The most common cause of PAH was congenital heart diseases (73.2%), and ventricular septal defect (VSD) was the most frequent congenital heart disease (19.5%). The most frequently encountered clinical symptoms were fatigue (65.9%) and exercise-induced cyanosis (63.4%). There were five patients with Down’s syndrome in the study cohort (12.2%).
Table 1.
Characteristics of patients at the time of diagnosis
| Age (months)& | 60.0 (2-240) |
| Female/Male | 21 (51.2%)/20 (48.8%) |
| Median follow-up (months)& | 60.0 (4-156) |
| Underlying etiology | |
| Congenital heart diseases | 30 (73.2%) |
| VSD | 8 (19.5%) |
| ASD+VSD | 7 (17.0%) |
| ASD+Agenesis of right pulmonary artery | 3 (7.3%) |
| DORV+VSD | 1 (2.4%) |
| TGA+VSD | 1 (2.4%) |
| cTGA+VSD | 1 (2.4%) |
| AVCD | 2 (4.9%) |
| TGA | 2 (4.9%) |
| TAPVR | 1 (2.4%) |
| Truncus arteriosus type 1 | 1 (2.4%) |
| Tricuspid atresia+ASD+VSD+PS | 1 (2.4%) |
| AP window | 2 (4.9%) |
| Residual PAH | 5 (12.2%) |
| Primary PAH | 4 (9.8%) |
| Chronic obstructive sleep apnea | 1 (2.4%) |
| Chronic pulmonary disease | 1 (2.4%) |
| Symptoms | |
| Fatigue | 27 (65.9%) |
| Exercise-induced cyanosis | 26 (63.4%) |
| Dyspnea | 3 (7.3%) |
| WHO Functional classification | |
| II | 7 (17.0%) |
| III | 29 (71.0%) |
| IV | 5 (12.0%) |
| Hemodynamic parameters& | |
| mPAP (mm Hg) | 66.0 (29-98) |
| PVRi (Wood Unit.m2) | 9.5 (1-64) |
| Rp/Rs | 0.58 (0.08-2.28) |
| Qp/Qs | 1.09 (0.48-6.40) |
| mRAP (mm Hg) | 5.0 (2-12) |
Data are presented as median (minimum-maximum).
VSD - ventricular septal defect; ASD - atrial septal defect; DORV - double outlet right ventricle; TGA - transposition of the great arteries; cTGA - corrected transposition of the great arteries; AVCD - atrioventricular canal defect; TAPVR - total anomalous pulmonary venous return; PS - pulmonary stenosis; AP window - aortapulmonary window; mPAP - mean pulmonary artery pressure; PVRi - pulmonary vascular resistance index; Rp/Rs - pulmonary resistance/systemic resistance; Qp/Qs - pulmonary flow/systemic flow; mRAP - mean right atrial pressure
Table 2 shows medical treatment protocols and their effects on clinical symptoms and findings during clinical follow-up. Bosentan was the most commonly administered monotherapy (48.8%), whereas bosentan+inhaled iloprost were the most frequently administered combination therapy (22.0%). The administration rate of combined therapy increased from 44% at the time of diagnosis to 73.5% at the last evaluation. Sildenafil and inhaled iloprost were the most commonly added drugs. The median time of adding a new medication to the therapy was 20 (range, 10–64) months. Compared with the time of diagnosis, fatigue and exercise-induced cyanosis were significantly less and serum proBNP level was significantly lower at the last evaluation (p=0.001, p=0.012, and p=0.037, respectively). Moreover, the 6MW distance and serum uric acid level was significantly higher at the last evaluation than at the time of diagnosis (p=0.012 and p=0.022, respectively).
Table 2.
Medical treatment and its effects determined during the clinical follow-up
| Time of diagnosis (n=41) | Last evaluation (n=41) | P | ||
|---|---|---|---|---|
| Treatment | ||||
| No treatment | 0 (0.0%) | 5 (12.2%) | ||
| Monotherapy | 23 (56.0%) | 11 (26.5%) | ||
| Bosentan | 20 (48.8%) | 10 (24.4%) | ||
| Sildenafil | 2 (4.9%) | 1 (2.4%) | ||
| Inhaled iloprost | 1 (2.4%) | 0 (0.0%) | ||
| Dual therapy | 18 (44.0%) | 19 (46.3%) | ||
| Bosentan+Sildenafil | 7 (17.1%) | 11 (26.8%) | ||
| Bosentan+Inhaled iloprost | 9 (22.0%) | 6 (14.6%) | ||
| Sildenafil+Inhaled iloprost | 2 (4.9%) | 2 (4.9%) | ||
| Triple therapy | 0 (0.0%) | 6 (14.6%) | ||
| Bosentan+Sildenafil+Inhaled iloprost | 0 (0.0%) | 3 (7.3%) | ||
| Bosentan+Tadalafil+Inhaled iloprost | 0 (0.0%) | 1 (2.4%) | ||
| Bosentan+Sildenafil+Treprostinil | 0 (0.0%) | 2 (4.9%) | ||
| Symptoms | ||||
| Fatigue | 27 (65.9%) | 13 (31.7%) | 0.001 | |
| Exercise-induced cyanosis | 26 (63.4%) | 17 (41.5%) | 0.012 | |
| 6MWT (m)& | 390 (134.0) | 480 (150.0) | 0.012 | |
| ProBNP (pg/mL)& | 280 (848.5) | 176.5 (646.6) | 0.037 | |
| Uric acid (mg/dL)& | 3.9 (1.5) | 4.4 (2.4) | 0.022 |
Data are presented as median (IQR).
Figure 1 points out that WHO-FC at the time of diagnosis significantly improved at the time of the last evaluation (p=0.007). The 6MW distance significantly increased, and the serum proBNP level significantly decreased in patients who had been undergoing monotherapy at the time of diagnosis (p=0.023 and p=0.031, respectively). However, there were no significant improvements in 6MWT and proBNP values of patients who were undergoing combined therapy at the time of diagnosis (Table 3).
Figure 1.
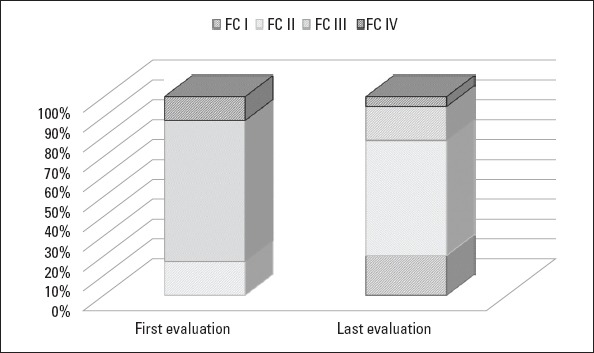
World Health Organization functional classification at the time of diagnosis and last evaluation
Table 3.
Effects of treatment modalities on clinical findings
| Monotherapy at the start of treatment | Combination therapy at the start of treatment | ||||||
|---|---|---|---|---|---|---|---|
| First | Last | P | First | Last | P | ||
| 6MWT (m) | 392 (136) | 486 (206) | 0.023 | 360 (204) | 441 (180) | 0.17 | |
| ProBNP (pg/mL) | 206 (783) | 151 (272) | 0.031 | 683 (1021.5) | 284 (1227.4) | 0.332 | |
Data are presented as median (IQR).
6MWT - 6-min walk test; proBNP - pro-brain natriuretic peptide
Four patients died during the clinical follow-up, while postoperative pulmonary hypertensive crisis following VSD+ASD surgery occurred in one patient and heart failure related to primary PAH affected one patient. In addition, infection developed in one patient who had PDA, and residual PAH was diagnosed in another patient. The 1- and 5-year survival rates were 94% and 86%, respectively (Fig. 2).
Figure 2.
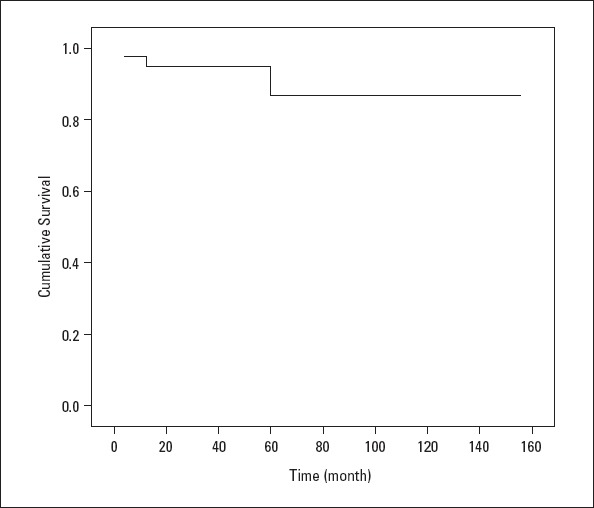
The Kaplan–Meier curve of the patients
Univariate Cox regression analysis showed that lower proBNP levels at the time of diagnosis and lower WHO-FC, higher 6MW distance, and lower proBNP and uric acid levels at the last evaluation were associated with survival (Table 4).
Table 4.
Parameters associated with the survival of patients
| n | Death n (%) | Median survival (months) | HR | Univariate analysis 95% CI | P | ||
|---|---|---|---|---|---|---|---|
| At diagnosis | |||||||
| WHO-FC | |||||||
| I-II | 7 | 1 (14.3) | 24 | 1 | |||
| III-IV | 34 | 3 (8.8) | 60 | 0.513 | 0.053 - 4.955 | 0.564 | |
| Treatment | |||||||
| Monotherapy | 23 | 2 (8.7) | 60 | 1 | |||
| Combination therapy | 18 | 2 (11.2) | 60 | 1.327 | 0.187 - 9.428 | 0.777 | |
| mPAP | 41 | 4 (9.8) | 60 | 1.010 | 0.960 - 1.063 | 0.694 | |
| PVRi | 41 | 4 (9.8) | 60 | 1.050 | 0.992 - 1.110 | 0.091 | |
| Rp/Rs | 41 | 4 (9.8) | 60 | 0.696 | 0.059 - 8.194 | 0.773 | |
| 6MWT | 41 | 4 (9.8) | 60 | 0.990 | 0.979 - 1.001 | 0.080 | |
| ProBNP | 41 | 4 (9.8) | 60 | 1.001 | 1.000 - 1.001 | 0.004 | |
| Uric acid | 41 | 4 (9.8) | 60 | 1.404 | 0.794 - 2.481 | 0.243 | |
| At last evaluation | |||||||
| WHO-FC | |||||||
| I-II | 32 | 1 (3.1) | 60 | 1 | |||
| III-IV | 9 | 3 (33.3) | 60 | 9.393 | 0.975 - 90.492 | 0.053 | |
| 6MWT | 41 | 4 (9.8) | 60 | 0.984 | 0.969 - 0.998 | 0.027 | |
| ProBNP | 41 | 4 (9.8) | 60 | 1.001 | 1.000 - 1.001 | 0.001 | |
| Uric acid | 41 | 4 (9.8) | 60 | 1.525 | 1.045 - 2.227 | 0.029 |
6MWT - 6-min walk test; proBNP - pro-brain natriuretic peptide; WHO-FC - WHO functional class; mPAP - mean pulmonary artery pressure; PVRi - pulmonary vascular resistance index; Rp/Rs - pulmonary resistance/systemic resistance; Qp/Qs - pulmonary flow/systemic flow
Discussion
PAH is a progressive disease that significantly impairs the functional status of the patient and even results in mortality. There is no scientific consensus on monitoring and prognostically assessing pediatric patients with PAH because of the limited number of related clinical studies. Similar to the management of adult patients, pediatric cardiologists dealing with PAH tend to use WHO-FC, 6MWT, and proBNP levels for the clinical follow-up of affected children (6).
At the time of diagnosis, the number of pediatric patients with WHO-FC III-IV disease was higher than expected, and this finding has been attributed to a delay in the diagnosis and rapid progression of the disease (11-14). Accordingly, at the time of diagnosis, WHO-FC III-IV was present in 83% patients reviewed in this study. As expected, shorter survival is associated with the presence of WHO-FC III-IV disease at the time of diagnosis (3, 15).
Balkin et al. (16) found a close correlation between WHO-FC at final evaluation and mortality, but they were unable to detect the same correlation between WHO-FC at the time of diagnosis and mortality. Similarly, in this study, WHO-FC at the last evaluation was an indicator for survival, but WHO-FC at the time of diagnosis did not indicate survival. Therefore, deteriorating WHO-FC during the clinical follow-up might have a greater prognostic value than WHO-FC at the time of diagnosis. Medical treatment should, thus, be renewed in patients who have worsening WHO-FC.
It is well-known that 6MWT has remarkable prognostic value in adults diagnosed with PAH (4, 17), but this test cannot be performed in small children because of a lack of cooperation. Although some studies have claimed that 6MWT is not beneficial in pediatric patients with PAH (18, 19), Lammers et al. (20) determined that 6MWT was associated with clinical outcomes in these patients. In this study, a significant improvement was achieved in 6MWT results of children with PAH at the last evaluation, and this improvement was significantly marked in pediatric patients who had been undergoing monotherapy. The 6MWT result at the last evaluation was also addressed as an indicator for survival.
It has been shown that the gradual increase in serum proBNP levels would help to predict the unfavorable prognosis of children diagnosed with PAH (3, 9, 21). A significant decrease was specified in serum proBNP levels at the last evaluation of the children with PAH and this decrease was significantly more prominent in pediatric patients who had been undergoing monotherapy. ProBNP values at the time of diagnosis and last evaluation were labeled as prognostic factors related to survival in pediatric patients with PAH.
Much like proBNP, an increase in serum uric acid levels can indicate an adverse outcome in children with PAH (19, 22). In this study, medical treatment was found to successfully decrease serum uric acid levels of pediatric patients with PAH. Moreover, serum uric acid levels at the last evaluation were found to predict survival.
Starting combination therapy at the time of diagnosis has been a controversial issue in the management of pediatric PAH. However, recent guidelines have suggested initiation of combined therapy in selected cases (23, 24). Therefore, the administration rate of combined therapy has increased in pediatric patients with PAH within the last decade (Table 5) (11, 13, 25).
Table 5.
Medical treatment in the current and previous studies
| Drugs | Current study n (%) | Fraise et al. (11) (%) | Roldan et al. (13) (%) | Favilli et al. (25) (%) | |
|---|---|---|---|---|---|
| Monotherapy | 11 (26.8) | 34.0 | 55.1 | 72.7 | |
| Combination therapy | 25 (60.9) | 44.0 | 45.9 | 27.3 | |
| Endothelin receptor antagonist | 33 (80.4) | 78.0 | 23.6 | 72.7 | |
| Phosphodiesterase inhibitor | 20 (48.7) | 34.0 | 58.0 | 24.2 | |
| Prostanoids | 14 (34.1) | 24.0 | 18.3 | 3.0 |
A thorough review of literature designates the endothelin receptor antagonist as the most frequently preferred monotherapy (11, 12, 25). In addition, sildenafil add-on to bosentan monotherapy reduces the severity of PAH in children (23). On the other hand, inhaled iloprost has been described as an efficient and safe therapy for PAH in pediatric patients. Most of the affected children can tolerate the combination of an endothelin receptor antagonist and phosphodiesterase inhibitor (24).
The present study identified significant improvements in patients who underwent monotherapy at the time of diagnosis, but these improvements were not detected in those who had been undergoing combination therapy at the beginning. This finding should not be interpreted as monotherapy being superior to combined therapy, rather it reflects the widespread approach of administering monotherapy to patients with early stage PAH (WHO-FC I-II) who are more likely to exhibit signs of improvement. This finding also emphasizes the importance of making an early diagnosis and initiating the optimal treatment.
The present study identifies bosentan+inhaled iloprost as the most frequently administered combination therapy at the time of diagnosis. This regimen has been changed to bosentan+sildenafil at the time of last evaluation. Such an alteration can be explained by the probably lower patient compliance associated with the administration of inhaled iloprost treatment. That is, oral ingestion of a drug twice or three times a day may be more easily performed and, thus, more frequently preferred than inhalation of a drug 6–9 times a day.
Because of PAH-specific drugs coming into routine use, the survival span of patients with PAH has been significantly prolonged (26). Complying with literature (11, 12, 19), in this study, 1- and 5-year survival rates are computed to be 94.5% and 86%, respectively. These relatively high survival rates can be attributed to the fact that majority study cohort is made up by children with congenital heart diseases. It has been well-established that idiopathic PAH has a worse course than congenital heart disease-related PAH; therefore, 1- and 5-year survival rates were 73% and 60%, respectively, for patients with idiopathic PAH (3, 26, 27).
Study limitations
Patients in this cohort had heterogeneous etiopathogenesis varying from primary to residual PAH.
A subgroup analysis based on PAH etiology could not be made because of the insufficient number of patients in etiological groups.
A subgroup analysis based on co-morbidities (i.e., Down’s syndrome) could not be made because of the relatively small cohort size.
Biochemical alterations related to pharmacological side effects could not be assessed.
Conclusion
Because of the latest advances in pharmacological treatment, functional status and survival rates of patients with PAH have significantly improved. Because PAH is a progressive disease, the prevention of rapid progression in affected children is particularly important. The 6MWT, proBNP and uric acid levels, and WHO-FC are prognostic factors that can be used for preventing rapid progression of pediatric PAH. Further research is warranted for clarifying prognostic factors and long-term efficacy of medical treatment in children diagnosed with PAH.
Acknowledgement
We thank Bülent Çelik PhD for statistical analysis.
Footnotes
Conflict of interest: None declared.
Peer-review: Externally peer-reviewed.
Authorship contributions: Concept – S.K.; Design – S.K.; Supervision – F.S.T.; Fundings – S.K.; Materials – F.C.; Data collection &/or processing – F.C.; Analysis &/or interpretation – S.K.; Literature search – V.A.; Writing – F.C.; Critical review – A.D.O.
References
- 1.Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease:pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8:443–55. doi: 10.1038/nrcardio.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ivy DD, Abman SH, Barst RJ, Berger RM, Bonnet D, Fleming TR, et al. Pediatric pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D117–26. doi: 10.1016/j.jacc.2013.10.028. [DOI] [PubMed] [Google Scholar]
- 3.Zijlstra WMH, Douwes JM, Rosenzweig EB, Schokker S, Krishnan U, Roofthooft MTR, et al. Survival differences in pediatric pulmonary arterial hypertension:clues to a better understanding of outcome and optimal treatment strategies. J Am Coll Cardiol. 2014;63:2159–69. doi: 10.1016/j.jacc.2014.02.575. [DOI] [PubMed] [Google Scholar]
- 4.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension:insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010;122:164–72. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 5.Nakau K, Sugimoto M, Oka H, Kajihama A, Maeda J, Yamagishi H, et al. Pharmacokinetics of drugs for pediatric pulmonary hypertension. Pediatr Int. 2016;58:1112–7. doi: 10.1111/ped.12997. [DOI] [PubMed] [Google Scholar]
- 6.Ploegstra MJ, Zijlstra WM, Douwes JM, Hillege HL, Berger RM. Prognostic factors in pediatric pulmonary arterial hypertension:A systematic review and meta-analysis. Int J Cardiol. 2015;184:198–207. doi: 10.1016/j.ijcard.2015.01.038. [DOI] [PubMed] [Google Scholar]
- 7.Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D42–50. doi: 10.1016/j.jacc.2013.10.032. [DOI] [PubMed] [Google Scholar]
- 8.Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, et al. Recommendations for cardiac chamber quantification by echocardiography in adults:an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2015;16:233–70. doi: 10.1093/ehjci/jev014. [DOI] [PubMed] [Google Scholar]
- 9.Barst RJ, McGoon MD, Elliott CG, Foreman AJ, Miller DP, Ivy DD. Survival in childhood pulmonary arterial hypertension:insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation. 2012;125:113–22. doi: 10.1161/CIRCULATIONAHA.111.026591. [DOI] [PubMed] [Google Scholar]
- 10.ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement:guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166:111–7. doi: 10.1164/ajrccm.166.1.at1102. [DOI] [PubMed] [Google Scholar]
- 11.Fraisse A, Jais X, Schleich JM, di Filippo S, Maragnes P, Beghetti M, et al. Characteristics and prospective 2-year follow-up of children with pulmonary arterial hypertension in France. Arch Cardiovasc Dis. 2010;103:66–74. doi: 10.1016/j.acvd.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Chung WJ, Park YB, Jeon CH, Jung JW, Ko KP, Choi SJ, et al. Baseline Characteristics of the Korean Registry of Pulmonary Arterial Hypertension. J Korean Med Sci. 2015;30:1429–38. doi: 10.3346/jkms.2015.30.10.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roldan T, Deiros L, Romero JA, Gutierrez-Larraya F, Herrero A, Del Cerro MJ. Safety and tolerability of targeted therapies for pulmonary hypertension in children. Pediatr Cardiol. 2014;35:490–8. doi: 10.1007/s00246-013-0811-4. [DOI] [PubMed] [Google Scholar]
- 14.Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension:results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med. 2012;186:790–6. doi: 10.1164/rccm.201203-0383OC. [DOI] [PubMed] [Google Scholar]
- 15.Park YM, Chung WJ, Choi DY, Baek HJ, Jung SH, Choi IS, et al. Functional class and targeted therapy are related to the survival in patients with pulmonary arterial hypertension. Yonsei Med J. 2014;55:1526–32. doi: 10.3349/ymj.2014.55.6.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balkin EM, Olson ED, Robertson L, Adatia I, Fineman JR, Keller RL. Change in Pediatric Functional Classification During Treatment and Morbidity and Mortality in Children with Pulmonary Hypertension. Pediatr Cardiol. 2016;37:756–64. doi: 10.1007/s00246-016-1347-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyamoto S, Nagaya N, Satoh T, Kyotani S, Sakamaki F, Fujita M, et al. Clinical correlates and prognostic significance of six-minute walk test in patients with primary pulmonary hypertension. Comparison with cardiopulmonary exercise testing. Am J Respir Crit Care Med. 2000;161:487–92. doi: 10.1164/ajrccm.161.2.9906015. [DOI] [PubMed] [Google Scholar]
- 18.Moledina S, Pandya B, Bartsota M, Mortensen KH, McMillan M, Quyam S, et al. Prognostic significance of cardiac magnetic resonance imaging in children with pulmonary hypertension. Circ Cardiovasc Imaging. 2013;6:407–14. doi: 10.1161/CIRCIMAGING.112.000082. [DOI] [PubMed] [Google Scholar]
- 19.Wagner BD, Takatsuki S, Accurso FJ, Ivy DD. Evaluation of circulating proteins and hemodynamics towards predicting mortality in children with pulmonary arterial hypertension. PLoS One. 2013;8:e80235. doi: 10.1371/journal.pone.0080235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lammers AE, Munnery E, Hislop AA, Haworth SG. Heart rate variability predicts outcome in children with pulmonary arterial hypertension. Int J Cardiol. 2010;142:159–65. doi: 10.1016/j.ijcard.2008.12.087. [DOI] [PubMed] [Google Scholar]
- 21.Chida A, Sato H, Shintani M, Nakayama T, Kawamura Y, Furutani Y, et al. Soluble ST2 and N-terminal pro-brain natriuretic peptide combination. Useful biomarker for predicting outcome of childhoodpulmonary arterial hypertension. Circ J. 2014;78:436–42. doi: 10.1253/circj.cj-13-1033. [DOI] [PubMed] [Google Scholar]
- 22.van Loon RL, Roofthooft MT, Delhaas T, van Osch-Gevers M, ten Harkel AD, Strengers JL, et al. Outcome of pediatric patients with pulmonary arterial hypertension in the era of new medical therapies. Am J Cardiol. 2010;106:117–24. doi: 10.1016/j.amjcard.2010.02.023. [DOI] [PubMed] [Google Scholar]
- 23.Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK, et al. American Heart Association Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation;Council on Clinical Cardiology;Council on Cardiovascular Disease in the Young;Council on Cardiovascular Radiology and Intervention;Council on Cardiovascular Surgery and Anesthesia;and the American Thoracic Society. Pediatric Pulmonary Hypertension:Guidelines From the American Heart Association and American Thoracic Society. Circulation. 2015;132:2037–99. doi: 10.1161/CIR.0000000000000329. [DOI] [PubMed] [Google Scholar]
- 24.Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. ESC Scientific Document Group. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension:The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS):Endorsed by:Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT) Eur Heart J. 2016;37:67–119. doi: 10.1093/eurheartj/ehv317. [DOI] [PubMed] [Google Scholar]
- 25.Favilli S, Spaziani G, Ballo P, Fibbi V, Santoro G, Chiappa E, et al. Advanced therapies in patients with congenital heart disease-related pulmonary arterial hypertension:results from a long-term, single center, real-world follow-up. Intern Emerg Med. 2015;10:445–50. doi: 10.1007/s11739-014-1185-1. [DOI] [PubMed] [Google Scholar]
- 26.Haworth SG, Hislop AA. Treatment and survival in children with pulmonary arterial hypertension:the UK Pulmonary Hypertension Service for Children 2001-2006. Heart. 2009;95:312–7. doi: 10.1136/hrt.2008.150086. [DOI] [PubMed] [Google Scholar]
- 27.van Loon RL, Roofthooft MT, Hillege HL, ten Harkel AD, van Osch-Gevers M, Delhaas T, et al. Pediatric pulmonary hypertension in the Netherlands:epidemiology and characterization during the period 1991 to 2005. Circulation. 2011;124:1755–64. doi: 10.1161/CIRCULATIONAHA.110.969584. [DOI] [PubMed] [Google Scholar]