

Despite the global awareness that mercury, and methylmercury in particular, is a neurotoxin to which millions of people continue to be exposed, there are sizable gaps in the understanding of the processes and organisms involved in methylmercury formation in aquatic ecosystems. In the present study, we shed light on the diversity of the microorganisms responsible for methylmercury formation in boreal lake sediments. All the microorganisms identified are associated with the processing of organic matter in aquatic systems. Moreover, our results show that the well-known mercury-methylating sulfate-reducing bacteria constituted only a minor portion of the potential mercury methylators. In contrast, methanogens and iron-reducing bacteria were important contributors to methylmercury formation, highlighting their role in mercury cycling in the environment.
KEYWORDS: mercury, methylation, hgcA gene, 16S rRNA gene, boreal lakes, methanogens, iron-reducing bacteria, sulfate-reducing bacteria
ABSTRACT
Methylmercury is a potent human neurotoxin which biomagnifies in aquatic food webs. Although anaerobic microorganisms containing the hgcA gene potentially mediate the formation of methylmercury in natural environments, the diversity of these mercury-methylating microbial communities remains largely unexplored. Previous studies have implicated sulfate-reducing bacteria as the main mercury methylators in aquatic ecosystems. In the present study, we characterized the diversity of mercury-methylating microbial communities of boreal lake sediments using high-throughput sequencing of 16S rRNA and hgcA genes. Our results show that in the lake sediments, Methanomicrobiales and Geobacteraceae also represent abundant members of the mercury-methylating communities. In fact, incubation experiments with a mercury isotopic tracer and molybdate revealed that only between 38% and 45% of mercury methylation was attributed to sulfate reduction. These results suggest that methanogens and iron-reducing bacteria may contribute to more than half of the mercury methylation in boreal lakes.
IMPORTANCE Despite the global awareness that mercury, and methylmercury in particular, is a neurotoxin to which millions of people continue to be exposed, there are sizable gaps in the understanding of the processes and organisms involved in methylmercury formation in aquatic ecosystems. In the present study, we shed light on the diversity of the microorganisms responsible for methylmercury formation in boreal lake sediments. All the microorganisms identified are associated with the processing of organic matter in aquatic systems. Moreover, our results show that the well-known mercury-methylating sulfate-reducing bacteria constituted only a minor portion of the potential mercury methylators. In contrast, methanogens and iron-reducing bacteria were important contributors to methylmercury formation, highlighting their role in mercury cycling in the environment.
INTRODUCTION
Mercury (Hg), a ubiquitous and naturally occurring element in the environment, is considered a priority hazardous substance because of its high toxicity (1). Humans have emitted Hg to the atmosphere for millennia (2). Long-range atmospheric transport of released Hg is accumulating in ecosystems globally, and this has led to increased Hg levels in surface (atmosphere, oceans, and terrestrial) reservoirs (3, 4). This is of special concern in the boreal biome, since a large pool of Hg has accumulated in soils following atmospheric deposition during the industrial era (5). As Hg binds strongly to organic matter (OM), the release of OM from soils to aquatic systems also affects the fate of Hg in boreal catchments (6). For example, increased import of terrigenous OM from surrounding catchments, due to ongoing climate change (7), may lead to higher Hg levels in boreal lakes (8, 9). As a result, millions of people are exposed to harmful levels of this potent neurotoxin via fish consumption (10, 11). This health hazard especially concerns organic methylmercury (MeHg) which bioaccumulates in organisms and is biomagnified in aquatic food webs (12, 13). A deeper understanding of the processes, organisms, and environmental conditions involved in MeHg production in aquatic ecosystems may thus help us develop more efficient management strategies for limiting human MeHg exposure.
In aquatic ecosystems, the methylation of divalent inorganic Hg [Hg(II)] to MeHg has mainly been attributed to the action of sulfate-reducing bacteria (SRB) (14–17) and, in some cases, to iron-reducing bacteria (FeRB) (18, 19), methanogens (20), and syntrophs (21). Previous studies have relied on inferences from soil phospholipid fatty acid analysis (22, 23), taxonomic markers (e.g., 16S rRNA genes [20, 24]), or specific functional genes involved in sulfate reduction (e.g., dsrAB) to indirectly describe the composition of putatively Hg(II)-methylating microbial communities (16, 25–27). However, the recent discovery of two functional genes, hgcA and hgcB, which play essential roles in Hg(II) methylation (28), opens the possibility to use direct markers for Hg(II)-methylating organisms in complex communities. Indeed, the recent use of hgcA and hgcB genes has greatly improved the ability to describe Hg(II)-methylating microbial communities at a higher taxonomic resolution (21, 29–31). Despite recent studies of Hg methylators in wetlands (21, 29, 30), the diversity of Hg(II)-methylating microbial communities and the factors shaping these communities in freshwater environments remain largely unexplored.
Temperature, redox, and pH are known to be important geochemical factors regulating Hg(II) methylation (32). Furthermore, a recent study showed that the molecular composition of organic matter (OM) controlled Hg(II) methylation in boreal lake sediments (9). This study revealed that sediments enriched in autochthonous plankton-derived OM presented high Hg(II) methylation rates, associated with enhanced bacterial activity. Specific OM compounds can also promote Hg(II) methylation by regulating Hg(II) speciation (33) and Hg(II) availability (34–36). OM can also facilitate Hg(II) methylation by enhancing mercury sulfide (HgS) dissolution or inhibiting HgS precipitation, thereby providing available Hg(II) for methylating microorganisms (37). In contrast, high OM concentrations might also decrease Hg methylation through the formation of high-molecular-mass complexes that limit Hg(II) availability (34). Altogether, these studies highlight that both geochemical conditions and OM composition are central regulators of microbial Hg(II) methylation. In the present study, we provide a detailed characterization of Hg(II)-methylating microbial communities of boreal lake sediments featuring contrasting OM molecular compositions (9). This was accomplished using a combination of experimental incubations and field surveys based on high-throughput sequencing of 16S rRNA and hgcA genes. In doing so, we implicate methanogens and iron-reducing bacteria as overlooked mediators of this process in boreal lake sediments, and we identified an important link between the composition of Hg(II)-methylating communities and the degradation status of phytoplankton-derived OM in 10 lakes with contrasting trophic statuses (Table 1).
TABLE 1.
Characteristics of the investigated lakes
| Lake | Sample code | Coordinates (°) |
Geochemical parameters of the sediment overlying watera |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | E | z (m) | pH | Temp (°C) | C (μS · s−1) | O2 concn (mg · liter−1) | DOC concn (mg · liter−1) | TP concn (μg · liter−1) | Chla concn (μg · liter−1) | SO42− concn (mg · liter−1) | SUVA254 (liters · mg−1 · m−1) | ||
| Lilla Sångaren | LS | 59.8996 | 15.3923 | 17.0 | 6.9 | 5 | 60 | 4.65 | 7.0 | 23 | 1.8 | 2.9 | 4.21 |
| Ljustjärn | LJU | 59.92375 | 15.453472 | 10 | 7.3 | 6.9 | 77 | 0.22 | 6.5 | 96 | 93 | 2.2 | 1.6 |
| Svarttjärn | S | 59.89073 | 15.2577 | 6.5 | 5.6 | 4.8 | 59 | 0.08 | 22.0 | 36 | 2.7 | 0.9 | 5.96 |
| Fälaren | F | 60.33656 | 17.79396 | 2.0 | 7.5 | 18.7 | 67 | 8.56 | 32.6 | 20 | 8.9 | 3.0 | 3.9 |
| Oppsveten | O | 59.98874 | 15.57562 | 10.0 | 6.3 | 8.6 | 30 | 0.79 | 16.7 | 14 | Udlb | 2.3 | 4.78 |
| Strandsjön | STR | 59.87099 | 17.168650 | 2.5 | 6.9 | 16.4 | 285 | 0.34 | 19.6 | 60 | 13.1 | 4.6 | 3.0 |
| Valloxen | V | 59.73846 | 17.83954 | 6.0 | 8.5 | 18.8 | 502 | 0.14 | 12.3 | 49 | 52 | 8.9 | 2.5 |
| Vallentunasjön | VALE | 59.50435 | 18.037083 | 4 | 7.1 | 17.2 | 469 | 0.24 | 14.0 | 77 | 58 | 16.2 | 1.4 |
| Marnästjärn | M | 60.14483 | 15.20714 | 2.0 | 7.2 | 17.8 | 185 | 6.30 | 9.2 | 185 | 190 | 3.6 | 1.8 |
| Lötsjön | LOTS | 59.86314 | 17.940110 | 7 | 6.8 | 11.5 | 288 | 0.3 | 13.3 | 65 | 18 | 1.60 | 1.7 |
C, conductivity; O2, oxygen concentration; DOC, dissolved organic carbon; TP, total phosphorus; Chla, chlorophyll a; SO42−, sulfate; SUVA254, specific UV absorbance of the water overlying the sediment (modified from reference 9).
Udl, under detection limit.
RESULTS
Hg(II)-methylating microbial communities.
The relative contribution of sulfate reducers to mercury methylation was assessed by adding molybdate and an enriched stable isotope (198HgCl2) to sediments of lakes known to have high Hg(II) methylation rate constants (km; lake M and lake V) (Table 2). The addition of molybdate inhibited the km by 38% and 45%, respectively (Fig. 1). This partial inhibition suggests that SRB were not the only significant Hg(II) methylators in the studied sediments.
TABLE 2.
Concentrations of Hg(II) and MeHg, Hg(II) methylation yields, total carbon, carbon to nitrogen ratio, total phosphorous, and bacterial production of the investigated lake sediments
| Lake | Sediment characteristicsa |
||||||
|---|---|---|---|---|---|---|---|
| Hg(II) (ng · g−1) | MeHg (ng · g−1) | km (day−1) | TC (%) | C/N | TP (%) | BP (μg C · liter−1 · day−1) | |
| LS | 231 ± 31 | 6.6 ± 0.9 | 0.0095 ± 0.0049 | 19.1 ± 1.0 | 15.1 ± 0.018 | 0.018 ± 0.0020 | 4.0 ± 0.44 |
| LJU | 261 ± 8 | 4.0 ± 0.3 | 0.0095 ± 0.0007 | 31.9 ± 0.2 | 12.9 ± 0.012 | 0.012 ± 0.0009 | 0.4 ± 0.66 |
| S | 373 ± 11 | 7.2 ± 1.1 | 0.0110 ± 0.0001 | 23.7 ± 0.8 | 16.0 ± 0.011 | 0.011 ± 0.0003 | 2.7 ± 0.04 |
| F | 235 ± 5 | 3.9 ± 0.2 | 0.0120 ± 0.0071 | 24.4 ± 0.3 | 12.5 ± 0.010 | 0.010 ± 0.0009 | 6.4 ± 1.87 |
| O | 253 ± 49 | 6.4 ± 0.4 | 0.0125 ± 0.0021 | 19.2 ± 0.3 | 17.5 ± 0.013 | 0.013 ± 0.0010 | 1.9 ± 0.37 |
| STR | 179 ± 1 | 1.4 ± 0.1 | 0.0130 ± 0.0028 | 11.7 ± 0.1 | 8.7 ± 0.014 | 0.014 ± 0.0008 | 16.6 ± 0.25 |
| V | 74 ± 1 | 2.5 ± 0.5 | 0.0775 ± 0.0007 | 13.9 ± 0.2 | 8.1 ± 0.015 | 0.015 ± 0.0000 | 23.6 ± 3.43 |
| VALE | 102 ± 8 | 2.0 ± 0.1 | 0.0590 ± 0.0099 | 18.9 ± 0.2 | 7.9 ± 0.018 | 0.018 ± 0.0001 | 21.3 ± 2.29 |
| M | 12,711 ± 158 | 100 ± 5.7 | 0.0385 ± 0.012 | 19.7 ± 0.3 | 9.1 ± 0.024 | 0.024 ± 0.0005 | 14.6 ± 1.93 |
| LOTS | 156 ± 8 | 2.6 ± 0.3 | 0.0780 ± 0.0014 | 14.3 ± 0.6 | 8.2 ± 0.021 | 0.021 ± 0.0004 | 17.2 ± 2.28 |
TC, total carbon; C/N, carbon-to-nitrogen ratio; TP, total phosphorus; BP, bacterial production. Data are means from two depths (0 to 1 cm and 1 to 2 cm). Modified from reference 9.
FIG 1.
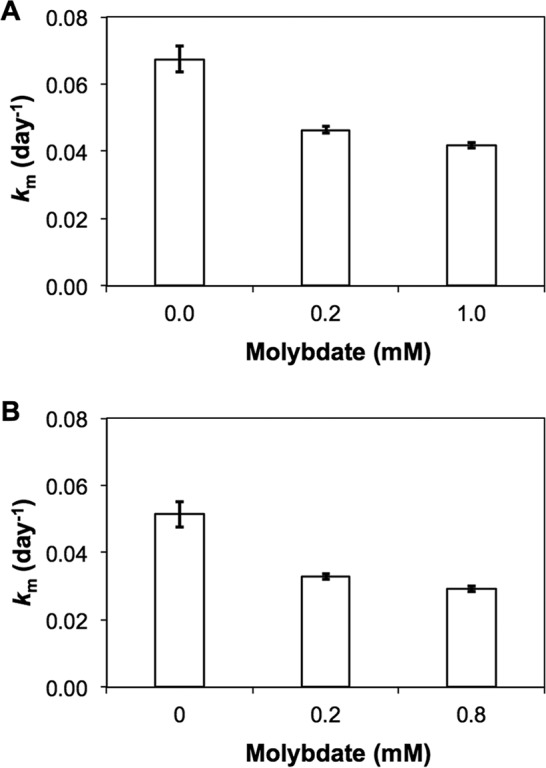
Hg(II) methylation (day−1) obtained in unamended (0 mM) and molybdate-amended sediments (3 replicate incubation experiments) for Valloxen (A) and Marnästjärn (B) lakes.
As our incubation experiments revealed Hg(II)-methylating activity beyond that of sulfate reducers, we focused on the identification of other organisms mediating this process. For this, we analyzed the compositions of the Hg(II)-methylating microbial communities by high-throughput sequencing of amplified hgcA genes. We detected the hcgA gene in nine samples from six of the ten lakes (Fig. 2), including samples from all four lakes with high Hg(II) methylation rate constants (VALE, V, LOTS, and M, km > 0.02 day−1) and two lakes with low kms (STR and LS, km < 0.02 day−1). The total microbial hgcA gene data set consisted of 78,642 reads distributed across 255 operational taxonomic units (OTUs). Of these, 224 were related to Bacteria, 25 to Archaea, and 6 remained unknown. Most of the Hg(II) methylator diversity targeted by the primers used was captured in our analyses (see Fig. S1 in the supplemental material). Deltaproteobacteria accounted for 48% of the reads, followed by Methanomicrobia with 42.1% of the reads and Clostridia with 0.82% of total reads (Fig. 2). Reads that could not be assigned to any known taxonomic group (unknown bacteria) comprised 7.2% of the total reads. Within Deltaproteobacteria, 30.5% of the reads were assigned as unknown Deltaproteobacteria, while 18% of the reads were affiliated with Desulfuromonadales. Approximately half of the Desulfuromonadales reads were affiliated with the FeRB Geobacteraceae (9.8% of total reads) and the other half affiliated with Pelobacteraceae (Desulfuromonadaceae; 7.2% of total reads) species. Well-known sulfate-reducing Hg(II) methylators such as Desulfovibrionales represented only 0.08% of the total reads despite the high efficiency of these PCR primers to amplify hgcA from cultured representatives within the Desulfovibrionales relative to that from other orders (29, 38).
FIG 2.

Hg(II)-methylating microbial community compositions of boreal lake sediments (two depths: 0 to 1 cm and 1 to 2 cm) based on the Hg(II) methylation gene (hgcA) sequencing at a highly resolved taxonomic level. The sizes of the symbols illustrate the relative abundance of each taxon. Shading of the symbols (gray scale) represents the number of individual operational taxonomic units (OTUs; 80% similarity threshold) within the taxa. By using hierarchical clustering, the dendrogram to the left demonstrates the community similarity between samples.
The phylogenetic analyses demonstrated that some of the unidentified OTUs in this study were closely related to Hg(II)-methylating methanogens (Fig. 3) and to Geobacteraceae and Desulfuromonadaceae families (Fig. 4). A substantial Hg(II) methylation activity could also be mediated by hitherto unknown Hg(II)-methylating organisms that could not be taxonomically resolved beyond the class level. This is the case for many of the OTUs classified merely as Deltaproteobacteria. To illustrate the lack of reference information, we placed the representative sequences of the most abundant unclassified hgcA gene OTUs in a phylogenetic tree to show their relationships to already described genes from genomes and isolates (see Fig. S2). These OTUs were abundant and also featured a high diversity. Several of the unknown Deltaproteobacteria OTUs found in our study were phylogenetically related to the SRB Desulfovibrio oxyclinae, indicating that unknown Deltaproteobacteria OTUs with Hg(II)-methylating capacity are also likely to be abundant in boreal lake sediments.
FIG 3.
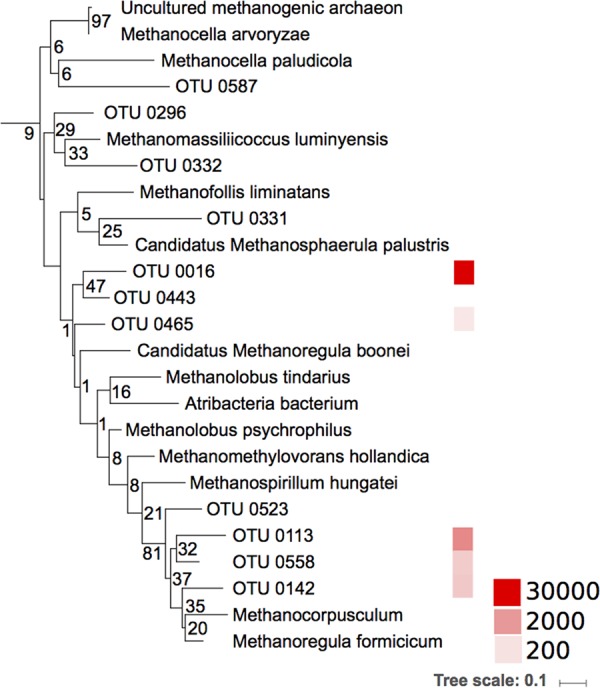
Phylogenetic distribution of the Hg(II) methylation gene (hgcA) sequences from archaeal methanogens, including both reference strains and environmental sequences from boreal lake sediments. The tree was generated using RAxML (version 8.2.4) with the PROTGAMMLG model and the autoMR to choose the number of necessary bootstraps (750). The colors to the right illustrate the abundance of the sequences (a total of 78,462 sequences). Scale bar represents estimated phylogenetic distance in substitutions per site, and the numbers at the branch points indicate the bootstrap values.
FIG 4.

Maximum likelihood tree and abundance of the Hg(II) methylation gene operational taxonomic units (OTUs) closely related to a Pelobacter sp. and Geobacter spp. from boreal lake sediments. The colors on the right illustrate the abundance of the sequences (a total of 78,462 sequences). Scale bar represents estimated phylogenetic distance in substitutions per site, and the numbers at the branch points indicate the bootstrap values.
Bacterial community composition.
As Hg(II)-methylating communities coexist with other microorganisms in sediments, we also described the compositions of the sediment bacterial communities to assess potential interactions and coupling between Hg(II)-methylating communities and the overall bacterial communities. The bacterial 16S rRNA gene data set contained 258,020 high-quality reads, which grouped into 38,681 OTUs at 97% sequence identity. The data were normalized to the lowest number of reads for any individual sample, resulting in 6,045 reads per sample and a total of 23,485 OTUs. In general, the bacterial communities of the samples collected in the same lake at different depths (0 to 1 cm and 1 to 2 cm) were highly similar (see Fig. S3). The most abundant phyla were Acidobacteria (15.0%), Proteobacteria (12.7%, almost exclusively represented by Deltaproteobacteria), Bacteroidetes (9.4%), and Chloroflexi (8.0%) (Fig. 5). These phyla are often seen in freshwater sediment communities (39–42). Except for lake LJU, the distributions of major taxa were similar across the full set of lakes, with phylum Acidobacteria and unknown bacteria exhibiting the largest variation in abundance (see Fig. S4). Bacterial alpha-diversity, measured as the number of observed OTUs, varied between samples (from 804 to 2,723 OTUs), and also between phyla (Fig. 5), with Acidobacteria (1,242 OTUs; 13.6% of total reads) representing a particularly abundant phylum (Fig. 5), followed by Bacteroidetes (1,610 OTUs; 9.2% of total reads), Chloroflexi (988 OTUs; 12.4% of total reads), and Deltaproteobacteria (1,944 OTUs; 7.9% of total reads). Within the Deltaproteobacteria, OTUs related to known Hg(II) methylators, i.e., orders Syntrophobacterales, Desulfobacterales, Desulfuromonadales, and Desulfovibrionales accounted for 4.7%, 1.0%, 0.8% and 0.01% of the reads, respectively, across the full set of samples. Families Syntrophaceae and Syntrophobacteraceae accounted for 4.2% and 0.4%, respectively, of Syntrophobacterales. Within the Firmicutes, the order Clostridiales contributed 0.7% of the total reads, half of them belonging to the family Ruminococcaceae.
FIG 5.

Compositions of sediment bacterial community across boreal lake sediments based on 16S rRNA gene amplicon analysis. Symbol sizes illustrate the abundance of the organisms at the phylum level, except for Proteobacteria (class level). Symbol shade (gray scale) represents the number of individual operational taxonomic units (OTUs; 97% similarity threshold) within each lineage. The dendrogram to the left shows the results of a cluster analysis and highlights community similarities between individual samples. Lakes with high Hg(II) methylation rate constants (km > 0.02 day−1) are highlighted in blue boldface font.
With regard to the overall composition of microbial communities, there seems to be coherence between hgcA and 16S rRNA genes (correlation in a symmetric Procrustes rotation, 0.67; P = 0.034). However, the community compositions between samples collected from the same lake were more uniform for 16S rRNA gene data than for the hgcA gene data (see Fig. S5). However, there was no direct link between the total bacterial community composition and Hg(II) methylation rate constants in the studied lake sediments (permutational multivariate analyses of variance [PerMANOVAs], P = 0.139, F = 1.46, R2 = 0.075). Indeed, lakes with highly similar bacterial community compositions, as for example, LS0-1 and M0-1 (Fig. 5), presented very different Hg(II) methylation rate constants (Table 2).
Effects of OM composition and resident bacterial communities on the composition of Hg(II)-methylating microorganisms.
In a previous study, Bravo et al. demonstrated that most of the variation in the OM composition was related to OM sources (terrestrial versus in-lake) and their degradation status (fresh versus degraded) (9). Here, we assessed the effect of OM molecular composition on the composition of Hg(II)-methylating microorganisms in boreal lake sediments by using the abundances of organic compounds previously identified by pyrolysis-gas chromatography-mass spectrometry (Py-GC/MS) (9). We used redundancy analysis (RDA), a well-recognized multivariate analysis method in microbial ecology (43), to regress a matrix of multiple response variables (i.e., the abundance of the OTUs for Hg(II)-methylating microorganisms) to a corresponding matrix of explanatory variables (i.e., the abundances of different OM compounds). However, RDA modeling has two prerequisites: (i) the number of pyrolytic organic compounds that can be included in the model must be lower than the number of observations (the 9 sediment samples), and (ii) the selected pyrolytic organic compounds should not be intercorrelated in order to optimize the precision in the prediction of the OTUs for Hg(II)-methylating microorganisms (43). In the final RDA model, the explanatory variables were composed of guaiacol, n-alkenes C11 to C14 (noted C11–14:1), prist-2-ene, phytene, and indoles (Fig. 6) (details on how these groups of pyrolytic organic compounds have been selected are provided in Materials and Methods). The RDA model (F = 1.5, P = 0.018) (Fig. 6) showed that those pyrolytic organic compounds explained 62% of the variation in the abundance of Hg(II)-methylating communities (RDA1 = 20%, RDA2 = 16%, RDA3 = 15%, RDA4 = 11%), with an associated R2 of 70%. More precisely, the Hg(II)-methylating community structure was significantly correlated with (listed in order of decreasing importance) indoles (P = 0.025), short n-alkenes (C11–14:1, P = 0.026), and prist-2-ene (P = 0.043). Both C11–14:1 and prist-2-ene are proxies of degraded OM. While the presence of indoles indicates protein processing, C11–14:1 and prist-2-ene denoted the presence of extensively degraded OM from lipids of multiple sources and chlorophylls or tocopherols (and thus from phytoplankton), respectively (44–46). In contrast, phytene and guaiacol are specific proxies for organic matter sources, namely, phytoplankton and terrestrially derived OM, respectively (44, 45, 47). Hence, the RDA suggests that the composition of Hg(II)-methylating communities is influenced by the degradation status of phytoplankton-derived OM. In contrast, the presence of terrigenous OM, for which guaiacol is a specific proxy, had smaller effects on the composition of Hg(II)-methylating communities in general (P > 0.05) (Fig. 6) and on the Hg(II) methylation rate constants of the studied lakes (Table 2) (9), as also suggested in the literature (48).
FIG 6.

RDA triplot (2 first components) of the Hg(II)-methylating microbial community (hgcA gene, OTU level) (response variable) constrained by specific OM compounds (predicting variable) and the studied sites. The color coding in the legend indicates the highly resolved taxonomic levels of the Hg(II)-methylating community. The shapes correspond to the order level. The arrows and text summarize explanatory variables, corresponding to the descriptors of OM as follows: phytene, fresh phytoplankton-derived OM; prist-2-ene, degraded phytoplankton-derived OM; guaiacol, terrestrial OM; C11–14:1, degraded lipids; indoles, processed proteins.
As sediments are habitats for a large number of organisms besides Hg(II) methylators, we also performed RDA to study the effect of the supporting and interacting bacterial communities on the composition of Hg(II) methylators (Fig. 7). The RDA identified a direct link between some specific groups of the sediment bacterial community and the composition of Hg(II)-methylating communities (F = 4.2, P = 0.005) (Fig. 7). The model explained 83.6% of the total variance (RDA1 = 30.1%, RDA2 = 26.8%, RDA3 = 16.1%, RDA4 = 10.6%) with an R2 of 96.7%. The percentage of accumulated constrained eigenvalues of the axes explained 31.1%, 27.7%, 16.6%, and 10.9% of the variance, respectively. The RDA showed that the Hg(II)-methylating community structure was significantly correlated to the presence of (listed in order of decreasing importance) Syntrophobacterales (P = 0.001), Acidobacteria_Gp15 (P = 0.003), Chlorobiales (P = 0.005), Fibrobacterales (P = 0.006), Holophagales (P = 0.014), Hydrogenophilales (P = 0.02), and Rhizobiales (P = 0.030). This suggests dependences or a mutualistic relationship between the methylators and these abundant community members.
FIG 7.

RDA triplot (2 first components) of the Hg(II)-methylating microbial community (hgcA gene, OTU level) (response variable) constrained by the resident bacterial community (16S rRNA gene) and the studied sites. The color coding in the legend indicates the highly resolved taxonomic levels of the Hg(II)-methylating community. The shapes correspond to the order level of the Hg(II)-methylating community. The arrows and text summarize explanatory variables, corresponding to taxa of resident bacterial community.
DISCUSSION
Relative contribution of different Hg(II)-methylating microbial guilds.
In this study, we found that SRB, considered primary Hg methylators in aquatic systems (14–16, 49), accounted for less than half of the Hg(II) methylation. Both the experiments with specific inhibitors and hgcA gene surveys demonstrated that the major part of the Hg(II) methylation (between 55% and 62%) km was most likely attributed to other functional groups, such as methanogenic Archaea and FeRB. The significance of methanogenic Archaea and FeRB as mediators for Hg(II) methylation has until recently been largely overlooked, but our incubations suggest that these metabolic guilds are important drivers of Hg(II) methylation. This is further supported by the abundance of hgcA reads affiliated with methanogens and FeRB (Fig. 1) in the data set. Methanogens were only recently revealed to play a role as Hg(II) methylators in experiments using pure cultures (50, 51), lake periphyton (20), and rice paddies (52) and by global hgcA gene inventories (29, 31, 53). The abundance of methanogens in the hgcA gene library (Fig. 1) and the phylogenetic analyses suggest that some of the unidentified OTUs in this study may represent hitherto unknown Hg(II)-methylating methanogens (Fig. 2).
It is also important to consider a potential indirect effect of molybdate amendments. Molybdate amendments might have decreased Hg(II) methylation rate constants not only by inhibiting the activity of Hg(II)-methylating SRB but also indirectly by interfering with syntrophic partnerships between Hg(II)-methylating methanogens and non-Hg(II)-methylating syntrophs (14), such as members of the family Syntrophaceae, that were abundant in the studied lakes.
Only a decade ago, FeRB were revealed as important mediators of Hg(II) methylation in sediments of Clear Lake (18) and, more recently, in sediments contaminated by sewage treatment plant effluents (19, 54), in paddy soils (30), and in wetlands (29). In addition to the reads specifically classified as belonging to Geobacteraceae, the phylogenetic analysis suggests that 11 OTUs (13% of the total reads) of the unknown Deltaproteobacteria may be close relatives of Desulfuromonadaceae and Geobacteraceae (Fig. 3). As many Geobacter spp. can methylate Hg(II) (53, 55), the abundance of Geobacteraceae, and relatives, in our hgcA data set suggests that FeRB are also putatively important members of Hg(II)-methylating communities of boreal lake sediments. The lack of specific inhibitors for Fe reduction limits our ability to quantify the specific contribution of FeRB to Hg(II) methylation rate constants relative to that from other groups and to determine whether Desulfuromonadaceae and Geobacteraceae methylate Hg(II) in Fe-reducing processes or might use alternative metabolic processes such as S0 reduction or syntrophic oxidation of OM (56).
Despite the expanded view that we provide on the diversity of Hg(II) methylators in boreal lake sediments, many of the putative Hg(II)-methylating microbial groups observed in the environment could not be robustly annotated according to metabolic type. Indeed, the SRB shown to mediate approximately 45% of MeHg formation were not robustly identified in the hgcA gene data set but were likely included in the large pool of OTUs classified merely as Deltaproteobacteria (see Fig. S2 in the supplemental material). Expanded genome-wide sequence databases for uncultured microbial lineages and further development and refinement of primers targeting hgcA and hgcB to specifically target different groups of Hg(II) methylators are thus needed to increase the comprehensive understanding of the distribution of Hg-methylating organisms in nature (38), including those in boreal lake sediments.
Factors affecting the composition of Hg(II) methylators: the role of organic matter.
In the studied lakes, nonmetric multidimensional scaling (NMDS) plots of 16S rRNA and hgcA genes indicated that there was more spatial variation in the diversity of the hgcA gene than of the general bacterial 16S rRNA gene (Fig. S5). This implies that Hg(II)-methylating microbial communities might be more endemic or at least less homogenous than the overall bacterial community, possibly responding more strongly to the different environmental conditions experienced in the different lakes. Furthermore, the amount of bioavailable Hg(II), temperature, pH, and redox (57) as well as the composition of OM strongly influenced Hg(II) methylation processes (9). RDA model 1 revealed that the degradation status of phytoplankton-derived OM was an important factor controlling the composition of Hg(II)-methylating microorganisms (Fig. 6). In lakes with the highest Hg(II) methylation rate constants (V and VALE) (Table 2), fresh phytoplankton-derived OM (i.e., phytene) affected the composition of the Hg(II)-methylating communities toward a higher contribution of Geobacteraceae, Methanomassiliicoccales and unknown Deltaproteobacteria (Fig. 6). In contrast, degraded phytoplankton-derived OM (e.g., prist-2-ene) exerted strong control on Hg(II)-methylating microbial communities in the lakes featuring intermediate to low Hg(II) methylation rate constants (M and LS). In this last case, Hg(II)-methylating microbial communities were more diverse (Fig. 6). As Hg(II)-methylating activities are strain specific (23, 58), it is not surprising to find different Hg(II)-methylating strains from the same order in the different lakes.
An increase in nutrients (and subsequently primary production and phytoplankton-derived OM) has previously been linked to an enhancement of the anaerobic microflora in sediments (59). However, anaerobic Hg(II)-methylating communities only represent a subset of a larger microbial community involved in the transformation and recycling of organic and inorganic compounds in these habitats. Our results hint at an important role of the resident bacterial community in shaping the composition of the Hg(II)-methylating community in boreal lake sediments (Fig. 7). The anaerobic oxidation of OM from complex organic compounds generally goes through different steps and processes (60). After an initial hydrolysis of large organic substances (by, for example, Rhizobiales and Fibrobacterales), the degradation intermediates are fermented into smaller organic molecules, such as lactate, propionate, butyrate, acetate, and formate, as well as CO2 and H2 (by, for example, Holophagaceae). These fermentation products might then be used as electron donors for Desulfovibrionales, Geobacteraceae, and Syntrophobacterales that are known to contain Hg(II) methylators. One of the dominant products of OM oxidation is H2, which can be used by, for example, Hydrogenophilales. Many groups characterized by their capability to degrade OM, and therefore featuring a potential to impact Hg(II) methylation, were indicated in the RDA model 2 (Fig. 7). Thus, our results suggest a potential role of the non-Hg(II)-methylating bacteria involved in the OM decomposition cascade to fuel Hg(II)-methylating microbial communities.
The understanding of mechanisms explaining why some strains occur in one location but are absent in others is still a fundamental challenge in ecology (61). It is becoming increasingly clear that in general, environmental conditions are important for structuring the biogeography of bacterial communities (62). Hence, the differences in the distributions of Hg(II)-methylating taxa among the studied lakes might derive primarily from different species of the same family having different niche requirements (61). Our results suggest that a high relative abundance of phytoplankton-derived OM and the presence of specific strains of non-Hg(II)-methylating bacteria involved in OM decomposition create a niche that promotes Hg(II) methylation. We are in need of a more complete understanding of the biological pathways involved in Hg(II) methylation and the environmental factors controlling the presence and activity of the organisms mediating this process. In this context, our study provides some insights about the hitherto overlooked role of methanogens and FeRB in Hg(II) methylation in boreal lakes.
MATERIALS AND METHODS
Study area.
In this study, we studied 10 lakes with contrasting trophic statuses and receiving different amounts of terrestrial OM (Table 1). According to a previous study (9) and to the OM molecular compositions, these 10 lakes were grouped into two types: Lötsjön (LOTS), Marnästjärn (M), Strandsjön (STR), Vallentunasjön (VALE), and Valloxen (V) were dominated by autochthonous OM, whereas sediments from Svarttjärn (S), Ljustjärn (LJU), Lilla Sångaren (LS), Oppsveten (O), and Fälaren (F) were characterized by a predominance of allochthonous terrestrially derived OM. The lakes ranged from clear water to humic, with average dissolved organic carbon (DOC) concentrations ranging from 6.5 to 32.6 mg · liter−1, and from oligotrophic to hypereutrophic systems with chlorophyll a from below detection (<0.5 μg · liter−1) to 190 μg · liter−1 (Table 1). Conductivity and pH varied between 30 μS · s−1 and 469 μS · s−1 and between 5.6 and 8.7, respectively. Hg(II) methylation rate constants, determined by incubations with enriched isotopic tracers, varied from 0.9% to 7.8% (Table 2) (9). The details about sampling and sample handling have been described previously (9). Briefly, vertical summer profiles of water samples for chemical characterization were collected using a GoFlo bottle (polyvinyl chloride [PVC]). Some of the lakes were thermally stratified and presented oxygen-depleted waters overlying the sediment (Table 1). The sediment characteristics are presented in Table 2.
Analytical methods.
Water samples were filtered through glass fiber filters (GF/F; Whatman, UK) and analyzed for sulfate, DOC, total phosphorus (TP), chlorophyll, and optical properties of the organic matter (Table 1). The DOC content from the water column was measured by high temperature catalytic oxidation (Shimadzu-TOC-L) (63). TP was analyzed according to Murphy and Riley (64). UV-visible absorbance spectra (200 to 800 nm) were measured with a Lambda 40 spectrophotometer (Perkin-Elmer) as previously described (65).
Sediment cores were collected using a 60-mm diameter gravity corer (Uwitec, Austria). Cores with approximately 40 cm of overlying water were kept upright at 4°C and processed within 12 h in a glove bag (Sigma-Aldrich, USA) under a N2 atmosphere. The water overlying the sediment was first retrieved with sterile syringes, and then the upper 2 cm of the sediment core (0 to 1 cm and 1 to 2 cm) were sliced using autoclaved sectioning tools. For each slice, a sediment subsample was immediately put in a sterile Cryotube and placed in liquid nitrogen for subsequent DNA extraction. Another subsample was immediately incubated for 1 h at in situ temperature after being spiked with 3H-labeled thymidine (Amersham, 1 mCi · ml−1, 80 Ci · mmol−1) to assess bacterial production (66). The last subsample was used to incubate sediments and determine Hg(II) methylation rate constants and Hg, MeHg, C, N, and P concentrations (Table 2) and to characterize OM molecular composition using pyrolysis-gas chromatography-mass spectrometry. A detailed description of the different analytical methods has been published elsewhere (9).
Determination of Hg(II) methylation rate constants in molybdate-amended sediments.
Sediment slurries were prepared by adding 30 ml of wet sediment to 30 ml of its overlying water with a range of different molybdate concentrations, from 0.1 mM to 1 mM. The concentration of molybdate was therefore set to be between 2 and 20 times higher than the prevailing sulfate concentration in the sediment overlying the water in order to approach complete inhibition of sulfate reduction. After 2 h of preequilibration, amended slurries were spiked with 198HgCl2 isotope tracer at close to ambient Hg(II) concentrations (19). One replicate was immediately frozen (t0) and another three replicates (tf) were incubated for 24 h in the glove box at 18°C and subsequently frozen. Hg(II) and MeHg were extracted from 200 mg of sediment using 7 ml of 6 N HNO3 with a 4-min microwave treatment at 80 W. The remaining particles were removed afterwards by centrifugation. Shortly thereafter, the extracts were buffered at pH 4, and isotopic-enriched Hg species Me199Hg and 201Hg(II) were added. The Hg(II) and MeHg species were ethylated with sodium tetraethyl borate, recovered in isooctane, and analyzed by species-specific isotope dilution using capillary gas chromatography-inductively coupled plasma mass spectrometry (GC-ICPMS) (67). Each sample was injected in triplicates, and blanks were used for contamination control. Hg(II) methylation rate constants (km) were calculated from the initial and final concentrations of the formed MeHg species derived from the enriched isotope 198Hg (Me198Hg) after isotopic pattern deconvolution (67) and assuming a pseudo-first-order rate law. The Isotope Program in the Office of Nuclear Physics of the United States Department of Energy, Office of Science, supplied the isotopes used in this study.
Bacterial community composition.
DNA was isolated from 0.2 g soil (wet weight) using the PowerSoil DNA isolation kit (Mo Bio Laboratories, Carlsbad, CA, USA). Bacterial primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGGTATCTAATCC-3′) targeting the 16S rRNA gene (68) were used for PCR amplification, where each sample was run in duplicates and 20 cycles were performed. The resulting PCR products were 100-fold diluted, and 1 μl of each of the diluted replicates was pooled and used for 10 additional cycles of amplification with barcoded primers as previously described (69). All PCRs were conducted in 20-μl volumes using 1.0 U Q5 high-fidelity DNA polymerase (New England BioLabs [NEB], UK), 0.25 μM primers, 200 μM deoxynucleoside triphosphate (dNTP) mix, and 0.4 μg bovine serum albumin (BSA). After amplicon purification with the Agencourt AMPure XP kit (Beckman Coulter, CA, USA), the final amplicon concentration was analyzed with PicoGreen as recommended by the manufacturer (Invitrogen, Carlsbad, CA, USA). Amplicons from 50 uniquely barcoded samples were pooled in equimolar concentrations, and amplicon sequencing was carried out using the Illumina MiSeq instrument with pair-end 300-bp read lengths at the SNP/SEQ SciLifeLab facility hosted by Uppsala University.
16S rRNA gene amplicon data were processed using mothur (70) according to a standard operation protocol (71). Operational taxonomic units (OTUs) were clustered at a 97% pairwise identity.
Targeting Hg-methylating microbial communities.
The isolated DNA used for PCR amplification of the 16S rRNA gene was also used in combination with the primer pair 261F/912R. For this PCR amplification targeting the hgcA gene (29), we used 50 μl master mix containing 1× Phusion GC buffer, 0.2 mM dNTP mix, 5% dimethyl sulfoxide (DMSO), 0.1 μM each primer with generic adaptors, 7 μg/μl BSA, 4 μl extracted DNA template, and 1.0 U Phusion high-fidelity DNA polymerase (NEB, UK). The PCR program started with an initial 2-min denaturation at 98°C followed by 35 amplification cycles (10 s at 96°C, 30 s 56.5°C, and 45 s at 72°C), and a final 7-min extension at 72°C. A second PCR was conducted to add sample-specific DNA barcodes (Table 3). These PCRs were performed in 20-μl volumes using 1× Q5 reaction buffer, 0.2 mM dNTP mix, 0.1 μM barcoded primers, diluted first PCR products, and 1.0 U Q5 high fidelity DNA polymerase (NEB, UK) with an initial denaturation of 30 s at 98°C followed by 18 cycles (10 s at 98°C, 30 s at 66°C, and 30 s at 72°C), and a final 2-min extension at 72°C. PCR products were then purified using Agencourt AMPure XP (Beckman Coulter, CA, USA) and quantified using PicoGreen according to the instructions from the manufacturer (Invitrogen, Carlsbad, CA, USA). The amplicons were then pooled in equimolar concentrations to obtain similar numbers of sequencing reads per sample. Amplicon sequencing was carried out using the Illumina MiSeq instrument with pair-end 300-bp read lengths at the SNP/SEQ SciLifeLab facility hosted by Uppsala University.
TABLE 3.
Barcoded adaptorsa
| Sample name in the database | Sample name in the main text | Index sequence |
|
|---|---|---|---|
| Forward | Reverse | ||
| VALE 01 | VALE 0–1 | TAGATCGC | AGGAGTCC |
| VALE 12 | VALE 1–2 | TAGATCGC | CATGCCTA |
| L 01 | LS 0–1 | TAGATCGC | AGCGTAGC |
| L 12 | LS 1–2 | TAGATCGC | CAGCCTCG |
| V 01 | V0_1 | TAGATCGC | TGCCTCTT |
| M 01 | M 0–1 | TAGATCGC | GGTATAAG |
| M 12 | M 1–2 | TAGATCGC | CAGCTAGA |
| LOTS 12 | LOTS 1–2 | TAGATCGC | TAGGCAAG |
| STRAN 01 | STR 0–1 | TAGATCGC | GTAGAGAG |
Used with the hgcA_261F (CGGCATCAAYGTCTGGTGYGC) and the reverse primer hgcA_912R (GGTGTAGGGGGTGCAGCCSGTRWARKT).
Since the length of the hgcA gene PCR product (680 bp) exceeded the total read length of the sequencing run (600 bp), only the forward amplicon was used for downstream data analysis. Bad-quality reads, adapters, and primer sequences were first removed (72, 73). Version 8.0 of the usearch software was used to truncate (-fastx_truncate), dereplicate (-derep_prefix), and then sort and remove singletons (-sort_by_size -minsize 2) (74). The obtained set of reads was then clustered into OTUs using cd-hit-est with an 80% similarity threshold (75). The original cleaned reads were then mapped to the representative sequences of the obtained clusters to generate a count table, using usearch (-usearch_global). The database used for the annotation of the sequences is based on the sequences published elsewhere (31).
Phylogenetic analyses.
The hgcA gene sequences of this study and those previously published (31) were used to generate a hidden Markov model (HMM) with HMMER (76), and this was used to mine Deltaproteobacteria from the Integrated Microbial Genomes (IMG) database of the Joint Genome Institute (JGI). The sequences where adequately curated and the taxonomy homogenized using taxtastic (https://github.com/fhcrc/taxtastic) and R-package taxize (77). The obtained protein sequences were then aligned with MUSCLE v3.8.1551 (78). The alignment was trimmed to the size of the amplicon, and a tree was generated using RAxML v.8.2.4 (79) with the PROTGAMMLGF model and autoMR to choose the necessary bootstrap number (750). Paralogs were manually removed. The tree and the corresponding alignment were used to generate a reference package for pplacer (80), and then guppy was used to classify the sequences with a likelihood threshold of 0.8.
Statistical analyses.
Statistical analyses were conducted using R 3.3.2 (https://www.r-project.org/). Samples were grouped with hierarchical clustering to visualize the relationships between community compositions of the samples, and the differences in community compositions observed with 16S rRNA gene and hgcA functional gene were compared with permutational multivariate analyses of variance (PerMANOVAs) using the function adonis and Procrustes tests of the package vegan (81).
Similarities between the bacterial community compositions and the Hg(II)-methylating community compositions of each lake at the two different depths were assessed by NMDS using the package vegan (81) and the function vegdist, with the Bray-Curtis coefficient used as a dissimilarity measure.
Redundancy analysis (RDA) was performed using the rda function in R package vegan (81) to explore the relationships between Hg(II)-methylating community composition and (i) OM composition, which was determined by Py-GC/MS, and (ii) bacterial community composition. Prior to this analysis, the relative abundances of pyrolytic compounds were log transformed in order to fit the model assumptions. The OTU tables (explanatory variables) were scaled and centered. RDA seeks a series of linear combinations of the explanatory variables that best explain the variation in the response matrix [i.e., Hg(II)-methylating bacteria] (82) but requires the number of explanatory variables to be below or equal to the number of observations (i.e., number of sediment samples, 9) and the explanatory variables to not be intercorrelated.
For the RDA of Hg(II)-methylating community composition (response matrix, 9 observations) and OM composition (explanatory matrix), the 110 identified pyrolytic organic compounds, previously characterized (9), were first grouped into 47 groups (Fig. S6) on the basis of similarity in the molecular structure within the 12 identified OM classes [i.e., carbohydrates, N compounds, chitin, n-alkenes, n-alkanes, alkan-2-ones, phenols, lignin, chlorophyll, steroids, hopanoids, and (poly)aromatics] (9). A subset of 9 of the 47 groups of pyrolytic organic compounds were then selected on the basis of two criteria: (i) to have a subset for the RDA which includes a group of OM compounds from the different and identified sources (terrestrial, phytoplanktonic, and microorganisms) and of different degradation statuses and (ii) to have limited intercorrelations within the subset of pyrolytic organic compound groups (intercorrelations between the 47 groups of pyrolytic organic compounds are shown in Fig. S6). RDA was performed using the subset of pyrolytic organic compounds, and intercorrelations between those variables were inspected by computing the variance inflation factor (VIF). The pyrolytic organic compounds with a VIF of >15 in the RDA model were excluded, leading to a final RDA model 1 including a subset of 5 pyrolytic organic compounds (phytene, prist-2-ene, guaiacol, indoles, and C11–14:1).
The correlation of NMDS structure of Hg(II)-methylating microbial communities and bacterial community composition (NMDS model 2) was tested by permutation tests (environmental fitting test, envfit function, 999 permutations) (data not shown). We selected among the most correlated explanatory bacterial OTUs to perform RDA model 2 (Syntrophobacterales, Acidobacteria_Gp15, Holophagales, Fibrobacterales, Rhizobiales, Chlorobiales, and Hydrogenophilales) (Fig. 7).
Graphics were built in R 3.2.4 (83) and modified to fit the journal requirements with Inkscape 0.92 (https://inkscape.org/es/).
Accession number(s).
All sequence data have been deposited to the ENA Sequence Read Archive under accession number PRJEB20960.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Johansson and Claudia Bergin for assistance in the field and in the laboratory.
This research was funded by Swedish Research Council (VR) grants to A.G.B. (project 2011-7192) and S. Bertilsson (grant 2012-3892), the Sino-Swedish Mercury Management Research Framework (grant 2013-6978), the Swedish Research Council Formas (grant 2012-986 to S. Bertilsson), the Academy of Finland (grant 265902 to S.P.), and the Kempe Foundation (grant SMK-2840 to S. Bouchet).
Sequencing was performed by the NGI SNP/SEQ facility at Science for Life Laboratory, Uppsala University. The computations were performed on resources provided by SNIC through the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX). NBIS (National Bioinformatics Infrastructure Sweden) is acknowledged for bioinformatics support.
A.G.B., S.P., and S. Bertilsson conceived and designed the study. A.G.B., A.M.-R., and S. Bouchet carried out the field campaigns. A.G.B. and A.M.-R., performed the experiments with molybdate. A.G.B. and S. Bouchet performed the GC-ICPMS analyses of mercury with guidance from E.B. A.M.-R. and S.H.O. performed the laboratory microbial analyses with significant help from O.A. and J.K.S. M.B., S.P., and A.G.B. processed the data. J.T. provided the data on OM molecular composition and helped A.G.B. with the related statistical analyses. A.G.B., S.P., J.K.S., and S. Bertilsson conducted all statistical analyses, built the figures, and wrote the manuscript with significant contributions from all the coauthors.
The authors declare no conflict of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01774-18.
REFERENCES
- 1.United Nations Environment Programme. 2013. Global mercury assessment: sources, emissions, releases and environmental transport. United Nations Environment Programme, Nairobi, Kenya. [Google Scholar]
- 2.Nriagu JO. 1993. Legacy of mercury pollution. Nature 363:589. doi: 10.1038/363589a0. [DOI] [Google Scholar]
- 3.Amos HM, Jacob DJ, Streets DG, Sunderland EM. 2013. Legacy impacts of all-time anthropogenic emissions on the global mercury cycle. Glob Biogeochem Cycles 27:410–421. doi: 10.1002/gbc.20040. [DOI] [Google Scholar]
- 4.Lamborg CH, Hammerschmidt CR, Bowman KL, Swarr GJ, Munson KM, Ohnemus DC, Lam PJ, Heimbürger L-E, Rijkenberg MJ, Saito MA. 2014. A global ocean inventory of anthropogenic mercury based on water column measurements. Nature 512:65–68. doi: 10.1038/nature13563. [DOI] [PubMed] [Google Scholar]
- 5.Johansson K, Tyler G. 2001. Impact of atmospheric long range transport of lead, mercury and cadmium on the Swedish forest environment. Water Air Soil Pollut Focus 1:279–297. doi: 10.1023/A:1017528826641. [DOI] [Google Scholar]
- 6.Grigal DF. 2003. Mercury sequestration in forests and peatlands: a review. J Environ Qual 32:393–405. doi: 10.2134/jeq2003.0393. [DOI] [PubMed] [Google Scholar]
- 7.Larsen S, Andersen T, Hessen DO. 2011. Climate change predicted to cause severe increase of organic carbon in lakes. Glob Chang Biol 17:1186–1192. doi: 10.1111/j.1365-2486.2010.02257.x. [DOI] [Google Scholar]
- 8.Isidorova A, Bravo AG, Riise G, Bouchet S, Björn E, Sobek S. 2016. The effect of lake browning and respiration mode on the burial and fate of carbon and mercury in the sediment of two boreal lakes. J Geophys Res Biogeosci 121:233–245. doi: 10.1002/2015JG003086,10.1002/2015JE004920. [DOI] [Google Scholar]
- 9.Bravo AG, Bouchet S, Tolu J, Björn E, Mateos-Rivera A, Bertilsson S. 2017. Molecular composition of organic matter controls methylmercury formation in boreal lakes. Nat Commun 8:14255. doi: 10.1038/ncomms14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mergler D, Anderson H a, Chan LHM, Mahaffey KR, Murray M, Sakamoto M, Stern AH, Panel on Health Risks and Toxicological Effects of Methylmercury. 2007. Methylmercury exposure and health effects in humans: a worldwide concern. Ambio 36:3–11. doi: 10.1579/0044-7447(2007)36[3:MEAHEI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 11.Bellanger M, Pichery C, Aerts D, Berglund M, Castaño A, Cejchanová M, Crettaz P, Davidson F, Esteban M, Fischer ME, Gurzau AE, Halzlova K, Katsonouri A, Knudsen LE, Kolossa-Gehring M, Koppen G, Ligocka D, Miklavèiè A, Reis MF, Rudnai P, Tratnik JS, Weihe P, Budtz-Jŗgensen E, Grandjean P. 2013. Economic benefits of methylmercury exposure control in Europe: monetary value of neurotoxicity prevention. Environ Health 12:3. doi: 10.1186/1476-069X-12-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morel FMM, Kraepiel AML, Amyot M. 1998. The chemical cycle and bioaccumulation of mercury. Annu Rev Ecol Syst 29:543–566. doi: 10.1146/annurev.ecolsys.29.1.543. [DOI] [Google Scholar]
- 13.Clarkson TW, Magos L. 2006. The toxicology of mercury and its chemical compounds. Crit Rev Toxicol 36:609–662. doi: 10.1080/10408440600845619. [DOI] [PubMed] [Google Scholar]
- 14.Pak K, Bartha R. 1998. Mercury methylation and demethylation in anoxic lake sediments and by strictly anaerobic bacteria. Appl Environ Microbiol 64:1013–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Achá D, Hintelmann H, Yee J. 2011. Importance of sulfate reducing bacteria in mercury methylation and demethylation in periphyton from Bolivian Amazon region. Chemosphere 82:911–916. doi: 10.1016/j.chemosphere.2010.10.050. [DOI] [PubMed] [Google Scholar]
- 16.Achá D, Hintelmann H, Pabón CA. 2012. Sulfate-reducing bacteria and mercury methylation in the water column of the lake 658 of the experimental lake area. Geomicrobiol J 29:667–674. doi: 10.1080/01490451.2011.606289. [DOI] [Google Scholar]
- 17.Bravo AG, Loizeau JL, Dranguet P, Makri S, Björn E, Ungureanu VG, Slaveykova VI, Cosio C. 2016. Persistent Hg contamination and occurrence of Hg-methylating transcript (hgcA) downstream of a chlor-alkali plant in the Olt River (Romania). Environ Sci Pollut Res Int 23:10529–10541. doi: 10.1007/s11356-015-5906-4. [DOI] [PubMed] [Google Scholar]
- 18.Fleming EJ, Mack EE, Green PG, Nelson DC. 2006. Mercury methylation from unexpected sources: molybdate-inhibited freshwater sediments and an iron-reducing bacterium. Appl Environ Microbiol 72:457–464. doi: 10.1128/AEM.72.1.457-464.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bravo AG, Bouchet S, Guédron S, Amouroux D, Dominik J, Zopfi J. 2015. High methylmercury production under ferruginous conditions in sediments impacted by sewage treatment plant discharges. Water Res 80:245–255. doi: 10.1016/j.watres.2015.04.039. [DOI] [PubMed] [Google Scholar]
- 20.Hamelin S, Amyot M, Barkay T, Wang Y, Planas D. 2011. Methanogens: principal methylators of mercury in lake periphyton. Environ Sci Technol 45:7693–7700. doi: 10.1021/es2010072. [DOI] [PubMed] [Google Scholar]
- 21.Bae HS, Dierberg FE, Ogram A. 2014. Syntrophs dominate sequences associated with the mercury methylation-related gene hgcA in the water conservation areas of the Florida Everglades. Appl Environ Microbiol 80:6517–6526. doi: 10.1128/AEM.01666-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macalady JL, Mack EE, Nelson DC. 2000. Sediment microbial community structure and mercury methylation in mercury-polluted Clear Lake, California. Appl Environ Microbiol 66:1479–1488. doi: 10.1128/AEM.66.4.1479-1488.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duran R, Ranchou-Peyruse M, Menuet V, Monperrus M, Bareille G, Goñi MS, Salvado JC, Amouroux D, Guyoneaud R, Donard OFX, Caumette P. 2008. Mercury methylation by a microbial community from sediments of the Adour Estuary (Bay of Biscay, France). Environ Pollut 156:951–958. doi: 10.1016/j.envpol.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 24.Ma M, Du H, Wang D, Kang S, Sun T. 2017. Biotically mediated mercury methylation in the soils and sediments of Nam Co Lake, Tibetan Plateau. Environ Pollut 227:243–251. doi: 10.1016/j.envpol.2017.04.037. [DOI] [PubMed] [Google Scholar]
- 25.Devereux R, Winfrey MR, Winfrey J, Stahl DA. 1996. Depth profile of sulfate-reducing bacterial ribosomal RNA and mercury methylation in an estuarine sediment. FEMS Microbiol Ecol 20:23–31. doi: 10.1111/j.1574-6941.1996.tb00301.x. [DOI] [Google Scholar]
- 26.King JK, Kostka JE, Frischer ME, Saunders FM, Jahnke RA. 2001. A quantitative relationship that demonstrates mercury methylation rates in marine sediments are based on the community composition and activity of sulfate-reducing bacteria. Environ Sci Technol 35:2491–2496. doi: 10.1021/es001813q. [DOI] [PubMed] [Google Scholar]
- 27.Yu R-Q, Flanders JR, Mack EE, Turner R, Mirza MB, Barkay T. 2012. Contribution of coexisting sulfate and iron reducing bacteria to methylmercury production in freshwater river sediments. Environ Sci Technol 46:2684–2691. doi: 10.1021/es2033718. [DOI] [PubMed] [Google Scholar]
- 28.Parks JM, Johs A, Podar M, Bridou R, Hurt RA, Smith SD, Tomanicek SJ, Qian Y, Brown SD, Brandt CC, Palumbo AV, Smith JC, Wall JD, Elias DA, Liang L. 2013. The genetic basis for bacterial mercury methylation. Science 339:1332–1335. doi: 10.1126/science.1230667. [DOI] [PubMed] [Google Scholar]
- 29.Schaefer JK, Kronberg R-M, Morel FMM, Skyllberg U. 2014. Detection of a key Hg methylation gene, hgcA, in wetland soils. Environ Microbiol Rep 6:441–447. doi: 10.1111/1758-2229.12136. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y-R, Yu R-Q, Zheng Y-M, He J-Z. 2014. Analysis of the microbial community structure by monitoring an Hg methylation gene (hgcA) in paddy soils along an Hg gradient. Appl Environ Microbiol 80:2874–2879. doi: 10.1128/AEM.04225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Podar M, Gilmour CC, Brandt CC, Soren A, Brown SD, Crable BR, Palumbo AV, Somenahally AC, Elias DA. 2015. Global prevalence and distribution of genes and microorganisms involved in mercury methylation. Sci Adv 1:e1500675. doi: 10.1126/sciadv.1500675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ullrich SM, Tanton TW, Abdrashitova SA, Svetlana A. 2001. Mercury in the aquatic environment: a review of factors affecting methylation. Crit Rev Environ Sci Technol 31:241–293. doi: 10.1080/20016491089226. [DOI] [Google Scholar]
- 33.Liem-Nguyen V, Skyllberg U, Björn E. 2017. Thermodynamic modeling of the solubility and chemical speciation of mercury and methylmercury driven by organic thiols and micromolar sulfide concentrations in boreal wetland soils. Environ Sci Technol 51:3678–3686. doi: 10.1021/acs.est.6b04622. [DOI] [PubMed] [Google Scholar]
- 34.Chiasson-Gould SA, Blais JM, Poulain AJ. 2014. Dissolved organic matter kinetically controls mercury bioavailability to bacteria. Environ Sci Technol 48:3153–3161. doi: 10.1021/es4038484. [DOI] [PubMed] [Google Scholar]
- 35.Schaefer JK, Morel FMM. 2009. High methylation rates of mercury bound to cysteine by Geobacter sulfurreducens. Nat Geosci 2:123–126. doi: 10.1038/ngeo412. [DOI] [Google Scholar]
- 36.Leclerc M, Planas D, Amyot M. 2015. Relationship between extracellular low-molecular-weight thiols and mercury species in natural lake periphytic biofilms. Environ Sci Technol 49:7709–7716. doi: 10.1021/es505952x. [DOI] [PubMed] [Google Scholar]
- 37.Graham AM, Aiken GR, Gilmour CC. 2013. Effect of dissolved organic matter source and character on microbial Hg methylation in Hg-S-DOM solutions. Environ Sci Technol 47:5746–5754. doi: 10.1021/es400414a. [DOI] [PubMed] [Google Scholar]
- 38.Christensen GA, Wymore AM, King AJ, Podar M, Hurt RA, Santillan EU, Soren A, Brandt CC, Brown SD, Palumbo AV, Wall JD, Gilmour CC, Elias DA. 2016. Development and validation of broad-range qualitative and clade-specific quantitative molecular probes for assessing mercury methylation in the environment. Appl Environ Microbiol 82:6068–6078. doi: 10.1128/AEM.01271-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ye W, Liu X, Lin S, Tan J, Pan J, Li D, Yang H. 2009. The vertical distribution of bacterial and archaeal communities in the water and sediment of Lake Taihu. FEMS Microbiol Ecol 70:263–276. doi: 10.1111/j.1574-6941.2009.00761.x. [DOI] [PubMed] [Google Scholar]
- 40.Duan D, Ran Y, Cheng H, Chen J, Wan G. 2014. Contamination trends of trace metals and coupling with algal productivity in sediment cores in Pearl River Delta, South China. Chemosphere 103:35–43. doi: 10.1016/j.chemosphere.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 41.Ruiz-González C, Niño-García JP, del Giorgio PA. 2015. Terrestrial origin of bacterial communities in complex boreal freshwater networks. Ecol Lett 18:1198–1206. doi: 10.1111/ele.12499. [DOI] [PubMed] [Google Scholar]
- 42.Haller L, Tonolla M, Zopfi J, Peduzzi R, Wildi W, Poté J. 2011. Composition of bacterial and archaeal communities in freshwater sediments with different contamination levels (Lake Geneva, Switzerland). Water Res 45:1213–1228. doi: 10.1016/j.watres.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 43.Ramette A. 2007. Multivariate analyses in microbial ecology. FEMS Microbiol Ecol 62:142–160. doi: 10.1111/j.1574-6941.2007.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishiwatari R, Morinaga S, Yamamoto S, Machihara T. 1991. Characteristics of kerogens from recent marine and lacustrine sediments: GC/MS analysis of alkaline permanganate oxidation products. J Southeast Asian Earth Sci 5:53–60. doi: 10.1016/0743-9547(91)90011-L. [DOI] [Google Scholar]
- 45.Nguyen RT, Harvey HR, Zang X, Van Heemst JDH, Hetényi M, Hatcher PG. 2003. Preservation of algaenan and proteinaceous material during the oxic decay of Botryococcus braunii as revealed by pyrolysis-gas chromatography/mass spectrometry and 13C NMR spectroscopy. Org Geochem 34:483–497. doi: 10.1016/S0146-6380(02)00261-9. [DOI] [Google Scholar]
- 46.Tolu J, Rydberg J, Meyer-Jacob C, Gerber L, Bindler R. 2017. Spatial variability of organic matter molecular composition and elemental geochemistry in surface sediments of a small boreal Swedish lake. Biogeosciences 14:1773–1792. doi: 10.5194/bg-14-1773-2017. [DOI] [Google Scholar]
- 47.Meyers PA, Ishiwatari R. 1993. Lacustrine organic geochemistry—an overview of indicators of organic-matter sources and diagenesis in lake-sediments. Org Geochem 20:867–900. doi: 10.1016/0146-6380(93)90100-P. [DOI] [Google Scholar]
- 48.Christensen GA, Somenahally AC, Moberly JG, Miller CM, King AJ, Gilmour CC, Brown SD, Podar M, Brandt CC, Brooks SC, Palumbo AV, Wall JD, Elias DA. 2018. Carbon amendments alter microbial community structure and net mercury methylation potential in sediments. Appl Environ Microbiol 84:e01049-17. doi: 10.1128/AEM.01049-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bravo AG, Cosio C, Amouroux D, Zopfi J, Chevalley P-A, Spangenberg JE, Ungureanu V-G, Dominik J. 2014. Extremely elevated methyl mercury levels in water, sediment and organisms in a Romanian reservoir affected by release of mercury from a chlor-alkali plant. Water Res 49:391–405. doi: 10.1016/j.watres.2013.10.024. [DOI] [PubMed] [Google Scholar]
- 50.Yu R-Q, Reinfelder JR, Hines ME, Barkay T. 2013. Mercury methylation by the methanogen Methanospirillum hungatei. Appl Environ Microbiol 79:6325–6330. doi: 10.1128/AEM.01556-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gilmour CC, Bullock AL, McBurney A, Podar M, Elias DA. 2018. Robust mercury methylation across diverse methanogenic Archaea. mBio 9:e02403-17. doi: 10.1128/mBio.02403-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vishnivetskaya TA, Hu H, Van Nostrand JD, Wymore AM, Xu X, Qiu G, Feng X, Zhou J, Brown SD, Brandt CC, Podar M, Gu B, Elias DA. 2018. Microbial community structure with trends in methylation gene diversity and abundance in mercury-contaminated rice paddy soils in Guizhou, China. Environ Sci Process Impacts 20:673–685. doi: 10.1039/C7EM00558J. [DOI] [PubMed] [Google Scholar]
- 53.Gilmour CC, Podar M, Bullock AL, Graham AM, Brown SD, Somenahally AC, Johs A, Hurt RA, Bailey KL, Elias DA. 2013. Mercury methylation by novel microorganisms from new environments. Environ Sci Technol 47:11810–11820. doi: 10.1021/es403075t. [DOI] [PubMed] [Google Scholar]
- 54.Bravo AG, Zopfi J, Buck M, Xu J, Bertilsson S, Schaefer JK, Poté J, Cosio C. 2018. Geobacteraceae are important members of mercury-methylating microbial communities of sediments impacted by wastewater releases. ISME J 12:802–812. doi: 10.1038/s41396-017-0007-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kerin EJ, Gilmour CC, Roden E, Suzuki MT, Coates JD, Mason RP. 2006. Mercury methylation by dissimilatory iron-reducing bacteria. Appl Environ Microbiol 72:7919–7921. doi: 10.1128/AEM.01602-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holmes DE, Shrestha PM, Walker DJF, Dang Y, Nevin KP, Woodard TL, Lovley DR. 2017. Metatranscriptomic evidence for direct interspecies electron transfer between Geobacter and Methanothrix species in methanogenic rice paddy soils. Appl Environ Microbiol 83:e00223-17. doi: 10.1128/AEM.00223-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paranjape AR, Hall BD. 2017. Recent advances in the study of mercury methylation in aquatic systems. Facets (Ott) 2:85–119. doi: 10.1139/facets-2016-0027. [DOI] [Google Scholar]
- 58.Graham AM, Bullock AL, Maizel AC, Elias DA, Gilmour CC. 2012. Detailed assessment of the kinetics of Hg-cell association, Hg methylation, and methylmercury degradation in several Desulfovibrio species. Appl Environ Microbiol 78:7337–7346. doi: 10.1128/AEM.01792-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Drake HL, Aumen NG, Kuhner C, Wagner C, Griesshammer A, Schmittroth M. 1996. Anaerobic microflora of everglades sediments: effects of nutrients on population profiles and activities. Appl Environ Microbiol 62:486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lovley DR, Chapelle FH. 1995. Deep subsurface microbial processes. Rev Geophys 33:365–381. doi: 10.1029/95RG01305. [DOI] [Google Scholar]
- 61.Logue JB, Mouquet N, Peter H, Hillebrand H, Metacommunity Working Group. 2011. Empirical approaches to metacommunities: a review and comparison with theory. Trends Ecol Evol 26:482–491. doi: 10.1016/j.tree.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 62.Andersson MGI, Berga M, Lindström ES, Langenheder S. 2014. The spatial structure of bacterial communities is influenced by historical environmental conditions. Ecology 95:1134–1140. doi: 10.1890/13-1300.1. [DOI] [PubMed] [Google Scholar]
- 63.Benner R, Strom M. 1993. A critical evaluation of the analytical blank associated with DOC measurements by high-temperature catalytic oxidation. Mar Chem 41:153–160. doi: 10.1016/0304-4203(93)90113-3. [DOI] [Google Scholar]
- 64.Murphy J, Riley JP. 1962. A modified single solution method for the determination of phosphate in natural waters. Anal Chim Acta 27:31–36. doi: 10.1016/S0003-2670(00)88444-5. [DOI] [Google Scholar]
- 65.Kothawala DN, von Wachenfeldt E, Koehler B, Tranvik LJ. 2012. Selective loss and preservation of lake water dissolved organic matter fluorescence during long-term dark incubations. Sci Total Environ 433:238–246. doi: 10.1016/j.scitotenv.2012.06.029. [DOI] [PubMed] [Google Scholar]
- 66.Smith DC, Azam F. 1992. A simple, economical method for measuring bacterial protein synthesis rates in seawater using 3H-leucine. Mar Microb Food Webs 6:107–114. [Google Scholar]
- 67.Rodriguez-Gonzalez P, Bouchet S, Monperrus M, Tessier E, Amouroux D. 2013. In situ experiments for element species-specific environmental reactivity of tin and mercury compounds using isotopic tracers and multiple linear regression. Environ Sci Pollut Res Int 20:1269–1280. doi: 10.1007/s11356-012-1019-5. [DOI] [PubMed] [Google Scholar]
- 68.Herlemann DPR, Labrenz M, Jürgens K, Bertilsson S, Waniek JJ, Andersson AF, Ju K, Bertilsson S, Waniek JJ, Andersson AF. 2011. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J 5:1571–1579. doi: 10.1038/ismej.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sinclair L, Osman OA, Bertilsson S, Eiler A. 2015. Microbial community composition and diversity via 16S rRNA gene amplicons: evaluating the Illumina platform. PLoS One 10:e0116955. doi: 10.1371/journal.pone.0116955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Joshi NA, Fass JN. 2011. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files (version 1.33). https://github.com/najoshi/sickle.
- 73.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 74.Edgar RC. 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 75.Fu L, Niu B, Zhu Z, Wu S, Li W. 2012. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comput Biol 7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chamberlain SA, Szöcs E. 2013. taxize: taxonomic search and retrieval in R. F1000Research 2:191. doi: 10.12688/f1000research.2-191.v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Matsen FA, Kodner RB, Armbrust EV. 2010. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11:538. doi: 10.1186/1471-2105-11-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oksanen AJ, Blanchet FG, Friendly M, Kindt R, Legendre P, Mcglinn D, Minchin PR, Hara RBO, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H. 2017. Community ecology package (Vegan). R package version 2.4-3. https://github.com/vegandevs/vegan.
- 82.Borcard D, Gillet F, Legendre P. 2018. Canonical ordination, p 203–297. In Numerical ecology with R. Springer International Publishing, Cham, Switzerland. [Google Scholar]
- 83.R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.