

Abstract
Glycosylation is one of the most prevalent posttranslational modifications that profoundly affects the structure and functions of proteins in a wide variety of biological recognition events. However, the structural complexity and heterogeneity of glycoproteins, usually resulting from the variations of glycan components and/or the sites of glycosylation, often complicates detailed structure— function relationship studies and hampers the therapeutic applications of glycoproteins. To address these challenges, various chemical and biological strategies have been developed for producing glycan-defined homogeneous glycoproteins. This review highlights recent advances in the development of chemoenzymatic methods for synthesizing homogeneous glycoproteins, including the generation of various glycosynthases for synthetic purposes, endoglycosidase-catalyzed glycoprotein synthesis and glycan remodeling, and direct enzymatic glycosylation of polypeptides and proteins. The scope, limitation, and future directions of each method are discussed.

1. INTRODUCTION
Glycoproteins are an important class of biomolecules that are involved in a wide variety of physiological and disease processes.1 The biological functions of protein glycosylation are truly multifaceted.1 It is well documented that glycosylation can profoundly affect a protein’s intrinsic properties such as the conformations, protease stability, antigenicity, and immunoge-nicity.2 The glycans of glycoproteins can also directly participate in a number of biological recognition processes including intracellular trafficking, cell adhesion, signaling, development, host-pathogen interactions, and immune responses, to name a few.1‘3–5 For example, N-glycosylation plays an important role in the lectin (calnexin/calreticulin)-mediated protein folding and the ER-associated degradation pathways in quality control;6,7 the mannose-6-phosphate (M6P)-tagged glycosylation of lysosomal enzymes is critical for successfully targeting the enzymes to lysosomes for degrading various dysfunctional biomolecules; ,9 cell surface glycans often serve as ligands for glycan-binding protein mediated host-pathogen interactions such as bacterial and viral infections; 10–14 and aberrant glycosylation is often associated with disease development and progression such as cancer and autoimmune disorders.15–17 A majority of therapeutic proteins, including monoclonal antibodies, are glycosylated, and the presence as well as the fine structures of the sugar chains are critical for the stability and biological functions. 8,19 Thus, understanding the structure and functions, as well as the control of the glycosylation status, is essential for the development and production of efficient protein-based therapeutics.20,21
A major challenging in dealing with glycoproteins comes from the structural heterogeneity of natural and recombinant glycoproteins usually aroused from the variations of glycan components and/or the sites of glycosylation. In fact, recombinant glycoproteins such as therapeutic antibodies are usually produced as mixtures of glycoforms that have the same protein backbone but differ in the pendent oligosaccharides, from which pure glycoforms are difficult to isolate using current chromatographic techniques. This situation significantly hampers a detailed understanding of the structure-function relationships and slows down the therapeutic and diagnostic applications of glycoproteins.18,21–27 Thus, efficient methods that allow controlling glycosylation during expression or permit in vitro construction of glycan-defined glycoproteins are urgently needed. Tremendous progress has been made in recent years for producing structurally well-defined, homogeneous glycoproteins, including tailor-made glycoforms of intact antibodies.28–31 These include total chemical synthesis,32–45 “tag and modify” approaches for site-selective protein-glycan conjugation,46–48 chemoenzymatic synthesis using enzymes for key modification andligation,28–31,49–62 and glycosylation engineering by manipulating the biosynthetic pathways in different host expression systems.63–69
The present review provides a survey of the chemoenzymatic methods that have been developed for the synthesis of glycopeptides and glycoproteins carrying defined oligosaccharides, with a focus on the advances in the past decade. Particular attention was turned to the following areas: the generation of various glycosynthases from glycosidases for synthetic purposes, the endoglycosidase-catalyzed glycoprotein synthesis and glycan remodeling of intact glycoproteins, and the direct enzymatic glycosylation of polypeptides and proteins using oligosacchar-yltransferase, N-glycosyltransferase, O-GlcNAc transferase and O-GalNAc transferase. Selected examples are presented to discuss the concept of the respective methods, while more indepth technical points can be found in the cited primary literature.
2. GENERAL ASPECTS OF THE STRUCTURE, FUNCTION, AND SYNTHESIS OF GLYCOPROTEINS
2.1. Structural Features of Glycans and Glycoproteins
The covalent attachment of mono- or oligosaccharide moieties to proteins, collectively called protein glycosylation, is one of the most prevalent posttranslational modifications (PTMs). Protein glycosylation was once considered as the event only reserved in eukaryotic systems. However, recent discoveries have shown that protein glycosylation is also a common phenomenon in some microorganisms, including bacteria, archaea, and fungi.70,71 In contrast to most other PTMs, such as phosphorylation, that usually involve a simple functional group transfer to one or a handful of amino acid residues, glycosylation can be much more complex and structurally and functionally diverse.1,72 So far, over 40 different types of the sugar-amino acid junctions have been identified that involve at least 8 different amino acid residues and 13 different proximal monosaccharides.73,74 While microorganisms are found to have diverse rare monosaccharide units in secondary metabolites, surprisingly only a dozen or so common monosaccharides are present in typical eukaryotic glycoproteins (Figure 1). Nevertheless, the limited numbers of building blocks can still form incredibly diverse structures due to the huge possibilities of linkage types, anomeric stereochemistry, and/or additional noncarbohydrate decorations of the sugar chains.
Figure 1.

Structures and symbols of common monosaccharide units found in eukaryotic systems.
For most glycoproteins found in eukaryotic systems, the glycans are attached to the proteins through three major types of linkages. One is the N-linked glycosylation, in which the oligosaccharide (N-glycan) is attached to the amide side chain of an asparagine residue in a consensus (Asn-Xaa-Ser/Thr) sequence, where Xaa could be any amino acids except proline, through the pGlcNAcβ1-Asn linkage; another is the O-linked glycosylation, where the sugar is linked to the hydroxyl group of a Ser or Thr residue in the protein backbone. The O-glycosylation is represented by the mucin type O-glycoproteins in which O-glycans are attached to protein through a GalNAcα1-Ser/Thr linkage and by proteoglycans in which the large glycosaminoglycans are attached to the proteins via a conserved GlcAα1,3-Galβ1,3-Galβ1,4-Xylβ1-Ser linkage. On the other hand, many proteins are anchored to the cell surface through a special glycosyl phosphatidylinositol (GPI)-anchor that locates the proteins at the cell surface for functions. In addition to the attachment of complex N- or O-glycans or GPI anchors, glycosylation with even a simple monosaccharide moiety has also been found to play important roles as represented by the O-GlcNAc glycosylation of many nuclear and cytoplasmic proteins in modulating signal transduction75,76 and the O-fucosylation and O-mannosylation in development.3 Typical structures of the glycans and glycan-protein linkages found in mammalian systems are depicted in Figure 2. It should be pointed out that each class of glycoproteins can have huge variations in the glycan structures attached. For example, the N-glycoproteins can be further categorized into high-mannose type, complex type, and hybrid type depending on the nature of the pendant N-glycans.
Figure 2.

Chemical structures and symbol representations of representative glycan-amino acid linkages found in eukaryotic glycoproteins: (a) complex type N-glycan; (b) core 2 O-glycan; (c) GlcNAc-glycosylation; (d) proteoglycan linkage; (e) linkage strcuture of GPI anchored proteins. In many cases, the monossaccharide units in the above structures can be subjected to further carbohydrate or various noncarbohydrate modifications.
2.2. Biosynthesis of Glycoproteins
Except for the attachment of simple monosaccharide moieties, such as O-GlcNAc glycosylation that involves a simple transfer of a monosaccharide to the protein by the GlcNAc transferase using UDP-GlcNAc as the substrate, the biosynthesis of typical N- and O-glycoproteins often consists of multiple steps involving a large panel of enzymes such as glycosyltransferases, glycosidases, and various carbohydrate modifying enzymes. For example, the biosynthesis of eukaryotic N-glycoproteins involves dozens of steps in two cellular compartments: the ER and the Golgi (Figure 3). First a large oligosaccharide precursor, Glc3Man9GlcNAc2, is assembled on a lipid (dolichol) carrier at the ER membrane that involves a large panel of glycosyl-transferases. Then the dolichol-linked oligosaccharide is transferred by a multisubunit oligosaccharyltransferase (OST) to the asparagine (Asn) side chain in a consensus sequence Asn-Xaa-Ser/Thr of a nascent polypeptide on the ribosome. Then the precursor is processed to a unique monoglucosylated glycoform (Glc1Man9GlcNAc2) that is a key intermediate involved in the calnexin/calreticulin chaperone-mediated protein folding in protein quality control. Once correctly folded, the precursor is trimmed further to Man8GlcNAc2-protein in the lumen of the ER and then translocated to the Golgi apparatus for further processing to produce different glycoforms, such as high-mannose, complex, and hybrid type glycoforms (Figure 3). The processing of the N-glycans is carried out by various glycosidases and glycosyltransferases in a species-, cell type-, protein-, and/or site-specific manner. Many of the enzymes are differentially expressed and sensitive to the physiological state of the cells, resulting in the production of diverse and heterogeneous glycoprotein glycoforms.
Figure 3.

Biosynthesis of eukaryotic N-glycoproteins.
In contrast to N-glycosylation that begins with the assembly of a large oligosaccharide precursor structure, the biosynthesis of O-glycans occurs in a stepwise manner in the Golgi apparatus, starting with the attachment of the first sugar, N-acetylgalactos-amine (GalNAc) to the hydroxyl group of a Ser or Thr residue in an a-glycosidic linkage under the catalysis of a polypeptide:- GalNAc transferase (ppGalNAcT). Then the sugar chain is further extended by sequential additions of monosaccharides under respective glycosyltransferase to form various O-glycan core structures (Figure 4). Although different ppGalNAc transferases show some preference to polypeptide sequence for attachment of the GalNAc moiety, there is no consensus sequence for O-glycosylation. All the enzymes involved in the N- and O-glycan biosynthesis can be explored for synthetic purposes. Recently, Moremen and co-workers have constructed an expression vector library encoding most of the known human carbohydrate enzymes, including glycosyltransferases, glycoside hydrolases, and sulfotransferases, as well as other glycan-modifying enzymes.77 This library provides a highly valuable resource for recombinant enzyme production useful for chemoenzymatic synthesis of oligosaccharides and glycoproteins.
Figure 4.

Biosynthesis of O-glycoproteins with typical O-glycan core structures.
2.3. Major Approaches for the Synthesis of Homogeneous Glycoproteins
Natural and recombinant glycoproteins are usually produced as mixtures of glycoforms that possess the same protein backbone but differ in the pendent glycans, from which pure glycoforms are extremely difficult to isolate. While the genetic approaches via glycosylation pathway engineering in different host expression systems have been able to produce certain controlled glycoforms of glycoproteins,63–67,78–84 the glycoforms that can be achieved by genetic approaches are limited and, in many cases, it is still difficult to obtain pure glycoforms through recombinant production. On the other hand, the chemical and chemoenzymatic synthesis of a given homogeneous glycoprotein usually has to deal with three major tasks: the production of the polypeptide or protein backbone; the preparation or building up of the oligosaccharide/glycan component; and the conjugation of the glycan to the protein/polypeptide backbone.
The past decade has witnessed tremendous progress in the method development for producing glycan-defined glycoproteins. Major synthetic strategies that are promising for the construction of homogeneous glycoproteins are listed Figure 5. These include the native chemical ligation of preassembled polypeptides and glycopeptides (Figure 5a); the chemoselective ligation of tagged proteins and activated glycans (Figure 5b); the endoglycosidase-catalyzed convergent glycosylation for N-glycoprotein synthesis and glycan remodeling (Figure 5c); the direct enzymatic glycosylation of intact proteins (Figure 5d); and the posttranslational glycan modifications of glycoproteins (Figure 5e).
Figure 5.
Major approaches for synthesizing homogeneous glycoproteins.
The total chemical synthesis ofglycoproteins involving native chemical ligation, as well as the “tag and modify” strategy (depicted in Figure 5a and Figure 5b) has been the focus of a number of excellent accounts.26,33,36,38,41,43,47,85 Indeed, the application and further development of the native chemical ligation concept, including expressed protein ligation (EPL), for ligating large glycopeptides and polypeptides have now made it possible to construct some large and complex intact glycoproteins such as the glycoprotein hormone α- and β- subunits,86,87 glycosylated human interferon-β,88 the glycosylated ribonuclease,89,90 and the fully glycosylated human erythropoietin (EPO).91–95 It should be pointed out that native chemical ligation involving large glycopeptides is not trivial, often leading to low yield, particularly when the bulky glycans are near the ligation sites. This was exemplified by the total synthesis of EPO, where the NCL protocol that was worked out for making the truncated EPO glycoform actually failed to produce the fully glycosylated EPO glycoform, which eventually required a new design of the synthetic scheme.92 In parallel, enzymatic and chemoenzymatic synthesis is emerging as an attractive approach that complements and expands the chemical methods for constructing homogeneous glycoproteins. For example, the discovery of an array of endoglycosynthases derived from the endo-β-N-acetylglucosaminidases has enabled a highly convergent synthesis of large glycopeptide and glycoproteins, and the combined use of the wild type endoglycosidases (for deglycosylation) and the mutant enzymes (for subsequent attachment of a large N-glycan en bloc) has been particularly useful for glycan remodeling of intact natural and recombinant glycoproteins including therapeutic antibodies (Figure 5c).29–31 Moreover, the direct enzymatic glycosylation of proteins is emerging as a practical approach to making homogeneous glycoproteins, thanks to the structural analysis and substrate specificity studies of related enzymes, including the bacterial oligosaccharyltransferase (PglB) that can transfer both N- and O-glycans and the N-glycosyltransferase (NGT) that can transfer a monosaccharide moiety to a consensus N-glycosylation site (Figure 5d).96–101 On the other hand, postglycosylational trimming and remodeling of glycoproteins, as depicted in Figure 5e, provides another chemoenzymatic approach to novel homogeneous glycoforms. In contrast to the above-mentioned endoglycosidase-catalyzed glycan remodeling that involves deglycosylation and endoglycosidase-catalyzed transglycosylation,29–31 glycosyltransferases can also be used to extend the sugar chain to give different glycoprotein glycoforms. As an example, Warnock and co-workers reported the in vitro enzymatic galactosylation of human serum IgG that contains truncated Fc glycans (mixtures of G0F, G1F, and G2F glycoforms) into mainly a homogeneous G2F glycoform of the antibody using a recombinant bovine galactosyltransferase (GalT).102 Under an optimized condition of substrate/enzyme concentrations, the human serum IgG was enzymatically remodeled to the G2F glycoform on a kilogram scale with a purity of 98% for the neutral fraction of Fc N-glycans. As another class example, Wong and co-workers demonstrated the glycan remodeling of bovine ribonuclease (RNase) B (a heterogeneous glycoprotein containing Man5-9GlcNAc2 high-mannose gly-cans) to a homogeneous glycoform carrying a novel N-linked sialyl Lewis X moiety.103 This was achieved by deglycosylation of RNase B with Endo-H, followed by sugar chain elongation by sequence additions of galactose, sialic acid, and fUcose using β1,4-galactosyltransferase, α2,3-sialyltransferase, and α1,3-fuco-syltransferase, respectively (Figure 5e). While diverse glyco-forms could be built up using different combinations of glycosyltransferases, a limitation of this approach is that sequential sugar chain extension by a combined use of several glycosyltransferases could not guarantee the homogeneity of the end product, as incomplete transformation at any of the enzymatic steps could end up with mixtures of truncated glycoforms.
In addition to direct enzymatic glycosylation of proteins and enzymatic sugar chain elongation, some other enzymes such as transpeptidases and transglutaminases have been explored for “native ligation” between preassembled peptides, glycopeptides, and/or oligosaccharides to make novel homogeneous glycopep-tide/glycoprotein conjugates.60,104 Sortases are a class of membrane-bound transpeptidases found in Gram-positive bacteria that are responsible for covalent anchoring of cell surface proteins to bacterial cell walls. Sortases such as the sortase A (SrtA) from Staphylococcus aureus recognize a conserved pentapeptide signal, LPXTG (where X is a variable amino acid), at the C-terminus of the target protein, that hydrolyzes the peptide bond between the T and G residues to form an enzyme thioester intermediate, and finally transfer the protein acyl group to the N-terminus of the oligoglycine side chain of cell wall peptidoglycan, resulting in the conjugation of the target protein to the cell wall. Interestingly, SrtA was found to have very promiscuous substrate recognition and was able to take various molecules with a flexible primary amine group as the acceptor substrate, making the SrtA catalyzed ligation a very promising method for site-specific bioconjugation. For example, Guo and co-workers have explored the SrtA-catalyzed trans-peptidation for chemoenzymatic synthesis of various glyco-sylphosphatidylinositol (GPI)-linked peptide and proteins,105–107 macrocyclic peptides and glycopeptides,108 and liposome-conjugated peptides;109 Roy and co-workers used the SrtA catalyzed reaction to ligate polypeptide and aminosugars to synthesize novel glycopeptides;110 Nishimura and co-workers synthesized large MUC1 type glycopeptides carrying O-glycan and N-glycan chains through a SrtA-mediated ligation between two glycopeptide fragments carrying the sorting signal sequence LPKTGLR and an Gly-Gly moiety, respectively;111 and Ploegh and co-workers successfully used a SrtA-catalyzed trans-peptidation reaction between a class II MHC-binding single domain antibody fragment (VHH7) and a synthetic MUC1-(Tn) fragment to create a two-component glycopeptide vaccine that could elicit a strong MUC1(Tn)-specific immune response in mice.112 On the other hand, microbial transglutaminase also had a broad acceptor substrate specificity and has been used for the chemoenzymatic synthesis of neoglycopeptides.113–115
Taken together, recent developments in chemical and chemoenzymatic methods have made it possible to obtain various homogeneous glycoprotein glycoforms. Further optimization of the existing methods, as well as continuous efforts in exploring new approaches, will provide efficient and new tools for making structurally well-defined glycoproteins for basic research and therapeutic explorations.
3. GLYCOSIDASE-CATALYZED TRANSGLYCOSYLATION AND THE GLYCOSYNTHASE CONCEPT
One unique feature of enzymatic glycosylations is their precise control of the anomeric configuration and regiochemistry without the need of protecting groups. Both glycosyltransferases and glycosidases have been vigorously studied and explored for synthetic purposes.28,116 Glycosyltransferases are the natural enzymes for constructing glycosidic bonds. However, a broad application of glycosyltransferases for practical and large scale synthesis ofglycoconjugates remains to be fulfilled because most membrane-associated glycosyltransferases are not easily available (not stable and difficult to express in most cases), have stringent substrate specificity, and usually require expensive sugar nucleotides (the Leloir pathway) or sugar phosphate (the non-Leloir pathway) as the donor substrates. In contrast, glycosidase-catalyzed glycosylation, usually in a transglycosylation mode, has several advantages, including the usually easy access to the enzymes, the use of readily available donor substrates, and the relaxed substrate specificity for acceptors in general. The typical mechanisms of glycosidases for hydrolysis and transglycosylation, as well as the mechanism-based approaches to generating synthetically more efficient glyco-synthase mutants are discussed in this section.
3.1. Common Mechanisms of Glycosidase-Catalyzed Hydrolysis and Transglycosylation
Glycoside hydrolase (GH), commonly called glycosidase, is a large group of enzymes, found in essentially all kingdoms of life. Naturally glycosidases are responsible for hydrolyzing distinct glycosidic bonds in glycans and glycoconjugates. Based on sequence and folding similarity, these enzymes have been classified into 148 families by the end of 2017 in the Carbohydrate-Active Enzyme database (CAZy, http://www.cazy.org/), which is coupled with a comprehensive encyclopedia (CAZYpedia, http://www.cazypedia.org) and has been frequently updated during the last two decades. The common mechanisms of glycosidase-catalyzed hydrolysis and trans-glycosylation are depicted in Figure 6, which were first proposed by Koshland in 1953.117 Most glycosidases undergo either a double displacement (retaining) or single displacement (inverting) catalytic mechanism. For a typical retaining glycosidase, the double displacement is facilitated by two key catalytic residues, a nucleophilic residue (the nucleophile) and a general acid/base residue, which are usually an aspartic or glutamic acid. During a retaining glycosidase-catalyzed reaction, the acid/base residue synergistically protonates the glycosidic oxygen to activate the glycosidic bond to facilitate the attack by the nucleophilic residue, leading to the formation of an enzyme—substrate covalent complex (intermediate). Subsequently the general acid/base residue activates a water molecule to attack the enzyme—substrate complex leading to hydrolysis or activates an alcoholic acceptor leading to the formation of a new glycosidic bond (transglycosylation) (Figure 6a). For an inverting glycosidase, the protonated glycosidic bond is directly replaced by the attack of a water molecule (for hydrolysis) or another acceptor (for transglycosylation) under the activation by the general base residue (Figure 6b). In the case of a substrate-assisted mechanism, the nucleophile comes from the 2-acetamido group to form an oxazolinium ion intermediate, which subsequently undergoes hydrolysis or transglycosylation (Figure 6c).
Figure 6.

Catalytic mechanisms of different types of glycosidases. R1 and R2 could be sugar or other moieties. In the case of the substrate-assisted mechanism, X is an essential residue to facilitate the formation or stability of reaction intermediate and could be an asparagine or aspartic acid residue.
One major drawback in glycosidase-catalyzed transglycosylation is the inherent product hydrolysis. Since the transfer product can also serve as a substrate and be rehydrolyzed by the glycosidase,62,118,119 thus, in using glycosidases for synthetic purposes, usually an excess of an easily synthesized glycoside is used as the donor substrate to drive the reaction to favor product formation (kinetic control).120,121 To overcome this problem, a major breakthrough in recent years is the invention of glycosynthases, a class of novel glycosidase mutants that are devoid of product hydrolysis activity but can use a suitable activated glycosyl donor for promoting glycosidic bond formation. 28,30,56,62,116,122–127
3.2. Development of the Glycosynthase Concept
In 1998, Withers and co-workers reported the first glycosynthase (Abg E358A) by engineering the β-glycosidase from Agrobacterium sp., which was generated by mutation at the nucleophile residue E358128 (Figure 7a). Shortly after this, Planas and co-workers reported the first endoglycosynthase through site-directed mutation of the nucleophile E134 of the retaining 1,3–1,4-β-glucanase from Bacillus licheniformis.129 In addition, Moracci and co-workers described another interesting glycosynthase approach, which used an activated glycosyl species as substrate but rescued the transglycosylation activity of the nonhydrolyzing glycosidase mutant with exogenous sodium formate or azide as an external nucleophile (Figure 7b).124,130,131 Glycosynthases derived from inverting glyco-sidases were also reported and similarly require an activated sugar donor with an opposite anomeric configuration to facilitate transglycosylation (Figure 7c).132–137
Figure 7.

Catalytic mechanisms of glycosynthases. R, R1, and R2 could be sugar or other moieties.
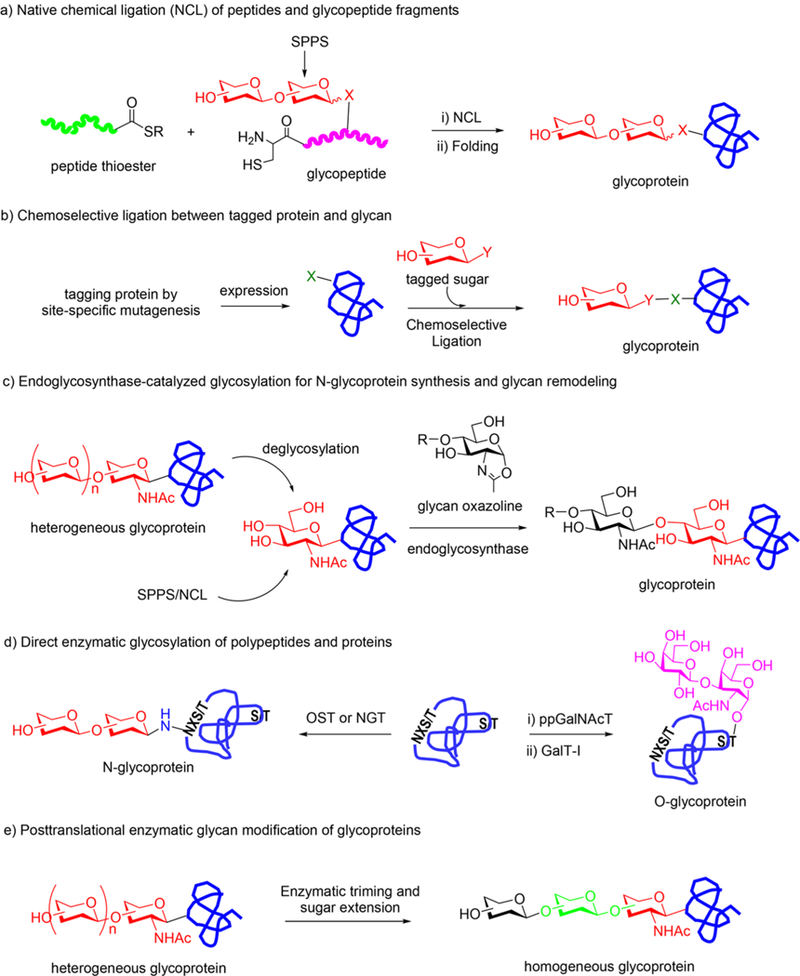
Following these pioneering studies, a number of glycosidases belonging to different glycoside hydrolase families and covering exo-, endo-, retaining, and inverting types have been successfully converted into glycosynthases.134,138–152 As an early example of applying glycosynthases for modifying glycopeptide and neoglycoproteins, Withers and co-workers have shown that the glycosynthases Abg E358G/S were capable of transferring a galactose moiety to solid-phase glycopeptide with high efficiency (>90%) (Scheme 1a).153 Later an improved mutant (2F6) to the parental glycosynthase (Abg E358G) was screened out and applied in a highly efficient synthesis of neoglycoprotein via chemical ligation and glycosynthase-catalyzed transglycosylation (Scheme 1b).154,155
Scheme 1.
Glycosynthase-Catalyzed Modification of Glycopeptide and Glycoprotein
To date, dozens of glycosynthases have been successfully developed to efficiently access valuable oligosaccharides and glycoconjugates.28,30,124 Protein engineering, including directed evolution coupled with various screening methods, has further expanded the scope of novel glycosynthases with either enhanced transglycosylation activity or altered substrate specificity.155–159 Recently, Rovira, Davis, and co-workers reported an unusual engineered glycosidase for efficient synthesis of disaccharide (78–100%), using an unexpected β-galactoside as donor substrate for a β-glycosynthase (SsβG E387Y) derived from Sulfolobus solfataricus.160 A front-side (same face) retaining mechanism was proposed based on structural and computational analysis for this special SNi-like glycosynthase-catalyzed transglycosylation.
Glycosynthases usually require a highly activated species as the glycosyl donor with an anomeric configuration opposite to that of the original substrate. For most β-glycosynthases, the relatively stable a-glycosyl fluoride is often the choice as the glycosyl donor substrate. But α-glycosynthases are less common, partly because the corresponding β-glycosyl fluoride is much less stable with a half-life of ca. 20 min in an aqueous buffer (pH 7).161 While exploiting a more suitable glycosyl donor such as the stable glycosyl azide to evolve α-glycosynthase is of great interest, 146,152,162 the glycoligase concept (see below) provides an attractive alternative approach.30 Considering the huge number of glycosidases characterized and classified to date, it is believed that more useful glycosynthases will be created to fulfill the urgent need in the synthesis of structurally well-defined biologically important glycans and glycoproteins.
3.3. The Glycoligase Concept
In contrast to typical glycosynthase generated by mutation at the critical nucleophilic residue, glycoligase is an alternative glycosidase mutant created by mutation at the general acid/ base residue of a retaining glycosidase (Figure 8). For glycoligase to work as a synthetic enzyme, a highly activated glycosyl donor such as a glycosyl fluoride with the same anomeric configuration as that of the substrate should be provided to allow a fast formation of enzyme—substrate complex without the help from the catalytic acid/base. Then the enzyme—substrate complex would be active enough and provide a suitable microenvironment to accommodate a suitable acceptor to act to form a new glycosidic bond instead of activation of water for hydrolysis.30 For a typical thio-glycoligase, a quickly formed intermediate is attacked by a deprotonated thio-sugar acceptor to give an S-linked glycoside (Figure 8a), while an O-linked product is generated when normal sugar acceptor is applied (Figure 8b).
Figure 8.

Catalytic mechanism of S-glycoligase and O-glycoligase.
In 2003, Withers and co-workers first developed two thio-glycoligases, Abg E171A and Man2A E429A using 2,4-dinitrophenyl β-glycosides as activated substrate to synthesize thio-disaccharides in high yield (around 70%).163 In a novel application, Withers and co-workers described protein modification using the thioglycoligase Abg E170G (Scheme 2).154 A metabolically stable thio-neoglycoprotein was synthesized by coupling chemical ligation and thioglycoligase-catalyzed trans-glycosylation. In 2006 the same group reported the first case of a-thioglycoligases, YicI D482A and MalA D416A, in the synthesis of thio-linked disaccharides using a-XylF and α-GlcF as substrate, respectively.164 In 2010, the same group found that the thioglycoligase YicI D482A can also form O-linked disaccharides.165 Following the pioneering work, several glycoligases derived from different GH families have been successfully created for the synthesis of functional oligosaccharides.154,166–169 Recently, Kim and co-workers developed several potent O-glycoligases in the synthesis of functional α-glycosides, including maltooligosaccharide, 3-O-maltosyl ascorbate, and various aryl glycosides.170–172 In 2017, Matsuo and co-workers reported an endo-a-mannosidase mutant (E407D), which is able to synthesize natural high-mannose-type glycan with low efficiency (4–42%) in a glycoligase manner.173 As the aspartic acid could still play a role of the glutamic acid residue as a general acid/base to promote substrate and/or product hydrolysis, it is possible that the glycosylation activity could be further improved by introducing some inert residues, like alanine, serine, or glycine.
Scheme 2.

Synthesis of Thio-neoglycoprotein by Chemical Ligation and Subsequent Transglycosylation via a Thioglycoligase
Most recently, Wang and co-workers generated a Lactobacillus casei a-fucoligases (AlfC E274A/G/S) that permits efficient direct core-fucosylation of glycopeptides (Scheme 3a) and glycoproteins, including intact antibodies (Scheme 3b).174 The discovery of the new α-fucoligases enables direct core-fucosylation of various mature intact glycoproteins that would not be achievable by the biosynthetic enzyme, FUT8, which has strict substrate specificity.175
Scheme 3.

Glycoligase-Catalyzed Direct Core Fucosylation of Glycopeptide and Glycoprotein
While much effort has been devoted into the development of novel glycosynthases in the last two decades, glycoligase strategy is emerging as an attractive approach to synthesis and modifications of oligosaccharides and glycoproteins, especially for those glycoconjugates containing a-glycosidic linkages.30 So far about ten S- and O-glycoligases have been successfully created, including both α- and β-types. However, the number of currently available glycoligases is still limited and there is no general rule about how to select promising candidates from the vast number of glycosidases in the database. Selection and evolution of those glycosidases with inherent transglycosylation activity could be a good starting point to generate new glycoligases and glycosynthases as well.170,174,176 The development of novel glycosynthase and glycoligases provides exciting new tools for oligosaccharide and glycoprotein synthesis.
3.4. Discovery of a New Class of Endoglycosynthases from endo-β-N-Acetylglucosaminidases (ENGases)
For those glycosidases such as the endo-β-N-acetylglucosami-nidases (ENGases) that proceed in a substrate-assisted mechanism, the conventional approach to generating glyco-synthase by mutating the nucleophilic residue of a retaining glycosidase would not work, as the nucleophile is the 2-acetamido group in the substrate instead of a residue in the enzyme. In 2008, Wang and co-workers reported the first example of ENGase-based glycosynthases by screening a series of Endo-M mutants using a Man9GlcNAc N-glycan oxazoline as the activated donor substrate.177 A special residue, N175, which promotes sugar oxazolinium ion intermediate formation, was identified, and mutation at this critical residue led to mutants (N175A and N175Q) that were devoid of hydrolysis activity but could use the activated glycan oxazoline as substrate for glycosylation (Figure 9a). This approach, involving mutation of a key residue that promotes the oxazolinium ion intermediate formation during the catalysis, has been expanded to several other GH family 85 ENGases, including Endo-A,178 Endo-D,179 and Endo-CC.180 These glycosynthases have been very useful for glycopeptide synthesis and glycoprotein glycan remodeling.31
Figure 9.

Catalytic mechanism of glycosylation via ENGase mutants derivated from distinct GH families. R could be sugar or other moieties. X could be alanine, glutamine, methionine, or histidine residue.
In 2012, Wang and co-workers described glycosynthase mutants (D233A and D233Q) from Endo-S, an endoglycosi-dase from Streptococcus pyogenes, which are particularly useful and specific for Fc antibody glycan remodeling.181 This was the first example of endoglycosynthases derived from the GH18 family ENGases. Later on, Wang and co-workers successfully generated glycosynthases such as the D184 M mutant from Endo-S2, a GH family 18 bacterial endoglycosidase from a serotype of S. pyogenes, that demonstrated much higher efficiency and more relaxed substrate specificity for antibody Fc glycan remodeling.182 More recently, the same research group reported glycosynthase mutants such as D165A from Endo-F3, another GH18 ENGase, which could transfer triantennary N-glycans to fucosylated GlcNAc-peptides and intact deglycosylated antibodies (Figure 9b).183
Related to the strategy to mutate the residue critical for promoting oxazolinium ion formation during catalysis, Fairbanks and co-workers reported that a different Endo-A mutant could be generated by site-directed mutation at the general acid/ base residue E173 and the resulting E173H and E173Qmutants were found to be capable of using the Man3GlcNAc-oxazoline for glycosylation of an GlcNAc moiety (Figure 9c).184 While the mechanism of the mutants to catalyze the glycosylation remained to be characterized, one possible explanation could be that the E173H or E173Q mutation might weaken the general acidity of this critical residue to reduce its hydrolytic activity on a grand-state glycosidic bond (e.g., the product), but the histidine or glutamate residue could still serve as a generate base to activate the acceptor for glycosylation when sugar oxazoline was used as an activated donor substrate. It would be interesting to see if this special mutation strategy could be extended to other ENGases to generate special glycosynthases. Taken together, the discovery of novel endoglycosynthases capable of using glycan oxazoline as donor substrate for enzymatic glycosylation of GlcNAc-peptides/proteins represents a landmark progress in the field.
4. ENDOGLYCOSIDASES AND ENDOGLYCOSYNTHASES FOR GLYCOPROTEIN SYNTHESIS AND GLYCAN REMODELING
4.1. Transglycosylation Activity of endo-β-N-Acetylglucosaminidases (ENGases)
The endo-β-N-acetylglucosaminidases (ENGases) are a class of endoglycosidases that can remove N-glycans from glycoproteins by hydrolyzing the β1,4-glycosidic bond in the N,N’- diacetylchitobiose core. They belong to the glycoside hydrolases (GH) family 85 or family 18 and are widely distributed in nature, ranging from bacteria, fungi, plants, and animals to humans.185–187 In addition to the inherent hydrolytic activity, some ENGases also possess transglycosylation activity, i.e., the ability to transfer the released oligosaccharide to a suitable acceptor such as a GlcNAc-peptide to form a new glycopeptide or a related glycoconjugate. These include Endo-A from Arthrobacter protophormiae,188–190 Endo-M from Mucor hiema-lis,191 Endo-CE from Caenorhabditis elegans,186 Endo-BH from alkaliphilic Bacillus halodurans C-125,192 Endo-D from Streptococcus pneumoniae,193 Endo-F1/F2/F3 from Flavobacterium meningosepticum,183‘194 Endo-S/S2 from Streptococcus pyogenes, 181,182,195, and Endo-CC from Coprinopsis cinerea.180,196 These enzymes show different substrate specificity in hydrolysis and transglycosylation. For example, Endo-A and Endo-CE are specific for high-mannose type N-glycans, while Endo-M can use both high-mannose type and complex type N-glycans. Table 1 provides a summary of the substrate specificity of various endoglycosidases in hydrolysis and potential transglycosylation.
Table 1.
Substrate Specificity of Endoglycosidases for Hydrolysis of N-Glycans and Glycoproteins
| Enzyme | Organisms | GH family | N-Glycan substrate | Core-fucose hydrolyzed? | Assistant residue | General acid/base | ref |
|---|---|---|---|---|---|---|---|
| Endo-A | Arthrobacter protophormiae | 85 | HM/Hyba | No | N171b | E173b | 197–199 |
| Endo-M | Mucor hiemalis | 85 | HM/Hyb/CTa | No | N175b | E177b | 200 |
| Endo-D | Streptococcus pneumoniae | 85 | truncated HM | Yes | N322 | E324 | 201, 202 |
| Endo-CE | Caenorhabditis elegans | 85 | HM/Hyba | No | N152 | E154 | 186 |
| Endo-BH | Bacillus halodurans | 85 | HM/Hyba | NDc | N170 | E172 | 192 |
| Endo-Om | Ogatea minuta | 85 | HM/Hyb/CTa | No | N194 | E196 | 203, 204 |
| Endo-CC1 | Coprinopsis cinerea | 85 | HM/Hyb/CTa | NDc | N180 | E182 | 180, 196 |
| Endo-CC2 | Coprinopsis cinerea | 85 | HM/Hyb/CTa | NDc | N186 | E188 | 196 |
| Endo-H | Streptomyces plicatus | 18 | HM/Hyba | Yes | D172 | E174 | 205 |
| Endo-F1 | Elizabethkingia meningoseptica | 18 | HM/Hyba | Yes | D180 | E182 | 206 |
| Endo-F2 | Elizabethkingia meningoseptica | 18 | CTa | Yes | D169 | E171 | 194, 206 |
| Endo-F3 | Elizabethkingia meningoseptica | 18 | CT/TCTa | Preferred | D165 | E167 | 194, 206 |
| Endo-BI1 | Bif idobacterium longum subsp. infantis | 18 | HM/Hyb/CTa | Yes | D184 | E186 | 207 |
| Endo-BI2 | Bif idobacterium longum subsp. infantis | 18 | HM/CT | NDc | NDc | NDc | 207 |
| Endo-S | Streptococcus pyogenes | 18 | CT | Yes | D233 | E235 | 195, 208–211 |
| Endo-S2 | Streptococcus pyogenes | 18 | HM/Hyb/CT | Yes | D184 | E186 | 210, 212 |
The abbreviations for N-glycans: HM, high-mannose type; Hyb, hybrid type; CT, biantennary complex type; TCT, triantennary complex type.
The numbering of amino acids refers to the peptide sequence after cleavage of an N-terminal signal peptide as described in the original publications.
Not determined.
In contrast to the method using glycosyltransferases that add monosaccharides one-by-one, the endoglycosidase-catalyzed method is highly convergent, involving the transfer of a large intact oligosaccharide en bloc to a preassembled GlcNAc-polypeptide to form a new glycopeptide in a regio- and stereospecific manner.55,57,213,214 The ENGase-catalyzed glycosylation is emerging as a general chemoenzymatic method for the synthesis of large homogeneous N-glycopeptides and glycoproteins and for glycan remodeling of intact glycoproteins including intact therapeutic antibodies.29–31 Among others, a recent review by Fairbanks gave an excellent account of the application of ENGases for glycopeptide and glycoprotein synthesis.31 Here we provide an overview of the development of the ENGase-based chemoenzymatic method with a focus on recent examples.
In 1995, Takegawa and co-workers reported the first example of using the Endo-A’s transglycosylation activity to remodel the glycans on bovine ribonuclease B, a natural glycoprotein of 124 amino acid carrying a heterogeneous high-mannose type N-glycan at the Asn-34.215 While the yield was estimated to be less than 5% by SDS-PAGE analysis, this study provided proof-of-concept data showing that it is possible to use the ENGase-catalyzed hydrolysis and transglycosylation to prepare neoglycoproteins via a glycan remodeling approach. The bacterial enzyme Endo-A and fungus enzyme Endo-M were the two ENGases extensively used for N-glycopeptide synthesis. In 1996, Inazu, Yamamoto, and co-workers reported the synthesis of an N-glycopeptide via the Endo-M catalyzed transglycosylation using a synthetic GlcNAc-peptide as the acceptor and a complex type Asn-linked N-glycan as the donor substrate.216 In the same year, Lee and co-workers reported the first example of Endo-A catalyzed synthesis of a C-linked glycopeptide as an inhibitor for peptide N-glycanase F and A (PNGase F and A) (by strict definition, they are N-glycosylamidases).217,218 Since then, a number of complex glycopeptides have been synthesized by the ENGase-catalyzed transglycosylation. These include glycosylated calcitonin,219,220 glycosylated fragments of the nicotinic acetylcholine receptor (nAChR),221 glycosylated substance P,222 large HIV-1 envelope glycoprotein fragments,223,224 and homogeneous CD52 antigens carrying full-size high-mannose and complex type N-glycans.225 This chemoenzymatic method was also used for the synthesis of multivalent glyco-polymers,226,227 natural and neo-glycopro-teins,215,228,229 glycosylated cyclodextrins,230 and glycosylated insulin.231
Despite these useful synthetic applications, a broader application of the ENGase-catalyzed transglycosylation has been hampered by several limitations, including product hydrolysis by the enzyme, the limitation to the use of only natural N-glycans/glycopeptides as donor substrates that are difficult to obtain in pure forms, and the relatively low transglycosylation yield (5–20%). Thanks to the concerted efforts from different research groups worldwide, remarkable progresses have been made in the past decade that have adequately addressed the major limitations of this chemo-enzymatic method. These include the exploration of synthetic glycan oxazolines as donor substrates for transglycosylation, which expands the substrate availability and dramatically enhances the glycosylation efficiency; the discovery of new enzymes with distinct substrate specificity for transglycosylation; and the generation of novel glycosynthase mutants that are devoid of product hydrolysis activity but can take the activated glycan oxazolines as donor substrate for enzymatic glycosylation.
4.2. Exploration of Sugar Oxazoline as Activated Substrates for ENGase-Catalyzed Glycosylation
In analogy to some GH family 18 chitinases232–234 and some GH family 20 β-N-acetylhexosaminidases235,236 that proceed via a substrate-assisted mechanism, ENGases were also implicated to follow the same substrate-assisted mechanism for catalyzing hydrolysis and transglycosylation via a sugar oxazolinium ion intermediate involving the participation of the 2-acetamido group. There were precedents that synthetic disaccharide oxazolines corresponding to polysaccharide subunits could serve as a substrate for enzymatic polymerization to polysaccharides by chitinases and related enzymes with trans-glycosylation activities.237–240 Logically, it would be interesting to examine if synthetic oligosaccharide oxazoline corresponding to N-glycans could serve as substrate of ENGases for transglycosylation. Nevertheless, an initial attempt to test this feasibility using wild type Endo-A and a semisynthesized Man6GlcNAc oxazoline failed to yield any detectable trans-glycosylation product, probably due to the very fast hydrolysis of the Man6GlcNAc oxazoline and/or the Man6GlcNAc2-peptide (if any) by the wild type Endo-A.213
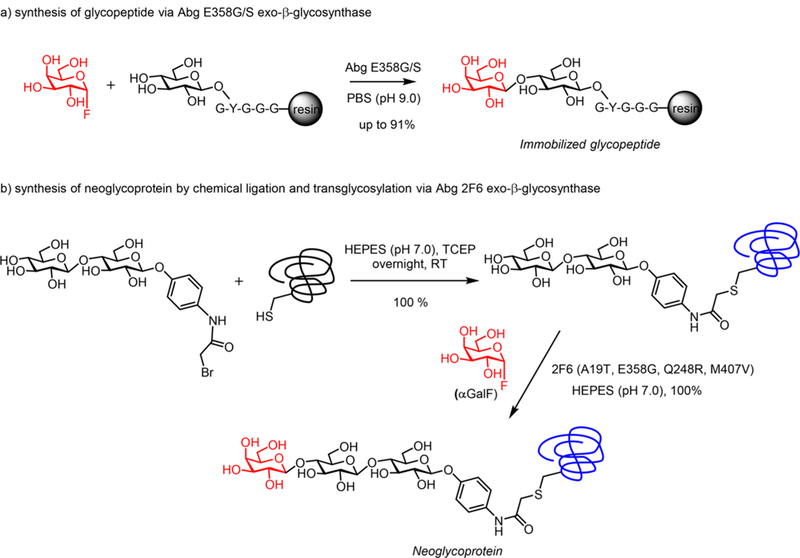
In 2001, Shoda and co-workers first reported that Endo-A and Endo-M could use a simple disaccharide oxazoline derived from Manβ1,4-GlcNAc for transglycosylation to form a p-nitrophenyl trisaccharide derivative, which was designed for the purpose of detecting the transglycosylation activity.241 The yield was about 50% when 2-fold excess of the sugar oxazoline was used, but it was found that the trisaccharide product Manβ1,4-GlcNAcβ1,4-GlcNAc-pNP was not hydrolyzed by Endo-A or Endo-M. This seminal study suggested that an activated sugar oxazoline corresponding to a truncated N-glycan could be recognized by the enzyme for transglycosylation but the resulting ground-state product would be resistant to enzymatic hydrolysis because of the truncated structures. In 2005, Wang and co-workers first reported that synthetic sugar oxazolines could serve as excellent donor substrates for ENGase-catalyzed N-glycopeptide synthesis.242,243 They demonstrated that using a GlcNAc-containing 34-mer peptide of HIV-1 gp41 as an acceptor, which was readily prepared by automated solid-phase peptide synthesis, the synthetic Man3GlcNAc oxazoline could serve as an excellent donor substrate for the Endo-A catalyzed glycosylation to give the corresponding N-glycopeptide carrying the Man3GlcNAc2 pentasaccharide core in 75% yield, when only 2-fold excess of the donor substrate was used (Scheme 4a). The enzymatic reaction proceeded quickly under very mild conditions (phosphate buffer, pH 6.5, 23 °C). It was found that the resulting Man3GlcNAc2-peptide product (ground state) could be hydrolyzed only very slowly by Endo-A under the reaction conditions, while the Man3GlcNAc-oxazoline was highly active for the Endo-A catalyzed glycosylation, thus permitting the accumulation of the transglycosylation product.
Scheme 4.
Sugar Oxazolines as Donor Substrates for ENGase-Catalyzed Synthesis of N-Glycopeptides
As another example, a cyclic 47-mer HIV-1 glycopeptide derived from the V3 domain of gp120 carrying two core N-linked pentasaccharide was also efficiently synthesized by the chemoenzymatic method.243 The synthesis was achieved by a concise two-step approach: the solid-phase synthesis of the 47-mer polypeptide that contains two GlcNAc moieties and the subsequent high-yield double glycosylation catalyzed by Endo-A, with Man3GlcNAc-oxazoline as the donor substrate (Scheme 4b). The synthetic V3 glycopeptides were successfully used for probing the effects of glycosylation on the global conformations of the V3 domain and on the protection of the polypeptide against protease digestion.243 Taken together, the results suggest that the use of synthetic sugar oxazolines as highly activated substrates not only expanded the substrate availability but also led to substantial enhancement of the overall synthetic efficiency for ENGase-catalyzed transglycosylation, permitting a high-yield assembly of large glycopeptides that are otherwise difficult to obtain by pure chemical methods.
Subsequently Wang and co-workers evaluated the donor substrate structural requirement in the enzymatic trans-glycosylation using an array of truncated and modified sugar oxazolines, including LacNAc-oxazoline, N,N’-diacetylchito-biose-oxazoline, Glcβ1,4-GlcNAc-oxazoline, and 6’-O-benzyl-Manβ1,4-GlcNAc-oxazoline, and tested their activity toward Endo-A.244 It appeared that the Manβ1,4-GlcNAc-oxazoline was the minimum structure recognized by Endo-A for an efficient transglycosylation. Interestingly, Endo-A was found to tolerate selective modification on the β-mannose moiety, such as an attachment of a tag or an additional sugar residue at the 6’-position, without significant loss of substrate activity.
Independently, Fairbanks and co-workers made key contributions to the early development of the sugar oxazoline based chemoenzymatic method.31,245–249 The group synthesized a series of truncated and modified N-glycan oxazolines and tested their activity for glycosylation with Endo-M, the fungus ENGase. A very interesting finding was that the glc-containing disaccharide oxazoline, Glcβ1,4-GlcNAc-oxazoline, showed a residual activity in Endo-M catalyzed transglycosylation, giving a 5% yield. However, when the disaccharide was extended to a trisaccharide derivative with an additional α1,3-linked mannosyl residue attached to the glucose moiety, the resulting trisaccharide oxazoline became an excellent substrate for Endo-M, giving a 91% yield of the glycosylation product, without product hydrolysis.
More recently, Matsuo and co-workers further demonstrated the structural requirement of the sugar oxazoline donors in the Endo-M catalyzed transglycosylation.250 A series of tetrasac-charide oxazoline derivatives was synthesized in which the innermost β-mannose moiety was replaced with other monosaccharides including β-glucose, β-galactose, and β-talose, as well as a 4-alkynylated derivative. The transglycosylation activity ofEndo-M and its two mutants, N175Qand N175A, on these tetrasaccharide donors was tested with p-nitrophenyl N-acetylglucosaminide (GlcNAc-pNP) as the acceptor. It was found that the β-mannose moiety could be replaced by glucose and talose without significant loss of the activity; however, substitution of the β-mannose moiety with a galactose resulted in almost abolishment of the acceptor activity to Endo-M and its mutants. Interestingly, the tetrasaccharide oxazoline could tolerate modification at the 4-hydroxy group of the β-mannose moiety with an alkyl functional group, which opens an avenue for further modification by click chemistry.
On the other hand, several studies were performed to evaluate the acceptor substrate specificity in the ENGase-catalyzed transglycosylation. Early study by Takegawa and co-workers demonstrated that both GlcNAc and glucose (Glc) were excellent substrates; mannose and some disaccharides such as N;N’-diacetylchitobiose and gentiobiose could also serve as acceptor at higher concentrations; but galactose could not act as an acceptor for the Endo-A catalyzed transglycosylation.189 Lee and co-workers further demonstrated that using Man9GlcNAc2Asn as the donor, L-fucose could serve as an acceptor for Endo-A catalyzed transglycosylation to form a novel Man9GlcNAc-Fuc conjugate in which the Man9GlcNAc was determined to be β1,2-linked to the L-fucose moiety.251 Later on, Inazu and co-workers reported that the 1,3-diol structure from the 4- to 6-hydroxy functions of GlcNAc appeared important as a substrate for the Endo-M catalyzed trans-glycosylation. Interestingly even an acyclic 1,3-diol structure consisting ofprimary and secondary hydroxyl groups could serve as an acceptor substrate.252 More recently, Manabe and coworkers studied the acceptor substrate specificity of Endo-CC N180H and Endo-M N175Qmutants using a range of modified GlcNAc and related derivatives.253 It was revealed that both mutants could accept compounds with a 1,3-diol structure consisting of primary and secondary hydroxyl groups for transglycosylation when a sialoglycopeptide (SGP) was used as the donor substrate. Interestingly, it was found that the Endo-CC N180H was able to take deglycosylated antibody as an acceptor for Fc glycan remodeling while the Endo-M N175Q mutant could not.253
Realizing the broad acceptor substrate specificity, Wang and co-workers tested an array of natural products containing a terminal glucose or GlcNAc moiety and found that those compounds could serve as an acceptor for the Endo-A catalyzed glycosylation with sugar oxazolines as donors to give a class of new glycosylated natural products carrying a novel glucose-linked N-glycan.254 Moreover, the ENGase-catalyzed trans-glycosylation was applied to the GlcNAc-terminated Asn-linked N-glycan core as an acceptor, leading to the synthesis of a class of novel N-glycan clusters that were successfully used for studying the ligand specificity of lectins.255 Furthermore, Wang and coworkers designed and synthesized a simple disaccharide oxazoline, Glcβ1,4-GlcNAc-oxazoline, and demonstrated that the glucose terminated sugar oxazoline could serve as both an acceptor and a donor substrate for Endo-A catalyzed reaction, leading to polymerization to form a novel hybrid of chitin and cellulose oligomers.256 This was the first example of an ENGase-catalyzed polymerization.
In 2006, Wang and co-workers first extended the sugar oxazoline method to glycosylation remodeling of glycoproteins, using bovine ribonuclease B (RNase B) as a model glycoprotein system.257 RNase B is a natural glycoprotein consisting of 124 amino acid residues. It carries a heterogeneous high-mannose type N-glycan (Man5—9GlcNAc2) at the Asn-34 site. In this approach, the heterogeneous N-glycan in RNase B was removed by enzymatic deglycosylation with Endo-H, leaving only the innermost GlcNAc being still attached at the Asn-34. Then a homogeneous N-glycan was installed at the glycosylation site by enzymatic glycosylation from a corresponding glycan oxazoline (Scheme 5). The initial test with Man3GlcNAc-oxazoline showed that Endo-A could efficiently glycosylate the GlcNAc-RNase to give a homogeneous RNase glycoform, Man3GlcNAc2-RNase, in 82% yield without the need of denaturing the protein (phosphate buffer, pH 6.5, 23 °C). A synthetic mimic of a complex type glycan oxazoline carrying two terminal β-galactose moieties was synthesized and found to also serve as excellent substrate for glycosylation remodeling of RNase B.257 Subsequent work showed that various truncated and modified N-glycans could be introduced by enzymatic glycosylation with respective synthetic glycan oxazolines via the glycan remodeling approach (Scheme 5a).258 Of particular interest was the site-specific introduction of an azide-tagged core pentasaccharide into glycoprotein, which permitted further site-specific modifications of the glycoproteins through click chemistry involving the azide-alkyne 1,3-dipolar cycloaddition.259–261 For example, Cu(I)-catalyzed click chemistry between an alkyne-containing α-Gal epitope and the azide-tagged RNase resulted in simultaneous introduction of two copies of a-Gal epitopes into RNase in excellent yield (Scheme 5b). The resulting glycoprotein could be specifically recognized by anti-a-Gal antibodies in human serum.258
Scheme 5.

Chemoenzymatic Synthesis of Homogeneous Glycoproteins by ENGase-Catalyzed Glcosylation Remodeling with Sugar Oxazolines
While the wild type ENGases were efficient to catalyze the transglycosylation with truncated N-glycan oxazolines due to the diminished hydrolysis activity of the enzymes toward the truncated N-glycan oxazolines and the corresponding glycoproteins, the use of larger natural sugar oxazolines corresponding to natural high-mannose or complex type N-glycans could be problematic, as the products would turn out to be excellent substrates of Endo-A or Endo-M and could be hydrolyzed quickly by the wild type enzymes.247 As discussed in the following sections, this problem was adequately addressed by the generation of novel endoglycosidase mutants (glycosyn-thases) that lack the product hydrolysis activity but can still use the activated sugar oxazoline as donor substrate for glycosylation.
4.3. Generation of ENGase-Based Glycosynthases with Diminished Product Hydrolysis Activity for Glycosylation
Despite the fact that the wild type enzymes could take modified N-glycan oxazoline as donor substrates for an efficient synthesis of various glycopeptides and glycoproteins carrying truncated and/or unnaturally modified N-glycans, the method would be problematic for the synthesis of natural glycoproteins carrying full-size natural N-glycans, as the wild type enzymes are expected to hydrolyze the product quickly. A potential solution to this problem is to generate novel glycosynthase mutants that could use glycan oxazoline as substrate for transglycosylation but lack product hydrolysis activity. However, the conventional approach116,122,128,262 to generating glycosynthases by mutating the key nucleophilic residue in a retaining glycosidase would not work, as the ENGase-catalyzed reaction proceeds via a substrate-assisted mechanism where the nucleophile is the 2-acetamido group in the substrate. The first example of ENGase-based glycosynthases was reported in 2008 by Wang, Yamamoto, and co-workers.177 By screening a series of mutants generated around the putative catalytic sites of Endo-M against a synthetic Man9GlcNAc oxazoline corresponding to the natural high-mannose N-glycan, they identified a mutant, N175A, that was able to catalyze the transglycosylation with the highly activated sugar oxazoline Man9GlcNAc-oxazoline but did not hydrolyze the glycopeptide product.177 It was proposed that the N175 residue functioned to orientate and promote the oxazoline ring formation during hydrolysis. Thus, mutation of this critical residue to an alanine rendered the enzyme incompetent of hydrolysis, but it could still accommodate the preassembled sugar oxazoline as a substrate for transglycosylation when a GlcNAc-peptide acceptor is available. Following this strategy, an analogous glycosynthase, EndoA-N171A, was generated from enzyme Endo-A, which could efficiently use high-mannose type glycan oxazolines for transglycosylation with diminished product hydrolytic activity.178 In addition, the Endo-M mutant was also efficient to transform complex type N-glycan oxazoline for glycoprotein synthesis (Scheme 6).178 Subsequent systematic mutagenesis on the N175 residue of Endo-M led to the discovery of additional glycosynthase mutants.263 In particular, one of the mutants, Endo-M N175Q, was identified to be much more efficient than other mutants for catalyzing trans-glycosylation to form homogeneous complex glycopeptides.263 This approach was also applied to Endo-D, leading to the discovery of N322A mutant as a glycosynthase that could use truncated high-mannose type N-glycan oxazoline as substrate for Fc N-glycan remodeling.179 More recently, a novel mutant (N180H) was generated from Endo-CC1 enzyme.180 The N180H mutant was capable of transferring biantennary complex type N-glycan, which might serve as a substitute to the Endo-M N175A or N175Qmutant.180
Scheme 6.

Chemoenzymatic Synthesis of Homogenous Glycoforms Ribonuclease B by ENGase-Derived Glycosynthases
In 2012, Davis and co-workers demonstrated that Endo-S, an ENGase from Streptococcus pyogenes of the GH18 family, possessed transglycosylation activity and was able to use Man3GlcNAc oxazoline for glycosylation of deglycosylated human IgG.195 While the yield was not reported, the transglycosylation product was assessed by mass spec analysis of the N-glycans released from the product. Later on it was verified that the wild type Endo-S catalyzed transglycosylation with Man3GlcNAc oxazoline ended up with very low yield, partly due to quick hydrolysis of the Man3GlcNAc oxazoline by the wild type enzyme.264 At about the same time, Wang and co-workers reported the first example of glycosynthases from the GH18 family ENGases. An array of mutants, including EndoS-D233A and D233Q, were generated by site-directed mutagenesis of Endo-S, an ENGase from Streptococcus pyogenes. The mutants were able to efficiently transfer synthetic biantennary complex type N-glycan oxazolines to the deglycosylated intact IgGs without product hydrolysis.181 Later on, Wang and co-workers performed a systematic mutagenesis on the D184 residue of Endo-S2, a GH family 18 bacterial endoglycosidase from a serotype of S. pyogenes. They replaced the D184 with the other 19 natural amino acids and tested the transglycosylation activity of the mutants.182 It was found that a number of mutants showed transglycosylation activity specific for glycosylation of the IgG-Fc domain. One of the mutants, D184M, was identified as the most efficient mutant for catalysis of IgG trans-glycosylation. In comparison, the Endo-S2 glycosynthases are much more efficient than the corresponding Endo-S mutants. Moreover, in contrast to the Endo-S enzyme that is limited to transferring of only biantennary complex type N-glycan on IgG-Fc, the Endo-S2 and its glycosynthase mutants have much more relaxed substrate specificity and are able to act on high-mannose type, complex type, and hybrid type Fc N-glycans, making them the most powerful enzymes so far discovered for Fc glycan remodeling of intact antibodies.182 In addition, Wang and co-workers also generated glycosynthase mutants from Endo-F3, another GH18 ENGase from Flavobacterium meningosepti- cum.183 The mutants (D165A and D165Q) could transfer both bi- and triantennary N-glycans to fucosylated GlcNAc-peptides and intact deglycosylated antibodies. The two mutants are the first glycosynthases capable of transferring triantennary complex type N-glycans. The glycosynthase mutants generated from various ENGases and their donor and acceptor substrate specificity are summarized in Table 2. Additional applications of the glycosynthase mutants for chemoenzymatic synthesis of complex N-glycopeptides and homogeneous glycoproteins are given in the following sections.
Table 2.
Substrate Specificity of Endoglycosynthase Mutants for Glycosylation with Glycan Oxazolines and Acceptors
| Enzyme | Donor (N-glycan oxazoline) | GlcNAc-Acceptor | Core-fucose accepted? | Mutants | ref |
|---|---|---|---|---|---|
| Endo-M | HM/CTa | Peptides/proteins | No | N175Ab | 177, 178, 263, 265 |
| No | N175Qb | ||||
| Yes | N175Q/W251Nb | 266 | |||
| Endo-A | HM/CTa | Peptides/proteins | No | N171Ab | 178,184 |
| No | E173Qb | 28 | |||
| No | E173Hb | 28 | |||
| Endo-D | Truncated HMa | Peptides/proteins | Yes | N322A | 179 |
| Yes | N322Q | ||||
| Endo-S | CTa | Antibodies | Yes | D233A | 181 |
| Yes | D233Q | ||||
| Endo-F3 | CT/TCTa | Peptides/Proteins/antibodies | Required | D165A | 183 |
| Required | D165Q | ||||
| Endo-CC1 | HM/CTa | Peptides/proteins | NDc | N180H | 180, 196 |
| NDc | N180Q | ||||
| Endo-S2 | HM/Hyb/CTa | Antibodies | Yes | D184M | 182 |
| Yes | D184Q | 182 | |||
| Yes | T138Q | 267 |
The abbreviations for N-glycans: HM, high-mannose type; Hyb, hybrid type; CT, biantennary complex type; TCT, triantennary complex type.
The numbering of amino acids refers to the peptide sequence after cleavage of an N-terminal signal peptide as described in the original publications.
Not determined.
In parallel to mutagenesis studies, important structural studies have also been performed on several ENGases from the GH85 and GH18 family. These include the crystal structures ofEndo-A complexed with an oxazoline analog, Man3GlcNAc-thiazo-line;199 the crystal structure of an Endo-A mutant, E173Q;198 the crystal structure of Endo-D from Streptococcus pneumoniae, another family GH85 ENGase;202 and the crystal structure of Endo-S.209 These structural analyses identified key residues involved in the mechanism of the substrate-assisted catalysis and also revealed the unusual folds such as the V-shape structure of the Endo-S enzyme. More recently, the crystal structure of full-length Endo-S in complex with a biantennary complex type N-glycan was solved, revealing how the enzyme interacts with the oligosaccharide.268 Taken together, the structural information obtained in these studies forms a basis for engineering of ENGases aiming to generate new glycosynthase mutants with improved catalysis efficiency or altered substrate specificity.
The discovery of these glycosynthases provides a unique opportunity to construct complex N-glycopeptides and glycoproteins that are otherwise difficult to obtain by other methods. This chemoenzymatic method permits independent manipulations of the sugar and protein portions and thus avoids the long-standing problem of incompatibility of protecting group manipulations on sugar and protein portions in chemical N-glycopeptide synthesis. Coupled with an ENGase-catalyzed enzymatic deglycosylation, the glycosynthase-catalyzed glyco-sylation also provides a unique approach for glycan remodeling of glycoproteins including therapeutic antibodies to produce homogeneous glycoforms valuable for functional studies and potentially for therapeutic applications. Selected examples were provided in the following section to showcase the applications.
4.4. ENGase-Catalyzed Synthesis of Biologically Interesting Glycopeptides and Glycoproteins
4.4.1. Synthesis of CD52 Antigen, Glycosylated CMV Antigen, and Glycosylated Pramlintide.
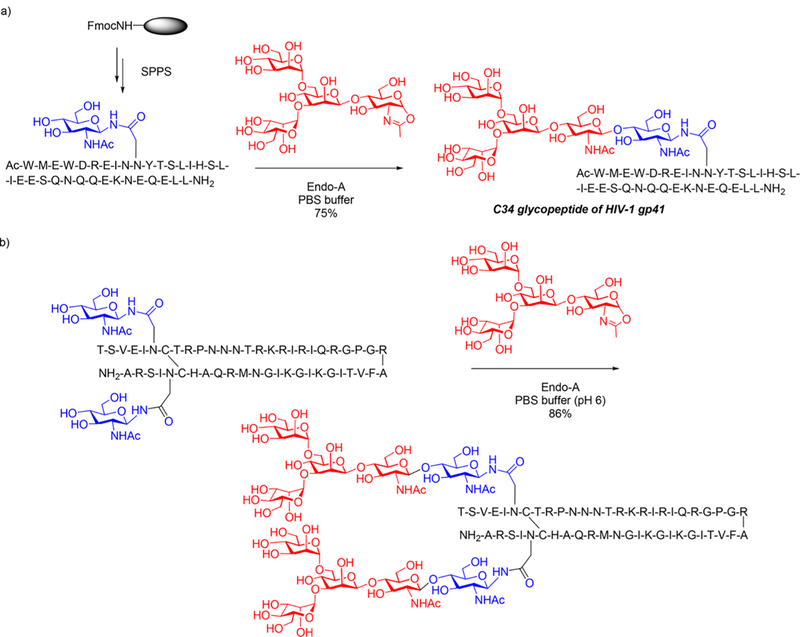
CD52 is a glycosylphosphatidylinositol (GPl)-anchored glycopeptide antigen that is present on sperm cells and human lymphocytes. Sperm-associated CD52 antigen carries both a complex type N-glycan and an O-glycan on the polypeptide backbone. However, isolation of structurally well-defined CD52 antigens from natural sources is extremely difficult as they are present in heterogeneous glycoforms. To obtain homogeneous materials for structural and functional studies, Wang and co-workers reported the first chemoenzymatic synthesis of homogeneous CD52 glycoforms that carry both N- and O-glycans at the predetermined sites269 (Scheme 7). The synthetic strategy consists of three key steps: (1) introduction of monosaccharide primers (Asn-linked GlcNAc and Thr-linked GalNAc) at the natural N- and O-glycosylation sites in the CD52 sequence by SPPS using glyco-amino acids as building blocks; (2) selective extension of the GlcNAc sugar chain by an Endo-M N175A catalyzed transglycosylation with a complex N-glycan, as the GalNAc moiety is not an acceptor for Endo-M N175A catalyzed transglycosylation; and (3) extension of the O-GalNAc sugar chain by T-synthase that specifically recognizes the GalNAc moiety to add a β1,3-linked Gal to the acceptor to afford the target CD52 antigen in high yield (Scheme 7).
Scheme 7.

Chemoenzymatic Synthesis of CD52 Glycopeptide Antigens Containing Both N- and O-Glycans
Core-fucosylated bi- and triantennary complex glycoforms of CD52 were also synthesized by using Endo-F3 and its glycosynthase mutants.183,194 Wang and co-workers first screened ENGases from Flavobacterium meningosepticum of the GH 1S family for transglycosylation and found that they possessed transglycosylation activity and were highly selective for the α1,6-fucosylated GlcNAc acceptor. Thus, reaction of a complex type N-glycan oxazoline and the Fucα1,6GlcNAc-CD52 under the catalysis of wild type Endo-F3 gave the corresponding core-fucosylated CD52 in 55% yield. Recently, Wang and co-workers reported glycosynthase mutants derived from Endo-F3, and the use of the D165A and D165Qmutants in place of the wild type Endo-F3 resulted in the synthesis of core-fucosylated, complex CD52 glycopeptide antigen in essentially quantitative yield when an excess of the donor substrate was used (Scheme 8).183 It should be mentioned that the Endo-F3 mutants could transfer triantennary complex type N-glycan, while other previously reported ENGases and their mutants, including Endo-A, Endo-M, Endo-D, Endo-S, and Endo-S2 could not recognize triantennary N-glycans.
Scheme 8.

Synthesis of CD52 Glycopeptide Carrying Fucosylated Bi- And Triantennary N-Glycan Using Endo-F3 D165A
Fairbanks and co-workers synthesized a series of glycosylated 19-mer peptide antigens derived from a human cytomegalovirus (CMV) by an ENGase-catalyzed approach (Figure 10).270 Subsequent cell based assay indicated that the glycosylation increased the binding of the peptide antigen to antigen presenting cells (APCs), probably through the interaction with the mannose receptor (MR) widely expressed on the APCs. Moreover, an embedded 9-mer peptide epitope was crosspresented by the APCs to a cytotoxic T-lymphocyte clone (CTL, CD8+). Thus, glycosylation with a high-mannose glycan may provide a way to augment the immunogenicity of a peptide vaccine candidate by improved presentation and processing of the antigen.
Figure 10.

Structures of the synthetic glycosylated CMV-peptides derived from a human cytomegalovirus (CMV).
Fairbanks and co-workers also reported the chemoenzymatic synthesis of an array of glycosylated variants of pramlintide, a peptide-based antidiabetes drug.271 The goal of this work was to improve the pharmacokinetic property of the drug, which itself is of low solubility and has a low in vivo half-life. In this study, the E173H mutant of Endo-A and the N175Qmutant of Endo-M were used to introduce the Man3 and a sialylated complex type N-glycan into the peptide from the respective glycan oxazoline as the donor substrate (Scheme 9). Subsequent structure— activity relationship studies indicated that most of the glycosylated pramlintide thus synthesized acted as potent receptor agonists. The results suggest that glycosylation provides a promising method for optimizing the pharmacokinetic properties of pramlintide, which may lead to more efficient agents for treating diabetes and obesity.272
Scheme 9.

Chemoenzymatic Synthesis of Glycopramlintides Using ENGase Glycosynthase Mutants
4.4.2. Synthesis of HIV-1 Glycopeptide Antigens.
The development of the ENGase-based chemoenzymatic methods has made it possible to efficiently synthesize various complex N-glycopeptides. In particular, this chemoenzymatic method has been useful for the construction of various homogeneous HIV-1 glycopeptide antigens to probe the glycan specificity and fine epitope structures of those glycan-reactive broadly neutralizing antibodies.29,273 As an approach to define the epitopes of the broadly HIV-neutralizing antibodies, PG9 and PG16 that target HIV-1 V1 V2 glycopeptide domains, Wang and co-workers designed and synthesized more than 25 homogeneous V1 V2 glycopeptides derived from the gp120 of two HIV-1 strains (CAP45 and ZM109).274 The glycopeptides carried different N-glycans at the conserved glycosylation sites (Asn160 and Asn156 or Asn173), and the varied glycosylation patterns were designed to probe the glycan specificity of the antibodies in antigen recognition. The chemoenzymatic synthesis was summarized in Scheme 10a. Briefly, the synthesis of glycopeptides carrying a single N-glycan was achieved by the synthesis of a common precursor, GlcNAc-peptide by SPPS. Then respective N-glycans were transferred to the GlcNAc-peptide to obtain the respective target glycopeptide.
Scheme 10.

Chemoenzymatic Synthesis of HIV-1 V1 V2 Glycopeptides Carrying Defined N-Glycans
However, the construction of glycopeptides carrying two different N-glycans would be more difficult, as the enzyme-catalyzed glycosylation normally would not show a clear site-selectivity. To address this problem, a limited amount (2.5 mol equiv) of the glycan oxazoline donor was used in the Endo-D N322Q-catalyzed transglycosylation in the first step, which led to the formation of ca. 1:1 mixture of the two monoglycosylated peptide intermediates (ZM-GP10 and ZM-GP11). After careful HPLC separation, the respective monoglycosylated peptide was subjected to the second enzymatic glycosylation with a complex type N-glycan to provide the target glycopeptides carrying two distinct N-glycans274 (Scheme 10b).
The availability of the synthetic V1 V2 glycopeptides with varied patterns of glycosylation permits a detailed surface plasmon resonance (SPR) evaluation of the antigen recognition by antibodies PG9 and PG16. The binding data indicated that a Man5GlcNAc2 glycan at Asn160 was essential for recognition by antibodies PG9 and PG16. Moreover, the SPR binding study further revealed that the presence of a sialylated N-glycan at the secondary glycosylation site (Asn156 or Asn173) significantly enhanced the affinity of the antibodies for the glycopeptide. This study led to the identification of a high affinity V1 V2 glycopeptide antigen that may serve as a valuable component of an HIV-1 vaccine.274
While the glycopeptides carrying two distinct N-glycans were synthesized by the above-described method, the dependence on HPLC separation of the monoglycosylated intermediates was a major limitation of the approach, which is tedious and might not be generally applicable to other glycopeptides. To address this issue, Wang and co-workers recently developed a selective protection group strategy that allows sequential enzymatic additions of different N-glycans in a polypeptide.275 For this purpose, they designed and synthesized two distinguishably protected GlcNAc-Asn building blocks: one carried the acidlabile O-DEIPS groups on the GlcNAc moiety, and the other building block had the base-labile O-acetyl groups on the GlcNAc moiety. After incorporation of the O-DEIPS protected and O-acetyl protected building blocks at the N160 and N173 sites, respectively, in the automated solid phase peptide synthesis (SPPS), the O-DEIPS protected groups were selectively removed during the acid-promoted global peptide deprotection to have a free GlcNAc moiety at the N160 site, to which a Man5 glycan was selectively introduced by an Endo-M N175Q catalyzed glycosylation. Then the O-acetyl protecting groups on the GlcNAc moiety at the N173 site were selectively deprotected by hydrazine treatment, and the resulting free GlcNAc could be elongated to different sugar chains by the second enzymatic glycosylation to generate a library of glycopeptides (Scheme 11). By using this approach, the efficiency of the synthesis of the identified V1 V2 glycopeptide antigens was significantly improved. SPR and ELISA binding analysis were performed to evaluate the affinity of antibodies PG9 and PG16. The results confirmed the essential role of a Man5GlcNAc2 glycan at the N160 for PG9 and PG16 binding. Interestingly, a sialylated N-glycan at the N173 site was found to also be critical for the high affinity binding to the antibodies, which was not revealed by the original crystal structural study of antibody PG9.276
Scheme 11.

Convergent Chemoenzymatic Installation of Two Distinct N-Glycans in the V1 V2 Cyclic Peptide
More recently, Wang and co-workers extended this chemo-enzymatic approach to the synthesis of libraries of HIV-1 V3 glycopeptide antigens for characterizing the neutralizing epitopes of the V3 glycan dependent broadly neutralizing antibodies, including PGT128, 10–1074, and PGT121.277 Using the improved chemoenzymatic method, a systematic synthesis of various homogeneous V3 glycopeptides was achieved, including the V3 glycopeptides derived from the HIV-1 JR-FL strain carrying defined N-glycans at the N332, N301, and N295 sites (Scheme 12). Antibody binding studies revealed that antibody PGT12S could recognize all the glycopeptides with a high-mannose N-glycan at the N332, N301, and/or N295 site, though with relatively low affinity for all of them, suggesting a glycosylation site promiscuity for PGT12S recognition; the PGT121 antibody demonstrated binding specificity for a sialylated N-glycan at the N3G1 site; and antibody 10–1074 was highly specific for glycopeptide carrying a high-mannose N-glycan at the conserved N332 site.277
Scheme 12.
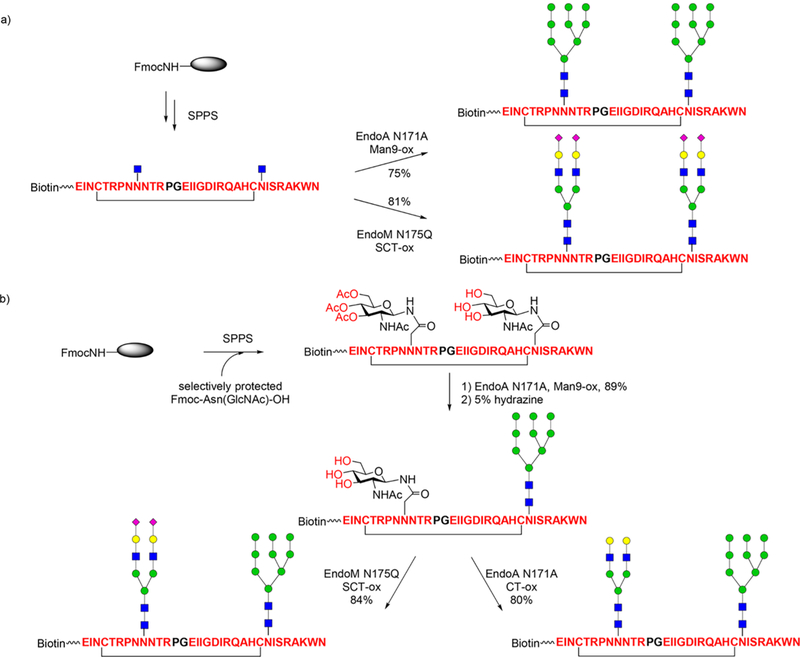
Chemoenzymatic Synthesis of V3 Glycopeptides Carrying Two N-Glycans
On the basis of the glycopeptide mapping results, Wang and co-workers recently designed and synthesized a novel three-component immunogen that consists of a 33-mer V3 glycopeptide epitope, a universal T helper epitope P30, and a lipopeptide (Pam3CSK4) that is a ligand of Toll-like receptor 2 (TLR2).278 A copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition (click chemistry) was applied to assemble the lipopeptide and HIV V3 glycopeptide (Scheme 13). First, an alkyne-tagged V3 glycopeptide was synthesized that carries the conserved Man9GlcNAc2 glycan at the N332 site; then the T cell epitope peptide P3G and the Pam3CSIK. lipopeptide were synthesized by stepwise SPPS using Fmoc chemistry to give the lipopeptide, in which a Lys(N3) residue was placed at the C-terminus of the P30 peptide. Finally, site-specific ligation between the alkyne-tagged V3 glycopeptide and the azide-containing lipopeptide using click reaction afforded the three-component V3 glycopeptide immunogen. The immunogenicity of the synthetic glycopeptide was evaluated by rabbit immunization. The results indicated that the self-adjuvanted glycopeptide could elicit substantial glycan-dependent antibodies that exhibited cross-reactivity toward various HIV-1 gp120 glycoproteins.278
Scheme 13.

Chemoenzymatic Synthesis of a Three-Component Glycopeptide Immunogen
HIV-1 envelope glycoproteins gp120 and gp41 are displayed as a trimer of heterodimer on the virus surface, which are the actual targets of broadly neutralizing antibodies. To mimic the trimeric display of the glycopeptide antigens on HIV-1 envelope trimer, Wang and co-workers designed and synthesized trivalent HIV-1 V3 domain glycopeptide.279 The target was constructed through combined chemoenzymatic glycopeptide synthesis and the click reaction. Antibody binding studies indicated that the broadly neutralizing antibody 10–1074 showed a remarkable multivalency in the glycopeptide antigen recognition, as reflected by the over 2G-fold enhanced affinity of 10–1074 for the trivalent glycopeptide construct over the monovalent highmannose type V3 glycopeptide. Interestingly, another V3 glycopeptide specific antibody, PGT12S, did not show apparent multivalent interaction. This study reveals a distinct antigen recognition mode by the two antibodies and suggests that synthetic multivalent glycopeptides might better mimic the neutralizing epitopes than the monomeric glycopeptide.279 More recently, Wang and co-workers included the multivalent glycopeptide antigen into a three-component HIV-1 vaccine design.280 Chemoenzymatic synthesis coupled with click chemistry was used to assemble the lipopeptide-glycopeptide carrying three copies of the high-mannose V3 glycopeptide derived from the HIV-1 JR-FL gp120. As revealed by rabbit immunization studies, the multivalent glycopeptide construct showed substantially enhanced immunogenicity over the corresponding monovalent V3 glycopeptide construct. The antisera was cross-reactive to heterologous HIV-1 gp120s andthe trimeric gp140s with higher affinity than those from the monovalent glycopeptide immunogen. It should be mentioned that the antisera from this preliminary immunization did not show neutralization activities against tier 1 and tier 2 HIV-1. This is probably due to the lack of somatic maturation. Since the glycopeptide construct could raise substantial glycan-dependent antibody responses, which have been difficult to achieve by most other HIV immunogens so far tested, the synthetic HIV-1 V3 glycopeptide could serve as a key component for further immunization studies together with other HIV-1 vaccine candidates.280
Independently, Danishefsky and co-workers have also made key contributions to the synthesis of HIV-1 glycopeptides and subsequent antigen binding and immunization studies.281–283 They took a multistep chemical synthesis approach to constructing the complex HIV-1 glycopeptides. In comparison, the chemoenzymatic synthesis using the ENGase-catalyzed glycosylation for attachment of N-glycans to a preassembled polypeptide precursor appears to be more flexible and actually more efficient because of its high convergence and efficiency.
4.4.3. Total Chemoenzymatic Synthesis of Saposin C.
The endoglycosidase-catalyzed glycosylation, when coupled with chemical protein synthesis, also provides a highly convergent approach to make full-size glycoproteins. In 2012, Nakahara and co-workers used this strategy to achieve the total synthesis of saposin C, a hydrophobic glycoprotein.284 Briefly, the GlcNAc-protein precursor was prepared by native chemical ligation of fragments synthesized by SPPS, followed by folding to provide the correctly folded GlcNAc-saposin C. Then a biantennary complex type oligosaccharide was transferred to the GlcNAc moiety by Endo-M N175Q, using the corresponding glycan oxazoline as the donor substrate to afford the full-size glycosylated saposin C (Scheme 14). This study demonstrates the feasibility of the chemoenzymatic approach for constructing large N-glycoproteins for structural and functional studies.
Scheme 14.

Chemoenzymatic Synthesis of Hydrophobic Glycoprotein Saposin C
4.4.4. Chemoenzymatic Synthesis of Mannose-6-phosphate (M6P)-Containing Glycoproteins.
Lysosomal storage diseases are caused by the deficiency of respective lysosomal hydrolases responsible for the degradation of substrates stored in lysosomes.8 More than 50 different human lysosomal storage diseases have been identified today, including the Gaucher, Fabry, and Pompe diseases, which affect 1 in 7000 newborns.8,285,286 Currently, the only method for treating these diseases is the so-called enzyme replacement therapy (ERT) by infusing directly exogenous recombinant lysosomal enzymes.287,288 However, a major issue in ERT is the relatively low cellular uptake of the recombinant enzymes. One way to address this problem is to tag the enzymes with a mannose-6-phosphate (M6P) oligosaccharide ligand, which can be recognized by cation-independent M6P receptor CI-MPR expressed on cell surface lysosome for receptor-facilitated cellular uptake.287,289,290 Several methods have been explored to M6P-oligosaccharide ligands into the target enzymes. These include chemical conjugation of natural or synthetic M6P-containing oligosaccharides to the target enzyme to make the conjugates, and the enzymatic phosphorylation of highmannose oligosaccharide containing enzymes to introduce the M6P moiety.291–298 Despite these efforts, the nonselective chemical conjugation usually leads to the formation of heterogeneous mixtures of neoglycoconjugates that may have different pharmacokinetic properties and potential issues of immunogenicity in humans because of the unnatural con-jugation.
In 2016, Fairbanks and co-workers reported the first chemoenzymatic synthesis of a phosphorylated glycoprotein containing the M6P moieties using the ENGase-catalyzed glycosylation.299 In this work, the authors chemically synthesized a M6P containing tetrasaccharide oxazoline and showed that wild type Endo-A could transfer it to the deglycosylated RNase B to form the corresponding phosphorylated glycoprotein (Scheme 15). Interestingly it was found that Endo-D had only low activity on the phosphorylated Man3GlcNAc oxazoline for transfer while Endo-M was unable to efficiently transfer the M6P glycan oxazoline under the same conditions.
Scheme 15.

Enzymatic Synthesis of Phosphorylated RNase B Containing M6P Moieties
Shortly afterward, Wang and co-workers reported an expanded study on the chemoenzymatic synthesis of M6P containing phosphorylated glycoproteins via the ENGase-catalyzed glycan remodeling.300 In this study, several phosphorylated high-mannose glycan oxazolines with different patterns of M6P substitution were designed and synthesized by multistep chemical synthesis, including the late stage introducing of the phosphate group during the oligosaccharide synthesis (Scheme 16). Using ribonuclease B as a model enzyme, it was shown that the Endo-A N171A mutant could efficiently transfer the large M6P-glycans to the deglycosylated ribonuclease B to afford homogeneous glycoproteins carrying M6P oligosaccharides (Scheme 17). A novel cyclic glycopeptide carrying two M6P-oligosaccahrides was also synthesized by enzymatic glycosylation.300 SPR binding studies revealed that a single M6P moiety located at the low α1,3-branch of the oligomannose context was sufficient for a high-affinity binding to receptor CI-MPR (KD = 61 nM). Interestingly, the presence of a M6P moiety located at the α1,6-branch of the oligomannose was found to be dispensable. The glycoprotein carrying the truncated M6P-containing pentasaccharide showed a KD of 168 nM. Interestingly, the synthetic cyclic bivalent M6P polypeptide showed the highest affinity to the M6P receptor with a KD of 2 nM, which was more than 20-fold more efficient in binding to the M6P receptor than the corresponding linear bivalent M6P glycopeptide. These data suggest that a close proximity of two M6P-oligosaccharide ligands in the peptide/ protein context is critical to achieve high affinity for the CI-MPR.300 This study identified the oligosaccharide containing a single M6P at the low a1,3 arm as an efficient MPR ligand. The present study suggests that the pattern of the M6P substitution, the oligosaccharide context, and the valency all are important for the high-affinity binding to the CI-MPR, the major M6P receptor. It is interesting to see if this chemoenzymatic method could also be efficiently applied for glycan remodeling of recombinant lysosomal enzymes, which usually contain multiple N-glycans. If successful, the present chemoenzymatic M6P glycan remodeling method would provide a unique approach for enhancing ERT.
Scheme 16.
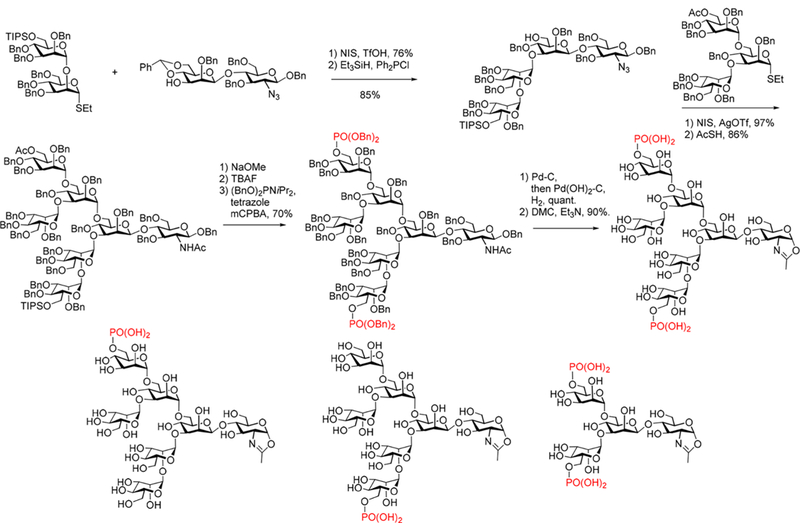
Chemical Synthesis of M6P-Containing High-Mannose N-Glycan Oxazolines as Enzyme Substrates
Scheme 17.

Chemoenzymatic Glycosyiation Remodeling of Ribonuclease B with M6P N-Glycans
4.4.5. Chemoenzymatic Glycan Remodeling of Human Erythropoietin (EPO).
The endoglycosynthase-catalyzed en bloc transfer of a large, preassembled glycan to a GlcNAc-protein acceptor (e.g., a deglycosylated protein) in a single step is particularly appealing for transforming recombinant heterogeneous glycoproteins to various homogeneous glycoforms. The effectiveness of this approach was demonstrated by the glycosylation remodeling of bovine ribonuclease B as a model natural N-glycoprotein. These include the remodeling of this glycoprotein into homogeneous glycoforms carrying well-defined high-mannose type, complex type, phosphorylated form, and GlqMan9GlcNAc2 glycans, proving unique tools for functional studies.178,258,299–302 However, RNase B carries only a single glycosylation site. Recently Wang and co-workers expanded this approach to glycan remodeling of human erythropoietin (EPO), a therapeutic glycoprotein that carries three N-glycans. For the glycan remodeling, a fully fucosylated Man5GlcNAc2 glycoform of EPO was expressed in an engineered HEK293 cell line (HEK293S GnT I−/−) concurrent with overexpression of FUT8, the enzyme responsible for core-fucosylation.303 Deglycosylation of the fucosylated Man5GlcNAc2 glycoform of EPO by Endo-H gave the EPO intermediate carrying three Fucα1,6-GlcNAc moieties at the original three glycosylation sites (Asn24, Asn38, and Asn83). It was found that Endo-F3 D126A mutant could transfer sialylated N-glycan to the intermediate to give fully glycosylated EPO when a large excess of sialylated N-glycan oxazoline was used. Surprisingly, a remarkable site selectivity was observed in this enzymatic transformation when the reaction was run with 6 equiv of sialyl glycan oxazoline, leading to selective glycosylation of two of the three sites. LC-MS/MS glycosylation site-profiling analysis of the glycosylation product indicated that the Asn38 and Asn83 sites were occupied while the Asn34 site was not transglycosylated.304
Transglycosylation of the rest of the GlcNAc at the Asn24 site (after defucosylation) with an excess of azide-tagged Man3GlcNAc oxazoline by Endo-A gave a site-selectively glycosylated EPO (Scheme 18).304 The observed site-selectivity of the enzymatic glycosylation in an intact glycoprotein suggests that the chemoenzymatic method may be further explored to achieve site-selective glycan engineering in a multiply glycosylated protein, which currently cannot be achieved by a genetic approach.
Scheme 18.

Glycan Remodeling of Human Erythropoietin (EPO)
4.5. ENGase-catalyzed Fc glycan remodeling of therapeutic monoclonal antibodies
Monoclonal antibodies (mAbs) of the IgG type are a major class of therapeutic proteins that are widely used for the treatment of cancer, autoimmune diseases, and infectious diseases. Ample studies have demonstrated that the presence and fine structures of Fc N-glycans can profoundly impact the effector functions of antibodies.18,305 Despite the importance of Fc glycosylation, controlling the patterns of glycosylation has been challenging in the production of antibodies. To explore the endoglycosidases for glycan remodeling on antibodies, Wang and co-workers reported in 2008 the first example showing that endoglycosidases can perform deglycosylation and reglycosylation on an intact Fc domain under mild conditions, without the need of denaturing the Fc domain (Scheme 19).306 This was exemplified by glycan remodeling on a recombinant human IgG1-Fc domain expressed in yeast Pichia pastoris. Briefly, the yeast heterogeneous oligomannose N-glycans were removed by Endo-H to give GlcNAc-Fc domain. Then various truncated N-glycans could be transferred to the GlcNAc moiety by Endo-A to give a panel of homogeneous glycoforms of IgG-Fc domain, under mild reaction conditions.306 The group further extended the glycan remodeling approach to synthesizing additional Fc glycoforms including those carrying bisecting monosaccharides for probing their effects on Fc/IIIa receptor binding.307 SPR binding studies indicated that the presence of a bisecting GlcNAc moiety did enhance the affinity of the antibody for the Fc/IIIa receptor.179,307 The high affinity of Fc for the Fc/IIIa receptor expressed on effector cells such as NK T-cells is often used as an indication of enhanced antibody-dependent cell-mediated cytotoxicity (ADCC) activity in vivo.
Scheme 19.

Chemeonzymatic Synthesis of Various IgG-Fc Glycoforms
A major breakthrough was made in 2012 by Wang and coworkers, who generated glycosynthase mutants from Endo-S, an endoglycosidase from Streptococcus pyogenes, which could efficiently perform Fc glycan remodeling on intact full-size antibodies. 81 They found that two mutants, Endo-S D233A and D233Q, were able to transfer complex type N-glycan from the corresponding glycan oxazoline to deglycosylated rituximab to form a homogeneous new glycoform of the antibody, without product hydrolysis (Scheme 20). A panel of homogeneous glycoforms, including the sialylated complex type, nonfucosylated complex type, and azide-tagged glycoforms, were synthesized efficiently. This chemoenzymatic method was also used by several research groups to synthesize specific glycoforms of antibodies for functional studies.308–314 For example, Wong and co-workers applied the Endo-S mutants (D233A and D233Q) for Fc glycan remodeling of an intact antibody to generate a series of antibody glycoforms. Subsequent binding studies and cell-based assays led to the identification of a sialylated biantennary N-glycan as a common and optimized structure for the enhancement of ADCC and complement-dependent cytotoxicity;308 Shirai and co-workers used Endo-S D233Q mutant and other glycosynthases to assemble a relatively large glycoform library of an anti-Her2 antibody, including both full-size and truncated Fc N-glycans, and performed Fc/ receptor binding and cell-based assays.309 The availability of the glycoform library enabled detailed structure—activity relationship studies and revealed how special Fc N-glycans would affect the receptor binding, the ADCC, and the complement dependent cytotoxicity (CDC) activities; Davis and co-workers evaluated the reaction conditions (ratio of substrates, substrate concentrations, temperature, and time) for the Endo-S mutant catalyzed transformation with an anti-Her2 antibody.311 The study led to an optimal condition that reduced the formation of byproducts caused by the potential reactions between the active sugar oxazoline and certain amino acid side chains. Moreover, through the use of modified sugar oxazolines containing non-natural functional groups, antibody variants containing defined numbers of selectively addressable chemical tags such as an alkyne moiety were generated that could be further explored for attachment of cargo molecules such as fluorescent probes or drug molecules.
Scheme 20.

Chemoenzymatic Glyco-Remodeling of Therapeutic Antibodies Using Endoglycosidases and Glycosynthase Mutants
As a continuous effort aiming to expand the scope of the glycan remodeling strategy, Wang and co-workers created novel glycosynthase mutants from Endo-S2, an endoglycosidase from Streptococcus pyogenes of serotype M49.210,315 In contrast to Endo-S that is specific for biantennary complex type Fc N-glycans, Endo-S2 demonstrates a much broader glycan substrate specificity than Endo-S, enabling an efficient deglycosylation of essentially all major types of Fc N-glycans.210 A systematic mutagenesis at D184 that was identified as an equivalent to the D233 of Endo-S by sequence alignment, followed by the evaluation of the 19 mutants, led to the discovery of several mutants, including the D184M, that not only had much higher catalytic efficiency than the corresponding Endo-S mutants but also had much more relaxed substrate specificity, capable of transferring complex type, high-mannose type, and hybrid type N-glycans (Scheme 20).182 This discovery significantly expanded the scope of the Fc N-glycan remodeling for glycoengineering of antibodies. Recently, Wong and co-workers generated additional mutants of Endo-S2 at other residues and found that some of the mutants such as T138Q also demonstrated reduced hydrolytic activity while maintaining the transglycosylation activity to generate new glycoforms of antibodies.267
As an application of the Endo-S2 mutants, Wang and coworkers constructed a focused library of structurally well-defined homogeneous glycoforms of antibody rituximab through the Endo-S2 catalyzed Fc glycan remodeling and used them to assess the effects of various combination of Fc glycosylation on effector functions through in vitro FcγR binding analyses, cell-based ADCC activity assays, and in vivo cellular depletion studies.316 This study revealed that core fucosylation significantly decreased the Fc/RIIIa binding and reduced the in vitro ADCC activity and the in vivo IgG-mediated cellular depletion, regardless of sialylation status. However, the effect of sialylation on ADCC was dependent on the status of core fucosylation. In the presence of core fucose, sialylation significantly decreased ADCC activity and suppressed antibody-mediated cell killing in vivo. In contrast, sialylation did not significantly impact ADCC activity in the absence of core fucosylation.
To streamline the chemoenzymatic glycan remodeling of antibodies, Wang and co-workers performed a site-specific covalent immobilization of the endoglycosidases (Endo-S2 and its glycosynthase mutant D184M) using a recombinant microbial transglutaminase (MTG) and evaluated the immobilized enzymes in deglycosylation and glycosylation of a therapeutic antibody.317 It was demonstrated that the immobilized Endo-S2 and its mutant, D184M, could be efficiently used for glycan remodeling to generate homogeneous antibody glycoforms without the need of intermediate purification, thus streamlining the chemoenzymatic Fc glycan remodeling of antibodies.
Wang and co-workers also generated glycosynthase mutants from Endo-F3, another GH18 family endoglycosidase.183 Two mutants, the D165A and D165Q mutants, were shown to be capable of efficiently transferring both bi- and triantennary complex type N-glycans (Scheme 20). The Endo-F3 mutants were the first glycosynthase mutants that were able to transfer triantennary complex type N-glycans, while the previously discovered glycosynthases from Endo-A, Endo-M, Endo-D, Endo-S, and Endo-S2 could not. Interestingly, these Endo-F3 mutants were highly specific for core-fucosylated N-glycans and were unable to attach glycans to nonfucosylated GlcNAc moieties.183
It should be pointed out that while pairing the glycosynthase mutants and the corresponding glycan oxazoline substrate provides a unique approach to glycoprotein synthesis and antibody Fc-glycan remodeling, some side reactions between the highly active sugar oxazolines and certain side chains on the protein might occur if the reaction conditions are not controlled and/or the enzyme activity is relatively low.301,311 Davis and co-workers performed a LC-MS analysis of possible side reactions when a large access of sugar oxazoline, relatively high temperature, and prolonged time were applied for the reaction between glycan oxazoline and the deglycosylated trastuzu-mab.311 The byproducts were proposed to be caused by the nonenzymatic glycation of glycan oxazoline with certain lysine residues, and the side-reactions were minimized by adding glycan oxazolines in portions and using more enzymes with a shortened reaction time.311 On the other hand, Wang and coworkers demonstrated that by using a highly active glycosynthase mutant, such as the Endo-S2 D184M, antibody glycan remodeling could be performed with a significantly reduced amount of glycan oxazoline and within a short reaction time (less than 1 h) to give essentially quantitative glycosylation of the antibody without any side reactions.182,316,317
Recently Ohno and co-workers reported that sugar oxazoline reacted with primary amines in water to produce sugar imidazolines and eventually sugar imidazoles when heated in water.318 In another model study, Ito and co-workers performed the reactions between sugar oxazoline and peptide under different conditions and identified disubstituted acetamidine as a byproduct.319 It was further proposed that the acetamidine derivative was formed via the attack of the oxazoline ring by the amino group of lysine in the peptide.319 These are interesting findings, but since the model reactions were performed under relatively harsh conditions (high temperature and/or high pH), it is still not clear how efficiently this side reaction might occur under the relatively mild enzymatic reaction conditions (at ambient temperature in a neutral buffer).
As an alternative approach to minimize these potential side reactions, several groups recently attempted to develop enzymatic methods that avoid the use of glycan oxazolines for antibody glycosylation.320,321 In one study, Wong and coworkers expressed trastuzumab in yeast P. pastoris and examined its deglycosylation with a panel of endoglycosidases and transglycosylation with wild type Endo-S2 using the sialoglyco-peptide (SGP) as the donor substrate instead of glycan oxazoline.320 Interestingly, when a large excess (over 1000 mol. equiv.) of SGP was used, Endo-S2 could catalyze the transglycosylation to give up to 80% of the glycoengineered antibody. Although using kinetic control at high donor substrate concentrations to drive a glycosidase-catalyzed glycosylation is not a new concept, this study presents an impressive example that “old concept” could be explored for a special application, and the use of the cheap, stable ground-state donor substrate (SGP) could avoid any potential side reactions even when it was used at molar concentrations. In another study, Iwamoto and coworkers generated improved mutants of Endo-S by introducing additional mutations to the D233Q mutant background.321 It was found that the double or triple mutants, including Endo-S D233Q/Q303L, D233Q/E350Q, and D233Q/Y402F/D405A, could perform transglycosylation with the stable SGP substrate at a relatively low donor/acceptor ratio to provide the target antibody carrying the sialylated Fc N-glycans in a high yield. Interestingly, the glycan remodeling reaction could be carried out in a one-pot manner when Endo-M or Endo-CC were included for in situ deglycosylation of the antibody without the need of isolating the deglycosylated antibody intermediate for transglycosylation. More recently, Manabe and co-workers reported that the Endo-CC mutant, N180H, could also perform transglycosylation of deglycosylated antibody using SGP as the donor substrate at a high concentration.253 Taken together, these recent discoveries have now made it possible to construct various homogeneous and specific antibody glycoforms, offering exciting new opportunities for probing the biological functions and for discovering novel antibody-based therapeutics.
4.6. Chemical and Chemoenzymatic Synthesis of N-Glycan Oxazolines
A broad application of the ENGase-catalyzed glycosylation for glycoprotein synthesis and glycan remodeling requires an easy access to respective glycan oxazolines as the activated donor substrates. In the past decade, a variety of oligosaccharide oxazolines corresponding to the natural N-glycans and their modified derivatives have been prepared either by total chemical synthesis or by semisynthesis using N-glycans isolated from natural sources. A review was recently published by Fairbanks that gave an excellent account on this topic.322 In this section, we provide a highlight on the common approaches to the synthesis of glycan oxazolines that are useful for the ENGase-catalyzed glycosylation.
4.6.1. Chemical Synthesis of Oligosaccharide Oxazolines Corresponding to N-Glycans.
Sugar oxazolines have been widely used as activated subunits for Chitinase- or hyaluronidase-catalyzed polymerization to form functionalized chitin and glycosaminoglycans.119,323,324 Early work on the synthesis of oligosaccharide oxazolines involved a stepwise chemical synthesis to build the oligosaccharide skeleton, changing all protecting groups into O-acetyl protecting groups, Lewis acid-catalyzed conversion of the peracetylated oligosaccharide into the corresponding sugar oxazoline, and finally the global de-O-acetylation to provide the free sugar oxazoline. Scheme 21 showed one of the early examples for the synthesis of tetrasaccharide oxazolines corresponding to the N-glycan core.242 In this synthesis, a key Manβ1,4-GlcNAc disaccharide was synthesized in which the 3- and 6-hydroxy groups of the mannose residue were selectively deprotected and were subject to a double glycosylation with a mannosyl trichloroacetimidate to form the tetrasaccharide structure. Then the 2-azide group was reduced and N-acetylated, and all other protecting groups were changed to O-acetyl groups. Initial treatment of the per-O-acetylated tetrasaccharide with Lewis acid such as TMSOTf in dichloroethane gave a mixture of the corresponding oxazoline derivative and the α-glycosyl acetate anomer that could not be easily converted into the sugar oxazoline under the condition.325 Fortunately, using a combination of TMSBr, BF3 Et2O, and 2,4,6-collidine in dichloroethane326 successfully converted the acetate into sugar oxazoline derivative in good yield. Finally, de-O-acetylation using a catalytic amount of MeONa in MeOH afforded the free tetrasaccharide oxazoline (Scheme 21a). As another example, azide groups were selectively introduced at the 6-position of the external mannose moieties by protecting group manipulations, and the corresponding azide-containing sugar oxazoline was prepared in a similar way (Scheme 21b).258 The introduction of azido functionality provides a means for further site-specific modifications of the glycoproteins through click chemistry.258
Scheme 21.

Chemical Synthesis of Oligosaccharide Oxazolines
While a number of sugar oxazolines were synthesized in this manner, the three-step procedures are tedious and, more importantly, hydrolysis of O-glycosidic linkages during a prolonged Lewis acid treatment would be problematic when large oligosaccharide is involved. An important development in this field came from the work of Shoda and co-workers in 2009, when they reported a single step conversion of unprotected oligosaccharides containing an N-acetyl aminosugar such as GlcNAc at the reducing end into free sugar oxazoline in water using an excess of 2-chloro-1,3-dimethylimidazolinium chloride (DMC) as a dehydrating reagent agent (Scheme 22).327 Under basic conditions in water, the anomeric hydroxyl group, which is more acidic than other hydroxyl groups, was selectively activated by DMC, and subsequent intramolecular reaction by the neighboring N-acetyl group participation resulted in the formation of sugar oxazoline. The yield was excellent in most cases and the free sugar oxazoline could be readily purified by a single size-exclusion chromatography if it is large enough. This important discovery significantly simplifies the preparation of N-glycan oxazolines which are an important donor substrate for the ENGase-catalyzed glycosylation for glycopeptide synthesis and glycoprotein glycan remodeling.29,31
Scheme 22.

Direct Conversion of Unprotected Oligosaccharide Containing a Reducing End GlcNAc Moiety to Oligosaccharide Oxazoline in Water
The development of this method allowed an efficient synthesis of even very large oligosaccharide oxazolines in excellent yield. Wang and co-workers reported a convergent chemoenzymatic synthesis of several monoglucosylated glycoprotein glycoforms, using bovine ribonuclease (RNase) as a model glycoprotein. These glycoforms were designed as specific ligands of calnexin (CNX) and calreticulin (CRT), two important lectins involved in protein folding and quality control (Scheme 23).302 That approach included the chemical synthesis of a selectively modified oligosaccharide, Gal1Glc1Man9GlcNAc, its single step conversion into the corresponding glycan oxazoline by DMC treatment (quantitative conversion), and the EndoA-N171A catalyzed transfer to GlcNAc-RNase to form the homogeneous glycoforms. The Glc1Man9GlcNAc2-RNase glycoform was synthesized by selective removal of the terminal galactose using β-galactosidase as the enzyme. The affinity of the glycoforms to the lectins was probed by SPR analysis. The results indicated that Glc1Man9GlcNAc2-RNase glycoform had high affinity to lectin CRT, while those glycoforms lacking the terminal glucose moiety did not show CRT-binding activity.302 The experimental data confirm the essential role of the glucose moiety in the glycoproteins in the lectin binding and specific recognition during the lectin-mediated protein folding.
Scheme 23.

Synthesis of Glucose-Containing High-Mannose N-Glycan Oxazoline
4.6.2. Semisynthesis of N-Glycan Oxazolines Using N-Glycans Isolated from NaturalSources.
The diversity of glycoforms of N-glycoproteins that can be reached by the chemoenzymatic method will also rely on the availability of diverse N-glycan structures. While chemical synthesis provides the flexibility to make both natural and selectively modified oligosaccharides, total chemical synthesis is tedious and technically demanding, particularly for construction of complex natural N-glycans. Fortunately, some natural N-glycans could be isolated from natural sources on a relatively large scale, which could serve as the starting materials for the preparation of a collection of full-size N-glycans and related truncated forms.
The chicken egg yolk contains a rich sialoglycopeptide (SGP) that can be isolated on large scale as a source of biantennary complex type N-glycans. It was demonstrated that the disialyl biantennary complex type N-glycan (SCT) can be readily prepared from chicken egg yolks through organic solvent precipitation, reverse-phase chromatography, Endo-M digestion, and final gel filtration chromatography of the released glycans (Scheme 24).29,194,255,301,328,329 Recently the isolation procedure of SCT glycoform was further optimized to a gramscale production by using a cheaper source, the egg yolk powder ($4.45/kg).330,331 The SCT glycan can be further trimmed by sialidases and β1,4-galactosidase to obtain additional structures.182,332 Finally, the free N-glycans could be converted to the corresponding glycan oxazolines in a single step DMC-promoted conversion (Scheme 24).
Scheme 24.

Semisynthesis of Sialyl Biantennary Complex Type N-Glycan Oxazoline (SCT-ox)
As another example, ultrapure high mannose N-glycan (Man9GlcNAc2) was readily obtained on a preparative scale (100 mg/batch) from soybean flour through fractional precipitation, sequential Pronase and Endo-A digestion of the crude soybean agglutinin glycoprotein fraction, and gel filtration chromatography of the released glycans (Scheme 25).29,177,178,333 The obtained Man9GlcNAc2 glycan can be further trimmed and extended by α1,2-mannosidase and glycosyltransferases for the synthesis of additional structures, including hybrid type N-glycans.182 Distinct high mannose N-glycans, including GlcMan9GlcNAc2, Man9GlcNAc, Man6GlcNAc, and Man5GlcNAc, could also be achieved from egg yolk and chicken ovalbumin.334–337
Scheme 25.

Semisynthesis of High Mannose Type N-Glycan (Man9-ox)
Recently, Wang and co-workers reported the synthesis of a triantennary complex type N-glycan oxazoline, starting with bovine fetuin, a natural glycoprotein carrying sialylated bi- and triantennary N-glycans.183 Briefly, the N-glycans were released from bovine fetuin by Endo-F3 treatment. After solid-phase extraction and ion-exchange chromatography, the sialylated triantennary N-glycan was separated. Sialidase-catalyzed desialylation gave the triantennary complex type glycan (TCT) on a preparative scale (25 mg/batch) with over 95% purity. Finally, the N-glycan was transformed into the glycan oxazoline by DMC treatment followed by size-exclusion chromatography (Scheme 26). The glycan oxazoline was successfully used to prepare novel glycoforms of therapeutic antibodies that are not readily available by recombinant expression.183
Scheme 26.

Semisynthesis of Triantennary Complex Type N-Glycan Oxazoline (TCT-ox)
In addition to total chemical synthesis and semisynthesis using N-glycans isolated from natural sources, tremendous progress has been made in the chemoenzymatic synthesis of complex N-glycans in recent years, as demonstrated by Boons, Peng George Wang, and other groups.338–341 Thus, the availability of diverse N-glycans and corresponding glycan oxazolines has further expanded the scope of the ENGase-catalyzed glycosylation for glycoprotein synthesis and glycan remodeling.
5. DIRECT ENZYMATIC GLYCOSYLATION OF POLYPEPTIDES AND PROTEINS
The ENGase-catalyzed glycosylation provides a very attractive method for the synthesis of complex glycopeptides and for global glycan remodeling of glycoproteins to provide homogeneous glycoforms. The advantages of this chemoenzymatic method are the high convergence of the approach, the nature of native ligation to restore the natural glycoside linkage, and the flexibility to manipulate the protein and oligosaccharide components independently. However, a prerequisite for this approach is the need of installation of a monosaccharide (a GlcNAc or Glc moiety) in the polypeptide or protein component to serve as a handle (acceptor) for enzymatic oligosaccharide transfer. Thus, an efficient method for direct introduction of a monosaccharide moiety to a preassembled polypeptide or protein would further expand the scope. Moreover, direct “native” ligation between a preassembled glycan and a free polypeptide or protein is becoming another attractive approach to homogeneous glycoproteins. This section highlights recent progress in this area.
5.1. Eukaryotic Oligosaccharyltransferase (OST) Catalyzed in Vitro Glycosylation of Polypeptides
In the biosynthesis of eukaryotic N-glycoproteins, the oligosaccharyltransferase (OST) is the sole enzyme responsible for transferring a large N-glycan precursor (Glc3Man9GlcNAc2) en bloc from a dolichol-phosphate glycolipid to a consensus sequon NXS/T on the nascent unfolded protein.342–344 Eukaryotic OST is in fact a multisubunit protein complex consisting of at least 9 subunits, and all of them are transmembrane proteins.345 Some early in vitro enzymatic studies from Coward, Imperiali, and several other laboratories have shown that OST is able to transfer truncated N-glycan core from the corresponding glycosyl-phosphate dolichol to synthetic peptides containing the consensus NXS/T sequence.346–355 However, a practical application of the eukaryotic OST for in vitro glycoprotein synthesis has not been fulfilled. Major hurdles include the instability of the eukaryotic enzyme complex, the complexity of the membrane-bound multiple protein subunits that are required for catalytic activity of the STT3 subunit, and the difficulty to obtain the dolichol-associated glycolipids as the substrates. More importantly, the eukaryotic OST is not suitable for transferring oligosaccharide to folded proteins. In fact, during biosynthesis, the precursor N-glycan is transferred to nascent protein (polypeptide) before folding.356,357
5.2. Prokaryotic Oligosaccharyltransferase-Catalyzed in Vitro Glycosylation of Polypeptides and Proteins
For decades, protein N-glycosylation was assumed to be an event occurring only in eukaryotic systems. However, this view was changed in 1999 when Campylobacter jejuni, a Gramnegative bacterium that is a human gut mucosal pathogen, was found to possess a protein N-glycosylation machine.358–360 The C. jejuni contains a gene cluster, termed the “Pgl gene cluster”, which is involved in the assembly of a novel heptasaccharide on an undecaprenyl-pyrophosphate carrier and its subsequent transfer to the asparagine side chain in a consensus NXS/T sequence of endogenous proteins at the bacterial periplasmic space.70,358,361,362 In contrast to the eukaryotic OST that consists of multisubunit proteins with the STT3 protein as the catalytic subunit, a single subunit protein, PglB, was found to be responsible for oligosaccharide transfer in C. jejuni and some other prokaryotic organisms.70,363
In 2005, Imperiali and co-workers reported the cloning, overexpression, and purification of four key Pgl glycosyltrans-ferases (PglA, PglH, PglI, and PglJ) and demonstrated an in vitro enzymatic assembly of the heptasaccharide, GalNAca1,4-GalNAcα1,4-(Glcβ1,3)-GalNAcα1,4-GalNAcα1,4-GalNAcα1,3-Bac, on a synthetic undecaprenyl-pyrophosphate, where Bac is bacillosamine (2,4-diacetamido-2,4,6-trideoxyglu-cose).364 The successful in vitro reconstitution of the sequential activities of the glycosyltransferases in the biosynthetic pathway provides a detailed characterization of the functions and biosynthetic sequence of these Pgl enzymes and provides important insights in manipulating the biosynthetic pathways of bacterial N-glycosylation. Further studies from the same group demonstrated that the C. jejuni oligosaccharyltransferase PglB had relatively relaxed substrate specificity on the undecaprenyl pyrophosphate-linked saccharide substrate for peptide glyco-sylation, and a wide variety of saccharides with different lengths and also modified subunits could be transferred by PglB from the undecaprenyl pyrophosphate-linked saccharide donor sub strates.98,365
More recently, Imperiali and co-workers evaluated the substrate specificity of Pgl glycosyltransferases and found that most of the Pgl enzymes could tolerate azide-modified UDP-sugar substrates, including the UDP derivatives of 2,4-diacetamidobacillosamine and N-acetylgalactosamine.366 Their experimental data indicated that PglC was quite flexible to add modified bacillosamine derivative to the lipid diphosphate carrier and the PglA and PglC were active for extending the sugar chain with N-azidoacetyl modified GalNAc derivative to form the respective undecaprenyl pyrophosphate-linked saccharides (Scheme 27). In addition, it was also demonstrated that the bacterial PglB was able to transfer the azide-tagged sugar from the glycolipid to a peptide containing the consensus N-glycosylation sequence to form the corresponding N-glycopeptides with site-specifically introduced azide functionality at the respective monosaccharide unit366 (Scheme 28). The azide-tagged glycolipids and glycopeptides would be very useful for studying the roles of C. jejuni cell surface glycoconjugates in host pathogen interactions, e.g., through biorthogonal reactions for tracking and cell surface imaging.
Scheme 27.

Chemoenzymatic Synthesis of Und-PP-Trisacchandes Carrying Site-Specific Azido Groups
Scheme 28.

In Vitro Enzymatic Synthesis of N-Glycopeptides Carrying Site-Specific Azido Groups Using C. jejuni PglB
In a separate study, Davis and co-workers investigated the lipid carrier specificity of the C. jejuni PglB by testing a series of well-designed, natural and unnatural C10, C20, C30, and C40 polyisoprenol sugar pyrophosphates, including those bearing repeating cis-prenyl units.367 The study revealed that some short, synthetically accessible C20 prenols could serve as excellent lipid carriers of oligosaccharides for PglB catalyzed glycosylation (Scheme 29). For example, the lipid-linked diphosphate GlcNAc with at least four prenyl units (ωEZZ) was an excellent substrate of PglB and the enzymatic glycosylation with a peptide containing a consensus sequon D/EZNXS/T could reach up to 92% yield, which was significantly higher than the yields previously reported (<20%).368,369 Interestingly, the modified GlcNAc derivatives, 6-azido-GlcNAc and GlcNAz linked to the lipid, were found to be inactive for PglB glycosylation. The sugar chain of the resulting GlcNAc-peptides was further extended by respective enzymatic glycosylation to give different glycopeptides (Scheme 29b). Impressively, AcrA, an endogenous C. jejuni protein, could be doubly glycosylated by PglB using this rationally designed lipid-linked GlcNAc donor substrate with a yield of 95%, showing the high efficiency of the PglB-catalyzed in vitro N-glycosylation with the synthetic donor substrate.
Scheme 29.

In Vitro Chemoenzymatic Synthesis of N-Glycopeptides Using PglB as the Enzyme and Synthetic Lipid-Linked GlcNAc as the Donor Substrates
In comparison with the eukaryotic OST that recognizes a conserved NXS/T sequence for N-glycosylation, PglB appears to have strict acceptor substrate specificity.370 It was shown that PglB required an extended N-glycosylation sequence, D/E-Z-N-X-S/T, in the polypeptide acceptor, where Z and X are any amino acids except proline, and it can transfer oligosaccharides to the extended sequence but only when it is located in a flexible region.371 In an in vitro protein N-glycosylation assay, PglB was able to efficiently glycosylate either folded or unfolded proteins, like native AcrA and active GFP protein, with an extended sequence (D/EZNXS/T) on a flexible region.372 However, it only showed marginal glycosylation activity toward fully folded RNase A, while its activity was significantly restored by partially or completely unfolding the acceptor protein. The in vitro N-glycosylation assay indicated that PglB has relatively strict acceptor substrate specificity, as it can only efficiently glycosylate the acceptors with an extended N-glycosylation consensus sequon on a flexible region. A similar result was reported recently by Imperiali and co-workers.373 In a quantitative radioactivity-based in vitro assay of a bacterial N-glycosylation system, PglB was able to glycosylate flexible peptides and full-length folded proteins with an extended consensus sequon.374 DQNAT was found to be the optimal sequon for PglB mediated N-glycosylation of a small bacterial model protein, Im7. Interestingly, the enzyme could not transfer the N,N’-diacetylchitobiose moiety, which is the core disaccharide found on eukaryotic N-glycoproteins. Recently an extensive study on the N-glycosylation site preferences of PglB homologues from different bacteria was performed by DeLisa and co-workers using an ectopic trans-complementation in vivo assay.375 Among 15 active PglB analogs, 5 of them exhibited a preference to noncanonical AQNAT motif, indicating that PglB homologues from different bacteria could have diverse determinants on their preferred N-glycosylation sites.
It should be pointed out that for in vitro studies, an efficient expression of recombinant PglB enzyme is required. Peng George Wang and co-workers reported that PglB was able to be overexpressed at a level of 1 mg/L culture in E. coli C43 (DE3), a host cell suitable for toxic protein expression.369 The enzyme activity was confirmed using a synthetic shorter Und-PP-GalNAc (2 cis, 2 trans) as donor substrate and polypeptide KDFNVSKA as an acceptor.
In 2002, Aebi and co-workers reported the first functional transfer of the C. jejuni protein N-glycosylation machinery (the pgl gene cluster) into E. coli.96 Later on, the same group demonstrated that the pgl gene cluster of Campylobacter lari encoding a functional glycosylation machinery could be reconstituted in E. coli and showed that the N-glycan produced in this system consisted of a linear hexasaccharide.376 The C. lari PglB also preferred a primary consensus of D/E-Z-N-X-S/T (where Z and X can be any amino acid but proline). Interestingly, the PglB exhibited a relaxed substrate specificity toward the acceptor site and could glycosylate the asparagine residues of a protein at sequences DANSG and NNNST.
In parallel to the biological and functional studies, there have been tremendous studies on the structures of the bacterial PglB. In 2011, Locher and co-workers reported the first crystal structure of PglB from C. lari that revealed the fold of PglB and related STT3 proteins.100 Similar structures were found in the related AglB protein from Archaeoglobus f ulgidus.377,378 These structural studies, together with more detailed biochemical analysis,379,380 provided insights into the molecular mechanism of sequon recognition and, particularly, the unexpected reactivity and mechanism of carboxamide activation in bacterial N-linked protein glycosylation. More recently, Locher and co-workers solved the crystal structure of C. lari PglB in complex with an acceptor peptide and a nonhydrolyzable lipid-linked oligosaccharide.381 This crystal allowed a ternary complex to be trapped and its structure to be determined at 2.7-Å resolution. The structure revealed the role of the external loop EL5, which is conserved in all OST enzymes, in substrate recognition. The N-terminal half of EL5 recognized the donor substrate lipid-linked oligosaccharide, while the C-terminal half interacted with the acceptor peptide. Taken together, these structural studies provided a molecular basis for rational engineering of PglB for improving the catalytic efficiency and/or for altering its substrate specificity to expand its synthetic applications.
5.3. Use of PglB-Catalyzed Glycosylation for the Synthesis of Glycoproteins Carrying Eukaryotic N-Glycans
Although the protein N-glycosylation machinery from the C. jejuni and C. Lari systems has been successfully transferred into E. coli, the biotechnology’s workhorse for protein expression, bacterial N-glycans are quite different from the mammalian N-glycans and are highly immunogenic. On the other hand, the ENGase-catalyzed glycosylation can conjugate preassembled N-glycans to proteins, but it requires the attachment of a GlcNAc moiety into a protein to serve as a primer for transglycosylation In 2010, Wang, Aebi, and co-workers reported a combined method to produce homogeneous glycoproteins carrying humanized N-glycans (Figure 11).382 The method involves the expression of a glycoprotein in an E. coli system that harbors a glycoengineered C. jejuni glycosylation machinery.371 In this glycoengineered E. coli system, the genes responsible for the biosynthesis of the bacillosamine (Bac) were deleted, enabling the replacement of the Asn-linked first sugar (Bac) by a GlcNAc moiety in the resulting recombinant glycoprotein. The outer GalNAc moieties were then trimmed off by the bacterial a-N- acetylgalactosaminidase to give the GlcNAc-protein, which served as the key intermediate for subsequent ENGase-catalyzed glycosylation to afford the target homogeneous eukaryotic glycoproteins (Figure 11). Several glycoproteins, including the complex type AcrA, glycosylated human IgG-Fc CH2 domain, and a single-chain antibody F8, were synthesized by this method. It should be pointed out that while this method combines the advantage of protein expression in E. coli and the flexibility of in vitro chemoenzymatic glycan remodeling, the yield for the in vivo initial glycosylation of the mammalian heterologous proteins was relatively low (5–40%). These problems should be addressed in future studies.
Figure 11.

Enzymatic synthesis of glycoproteins carrying eukaryotic N-glycans by coupling engineered C. jejuni PglB N-glycosylation pathway in E. coli with in vitro enzymatic glycan-remodeling.
In 2012, DeLisa and co-workers reported a bottom-up glycoengineering method for producing eukaryotic N-glyco-proteins in E. coli (Figure 12).383 In this approach, the yeast glycosyltransferases responsible for constructing the pentasaccharide N-glycan core (Man3GlcNAc2) were engineered in E. coli to build up the N-glycan glycolipid precursor. When coexpressed with PglB, the bacterial oligosaccharyltransferase PglB from C. jejuni, the production of a glycosylated protein carrying a Man3GlcNAc2 glycan was detected. Unfortunately, the glycosylation yield was very low (ca. 1%). This was understandable, as previous studies indicated that the lipid-linked eukaryotic oligosaccharides were poor substrates in in vitro assays.374 It should be noted that although the core structure, Man3GlcNAc2, was the primary oligosaccharide formed in this engineered biosynthetic pathway, other truncated N-glycans, including Man1GlcNAc2, Man2GlcNAc2, and Man4GlcNAc2, were also detected. The efficiency of this N-glycoprotein expression system could be improved if an engineered PglB with enhanced activity on the lipid-linked eukaryotic oligosaccharide could be generated. Crystal structures ofPglB were solved,100,381 which provide a molecular basis for rational engineering and or directed evolution for broadening its substrate specificity and catalytic efficiency.
Figure 12.

Enzymatic protein glycosylation by engineeering eukaryotic N-glycan assembly coupled with PglB-catalyzed N-glycan transfer in E. coli.
5.4. Use of PglB-Catalyzed Glycosylation for Producing Glycoconjugate Vaccines
Although PglB cannot efficiently transfer eukaryotic N-glycans from the corresponding the lipid-linked eukaryotic oligosaccharide donors, the enzyme actually could accommodate a wide variety of lipid-linked oligosaccharides and even polysaccharides so long as the reducing end monosaccharide is a bacillosamine or another 2-acetamino-2-deoxy-monosaccharide moiety, in which the 4-position of the reducing end monosaccharide is not modified.97,384 In 2005, Aebi and co-workers first explored this unique property of PglB to produce glycoconjugate vaccine in E. coli.97 The authors engineered E. coli cells to merge the lipopolysaccharide (LPS) biosynthesis and the protein N-glycosylation by PglB, which was able to transfer the O-polysaccharide from a lipid carrier (undecaprenyl pyrophosphate) to an acceptor protein (Figure 13). The relaxed specificity of the PglB oligosaccharyltransferase toward the polysaccharide structures enabled the production of poly-saccharide-protein conjugates containing two distinct E. coli or Pseudomonas aeruginosa O-antigens, which could serve as candidate vaccines. Following this concept, a number of bacterial glycoconjugate vaccines have been synthesized by hijacking the biosynthetic pathways and PglB catalyzed polysaccharide transfer. Some of them have been successfully applied in clinical trials.385–391
Figure 13.

C. jejuni PglB mediated enzymatic synthesis of glycoconjugate vaccines carrying bacterial O-antigens.
5.5. Bacterial N-Glycosyltransferase (NGT)-Catalyzed Glycosylation of Proteins
The activity of N-glycosyltransferase (NGT) was found more than three decades ago, but only until 2000 was the enzyme first isolated and the activity was confirmed.392 The purified enzyme from Saccharothrix aerocolonigenes was found to be able to transfer a glucose moiety to the derivatives of an indolocarbazole with an N-linked glycosidic bond.392 In 2003, St Geme and coworkers reported that HMW1C was an essential enzyme for the glycosylation of HMW1 adhesion, which is involved in the attachment of respiratory pathogen Haemophilus inf luenza to human epithelial cells.393,394 The HMW1C was confirmed to be an N-glycosyltransferase to attach glucose or galactose to the asparagine site of HMW1 adhesion at a consensus sequence of N-X-S/T similar to OST.395 Independently, Yeo and co-workers demonstrated that a novel protein from Actinobacillus pleuro-pneumoniae (ApHMW1C), sharing a high-level homology with HMW1C, was able to regulate the N-glycosylation of HMW1 in a similar manner.396 Shortly afterward the same group solved the crystal structure of ApHMW1C and found that this HMW1C-like enzyme shared the same structure with HMW1C but harbored a unique catalytic activity different from other members in the GT41 family.397
Aebi and co-workers reported the expression of the HMW1C-like N-glycosyltransferase (ApNGT) in E. coli and performed a detailed biochemical analysis of the enzyme.99,101 These studies confirmed that ApNGT could transfer a glucose or galactose moiety from the corresponding UDP-sugars to a peptide containing the N-X-S/T sequence to form an N-linked Glc or Gal-peptide. Later on, the same group successfully transferred the ApNGT N-glycosylation system from A. pleuropneumoniae to E. coli.398 When coexpressed, the ApNGT was able to perform N-glycosylation of endogenous and/or recombinant protein in the cytoplasm of E. coli.
The ability of ApNGT to directly introduce a monosaccharide moiety in a polypeptide/protein provides an excellent starting point to produce full-size glycopeptides/glycoproteins when it is combined with an endoglycosidase-catalyzed glycosylation. In 2013, Wang and co-workers reported a two-step enzymatic glycosylation strategy that involves the introduction of a glucose moiety in a polypeptide at the consensus N-glycosylation sequence (NXS/T) by N-glycosyltransferase (NGT), followed by an ENGase-catalyzed transfer of N-glycans.399 The glucose moiety could be efficiently introduced by NGT and the glucose moiety was an excellent acceptor for the Endo-M N175A catalyzed transfer of both high-mannose and complex type N-glycans, as exemplified by the synthesis of several HIV-1 gp41 glycopeptides (Scheme 30). Interestingly, it was found that in contrast to the natural GlcNAc-Asn linkage, the Glc-Asn linked glycopeptide was resistant to the hydrolysis by PNGase F. Moreover, the Glc-Asn linked glycopeptide demonstrated more than 10-fold lower hydrolytic activity toward Endo-M than the natural GlcNAc-Asn linked glycopeptide. This chemoenzymatic method should be generally applicable to other target glycopeptides, and the new properties of the Glc-Asn linked glycopeptides might be explored for different applications such as the synthesis of glycopeptide analogs with improved stability against PNGases and endoglycosidases.
Scheme 30.

Enzymatic Synthesis of Glycopeptidesbearing Complex N-Glycans by Coupling ApNGT and Endo-glycosidases
The crystal structure of the NGT from A. pleuropneumoniae (ApNGT) was recently solved, which shows an N-terminal a- helical domain fold and a C-terminal GT-B fold with two Rossmann-like domains.397 Based on this structure, Peng George Wang and co-workers carried out site-directed mutagenesis around the domain involved in the recognition of the N-glycosylation site aiming to broaden the acceptor substrate specificity of ApNGT.400 Screening of the mutants against a small library of synthetic peptides revealed that the introduction of small hydrophobic residues at a critical residue, Q469 of ApNGT, could weaken the stringency of ApNGT and led to significant enhancement of the glycosylation efficiency for both short peptides and proteins. The usefulness of the identified Q469A mutant was exemplified by the glycosylation of a HMW1 adhesion fragment, carrying 12 NXS/T consensus sequons, and 3 of them are potential sites for wild type ApNGT. It was shown that the mutant could attach glucose moiety to 7 of those sites. Moreover, coexpression of the Q469A mutant with a protein bearing 10 optimized N-glycosylation sites resulted in homogeneous N-glycosylation, which could not be achieved by the wild type ApNGT.
In another related study, Peng George Wang and co-workers demonstrated that the ApNGT mutant Q469A also exhibited a broader donor substrate specificity than wild type ApNGT and was able to transfer a glucosamine (GlcN) residue from UDP-GlcN to a peptide (Scheme 31).401 After conversion of the GlcN to GlcNAc by a glucosamine N-acetyltransferase (GlmA), the resulting GlcNAc-peptide was extended in the sugar chain by an endo-β-N-acetylglucosaminidase M mutant (Endo-M N175Q) catalyzed glycosylation to provide a glycopeptide carrying a natural N-glycan. This is a remarkable study, and it is expected that the method will be applicable for synthesizing other biologically interesting N-glycopeptides or N-glycoproteins for various applications. More recently, Peng George Wang and coworkers expressed and characterized the N-glycosyltransferase from Aggregatibacter aphrophilus.402 In contrast to ApNGT that is restricted to transfer Glc and Gal, the new NGT, named AaNGT, was able to utilize a variety of nucleotide-activated sugar donors, including UDP-Glc, UDP-Gal, UDP-Xyl, GDP-Glc, dGDP-Glc, and UDP-GlcN, to glycosylate peptides with the consensus N-glycosylation sequence. Moreover, the AaNGT was able to transfer glucosamine from UDP-GlcN, making it particularly synthetically useful when coupled with the glucosamine N-acetyltransferase (GlmA) to convert the GlcN moiety to a GlcNAc residue for subsequent sugar chain elongation.402
Scheme 31.

Enzymatic Synthesis of Glycopeptides Carrying Natural Eukaryotic N-Glycans Scheme
5.6. Enzymatic O-GlcNAc Glycosylation of Proteins and Sugar Chain Elongation
O-linked GlcNAc modification of protein is an important dynamic glycosylation process occurring in nuclear, mitochondrial, and cytoplasmic compartments of cells.75,403 The monosaccharide is attached to proteins by an O-GlcNAc transferase (OGT) at the Ser or Thr residues in a protein. Recently, Peng George Wang and co-workers described an efficient site-directed glycosylation of peptide/protein with homogeneous O-linked N-glycans using OGT transferase coupled with an endoglycosidase mutant-catalyzed sugar chain extension.404 It was achieved by transferring an O-GlcNAc to chemically synthesized peptide sequences, derived from bioactive proteins (Scheme 32a). The method was further expanded to an in vivo O-GlcNAc modification of a bovine protein, α-Crystallin mutant (Crys-A), by coexpressing OGT and Crys-A in E. coli and a subsequent N-glycosylation on the GlcNAc site using Endo-M N175Qin vitro (Scheme 32b). The GlcNAc-O-Crys-A protein was produced on a milligram-scale and could be remodeled with a sialo biantennary complex type N-glycan using Endo-M mutant with approx. 30% conversion yield. Several specific O-GlcNAc peptides derived from human proteins were also successfully converted into N-glycosylated O-linked glycopeptides by using the Endo-M mutant. It was observed that by introducing N-glycan on the O-GlcNAc site of peptides, the N-glycosylated O-GlcNAc peptides were resistant to OGA-catalyzed hydrolysis.
Scheme 32.

Site-Specific Enzymatic Synthesis of Glyco-Peptide/Protein Carrying O-Linked Eukaryotic N-Glycans
5.7. Enzymatic O-GalNAc Glycosylation of Proteins
O-Glycoproteins are another important class of glycoproteins in which the carbohydrate component is linked to the hydroxyl group of amino acids such as serine, threonine, tyrosine, or 5-hydroxylysine in the protein through an O-glycosidic bond.73,405 Although O-glycosylation can involve the attachment of a range of monosaccharides, including GalNAc, galactose, GlcNAc, mannose, fucose, glucose, or xylose, the most prevalent O-linked glycoprotein is the mucin-type glycoproteins, where the GalNAc moiety is attached to a Ser or Thr residue in an a-glycosidic bond.405 Mucins are glycoproteins with dense O-glycan clusters in which the carbohydrate can account for up to 70% by mass. Mucin glycoproteins play important roles in normal physiological processes such as embryo development, organogenesis, and tissue homeostasis.406 On the other hand, aberrant glycosylation in mucins is often associated with cancer metastasis and progression.407 For example, much smaller, truncated O-glycans including the GalNAc (Tn antigen), Gal^1,3-GalNAc (T antigen), or sialylated GalNAc (STn antigen) moieties are often found on the type 1 mucin glycoprotein (MUC1) in various cancer cells, which are an important class of tumor-associated carbohydrate antigens.407 The structural and functional diversity and the potential in use of the tumor-associated carbohydrate antigens as novel biomarkers for diagnosis and for cancer vaccines have driven the interest in synthesis of mucin O-glycopeptides and O-glycoproteins.42,408–411
The extracellular domains ofMUC1 contain tandem repeats of a polypeptide consisting of 20 amino acid residues, HGVTSAPDTRPAPGSTAPPA. This sequence contains five potential O-glycosylation sites, including Thr-4, Ser-5, Thr-9, Ser-15, and Thr-16. Tremendous work has been carried out in chemical synthesis of mucin glycopeptides with a focus on development of carbohydrate-based cancer vaccines.33,42,408–412 The general approach is to synthesize different glycoamino acid building blocks and then to incorporate them in the solid-phase peptide synthesis SPPS).42,411 A major advantage of the glycoamino acid building block-based strategy is its flexibility to install the O-glycans at any sites along the peptide sequence.413–417 However, it remains a big challenge to use pure chemical synthesis to build large mucin glycopeptides or glycoproteins with multiple domain repeats, particularly when multiple O-linked oligosaccharides are introduced and the acidlabile sialic acid residues are involved.
Chemoenzymatic synthesis that combines chemical manipulations and enzymatic transformations provides a promising alternative approach to large O-glycopeptides and glycoproteins. In this approach, primer sugars such as the Thr/Ser-linked GalNAc moieties are usually introduced into the polypeptide first using SPPS and then the sugar chain is extended by different glycosyltransferases to achieve the targets.418–424 In particular, the early work on the successful chemoenzymatic assembly of the sulfate- and sialyl Lewis X containing PSGL-1 glycopeptides and subsequent P-selectin binding study, reported by Cummings, Wong, and co-workers, showcases the power of chemoenzymatic synthesis for making h-eavily modified O-glycopeptides for biological evaluation. 418–421
Alternatively, direct introduction of GalNAc moieties coupled with subsequent sugar chain elongation could also be achieved by exploring the biosynthetic pathways for O-glycoprotein assembly. As an example, Clausen and co-workers described an impressive chemoenzymatic synthesis of large MUC1 glyco-peptides carrying Tn and STn motifs on a 60-mer mucin polypeptide backbone.425 Briefly, the 60-mer MUC1 polypeptide was synthesized by SPPS. Then the Tn antigens (GalNAc-Ser/Thr) were introduced into the peptide by sequential glycosylation with an array of recombinant polypeptide GalNAc transferases by taking advantage of their unique site-selectivity. Then, the sugar chains were extended through glycosylations with the recombinant core 1 β3Gal transferase (C1Gal-T1) and the recombinant murine sialyltransferase (ST6GalNAc-I) to provide the target O-glycoprotein carrying multiple T and STn antigens (Scheme 33). The structure and site selectivity of glycosylation in the intermediates and final glycopeptide were verified by mass spectroscopic analysis. As an application for cancer vaccine, the synthetic glycopeptides were conjugated to a carrier protein, KLH, and the glycopeptide-KLH conjugates were immunized in mice to evaluate the different glycosylation patterns on immunogenicity. The results indicated that a higher dense glycosylation pattern with the Tn/STn antigens gave much stronger immune responses than the lower substituted glycopeptide, and the elicited antibodies could recognize specifically cancer cells.425 This study showcases the power of the chemoenzymatic synthesis as a tool in design, construction, and evaluation of mucin glycopeptide-based cancer vaccines.
Scheme 33.

Chemoenzymatic Synthesis of MUC1 Glycoproteins Carrying Multiple Tn, T, and STn O-Glycans
Compared with N-glycosylation, mucin O-glycosylation does not have a strict consensus sequence for signaling the attachment of GalNAc. Nevertheless, various GalNAc transferase isoforms involved in the biosynthesis do have sequence preference. Indeed, systematic studies of the substrate specificity of various GalNAc transferase isoforms reveal a complex balance between redundancy and hierarchy within this family of enzymes.426 So far, about 20 peptide-specific GalNAc transferase isoforms have been found and classified into two subfamilies, I and II.426 Members (GalNAc-T1–3, 5, 6, 8, 9, 11, 13–16, and 18–20) from subfamily I prefer peptide substrates, called “peptide-preferring” isoforms, while GalNAc-T4, 7, 10, 12, and 17 from subfamily II favor glycopeptide substrates, called “glycopeptide-preferring” isoforms.427–429 For example, GalNAcT-1, 2, and 5 prefer non- and monoglycosylated peptide substrates, and they usually participate in the initial step of peptide glycosylation; GalNAcT-3 and 4 prefer GalNAc glycosylation of protein substrates carrying 1 or 2 neighboring glycosylated sites; and GalNAcT-10 can be used to further glycosylate the resting site with high density of existing O-GalNAc glycosylation.426 After introduction of the primer GalNAc moieties, the sugar chain could be extended by other glycosyltransferases such as different galactosyltransferases and sialyltransferases to diversify the glycan structures. Future studies should be directed to further characterization and improved expression of these GalNAc transferase isoforms as well as related enzymes aiming to explore the novel substrate specificity of these enzymes for in vitro chemoenzymatic synthesis of O-glycoproteins with varied density and patterns of glycosylation.
6. CONCLUDING REMARKS
Glycoproteins, the conjugates of glycans and proteins, are an important class of biomolecules that demonstrate unique structural features and diverse biological functions. Yet, a detailed understanding of the structure—activity relationships of glycoproteins, as well as their therapeutic and diagnostic applications, has been hampered by the structural heterogeneity of glycoprotein glycoforms that are formidable to isolate by current chromatographic techniques and are also difficult to control during recombinant production.
To address this urgent need, both chemical and enzymatic methods have been remarkably progressed in the past decade and, as surveyed in this review, the chemoenzymatic methods that combine the flexibility of chemical synthesis and the specificity ofenzymatic transformation are emerging as the most promising approaches that have now made it possible to assemble various natural and tailor-made glycoproteins including intact therapeutic antibodies.
Despite this tremendous progress, the synthetic methods for constructing homogeneous glycoproteins are still far from maturation. For chemical synthesis, the use of the prototype native chemical ligation and more recently developed auxiliary-based ligation has now made it possible to construct some large natural glycoproteins such as the fully glycosylated human erythropoietin (EPO). Nevertheless, each target glycoprotein may pose special challenges, and a careful choice of ligation strategy and a thoughtful design of the synthetic scheme would be essential. For the chemoenzymatic methods, one bottleneck is the strict substrate specificity of many enzymes that limits a wider application of a specific enzymatic transformation. Novel and more efficient enzymes such as enabling oligosaccharyl-transferases and N-glycosyltransferase are needed for site-specific direct enzymatic glycosylation of folded recombinant proteins so that oligosaccharide components can be directly conjugated to proteins with native glyco-amino acid linkages. Another challenge in the field is how to achieve site-specific posttranslational “mutagenesis” on glycans, i.e., the alteration or modification of glycans at predetermined sites in a multiply glycosylated glycoprotein. Continuous efforts in exploring new synthetic methods and concepts are needed to assemble more complex natural and/or tailor-made glycoproteins for addressing structural and biological problems. The ability to produce homogeneous natural and tailor-made glycoproteins, such as cytokines, glycoprotein hormones, glycoengineered therapeutic antibodies, and structurally well-defined antibody-drug conjugates, will certainly speed up the development of more effective glycoprotein-based therapeutics in the years ahead.
ACKNOWLEDGMENTS
We thank other members of the Wang Lab for stimulating discussions during the development of this project. The National Institutes of Health is acknowledged for financial support (grant numbers R01 GM080374, R01 GM096973, and R01 AI113896).
Biographies
Chao Li received his B.S. in chemistry and Ph.D. in organic chemistry under the supervision of Professor Xiao-Qi Yu and Professor Na Wang from Sichuan University, China. His doctoral thesis focused on biocatalytic promiscuity for asymmetric carbon-carbon bond formation. He joined Korea University in 2012 as a visiting scholar, where he worked with Professor Young-Wan Kim on engineering glycosidases and glycosyltransferases for the synthesis ofbioactive oligosaccharides. He is currently a postdoctoral research associate in Professor Lai-Xi Wang’s group at University of Maryland, where his work entails the development of novel glycosidase mutants for glycoengineering of proteins and therapeutic antibodies. His research interest is in the areas of chemoenzymatic synthesis of biologically important oligosaccharides and glycoconjugates, mechanistic enzymology, and protein engineering.
Lai-Xi Wang is a professor of chemistry and biochemistry at University of Maryland, College Park. He received his B.Sc. in chemistry from Jiangxi Normal University and his Ph.D. in organic chemistry from Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences. During his graduate study, he spent three years in RIKEN, Japan, under a joint graduate training program. After postdoctoral studies in glycobiology and molecular biology at Johns Hopkins University and Massachusetts Institute of Technology (MIT), respectively, he joined the faculty of University of Maryland as a Tenure-Track Assistant Professor in 2000, promoted to Associate Professor with tenure in 2005, and to Full Professor with tenure in 2009 in the Institute of Human Virology and the Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine. In 2015, he was recruited to University of Maryland College Park to lead efforts in developing new research programs centered on chemical biology. Dr. Wang was the recipient of the 2004 Young Investigator Award and the 2014 Melville L. Wolfrom Award, both from the American Chemical Society Division of Carbohydrate Chemistry. He was inducted to the Johns Hopkins University Society of Scholars in 2009 and was elected as AAAS Fellow in 2014. Dr. Wang was the Vice Chair and the Chair of the 2015 and 2017 Gordon Research Conferences (GRC) in Carbohydrates, respectively. He served as the Chair-Elect (2015) and the Chair (2016) of the American Chemistry Society Division of Carbohydrate Chemistry. Dr. Wang’s research interests are in the areas of synthetic carbohydrate chemistry, bioorganic chemistry, enzymology, glycobiology, and immunology, including development of chemoenzymatic methods for oligosaccharide and glycoprotein synthesis, glycoengineering of therapeutic antibodies, chemical biology of protein glycosylation, and the design and development of an effective HIV vaccine.
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Varki A Biological Roles of Glycans. Glycobiology 2017,27,3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Petrescu AJ; Wormald MR; Dwek RA Structural Aspects of Glycomes with a Focus on N-Glycosylation and Glycoprotein Folding. Curr. Opin. Struct. Biol. 2006, 16, 600–607. [DOI] [PubMed] [Google Scholar]
- (3).Haltiwanger RS; Lowe JB Role of Glycosylation in Development. Annu. Rev. Biochem. 2004, 73, 491–537. [DOI] [PubMed] [Google Scholar]
- (4).Hart GW; Copeland RJ Glycomics Hits the Big Time. Cell 2010, 143, 672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Slawson C; Hart GW O-Glcnac Signalling: Implications for Cancer Cell Biology. Nat. Rev. Cancer 2011, 11, 678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Helenius A; Aebi M Intracellular Functions of N-Linked Glycans. Science 2001, 291 , 2364–2369. [DOI] [PubMed] [Google Scholar]
- (7).Roth J; Zuber C Quality Control of Glycoprotein Folding and Erad: The Role of N-Glycan Handling, Edem1 and Os-9. Histochem. Cell Biol 2017, 147, 269–284. [DOI] [PubMed] [Google Scholar]
- (8).Coutinho MF; Prata MJ; Alves S Mannose-6-Phosphate Pathway: A Review on Its Role in Lysosomal Function and Dysfunction. Mol. Genet. Metab. 2012, 105, 542–550. [DOI] [PubMed] [Google Scholar]
- (9).Gary-Bobo M; Nirde P; Jeanjean A; Morere A; Garcia M Mannose 6-Phosphate Receptor Targeting and Its Applications in Human Diseases. Curr. Med. Chem. 2007, 14, 2945–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Valguarnera E; Kinsella RL; Feldman MF Sugar and Spice Make Bacteria Not Nice: Protein Glycosylation and Its Influence in Pathogenesis. J. Mol. Biol. 2016, 428, 3206–3220. [DOI] [PubMed] [Google Scholar]
- (11).Tytgat HL; de Vos WM Sugar Coating the Envelope: Glycoconjugates for Microbe-Host Crosstalk. Trends Microbiol. 2016, 24, 853–861. [DOI] [PubMed] [Google Scholar]
- (12).Tytgat HL; Lebeer S The Sweet Tooth of Bacteria: Common Themes in Bacterial Glycoconjugates. Microbiol. Mol. Biol. Rev. 2014, 78, 372–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Raman R; Tharakaraman K; Sasisekharan V; Sasisekharan R Glycan-Protein Interactions in Viral Pathogenesis. Curr. Opin. Struct. Biol. 2016, 40, 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Stencel-Baerenwald JE; Reiss K; Reiter DM; Stehle T; Dermody TS The Sweet Spot: Defining Virus-Sialic Acid Interactions. Nat. Rev. Microbiol. 2014, 12, 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Reis CA; Osorio H; Silva L; Gomes C; David L Alterations in Glycosylation as Biomarkers for Cancer Detection. J. Clin. Pathol. 2010, 63, 322–329. [DOI] [PubMed] [Google Scholar]
- (16).Zhao J; Patwa TH; Lubman DM; Simeone DM Protein Biomarkers in Cancer: Natural Glycoprotein Microarray Approaches. Curr. Opin Mol. Ther 2008, 10, 602–610. [PMC free article] [PubMed] [Google Scholar]
- (17).Dube DH; Bertozzi CR Glycans in Cancer and Inflammation-Potential for Therapeutics and Diagnostics. Nat. Rev. Drug Discovery 2005, 4, 477–488. [DOI] [PubMed] [Google Scholar]
- (18).Jefferis R Glycosylation as a Strategy to Improve Antibody-Based Therapeutics. Nat. Rev. Drug Discovery 2009, 8, 226–234. [DOI] [PubMed] [Google Scholar]
- (19).Walsh G; Jefferis R Post-Translational Modifications in the Context of Therapeutic Proteins. Nat. Biotechnol. 2006, 24, 1241–1252. [DOI] [PubMed] [Google Scholar]
- (20).Dalziel M; Crispin M; Scanlan CN; Zitzmann N; Dwek RA Emerging Principles for the Therapeutic Exploitation of Glyco-sylation. Science 2014, 343, 1235681. [DOI] [PubMed] [Google Scholar]
- (21).Hudak JE; Bertozzi CR Glycotherapy: New Advances Inspire a Reemergence of Glycans in Medicine. Chem. Biol. 2014, 21, 16–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jenkins N; Parekh RB; James DC Getting the Glycosylation Right: Implications for the Biotechnology Industry. Nat. Biotechnol. 1996, 14,975–981. [DOI] [PubMed] [Google Scholar]
- (23).Kiessling LL; Splain RA Chemical Approaches to Glycobiology. Annu. Rev. Biochem. 2010, 79, 619–653. [DOI] [PubMed] [Google Scholar]
- (24).Rillahan CD; Paulson JC Glycan Microarrays for Decoding the Glycome. Annu. Rev. Biochem. 2011, 80, 797–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wolfert MA; Boons GJ Adaptive Immune Activation: Glycosylation Does Matter. Nat. Chem. Biol. 2013, 9, 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wang LX; Davis BG Realizing the Promise of Chemical Glycobiology. Chem. Sci. 2013, 4, 3381–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lowary T Context and Complexity: The Next Big Thing in Synthetic Glycobiology. Curr. Opin. Chem. Biol. 2013, 17, 990–996. [DOI] [PubMed] [Google Scholar]
- (28).Schmaltz RM; Hanson SR; Wong CH Enzymes in the Synthesis of Glycoconjugates. Chem. Rev. 2011, 111, 4259–4307. [DOI] [PubMed] [Google Scholar]
- (29).Wang LX; Amin MN Chemical and Chemoenzymatic Synthesis of Glycoproteins for Deciphering Functions. Chem. Biol. 2014,21,51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Danby PM; Withers SG Advances in Enzymatic Glycoside Synthesis. ACS Chem. Biol. 2016, 11, 1784–1794. [DOI] [PubMed] [Google Scholar]
- (31).Fairbanks AJ The ENGases: Versatile Biocatalysts for the Production of Homogeneous N-Linked Glycopeptides and Glycoroteins. Chem. Soc. Rev. 2017, 46, 5128–5146. [DOI] [PubMed] [Google Scholar]
- (32).Grogan MJ; Pratt MR; Marcaurelle LA; Bertozzi CR Homogeneous Glycopeptides and Glycoproteins for Biological Investigation. Annu. Rev. Biochem. 2002, 71, 593–634. [DOI] [PubMed] [Google Scholar]
- (33).Pratt MR; Bertozzi CR Synthetic Glycopeptides and Glycoproteins as Tools for Biology. Chem. Soc. Rev. 2005, 34, 58–68. [DOI] [PubMed] [Google Scholar]
- (34).Guo Z; Shao N Glycopeptide and Glycoprotein Synthesis Involving Unprotected Carbohydrate Building Blocks. Med. Res. Rev. 2005,25, 655–678. [DOI] [PubMed] [Google Scholar]
- (35).Buskas T; Ingale S; Boons GJ Glycopeptides as Versatile Tools for Glycobiology. Glycobiology 2006, 16, 113R–136R. [DOI] [PubMed] [Google Scholar]
- (36).Gamblin DP; Scanlan EM; Davis BG Glycoprotein Synthesis: An Update. Chem. Rev. 2009, 109, 131–163. [DOI] [PubMed] [Google Scholar]
- (37).Yuan Y; Chen J; Wan Q; Wilson RM; Danishefsky SJ Toward Fully Synthetic, Homogeneous Glycoproteins: Advances in Chemical Ligation. Biopolymers 2010, 94, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Payne RJ; Wong CH Advances in Chemical Ligation Strategies for the Synthesis ofGlycopeptides and Glycoproteins. Chem. Commun. (Cambridge, U. K.) 2010, 46, 21–43. [DOI] [PubMed] [Google Scholar]
- (39).Kajihara Y; Yamamoto N; Okamoto R; Hirano K; Murase T Chemical Synthesis of Homogeneous Glycopeptides and Glycoproteins. Chem. Rec 2010, 10, 80–100. [DOI] [PubMed] [Google Scholar]
- (40).Swarts BM; Guo Z Chemical Synthesis of Glycosylphospha-tidylinositol Anchors. Adv. Carbohydr. Chem. Biochem. 2012, 67, 137–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wilson RM; Dong S; Wang P; Danishefsky SJ The Winding Pathway to Erythropoietin Along the Chemistry-Biology Frontier: A Success at Last. Angew. Chem., Int. Ed. 2013, 52, 7646–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gaidzik N; Westerlind U; Kunz H The Development of Synthetic Antitumour Vaccines from Mucin Glycopeptide Antigens. Chem. Soc. Rev. 2013, 42, 4421–4442. [DOI] [PubMed] [Google Scholar]
- (43).Unverzagt C; Kajihara Y Chemical Assembly of N-Glycoproteins: A Refined Toolbox to Address a Ubiquitous Posttranslational Modification. Chem. Soc. Rev. 2013, 42, 4408–4420. [DOI] [PubMed] [Google Scholar]
- (44).Fernandez-Tejada A; Brailsford J; Zhang Q; Shieh JH; Moore MA; Danishefsky SJ Total Synthesis of Glycosylated Proteins. Top. Curr. Chem. 2014, 362, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Seeberger PH; Overkleeft HS Chemical Synthesis of Glycans and Glycoconjugates. In Essentials of Glycobiology, 3rd ed.; Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, et al. , Eds.; Cold Spring Harbor: New York, 2015; Chapter 53. [PubMed] [Google Scholar]
- (46).Hang HC; Bertozzi CR Chemoselective Approaches to Glycoprotein Assembly. Acc. Chem. Res. 2001, 34, 727–736. [DOI] [PubMed] [Google Scholar]
- (47).Chalker JM; Bernardes GJ; Davis BGA “Tag-and-Modify” Approach to Site-Selective Protein Modification. Acc. Chem. Res. 2011, 44, 730–741. [DOI] [PubMed] [Google Scholar]
- (48).Wright TH; Vallee MR; Davis BG From Chemical Mutagenesis to Post-Expression Mutagenesis: A 50 Year Odyssey. Angew. Chem., Int. Ed. 2016, 55, 5896–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Gijsen HJ; Qiao L; Fitz W; Wong CH Recent Advances in the Chemoenzymatic Synthesis of Carbohydrates and Carbohydrate Mimetics. Chem. Rev. 1996, 96, 443–474. [DOI] [PubMed] [Google Scholar]
- (50).Koeller KM; Wong CH Synthesis of Complex Carbohydrates and Glycoconjugates: Enzyme-Based and Programmable One-Pot Strategies. Chem. Rev. 2000, 100, 4465–4494. [DOI] [PubMed] [Google Scholar]
- (51).Hanson S; Best M; Bryan MC; Wong CH Chemo-enzymatic Synthesis of Oligosaccharides and Glycoproteins. Trends Biochem. Sci. 2004, 29, 656–663. [DOI] [PubMed] [Google Scholar]
- (52).Bennett CS; Wong CH Chemoenzymatic Approaches to Glycoprotein Synthesis. Chem. Soc. Rev. 2007, 36, 1227–1238. [DOI] [PubMed] [Google Scholar]
- (53).Koeller KM; Wong CH Emerging Themes in Medicinal Glycoscience. Nat. Biotechnol. 2000, 18, 835–841. [DOI] [PubMed] [Google Scholar]
- (54).Muthana S; Cao H; Chen X Recent Progress in Chemical and Chemoenzymatic Synthesis of Carbohydrates. Curr. Opin. Chem. Biol. 2009, 13, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Wang LX Chemoenzymatic Synthesis of Glycopeptides and Glycoproteins through Endoglycosidase-Catalyzed Transglycosylation. Carbohydr. Res. 2008, 343, 1509–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Rich JR; Withers SG Emerging Methods for the Production of Homogeneous Human Glycoproteins. Nat. Chem. Biol. 2009, 5, 206–215. [DOI] [PubMed] [Google Scholar]
- (57).Wang LX; Huang W Enzymatic Transglycosylation for Glycoconjugate Synthesis. Curr. Opin. Chem. Biol. 2009, 13, 592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Bernardes GJ; Castagner B; Seeberger PH Combined Approaches to the Synthesis and Study of Glycoproteins. ACS Chem. Biol. 2009, 4, 703–713. [DOI] [PubMed] [Google Scholar]
- (59).Wang LX; Lomino JV Emerging Technologies for Making Glycan-Defined Glycoproteins. ACS Chem. Biol. 2012, 7, 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Wu Z; Guo Z Sortase-Mediated Transpeptidation for Site-Specific Modification of Peptides, Glycopeptides, and Proteins. J. Carbohydr. Chem. 2012, 31, 48–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Overkleeft HS; Seeberger P Chemoenzymatic Synthesis of Glycans and Glycoconjugates In Essentials of Glycobiology, 3rd ed.; Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, et al. , Eds.; Cold Spring Harbor: New York, 2015; Chapter 54. [PubMed] [Google Scholar]
- (62).Li C; Wang LX Endoglycosidases for the Synthesis of Polysaccharides and Glycoconjugates. Adv. Carbohydr. Chem. Biochem. 2016, 73, 73–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Stanley P Glycosylation Engineering. Glycobiology 1992, 2, 99–107. [DOI] [PubMed] [Google Scholar]
- (64).Wildt S; Gerngross TU The Humanization of N-Glycosylation Pathways in Yeast. Nat. Rev. Microbiol. 2005,3,119–128. [DOI] [PubMed] [Google Scholar]
- (65).Hamilton SR; Gerngross TU Glycosylation Engineering in Yeast: The Advent of Fully Humanized Yeast. Curr. Opin. Biotechnol. 2007, 18, 387–392. [DOI] [PubMed] [Google Scholar]
- (66).Jacobs PP; Geysens S; Vervecken W; Contreras R; Callewaert N Engineering Complex-Type N-Glycosylation in Pichia Pastoris Using Glycoswitch Technology. Nat. Protoc. 2009, 4, 58–70. [DOI] [PubMed] [Google Scholar]
- (67).Castilho A; Steinkellner H Glyco-Engineering in Plants to Produce Human-Like N-Glycan Structures. Biotechnol. J. 2012, 7, 1088–1098. [DOI] [PubMed] [Google Scholar]
- (68).Vogl T; Hartner FS; Glieder A New Opportunities by Synthetic Biology for Biopharmaceutical Production in Pichia Pastoris. Curr. Opin. Biotechnol. 2013, 24, 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Clausen H; Wandall HH; Steentoft C; Stanley P; Schnaar RL Glycosylation Engineering In Essentials of Glycobiology, 3rd ed.; Varki A, Cummings RD, Esko JD, Stanley P., Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, et al. , Eds.; Cold Spring Harbor: New York, 2015; Chapter 56. [Google Scholar]
- (70).Weerapana E; Imperiali B Asparagine-Linked Protein Glycosylation: From Eukaryotic to Prokaryotic Systems. Glycobiology 2006, 16, 91R–101R. [DOI] [PubMed] [Google Scholar]
- (71).Abu-Qarn M; Eichler J; Sharon N Not Just for Eukarya Anymore: Protein Glycosylation in Bacteria and Archaea. Curr. Opin. Struct. Biol. 2008, 18, 544–550. [DOI] [PubMed] [Google Scholar]
- (72).Li Y; Li X; Chen X In Carbohydrate Chemistry: State oftheArt and Challenges for Drug Development; Cipolla L, Ed.; World Scientific Publishing Co.: UK, 2015; Chapter 1. [Google Scholar]
- (73).Spiro RG Protein Glycosylation: Nature, Distribution, Enzymatic Formation, and Disease Implications of Glycopeptide Bonds. Glycobiology 2002, 12, 43R–56R [DOI] [PubMed] [Google Scholar]
- (74).Dutta D; Mandal C; Mandal C Unusual Glycosylation of Proteins: Beyond the Universal Sequon and Other Amino Acids. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 3096–3108. [DOI] [PubMed] [Google Scholar]
- (75).Hart GW; Slawson C; Ramirez-Correa G; Lagerlof O Cross Talk between O-Glcnacylation and Phosphorylation: Roles in Signaling, Transcription, and Chronic Disease. Annu. Rev. Biochem. 2011, 80, 825–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Zachara NE; Hart GW The Emerging Significance of O-Glcnac in Cellular Regulation. Chem. Rev. 2002, 102, 431–438. [DOI] [PubMed] [Google Scholar]
- (77).Moremen KW; Ramiah A; Stuart M; Steel J; Meng L; Forouhar F; Moniz HA; Gahlay G; Gao Z; Chapla D; et al. Expression System for Structural and Functional Studies of Human Glycosylation Enzymes. Nat. Chem. Biol. 2018, 14, 156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Umana P .; Jean-Mairet J; Moudry R; Amstutz H; Bailey J Engineered Glycoforms of an Antineuroblastoma Iggl with Optimized Antibody-Dependent Cellular Cytotoxic Activity. Nat. Biotechnol 1999, 17, 176–180. [DOI] [PubMed] [Google Scholar]
- (79).Yamane-Ohnuki N; Kinoshita S; Inoue-Urakubo M; Kusunoki M; Iida S; Nakano R; Wakitani M; Niwa R; Sakurada M; Uchida K; et al. Establishment of Fut8 Knockout Chinese Hamster Ovary Cells: An Ideal Host Cell Line for Producing Completely Defucosylated Antibodies with Enhanced Antibody-Dependent Cellular Cytotoxicity. Biotechnol. Bioeng. 2004, 87, 614–622. [DOI] [PubMed] [Google Scholar]
- (80).Stanley P; Sundaram S; Tang J; Shi S Molecular Analysis of Three Gain-of-Function CHO Mutants That Add the Bisecting GlcNAc to N-Glycans. Glycobiology 2005, 15, 43–53. [DOI] [PubMed] [Google Scholar]
- (81).Cox KM; Sterling JD; Regan JT; Gasdaska JR; Frantz KK; Peele CG; Black A; Passmore D; Moldovan-Loomis C; Srinivasan M; et al. Glycan Optimization of a Human Monoclonal Antibody in the Aquatic Plant Lemna Minor. Nat. Biotechnol. 2006, 24, 1591–1597. [DOI] [PubMed] [Google Scholar]
- (82).Strasser R; Castilho A; Stadlmann J; Kunert R; Quendler H; Gattinger P; Jez J; Rademacher T; Altmann F; Mach L; et al. Improved Virus Neutralization by Plant-Produced Anti-Hiv Antibodies with a Homogeneous Beta1,4-Galactosylated N-Glycan Profile. J. Biol. Chem. 2009, 284, 20479–20485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Li H; Sethuraman N; Stadheim TA; Zha D; Prinz B; Ballew N; Bobrowicz P; Choi BK; Cook WJ; Cukan M; et al. Optimization of Humanized Iggs in Glycoengineered Pichia Pastoris. Nat. Biotechnol. 2006, 24, 210–215. [DOI] [PubMed] [Google Scholar]
- (84).Zhou Q; Shankara S; Roy A; Qiu H; Estes S; McVie-Wylie A; Culm-Merdek K; Park A; Pan C; Edmunds T Development of a Simple and Rapid Method for Producing Non-Fucosylated Oligoman-nose Containing Antibodies with Increased Effector Function. Biotechnol. Bioeng. 2008, 99, 652–665. [DOI] [PubMed] [Google Scholar]
- (85).Spicer CD; Davis BG Selective Chemical Protein Modification. Nat. Commun. 2014, 5, 4740. [DOI] [PubMed] [Google Scholar]
- (86).Aussedat B; Fasching B; Johnston E; Sane N; Nagorny P; Danishefsky SJ Total Synthesis of the Alpha-Subunit of Human Glycoprotein Hormones: Toward Fully Synthetic Homogeneous Human Follicle-Stimulating Hormone. J. Am. Chem. Soc. 2012, 134, 3532–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Nagorny P; Sane N; Fasching B; Aussedat B; Danishefsky SJ Probing the Frontiers of Glycoprotein Synthesis: The Fully Elaborated Beta-Subunit of the Human Follicle-Stimulating Hormone. Angew. Chem., Int. Ed. 2012, 51, 975–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Sakamoto I; Tezuka K; Fukae K; Ishii K; Taduru K; Maeda M; Ouchi M; Yoshida K; Nambu Y; Igarashi J; et al. Chemical Synthesis of Homogeneous Human Glycosyl-Interferon-Beta That Exhibits Potent Antitumor Activity in Vivo. J.Am. Chem. Soc. 2012,134, 5428–5431. [DOI] [PubMed] [Google Scholar]
- (89).Piontek C; Varon Silva D; Heinlein C; Pohner C; Mezzato S; Ring P; Martin A; Schmid FX; Unverzagt C Semisynthesis of a Homogeneous Glycoprotein Enzyme: Ribonuclease C: Part 2. Angew. Chem., Int. Ed. 2009, 48, 1941–1945. [DOI] [PubMed] [Google Scholar]
- (90).Piontek C; Ring P; Harjes O; Heinlein C; Mezzato S; Lombana N; Pohner C; Puttner M; Varon Silva D; Martin A; et al. Semisynthesis of a Homogeneous Glycoprotein Enzyme: Ribonuclease C: Part 1. Angew. Chem., Int. Ed. 2009, 48, 1936–1940. [DOI] [PubMed] [Google Scholar]
- (91).Hirano K; Macmillan D; Tezuka K; Tsuji T; Kajihara Y Design and Synthesis of a Homogeneous Erythropoietin Analogue with Two Human Complex-Type Sialyloligosaccharides: Combined Use of Chemical and Bacterial Protein Expression Methods. Angew. Chem., Int. Ed. 2009, 48, 9557–9560. [DOI] [PubMed] [Google Scholar]
- (92).Wang P; Dong S; Shieh JH; Peguero E; Hendrickson R; Moore MA; Danishefsky SJ Erythropoietin Derived by Chemical Synthesis. Science 2013, 342, 1357–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Wang P; Dong S; Brailsford JA; Iyer K; Townsend SD; Zhang Q; Hendrickson RC; Shieh J; Moore MA; Danishefsky SJ At Last: Erythropoietin as a Single Glycoform. Angew. Chem., Int. Ed. 2012, 51, 11576–11584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Murakami M; Okamoto R; Izumi M; Kajihara Y Chemical Synthesis ofan Erythropoietin Glycoform Containing a Complex-Type Disialyloligosaccharide. Angew. Chem., Int. Ed. 2012, 51, 3567–3572. [DOI] [PubMed] [Google Scholar]
- (95).Murakami M; Kiuchi T; Nishihara M; Tezuka K; Okamoto R; Izumi M; Kajihara Y Chemical Synthesis of Erythropoietin Glycoforms for Insights into the Relationship between Glycosylation Pattern and Bioactivity. Sci. Adv 2016, 2, e1500678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Wacker M; Linton D; Hitchen PG; Nita-Lazar M; Haslam SM; North SJ; Panico M; Morris HR; Dell A; Wren BW; et al. N-Linked Glycosylation in Campylobacter Jejuni and Its Functional Transfer into E. Coli. Science 2002, 298, 1790–1793. [DOI] [PubMed] [Google Scholar]
- (97).Feldman MF; Wacker M; Hernandez M; Hitchen PG; Marolda CL; Kowarik M; Morris HR; Dell A; Valvano MA; Aebi M Engineering N-Linked Protein Glycosylation with Diverse O Antigen Lipopolysaccharide Structures in Escherichia Coli. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 3016–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Glover KJ; Weerapana E; Numao S; Imperiali B Chemoenzymatic Synthesis of Glycopeptides with Pglb, a Bacterial Oligosaccharyl Transferase from Campylobacter Jejuni. Chem. Biol. 2005, 12, 1311–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Schwarz F; Fan YY; Schubert M; Aebi M Cytoplasmic N-Glycosyltransferase of Actinobacillus Pleuropneumoniae Is an Inverting Enzyme and Recognizes the Nx(S/T) Consensus Sequence. J. Biol. Chem. 2011, 286, 35267–35274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Lizak C; Gerber S; Numao S; Aebi M; Locher KP X-Ray Structure of a Bacterial Oligosaccharyltransferase. Nature 2011, 474, 350–355. [DOI] [PubMed] [Google Scholar]
- (101).Naegeli A; Michaud G; Schubert M; Lin CW; Lizak C; Darbre T; Reymond JL; Aebi M Substrate Specificity of Cytoplasmic N-Glycosyltransferase. J. Biol. Chem. 2014, 289, 24521–24532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Warnock D; Bai X; Autote K; Gonzales J; Kinealy K; Yan B; Qian J; Stevenson T; Zopf D; Bayer RJ In Vitro Galactosylation of Human Igg at 1 kg Scale Using Recombinant Galactosyltransferase. Biotechnol. Bioeng. 2005, 92, 831–842. [DOI] [PubMed] [Google Scholar]
- (103).Witte K; Sears P; Martin R; Wong CH Enzymatic Glycoprotein Synthesis: Preparation of Ribonuclease Glycoforms Via Enzymatic Glycopeptide Condensation and Glycosylation. J. Am. Chem. Soc. 1997, 119, 2114–2118. [Google Scholar]
- (104).Gundersen MT; Keillor JW; Pelletier JN Microbial Transglutaminase Displays Broad Acyl-Acceptor Substrate Specificity. Appi. Microbiol. Biotechnol. 2014, 98, 219–230. [DOI] [PubMed] [Google Scholar]
- (105).Wu Z; Guo X; Gao J; Guo Z Sortase a-Mediated Chemoenzymatic Synthesis of Complex Glycosylphosphatidylinosi-tol-Anchored Protein. Chem. Commun. {Cambridge, U. K.) 2013, 49, 11689–11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Wu Z; Guo X; Wang Q; Swarts BM; Guo Z Sortase a-Catalyzed Transpeptidation of Glycosylphosphatidylinositol Derivatives for Chemoenzymatic Synthesis of Gpi-Anchored Proteins. J. Am. Chem. Soc. 2010, 132, 1567–1571. [DOI] [PubMed] [Google Scholar]
- (107).Guo X; Wang Q; Swarts BM; Guo Z Sortase-Catalyzed Peptide-Glycosylphosphatidylinositol Analogue Ligation. J. Am. Chem. Soc. 2009, 131, 9878–9879. [DOI] [PubMed] [Google Scholar]
- (108).Wu Z; Guo X; Guo Z Sortase a-Catalyzed Peptide Cyclization for the Synthesis of Macrocyclic Peptides and Glycopep-tides. Chem. Commun. {Cambridge, U. K.) 2011, 47, 9218–9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Guo X; Wu Z; Guo Z New Method for Site-Specific Modification of Liposomes with Proteins Using Sortase a-Mediated Transpeptidation. Bioconjugate Chem. 2012, 23, 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Samantaray S; Marathe U; Dasgupta S; Nandicoori VK; Roy RP Peptide-Sugar Ligation Catalyzed by Transpeptidase Sortase: A Facile Approach to Neoglycoconjugate Synthesis. J. Am. Chem. Soc. 2008, 130,2132–2133. [DOI] [PubMed] [Google Scholar]
- (111).Matsushita T; Sadamoto R; Ohyabu N; Nakata H; Fumoto M; Fujitani N; Takegawa Y; Sakamoto T; Kurogochi M; Hinou H; et al. Functional Neoglycopeptides: Synthesis and Characterization of a New Class of Muc1 Glycoprotein Models Having Core 2-Based O-Glycan and Complex-Type N-Glycan Chains. Biochemistry 2009, 48, 11117–11133. [DOI] [PubMed] [Google Scholar]
- (112).Fang T; Van Elssen C; Duarte JN; Guzman JS; Chahal JS; Ling J; Ploegh HL Targeted Antigen Delivery by an Anti-Class Ii Mhc Vhh Elicits Focused Alphamuc1(Tn) Immunity. Chem. Sci 2017, 8, 5591–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Ramos D; Rollin P; Klaffke W Chemoenzymatic Synthesis of Neoglycopeptides: Application to an Alpha-Gal-Terminated Neo-glycopeptide. J. Org. Chem. 2001, 66, 2948–2956. [DOI] [PubMed] [Google Scholar]
- (114).Hong PK; Gottardi D; Ndagijimana M; Betti M Glycation and Transglutaminase Mediated Glycosylation of Fish Gelatin Peptides with Glucosamine Enhance Bioactivity. Food Chem. 2014, 142, 285–293. [DOI] [PubMed] [Google Scholar]
- (115).Ramos D; Rollin P; Klaffke W Enzymatic Synthesis of Neoglycopeptide Building Blocks. Angew. Chem., Int. Ed. 2000, 39, 396–39S. [PubMed] [Google Scholar]
- (116).Hancock SM; Vaughan MD; Withers SG Engineering of Glycosidases and Glycosyltransferases. Curr. Opin. Chem. Biol. 2006, 10,509–519. [DOI] [PubMed] [Google Scholar]
- (117).Koshland DE Stereochemistry and the Mechanism of Enzymatic Reactions. Biological Reviews 1953, 28, 416–436. [Google Scholar]
- (118).Kobayashi S; Kashiwa K; Kawasaki T; Shoda S Novel Method for Polysaccharide Synthesis Using an Enzyme: The First in Vitro Synthesis of Cellulose Via a Nonbiosynthetic Path Utilizing Cellulase as Catalyst. J. Am. Chem. Soc. 1991, 113, 3079–3084 [Google Scholar]
- (119).Shoda S; Uyama H; Kadokawa J; Kimura S; Kobayashi S Enzymes as Green Catalysts for Precision Macromolecular Synthesis. Chem. Rev. 2016, 116, 2307–2413. [DOI] [PubMed] [Google Scholar]
- (120).Crout DH; Vic G Glycosidases and Glycosyl Transferases in Glycoside and Oligosaccharide Synthesis. Curr. Opin. Chem. Biol. 1998, 2, 98–111. [DOI] [PubMed] [Google Scholar]
- (121).Scigelova M; Singh S; Crout DHG Glycosidases-a Great Synthetic Tool. J. Mol. Catal. B: Enzym. 1999, 6, 4S3–494. [Google Scholar]
- (122).Perugino G; Trincone A; Rossi M; Moracci M Oligosaccharide Synthesis by Glycosynthases. Trends Biotechnol. 2004, 22, 31–37. [DOI] [PubMed] [Google Scholar]
- (123).Shaikh FA; Withers SG Teaching Old Enzymes New Tricks: Engineering and Evolution of Glycosidases and Glycosyl Transferases for Improved Glycoside Synthesis. Biochem. Cell Biol. 2008, 86, 169–177. [DOI] [PubMed] [Google Scholar]
- (124).Cobucci-Ponzano B; Moracci M Glycosynthases as Tools for the Production of Glycan Analogs of Natural Products. Nat. Prod. Rep. 2012, 29, 697–709. [DOI] [PubMed] [Google Scholar]
- (125).Armstrong Z; Withers SG Synthesis of Glycans and Glycopolymers through Engineered Enzymes. Biopolymers 2013, 99, 666–674. [DOI] [PubMed] [Google Scholar]
- (126).Cobucci-Ponzano B; Strazzulli A; Rossi M; Moracci M Glycosynthases in Biocatalysis. Adv. Synth. Catal 2011, 353, 2284–2300. [Google Scholar]
- (127).Bojarova P; Kren V Glycosidases: A Key to Tailored Carbohydrates. Trends Biotechnol. 2009, 27, 199–209. [DOI] [PubMed] [Google Scholar]
- (128).MacKenzie LF; Wang Q; Warren RAJ; Withers SG Glycosynthases: Mutant Glycosidases for Oligosaccharide Synthesis. J. Am. Chem. Soc. 1998, 120, 5583–5584. [Google Scholar]
- (129).Malet C; Planas A From Beta-Glucanase to Beta-Glucansynthase: Glycosyl Transfer to Alpha-Glycosyl Fluorides Catalyzed by a Mutant Endoglucanase Lacking Its Catalytic Nucleophile. FEBS Lett. 1998, 440, 208–212. [DOI] [PubMed] [Google Scholar]
- (130).Moracci M; Trincone A; Perugino G; Ciaramella M; Rossi M Restoration of the Activity of Active-Site Mutants of the Hyperthermophilic Beta-Glycosidase from Sulfolobus Solfataricus: Dependence of the Mechanism on the Action of External Nucleophiles. Biochemistry 1998, 37, 17262–17270. [DOI] [PubMed] [Google Scholar]
- (131).Wang LX Expanding the Repertoire ofGlycosynthases. Chem. Biol. 2009, 16, 1026–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Honda Y; Kitaoka M The First Glycosynthase Derived from an Inverting Glycoside Hydrolase. J. Biol. Chem. 2006, 281 , 1426–1431. [DOI] [PubMed] [Google Scholar]
- (133).Honda Y; Fushinobu S; Hidaka M; Wakagi T; Shoun H; Taniguchi H; Kitaoka M Alternative Strategy for Converting an Inverting Glycoside Hydrolase into a Glycosynthase. Glycobiology 2008, 18, 325–330. [DOI] [PubMed] [Google Scholar]
- (134).Ohnuma T; Fukuda T; Dozen S; Honda Y; Kitaoka M; Fukamizo T A Glycosynthase Derived from an Inverting Gh19 Chitinase from the Moss Bryum Coronatum. Biochem. J. 2012, 444, 437–443. [DOI] [PubMed] [Google Scholar]
- (135).Wada J; Honda Y; Nagae M; Kato R; Wakatsuki S; Katayama T; Taniguchi H; Kumagai H; Kitaoka M; Yamamoto K 1,2-Alpha-L-Fucosynthase: A Glycosynthase Derived from an Inverting Alpha-Glycosidase with an Unusual Reaction Mechanism. FEBS Lett. 2008, 582, 3739–3743. [DOI] [PubMed] [Google Scholar]
- (136).Sugiyama Y; Katoh T; Honda Y; Gotoh A; Ashida H; Kurihara S; Yamamoto K; Katayama T Application Study of 1,2-Alpha-L-Fucosynthase: Introduction of Fucalpha1–2 gal Disaccharide Structures on N-Glycan, Ganglioside, and Xyloglucan Oligosaccharide. Biosci.j Biotechnol., Biochem 2017, 81, 283–291. [DOI] [PubMed] [Google Scholar]
- (137).Sugiyama Y; Gotoh A; Katoh T; Honda Y; Yoshida E; Kurihara S; Ashida H; Kumagai H; Yamamoto K; Kitaoka M; et al. Introduction of H-Antigens into Oligosaccharides and Sugar Chains of Glycoproteins Using Highly Efficient 1,2-Alpha-L-Fucosynthase. Glycobiology 2016, 26, 1235–1247. [DOI] [PubMed] [Google Scholar]
- (138).Nashiru O; Zechel DL; Stoll D; Mohammadzadeh T; Warren RA; Withers SG Beta-Mannosynthase: Synthesis of Beta-Mannosides with a Mutant Beta-Mannosidase We Thank the Protein Engineering Network of Centres of Excellence of Canada and the Natural Sciences and Engineering Research Council of Canada for Support. D.L.Z. Thanks the Izaak Walton Killam Foundation for a Fellowship. Angew. Chem., Int. Ed. 2001, 40, 417–420. [PubMed] [Google Scholar]
- (139).Jakeman DL; Sadeghi-Khomami A A Beta-(1,2)-Glycosynthase and an Attempted Selection Method for the Directed Evolution of Glycosynthases. Biochemistry 2011, 50, 10359–10366. [DOI] [PubMed] [Google Scholar]
- (140).Vaughan MD; Johnson K; DeFrees S; Tang X; Warren RA ; Withers SG Glycosynthase-Mediated Synthesis of Glycosphingo-lipids. J. Am. Chem. Soc. 2006, 128, 6300–6301. [DOI] [PubMed] [Google Scholar]
- (141).Spadiut O; Ibatullin FM; Peart J; Gullfot F; Martinez-Fleites C; Ruda M; Xu C; Sundqvist G; Davies GJ; Brumer H Building Custom Polysaccharides in Vitro with an Efficient, Broad-Specificity Xyloglucan Glycosynthase and a Fucosyltransferase. J. Am. Chem. Soc. 2011, 133, 10892–10900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Fort S; Boyer V; Greffe L; Davies GJ; Moroz O; Christiansen L; Schulein M; Cottaz S; Driguez H Highly Efficient Synthesis of B(1 4)-Oligo- and -Polysaccharides Using a Mutant Cellulase. J.Am. Chem. Soc. 2000, 122, 5429–5437. [Google Scholar]
- (143).Yang M; Davies GJ; Davis BG A Glycosynthase Catalyst for the Synthesis of Flavonoid Glycosides. Angew. Chem., Int. Ed. 2007, 46, 3885–388S. [DOI] [PubMed] [Google Scholar]
- (144).Vasur J; Kawai R; Jonsson KHM; Widmalm G; Engstrom A; Frank M; Andersson E; Hansson H; Forsberg Z; Igarashi K; et al. Synthesis of Cyclic Beta-Glucan Using Laminarinase 16a Glycosynthase Mutant from the Basidiomycete Phanerochaete Chrysosporium.J.Am. Chem. Soc. 2010, 132, 1724–1730. [DOI] [PubMed] [Google Scholar]
- (145).Jahn M; Stoll D; Warren RA; Szabo L; Singh P; Gilbert HJ; Ducros VM; Davies GJ; Withers SG Expansion of the Glycosynthase Repertoire to Produce Defined Manno-Oligosacchar-ides. Chem. Commun. (Cambridge, U. K.) 2003, 1327–1329. [DOI] [PubMed] [Google Scholar]
- (146).Cobucci-Ponzano B; Conte F; Bedini E; Corsaro MM; Parrilli M; Sulzenbacher G; Lipski A; Dal Piaz F; Lepore L; Rossi M; et al. Beta-Glycosyl Azides as Substrates for Alpha-Glycosynthases: Preparation of Efficient Alpha-L-Fucosynthases. Chem. Biol. 2009, 16, 1097–1108. [DOI] [PubMed] [Google Scholar]
- (147).Yamamoto K; Davis BG Creation of an Alpha-Mannosynthase from a Broad Glycosidase Scaffold. Angew. Chem., Int. Ed. 2012, 51, 7449–7453. [DOI] [PubMed] [Google Scholar]
- (148).Rich JR; Withers SG A Chemoenzymatic Total Synthesis of the Neurogenic Starfish Ganglioside Llg-3 Using an Engineered and Evolved Synthase. Angew. Chem., Int. Ed. 2012, 51, 8640–8643. [DOI] [PubMed] [Google Scholar]
- (149).Li C; Kim YW Characterization of a Galactosynthase Derived from Bacillus Circulans Beta-Galactosidase: Facile Synthesis of D-Lacto- and D-Galacto-N-Bioside. ChemBioChem 2014,15,522–526. [DOI] [PubMed] [Google Scholar]
- (150).Henze M; Schmidtke S; Hoffmann N; Steffens H; Pietruszka J; Elling L Combination of Glycosyltransferases and a Glycosynthase in Sequential and One-Pot Reactions for the Synthesis of Type 1 and Type 2 N-Acetyllactosamine Oligomers. ChemCatChem 2015, 7, 3131–3139. [Google Scholar]
- (151).Perugino G; Trincone A; Giordano A; van der Oost J; Kaper T; Rossi M; Moracci M Activity of Hyperthermophilic Glycosynthases Is Significantly Enhanced at Acidic Ph. Biochemistry 2003, 42, 8484–8493. [DOI] [PubMed] [Google Scholar]
- (152).Cobucci-Ponzano B; Zorzetti C; Strazzulli A; Carillo S; Bedini E; Corsaro MM; Comfort DA; Kelly RM; Rossi M; Moracci M A Novel Alpha-D-Galactosynthase from Thermotoga Maritima Converts Beta-D-Galactopyranosyl Azide to Alpha-Galacto-Oligosaccharides. Glycobiology 2011, 21, 448–456. [DOI] [PubMed] [Google Scholar]
- (153).Tolborg JF; Petersen L; Jensen KJ; Mayer C; Jakeman DL; Warren RA; Withers SG Solid-Phase Oligosaccharide and Glycopeptide Synthesis Using Glycosynthases. J. Org. Chem. 2002, 67, 4143–4149. [DOI] [PubMed] [Google Scholar]
- (154).Mullegger J; Chen HM; Warren RA; Withers SG Glycosylation of a Neoglycoprotein by Using Glycosynthase and Thioglycoligase Approaches: The Generation of a Thioglycoprotein. Angew. Chem., Int. Ed. 2006, 45, 2585–2588. [DOI] [PubMed] [Google Scholar]
- (155).Kim YW; Lee SS; Warren RA; Withers SG Directed Evolution of a Glycosynthase from Agrobacterium Sp. Increases Its Catalytic Activity Dramatically and Expands Its Substrate Repertoire. J. Biol. Chem. 2004, 279, 42787–42793. [DOI] [PubMed] [Google Scholar]
- (156).Lin H; Tao H; Cornish VW Directed Evolution of a Glycosynthase Via Chemical Complementation. J. Am. Chem. Soc. 2004, 126, 15051–15059. [DOI] [PubMed] [Google Scholar]
- (157).Ben-David A; Shoham G; Shoham Y A Universal Screening Assay for Glycosynthases: Directed Evolution of Glycosynthase Xynb2(E335g) Suggests a General Path to Enhance Activity. Chem. Biol. 2008, 15, 546–551. [DOI] [PubMed] [Google Scholar]
- (158).Kone FM; Le Bechec M; Sine JP; Dion M; Tellier C Digital Screening Methodology for the Directed Evolution of Transglycosidases. Protein Eng. Des. Sel. 2009, 22, 37–44. [DOI] [PubMed] [Google Scholar]
- (159).Hancock SM; Rich JR; Caines ME; Strynadka NC; Withers SG Designer Enzymes for Glycosphingolipid Synthesis by Directed Evolution. Nat. Chem. Biol. 2009, 5, 508–514. [DOI] [PubMed] [Google Scholar]
- (160).Iglesias-Fernandez J; Hancock SM; Lee SS; Khan M; Kirkpatrick J; Oldham NJ; McAuley K; Fordham-Skelton A; Rovira C; Davis BG A Front-Face ‘Sni Synthase’ Engineered from a Retaining ‘Double-Sn2’ Hydrolase. Nat. Chem. Biol. 2017, 13, 874–881. [DOI] [PubMed] [Google Scholar]
- (161).Sakurama H; Fushinobu S; Hidaka M; Yoshida E; Honda Y; Ashida H; Kitaoka M; Kumagai H; Yamamoto K; Katayama T 1,3– 1,4-Alpha-L-Fucosynthase That Specifically Introduces Lewis a/X Antigens into Type-1/2 Chains. J. Biol. Chem. 2012, 287, 16709–16719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Cobucci-Ponzano B; Zorzetti C; Strazzulli A; Bedini E; Corsaro MM; Sulzenbacher G; Rossi M; Moracci M Exploitation of B-Glycosyl Azides for the Preparation of A-Glycosynthases. Biocatal. Biotransform. 2012, 30, 288–295. [Google Scholar]
- (163).Jahn M; Marles J; Warren RA; Withers SG Thioglycoligases: Mutant Glycosidases for Thioglycoside Synthesis. Angew. Chem., Int. Ed. 2003, 42, 352–354. [DOI] [PubMed] [Google Scholar]
- (164).Kim YW; Lovering AL; Chen H; Kantner T; McIntosh LP; Strynadka NC; Withers SG Expanding the Thioglycoligase Strategy to the Synthesis of Alpha-Linked Thioglycosides Allows Structural Investigation of the Parent Enzyme/Substrate Complex. J. Am. Chem. Soc. 2006, 128, 2202–2203. [DOI] [PubMed] [Google Scholar]
- (165).Kim YW; Zhang R; Chen H; Withers SG O-Glycoligases, a New Category of Glycoside Bond-Forming Mutant Glycosidases, Catalyse Facile Syntheses of Isoprimeverosides. Chem. Commun. (Cambridge, U. K.) 2009, 46, 8725–8727. [DOI] [PubMed] [Google Scholar]
- (166).Mullegger J; Jahn M; Chen HM; Warren RA; Withers SG Engineering of a Thioglycoligase: Randomized Mutagenesis of the Acid-Base Residue Leads to the Identification of Improved Catalysts. Protein Eng. Des. Sel. 2005, 18, 33–40. [DOI] [PubMed] [Google Scholar]
- (167).Mullegger J; Chen HM; Chan WY; Reid SP; Jahn M; Warren RA; Salleh HM; Withers SG Thermostable Glycosynthases and Thioglycoligases Derived from Thermotoga Maritima Beta-Glucuronidase. ChemBioChem 2006, 7, 1028–1030. [DOI] [PubMed] [Google Scholar]
- (168).Armstrong Z; Reitinger S; Kantner T; Withers SG Enzymatic Thioxyloside Synthesis: Characterization ofThioglycoligase Variants Identified from a Site-Saturation Mutagenesis Library of Bacillus Circulans Xylanase. ChemBioChem 2010, 11, 533–538. [DOI] [PubMed] [Google Scholar]
- (169).Tshililo NO; Strazzulli A; Cobucci-Ponzano B; Maurelli L; Iacono R; Bedini E; Corsaro MM; Strauss E; Moracci M The A-Thioglycoligase Derived from a Gh89 A-N-Acetylglucosaminidase Synthesises A-N-Acetylglucosamine-Based Glycosides of Biomedical Interest. Adv. Synth. Catal. 2017, 359, 663–676. [Google Scholar]
- (170).Li C; Ahn HJ; Kim JH; Kim YW Transglycosylation of Engineered Cyclodextrin Glucanotransferases as O-Glycoligases. Carbohydr. Polym. 2014, 99, 39–46. [DOI] [PubMed] [Google Scholar]
- (171).Li C; Kim J-H; Kim Y-W Alpha-Thioglycoligase-Based Synthesis of O-Aryl Alpha-Glycosides as Chromogenic Substrates for Alpha-Glycosidases. J. Mol. Catal. B: Enzym. 2013, 87, 24–29. [Google Scholar]
- (172).Ahn HJ; Li C; Cho HB; Park S; Chang PS; Kim YW Enzymatic Synthesis of 3-O-Alpha-Maltosyl-L-Ascorbate Using an Engineered Cyclodextrin Glucanotransferase. Food Chem. 2015, 169, 366–371. [DOI] [PubMed] [Google Scholar]
- (173).Iwamoto S; Kasahara Y; Yoshimura Y; Seko A; Takeda Y; Ito Y; Totani K; Matsuo I Endo-Alpha-Mannosidase-Catalyzed Transglycosylation. ChemBioChem 2017, 18, 1376–1378. [DOI] [PubMed] [Google Scholar]
- (174).Li C; Zhu S; Ma C; Wang LX Designer Alpha1,6-Fucosidase Mutants Enable Direct Core Fucosylation of Intact N-Glycopeptides and N-Glycoproteins. J. Am. Chem. Soc. 2017, 139, 15074–15087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Yang Q; Zhang R; Cai H; Wang LX Revisiting the Substrate Specificity of Mammalian Alpha1,6-Fucosyltransferase Reveals That It Catalyzes Core Fucosylation of N-Glycans Lacking Alpha1,3-Arm Glcnac. J. Biol Chem. 2017, 292, 14796–14803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Ducros VM; Tarling CA; Zechel DL; Brzozowski AM; Frandsen TP; von Ossowski I; Schulein M; Withers SG; Davies GJ Anatomy of Glycosynthesis: Structure and Kinetics of the Humicola Insolens Cel7b E197a and E197s Glycosynthase Mutants. Chem. Biol. 2003, 10, 619–628. [DOI] [PubMed] [Google Scholar]
- (177).Umekawa M; Huang W; Li B; Fujita K; Ashida H; Wang LX; Yamamoto K Mutants of Mucor Hiemalis Endo-Beta-N-Acetylglucosaminidase Show Enhanced Transglycosylation and Gly-cosynthase-Like Activities. J. Biol. Chem. 2008, 283, 4469–4479. [DOI] [PubMed] [Google Scholar]
- (178).Huang W; Li C; Li B; Umekawa M; Yamamoto K; Zhang X; Wang LX Glycosynthases Enable a Highly Efficient Chemo-enzymatic Synthesis of N-Glycoproteins Carrying Intact Natural N-Glycans. J. Am. Chem. Soc. 2009, 131, 2214–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Fan SQ; Huang W; Wang LX Remarkable Trans-glycosylation Activity of Glycosynthase Mutants of Endo-D, an Endo-Beta-N-Acetylglucosaminidase from Streptococcus Pneumoniae. J. Biol. Chem. 2012, 287, 11272–11281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Higuchi Y; Eshima Y; Huang Y; Kinoshita T; Sumiyoshi W; Nakakita SI; Takegawa K Highly Efficient Transglycosylation of Sialo-Complex-Type Oligosaccharide Using Coprinopsis Cinerea Endoglycosidase and Sugar Oxazoline. Biotechnol. Lett. 2017, 39, 157–162. [DOI] [PubMed] [Google Scholar]
- (181).Huang W; Giddens J; Fan SQ; Toonstra C; Wang LX Chemoenzymatic Glycoengineering of Intact Igg Antibodies for Gain of Functions. J. Am. Chem. Soc. 2012, 134, 12308–12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Li T; Tong X; Yang Q; Giddens JP; Wang LX Glycosynthase Mutants of Endoglycosidase S2 Show Potent Trans-glycosylation Activity and Remarkably Relaxed Substrate Specificity for Antibody Glycosylation Remodeling. J. Biol Chem. 2016, 291, 165GS–1651S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Giddens JP; Lomino JV; Amin MN; Wang LX Endo-F3 Glycosynthase Mutants Enable Chemoenzymatic Synthesis of Core-Fucosylated Triantennary Complex Type Glycopeptides and Glycoproteins. J. Biol. Chem. 2016, 291, 9356–937G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Heidecke CD; Ling Z; Bruce NC; Moir JW; Parsons TB ; Fairbanks AJ Enhanced Glycosylation with Mutants of Endohexosaminidase a (Endo a). ChemBioChem 2008, 9, 2G45–2G51. [DOI] [PubMed] [Google Scholar]
- (185).Tarentino AL; Plummer TH Jr. Enzymatic Deglycosylation of Asparagine-Linked Glycans: Purification, Properties, and Specificity of Oligosaccharide-Cleaving Enzymes from Flavobacterium Meningo-septicum. Methods Enzymol 1994, 230, 44–57. [DOI] [PubMed] [Google Scholar]
- (186).Kato T; Fujita K; Takeuchi M; Kobayashi K; Natsuka S; Ikura K; Kumagai H; Yamamoto K Identification of an Endo-Beta-N-Acetylglucosaminidase Gene in Caenorhabditis Elegans and Its Expression in Escherichia Coli. Glycobiology 2002, 12, 5S1–5S7. [DOI] [PubMed] [Google Scholar]
- (187).Suzuki T; Yano K; Sugimoto S; Kitajima K; Lennarz WJ; Inoue S; Inoue Y; Emori Y Endo-Beta-N-Acetylglucosaminidase, an Enzyme Involved in Processing ofFree Oligosaccharides in the Cytosol. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 9691–9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Takegawa K; Yamaguchi S; Kondo A; Iwamoto H; Nakoshi M; Kato I; Iwahara S Transglycosylation Activity of Endo-Beta-N-Acetylglucosaminidase from Arthrobacter Protophormiae. Biochem Int. 1991, 24, S49–S55. [PubMed] [Google Scholar]
- (189).Takegawa K; Yamaguchi S; Kondo A; Kato I; Iwahara S Enzymatic Synthesis of Novel Oligosaccharides by Use of Trans-glycosylation Activity of Endo-Beta-N-Acetylglucosaminidase from Arthrobacter Protophormiae. Biochem. Int. 1991, 25, S29–S35. [PubMed] [Google Scholar]
- (190).Fan JQ; Takegawa K; Iwahara S; Kondo A; Kato I; Abeygunawardana C; Lee YC Enhanced Transglycosylation Activity of Arthrobacter Protophormiae Endo-Beta-N-Acetylglucosaminidase in Media Containing Organic Solvents. J. Biol. Chem. 1995, 270, 17723–17729. [DOI] [PubMed] [Google Scholar]
- (191).Yamamoto K; Kadowaki S; Watanabe J; Kumagai H Transglycosylation Activity of Mucor Hiemalis Endo-Beta-N-Acetyl-Glucosaminidase Which Transfers Complex Oligosaccharides to the N-Acetylglucosamine Moieties of Peptides. Biochem. Biophys. Res. Commun. 1994, 203, 244–252. [DOI] [PubMed] [Google Scholar]
- (192).Fujita K; Takami H; Yamamoto K; Takegawa K Characterization of Endo-Beta-N-Acetylglucosaminidase from Alka-liphilic Bacillus Halodurans C-125. Biosci., Biotechnol., Biochem. 2004, 68, 1G59–1G66. [DOI] [PubMed] [Google Scholar]
- (193).Parsons TB; Patel MK; Boraston AB; Vocadlo DJ; Fairbanks AJ Streptococcus Pneumoniae Endohexosaminidase D; Feasibility of Using N-Glycan Oxazoline Donors for Synthetic Glycosylation of a Glcnac-Asparagine Acceptor. Org. Biomol Chem. 2010, 8, 1S61–1S69. [DOI] [PubMed] [Google Scholar]
- (194).Huang W; Li J; Wang LX Unusual Transglycosylation Activity of Flavobacterium Meningosepticum Endoglycosidases Enables Convergent Chemoenzymatic Synthesis of Core Fucosylated Complex N-Glycopeptides. ChemBioChem 2011, 12, 932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Goodfellow JJ; Baruah K; Yamamoto K; Bonomelli C; Krishna B; Harvey DJ; Crispin M; Scanlan CN; Davis BG An Endoglycosidase with Alternative Glycan Specificity Allows Broadened Glycoprotein Remodelling. J. Am. Chem. Soc. 2012, 134, SG3G–SG33. [DOI] [PubMed] [Google Scholar]
- (196).Eshima Y; Higuchi Y; Kinoshita T; Nakakita S; Takegawa K Transglycosylation Activity of Glycosynthase Mutants ofEndo-Beta-N-Acetylglucosaminidase from Coprinopsis Cinerea. PLoS One 2015, 10, eG132S59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Takegawa K; Nakoshi M; Iwahara S; Yamamoto K; Tochikura T Induction and Purification of Endo-Beta-N-Acetylglu-cosaminidase from Arthrobacter Protophormiae Grown in Ovalbumin. Appl. Environ. Microbiol. 1989, 55, 31G7–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Ling Z; Suits MD; Bingham RJ; Bruce NC; Davies GJ; Fairbanks AJ; Moir JW; Taylor EJ The X-Ray Crystal Structure of an Arthrobacter Protophormiae Endo-Beta-N-Acetylglucosaminidase Reveals a (Beta/Alpha)(S) Catalytic Domain, Two Ancillary Domains and Active Site Residues Key for Transglycosylation Activity. J. Mol. Biol. 2009, 389, 1–9. [DOI] [PubMed] [Google Scholar]
- (199).Yin J; Li L; Shaw N; Li Y; Song JK; Zhang W; Xia C; Zhang R; Joachimiak A; Zhang HC; et al. Structural Basis and Catalytic Mechanism for the Dual Functional Endo-Beta-N-Acetylglu-cosaminidase A. PLoS One 2009, 4, e4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Kadowaki S; Yamamoto K; Fujisaki M; Izumi K; Tochikura T; Yokoyama T Purification and Characterization of a Novel Fungal Endo-Beta-N-Acetylglucosaminidase Acting on Complex Oligosaccharides of Glycoproteins. Agric. Biol. Chem. 1990, 54, 97–106. [PubMed] [Google Scholar]
- (201).King SJ; Hippe KR; Weiser JN Deglycosylation of Human Glycoconjugates by the Sequential Activities of Exoglycosi-dases Expressed by Streptococcus Pneumoniae. Mol. Microbiol. 2006, 59, 961–974. [DOI] [PubMed] [Google Scholar]
- (202).Abbott DW; Macauley MS; Vocadlo DJ; Boraston AB Streptococcus Pneumoniae Endohexosaminidase D, Structural and Mechanistic Insight into Substrate-Assisted Catalysis in Family 85 Glycoside Hydrolases. J. Biol. Chem. 2009, 284, 11676–11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Murakami S; Takaoka Y; Ashida H; Yamamoto K; Narimatsu H; Chiba Y Identification and Characterization of Endo-Beta-N-Acetylglucosaminidase from Methylotrophic Yeast Oga-taea Minuta. Glycobiology 2013, 23, 736–744. [DOI] [PubMed] [Google Scholar]
- (204).Kitajima T; Jia Y; Komatsuzaki A; Cui J; Matsuzawa F; Aikawa SI; Gao XD; Chiba Y Structural Modeling and Mutagenesis of Endo-Beta-N-Acetylglucosaminidase from Ogataea Minuta Identifies the Importance of Trp295 for Hydrolytic Activity. J. Biosci Bioeng 2018, 125, 168–174. [DOI] [PubMed] [Google Scholar]
- (205).Tarentino AL; Plummer TH Jr.; Maley F The Release of Intact Oligosaccharides from Specific Glycoproteins by Endo-Beta-N-Acetylglucosaminidase H. J. Biol. Chem. 1974, 249, 818–824. [PubMed] [Google Scholar]
- (206).Trimble RB; Tarentino AL Identification of Distinct Endoglycosidase (Endo) Activities in Flavobacterium Meningosepticum: Endo F1, Endo F2, andEndo F3. Endo F1 andEndo H Hydrolyze Only High Mannose and Hybrid Glycans. J. Biol. Chem. 1991, 266, 1646–1651. [PubMed] [Google Scholar]
- (207).Garrido D; Nwosu C; Ruiz-Moyano S; Aldredge D; German JB; Lebrilla CB; Mills DA Endo-Beta-N-Acetylglucosaminidases from Infant Gut-Associated Bifidobacteria Release Complex N-Glycans from Human Milk Glycoproteins. Mol. Cell. Proteomics 2012, 11, 775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Collin M; Olsen A Endos, a Novel Secreted Protein from Streptococcus Pyogenes with Endoglycosidase Activity on Human Igg. EMBOJ. 2001, 20, 3046–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Trastoy B; Lomino JV; Pierce BG; Carter LG; Gunther S; Giddens JP; Snyder GA; Weiss TM; Weng Z; Wang LX; et al. Crystal Structure of Streptococcus Pyogenes Endos, an Immunomodulatory Endoglycosidase Specific for Human Igg Antibodies. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 6714–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Sjogren J; Cosgrave EF; Allhorn M; Nordgren M; Bjork S; Olsson F; Fredriksson S; Collin M Endos and Endos2 Hydrolyze Fc-Glycans on Therapeutic Antibodies with Different Glycoform Selectivity and Can Be Used for Rapid Quantification ofHigh-Mannose Glycans. Glycobiology 2015, 25, 1053–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Tersa M; Raich L; Albesa-Jove D; Trastoy B; Prandi J; Gilleron M; Rovira C; Guerin ME The Molecular Mechanism of Substrate Recognition and Catalysis of the Membrane Acyltransferase Pata from Mycobacteria. ACS Chem. Biol. 2018, 13, 131–140. [DOI] [PubMed] [Google Scholar]
- (212).Sjogren J; Struwe WB; Cosgrave EF; Rudd PM; Stervander M; Allhorn M; Hollands A; Nizet V; Collin M Endos2 Is a Unique and Conserved Enzyme of Serotype M49 Group a Streptococcus That Hydrolyses N-Linked Glycans on Igg and Alpha1-Acid Glycoprotein. Biochem. J. 2013, 455, 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Wang LX The Amazing Transglycosylation Activity ofEndo-Beta-N-Acetylglucosaminidases. Trends Glycosci. Glycotechnol. 2011, 23, 33–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Yamamoto K; Takegawa K Transglycosylation Activity of Endoglycosidases and Its Application. Trends Glycosci. Glycotechnol. 1997, 9, 339–354. [Google Scholar]
- (215).Takegawa K; Tabuchi M; Yamaguchi S; Kondo A; Kato I; Iwhara S Synthesis of Neoglycoproteins Using Oligosaccharide Transfer Activity with Endo-Beta-N-Acetylglucosaminidase. J. Biol. Chem. 1995, 270, 3094–3099. [DOI] [PubMed] [Google Scholar]
- (216).Haneda K; Inazu T; Yamamoto K; Kumagai H; Nakahara Y; Kobata A Transglycosylation of Intact Sialo Complex-Type Oligosaccharides to the N-Acetylglucosamine Moieties of Glycopep-tides by Mucor Hiemalis Endo-Beta-N-Acetylglucosaminidase. Carbohydr. Res. 1996, 292, 61–70. [DOI] [PubMed] [Google Scholar]
- (217).Wang LX; Fan JQ; Lee YC Chemoenzymatic Synthesis of a High-Mannose Type N-Glycopeptide Analog with C-Glycosidic Linkage. Tetrahedron Lett. 1996, 37, 1975–1978. [Google Scholar]
- (218).Wang LX; Tang M; Suzuki T; Kitajima K; Inoue Y; Inoue S; Fan JQ; Lee YC Combined Chemical and Enzymic Synthesis of a C-Glycopeptide and Its Inhibitory Activity toward Glycoamidases. J. Am. Chem. Soc. 1997, 119, 11137–11146. [Google Scholar]
- (219).Haneda K; Inazu T; Mizuno M; Iguchi R; Yamamoto K; Kumagai H; Aimoto S; Suzuki H; Noda T Chemo-Enzymatic Synthesis of Calcitonin Derivatives Containing N-Linked Oligosaccharides. Bioorg. Med. Chem. Lett. 1998, 8, 1303–1306. [DOI] [PubMed] [Google Scholar]
- (220).Mizuno M; Haneda K; Iguchi R; Muramoto I; Kawakami T; Aimoto S; Yamamoto K; Inazu T Synthesis of a Glycopeptide Containing Oligosaccharides: Chemoenzymatic Synthesis of Eel Calcitonin Analogues Having Natural N-Linked Oligosaccharides. J. Am. Chem. Soc. 1999, 121, 284–290. [Google Scholar]
- (221).O’Connor SE; Pohlmann J; Imperiali B; Saskiawan I; Yamamoto K Probing the Effect of the Outer Saccharide Residues of N-Linked Glycans on Peptide Conformation. J. Am. Chem. Soc. 2001, 123, 6187–6188. [DOI] [PubMed] [Google Scholar]
- (222).Haneda K; Inazu T; Mizuno M; Iguchi R; Tanabe H; Fujimori K; Yamamoto K; Kumagai H; Tsumori K; Munekata E Chemo-Enzymatic Synthesis of a Bioactive Peptide Containing a Glutamine-Linked Oligosaccharide and Its Characterization. Biochim. Biophys. Acta, Gen. Subj 2001, 1526, 242–248. [DOI] [PubMed] [Google Scholar]
- (223).Singh S; Ni J; Wang LX Chemoenzymatic Synthesis of High-Mannose Type Hiv-1 Gp120 Glycopeptides. Bioorg. Med. Chem. Lett. 2003, 13, 327–330. [DOI] [PubMed] [Google Scholar]
- (224).Wang LX; Song H; Liu S; Lu H; Jiang S; Ni J; Li H Chemoenzymatic Synthesis of Hiv-1 Gp41 Glycopeptides: Effects of Glycosylation on the Anti-Hiv Activity and Alpha-Helix Bundle-Forming Ability of Peptide C34. ChemBioChem 2005, 6, 1068–1074. [DOI] [PubMed] [Google Scholar]
- (225).Li H; Singh S; Zeng Y; Song H; Wang LX Chemoenzymatic Synthesis of Cd52 Glycoproteins Carrying Native N-Glycans. Bioorg. Med. Chem. Lett. 2005, 15, 895–898. [DOI] [PubMed] [Google Scholar]
- (226).Fan JQ; Quesenberry MS; Takegawa K; Iwahara S; Kondo A; Kato I; Lee YC Synthesis of Neoglycoconjugates by Transglycosylation with Arthrobacter Protophormiae Endo-Beta-N-Acetylglucosaminidase. Demonstration of a Macro-Cluster Effect for Mannose-Binding Proteins. J. Biol. Chem. 1995, 270, 17730–17735. [DOI] [PubMed] [Google Scholar]
- (227).Makimura Y; Watanabe S; Suzuki T; Suzuki Y; Ishida H; Kiso M; Katayama T; Kumagai H; Yamamoto K Chemoenzymatic Synthesis and Application of a Sialoglycopolymer with a Chitosan Backbone as a Potent Inhibitor of Human Influenza Virus Hemagglutination. Carbohydr. Res. 2006, 341, 1803–1808. [DOI] [PubMed] [Google Scholar]
- (228).Fujita K; Takegawa K Chemoenzymatic Synthesis of Neoglycoproteins Using Transglycosylation with Endo-Beta-N-Acetyl-glucosaminidase A. Biochem. Biophys. Res. Commun. 2001, 282, 678–682. [DOI] [PubMed] [Google Scholar]
- (229).Fujita K; Yamamoto K A Remodeling System for the Oligosaccharide Chains on Glycoproteins with Microbial Endo-Beta-N-Acetylglucosaminidases. Biochim. Biophys. Acta, Gen. Subj 2006, 1760, 1631–1635. [DOI] [PubMed] [Google Scholar]
- (230).Yamanoi T; Yoshida N; Oda Y; Akaike E; Tsutsumida M; Kobayashi N; Osumi K; Yamamoto K; Fujita K; Takahashi K; et al. Synthesis of Mono-Glucose-Branched Cyclodextrins with a High Inclusion Ability for Doxorubicin and Their Efficient Glycosylation Using Mucor Hiemalis Endo-Beta-N-Acetylglucosaminidase. Bioorg. Med. Chem. Lett. 2005, 15, 1009–1013. [DOI] [PubMed] [Google Scholar]
- (231).Tomabechi Y; Suzuki R; Haneda K; Inazu T Chemo-Enzymatic Synthesis of Glycosylated Insulin Using a Glcnac Tag. Bioorg. Med. Chem. 2010, 18, 1259–1264. [DOI] [PubMed] [Google Scholar]
- (232).Terwisscha van Scheltinga AC; Armand S; Kalk KH; Isogai A; Henrissat B; Dijkstra BW Stereochemistry of Chitin Hydrolysis by a Plant Chitinase/Lysozyme and X-Ray Structure of a Complex with Allosamidin: Evidence for Substrate Assisted Catalysis. Biochemistry 1995, 34, 15619–15623. [DOI] [PubMed] [Google Scholar]
- (233).Tews I; Terwisscha van Scheltinga AC; Perrakis A; Wilson KS; Dijkstra BW Substrate-Assisted Catalysis Unifies Two Families of Chitinolytic Enzymes. J. Am. Chem. Soc. 1997, 119, 7954–7959. [Google Scholar]
- (234).Brameld KA; Shrader WD; Imperiali B; Goddard WA 3rd Substrate Assistance in the Mechanism of Family 18 Chitinases: Theoretical Studies of Potential Intermediates and Inhibitors. J. Mol. Biol. 1998, 280, 913–923. [DOI] [PubMed] [Google Scholar]
- (235).Mark BL; Vocadlo DJ; Knapp S; Triggs-Raine BL; Withers SG; James MN Crystallographic Evidence for Substrate-Assisted Catalysis in a Bacterial Beta-Hexosaminidase. J. Biol. Chem. 2001, 276, 10330–10337. [DOI] [PubMed] [Google Scholar]
- (236).Williams SJ; Mark BL; Vocadlo DJ; James MN; Withers SG Aspartate 313 in the Streptomyces Plicatus Hexosaminidase Plays a Critical Role in Substrate-Assisted Catalysis by Orienting the 2-Acetamido Group and Stabilizing the Transition State. J. Biol. Chem. 2002, 277, 40055–40065. [DOI] [PubMed] [Google Scholar]
- (237).Kobayashi S; Kiyosada T; Shoda S Synthesis of Artificial Chitin: Irreversible Catalytic Behavior of a Glycosyl Hydrolase through a Transition State Analogue Substrate. J. Am. Chem. Soc. 1996, 118, 13113–13114. [Google Scholar]
- (238).Shoda S; Kiyosada T; Mori H; Kobayashi S Chitinase-Catalyzed Synthesis of Oligosaccharides by Using a Sugar Oxazoline as Glycosyl Donor. Heterocycles 2000, 52, 599–602. [Google Scholar]
- (239).Kobayashi S; Fujikawa S; Ohmae M Enzymatic Synthesis of Chondroitin and Its Derivatives Catalyzed by Hyaluronidase. J. Am. Chem. Soc. 2003, 125, 14357–14369. [DOI] [PubMed] [Google Scholar]
- (240).Ochiai H; Ohmae M; Kobayashi S Enzymatic Glycosidation of Sugar Oxazolines Having a Carboxylate Group Catalyzed by Chitinase. Carbohydr. Res. 2004, 339, 2769–2788. [DOI] [PubMed] [Google Scholar]
- (241).Fujita M; Shoda S; Haneda K; Inazu T; Takegawa K; Yamamoto K A Novel Disaccharide Substrate Having 1,2-Oxazoline Moiety for Detection of Transglycosylating Activity of Endoglycosi-dases. Biochim. Biophys. Acta, Gen. Subj. 2001, 1528, 9–14. [DOI] [PubMed] [Google Scholar]
- (242).Li B; Zeng Y; Hauser S; Song H; Wang LX Highly Efficient Endoglycosidase-Catalyzed Synthesis of Glycopeptides Using Oligosaccharide Oxazolines as Donor Substrates. J. Am. Chem. Soc. 2005, 127,9692–9693. [DOI] [PubMed] [Google Scholar]
- (243).Li H; Li B; Song H; Breydo L; Baskakov IV; Wang LX Chemoenzymatic Synthesis of Hiv-1 V3 Glycopeptides Carrying Two N-Glycans and Effects of Glycosylation on the Peptide Domain. J. Org. Chem. 2005, 70, 9990–9996. [DOI] [PubMed] [Google Scholar]
- (244).Zeng Y; Wang J; Li B; Hauser S; Li H; Wang LX Glycopeptide Synthesis through Endo-Glycosidase-Catalyzed Oligosaccharide Transfer of Sugar Oxazolines: Probing Substrate Structural Requirement. Chem. - Eur. J. 2006, 12, 3355–3364. [DOI] [PubMed] [Google Scholar]
- (245).Rising TW; Claridge TD; Davies N; Gamblin DP; Moir JW; Fairbanks AJ Synthesis of N-Glycan Oxazolines: Donors for Endohexosaminidase Catalysed Glycosylation. Carbohydr. Res. 2006, 341, 1574–1596. [DOI] [PubMed] [Google Scholar]
- (246).Rising TW; Claridge TD; Moir JW; Fairbanks AJ Endohexosaminidase M: Exploring and Exploiting Enzyme Substrate Specificity. ChemBioChem 2006, 7, 1177–1180. [DOI] [PubMed] [Google Scholar]
- (247).Rising TW; Heidecke CD; Moir JW; Ling Z; Fairbanks AJ Endohexosaminidase-Catalysed Glycosylation with Oxazoline Donors: Fine Tuning of Catalytic Efficiency and Reversibility. Chem. - Eur. J. 2008, 14, 6444–6464. [DOI] [PubMed] [Google Scholar]
- (248).Heidecke CD; Parsons TB; Fairbanks AJ Endohex-osaminidase-Catalysed Glycosylation with Oxazoline Donors: Effects of Organic Co-Solvent and Ph on Reactions Catalysed by Endo a and Endo M. Carbohydr. Res. 2009, 344, 2433–2438. [DOI] [PubMed] [Google Scholar]
- (249).Parsons TB; Moir JW; Fairbanks AJ Synthesis of Truncated Bi-Antennary Complex-Type N-Glycan Oxazoline: Glycosylation Catalysed by the Endohexosaminidases Endo a and Endo M. Org. Biomol. Chem. 2009, 7, 3128–3140. [Google Scholar]
- (250).Ishii N; Ogiwara K; Sano K; Kumada J; Yamamoto K; Matsuzaki Y; Matsuo I Specificity of Donor Structures forEndo-Beta-N-Acetylglucosaminidase-Catalyzed Transglycosylation Reactions. ChemBioChem 2018, 19, 136–141. [DOI] [PubMed] [Google Scholar]
- (251).Fan JQ; Huynh LH; Reinhold BB; Reinhold VN; Takegawa K; Iwahara S; Kondo A; Kato I; Lee YC Transfer of Man9glcnac to L-Fucose by Endo-Beta-N-Acetylglucosaminidase from Arthrobacter Protophormiae. Glycoconjugate J. 1996, 13, 643–652. [DOI] [PubMed] [Google Scholar]
- (252).Tomabechi Y; Odate Y; Izumi R; Haneda K; Inazu T Acceptor Specificity in the Transglycosylation Reaction Using Endo-M. Carbohydr. Res. 2010, 345, 2458–2463. [DOI] [PubMed] [Google Scholar]
- (253).Manabe S; Yamaguchi Y; Abe J; Matsumoto K; Ito Y Acceptor Range ofEndo-Beta-N-Acetylglucosaminidase Mutant Endo-Cc N180h: From Monosaccharide to Antibody. R. Soc. Open Sci. 2018, 5, 171521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Huang W; Ochiai H; Zhang X; Wang LX Introducing N-Glycans into Natural Products through a Chemoenzymatic Approach. Carbohydr. Res. 2008, 343, 2903–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Huang W; Wang D; Yamada M; Wang LX Chemo-enzymatic Synthesis and Lectin Array Characterization of a Class of N-Glycan Clusters. J. Am. Chem. Soc. 2009, 131, 17963–17971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Ochiai H; Huang W; Wang LX Endo-Beta-N-Acetylglucosaminidase-Catalyzed Polymerization of Beta-Glcp-(1->4)-Glcpnac Oxazoline: A Revisit to Enzymatic Transglycosylation. Carbohydr. Res. 2009, 344, 592–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Li B; Song H; Hauser S; Wang LX A Highly Efficient Chemoenzymatic Approach toward Glycoprotein Synthesis. Org. Lett. 2006, 8, 3081–3084. [DOI] [PubMed] [Google Scholar]
- (258).Ochiai H; Huang W; Wang LX Expeditious Chemo-enzymatic Synthesis of Homogeneous N-Glycoproteins Carrying Defined Oligosaccharide Ligands. J. Am. Chem. Soc. 2008, 130, 13790–13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Kolb HC; Sharpless KB The Growing Impact of Click Chemistry on Drug Discovery. Drug Discovery Today 2003, 8, 1128–1137. [DOI] [PubMed] [Google Scholar]
- (260).Best MD Click Chemistry and Bioorthogonal Reactions: Unprecedented Selectivity in the Labeling of Biological Molecules. Biochemistry 2009, 48, 6571–6584. [DOI] [PubMed] [Google Scholar]
- (261).Jewett JC; Bertozzi CR Cu-Free Click Cycloaddition Reactions in Chemical Biology. Chem. Soc. Rev. 2010, 39, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Ly HD; Withers SG Mutagenesis of Glycosidases. Annu. Rev. Biochem. 1999, 68, 487–522. [DOI] [PubMed] [Google Scholar]
- (263).Umekawa M; Li C; Higashiyama T; Huang W; Ashida H; Yamamoto K; Wang LX Efficient Glycosynthase Mutant Derived from Mucor Hiemalis Endo-Beta-N-Acetylglucosaminidase Capable of Transferring Oligosaccharide from Both Sugar Oxazoline and Natural N-Glycan. J. Biol. Chem. 2010, 285, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Tong X; Li T; Orwenyo J; Toonstra C; Wang LX One-Pot Enzymatic Glycan Remodeling of a Therapeutic Monoclonal Antibody by Endoglycosidase S (Endo-S) from Streptococcus Pyogenes. Bioorg. Med. Chem. 2018, 26, 1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Umekawa M; Higashiyama T; Koga Y; Tanaka T; Noguchi M; Kobayashi A; Shoda S; Huang W; Wang LX; Ashida H; et al. Efficient Transfer of Sialo-Oligosaccharide onto Proteins by Combined Use of a Glycosynthase-Like Mutant of Mucor Hiemalis Endoglyco-sidase and Synthetic Sialo-Complex-Type Sugar Oxazoline. Biochim. Biophys. Acta, Gen. Subj. 2010, 1800, 1203–1209. [DOI] [PubMed] [Google Scholar]
- (266).Katoh T; Katayama T; Tomabechi Y; Nishikawa Y; Kumada J; Matsuzaki Y; Yamamoto K Generation of a Mutant Mucor Hiemalis Endoglycosidase That Acts on Core-Fucosylated N-Glycans. J. Biol. Chem. 2016, 291, 23305–23317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Shivatare SS; Huang LY; Zeng YF; Liao JY; You TH; Wang SY; Cheng T; Chiu CW; Chao P; Chen LT; et al. Development of Glycosynthases with Broad Glycan Specificity for the Efficient Glyco-Remodeling of Antibodies. Chem. Commun. (Cambridge, U. K.) 2018, 54, 6161–6164. [DOI] [PubMed] [Google Scholar]
- (268).Trastoy B; Klontz E; Orwenyo J; Marina A; Wang LX; Sundberg EJ; Guerin ME Structural Basis for the Recognition of Complex-Type N-Glycans by Endoglycosidase S. Nat. Commun. 2018, 9, 1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Huang W; Zhang X; Ju T; Cummings RD; Wang LX Expeditious Chemoenzymatic Synthesis of Cd52 Glycopeptide Antigens. Org. Biomol. Chem. 2010, 8, 5224–5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).McIntosh JD; Brimble MA; Brooks AES; Dunbar PR; Kowalczyk R; Tomabechi Y; Fairbanks AJ Convergent Chemo-Enzymatic Synthesis of Mannosylated Glycopeptides; Targeting of Putative Vaccine Candidates to Antigen Presenting Cells. Chem. Sci. 2015,6, 4636–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Tomabechi Y; Krippner G; Rendle PM; Squire MA; Fairbanks AJ Glycosylation of Pramlintide: Synthetic Glycopeptides That Display in Vitro and in Vivo Activities as Amylin Receptor Agonists. Chem. - Eur. J. 2013, 19, 15084–15088. [DOI] [PubMed] [Google Scholar]
- (272).Kowalczyk R; Brimble MA; Tomabechi Y; Fairbanks AJ; Fletcher M; Hay DL Convergent Chemoenzymatic Synthesis of a Library of Glycosylated Analogues of Pramlintide: Structure-Activity Relationships for Amylin Receptor Agonism. Org. Biomol. Chem. 2014, 12, 8142–8151. [DOI] [PubMed] [Google Scholar]
- (273).Wang LX Synthetic Carbohydrate Antigens for Hiv Vaccine Design. Curr. Opin. Chem. Biol. 2013, 17, 997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Amin MN; McLellan JS; Huang W; Orwenyo J; Burton DR; Koff WC; Kwong PD; Wang LX Synthetic Glycopeptides Reveal the Glycan Specificity of Hiv-Neutralizing Antibodies. Nat. Chem. Biol. 2013, 9, 521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Toonstra C; Amin MN; Wang LX Site-Selective Chemoenzymatic Glycosylation of an Hiv-1 Polypeptide Antigen with Two Distinct N-Glycans Via an Orthogonal Protecting Group Strategy. J. Org. Chem. 2016, 81, 6176–6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).McLellan JS; Pancera M; Carrico C; Gorman J; Julien JP; Khayat R; Louder R; Pejchal R; Sastry M; Dai K; et al. Structure of Hiv-1 Gp120 V1/V2 Domain with Broadly Neutralizing Antibody Pg9. Nature 2011, 480, 336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Orwenyo J; Cai H; Giddens J; Amin MN; Toonstra C; Wang LX Systematic Synthesis and Binding Study of Hiv V3 Glycopeptides Reveal the Fine Epitopes of Several Broadly Neutralizing Antibodies. ACS Chem. Biol. 2017, 12, 1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Cai H; Orwenyo J; Giddens JP; Yang Q; Zhang R; LaBranche CC; Montefiori DC; Wang LX Synthetic Three-Component Hiv-1 V3 Glycopeptide Immunogens Induce Glycan-Dependent Antibody Responses. Cell Chem. Biol. 2017,24,1513–1522 (e1514).. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Cai H; Orwenyo J; Guenaga J; Giddens J; Toonstra C; Wyatt RT; Wang LX Synthetic Multivalent V3 Glycopeptides Display Enhanced Recognition by Glycan-Dependent Hiv-1 Broadly Neutralizing Antibodies. Chem. Commun. (Cambridge, U. K.) 2017, 53, 5453–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Cai H; Zhang R; Orwenyo J; Giddens J; Yang Q; LaBranche CC; Montefiori DC; Wang LX Multivalent Antigen Presentation Enhances the Immunogenicity of a Synthetic Three-Component Hiv-1 V3 Glycopeptide Vaccine. ACS Cent. Sci. 2018, 4, 582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Aussedat B; Vohra Y; Park PK; Fernandez-Tejada A; Alam SM; Dennison SM; Jaeger FH; Anasti K; Stewart S; Blinn JH; et al. Chemical Synthesis of Highly Congested Gp120 V1v2 N-Glycopeptide Antigens for Potential Hiv-1-Directed Vaccines. J. Am. Chem. Soc. 2013, 135, 13113–13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Fernandez-Tejada A; Haynes BF; Danishefsky SJ Designing Synthetic Vaccines for Hiv. Expert Rev. Vaccines 2015, 14, 815–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Alam SM; Dennison SM; Aussedat B; Vohra Y; Park PK; Fernandez-Tejada A; Stewart S; Jaeger FH; Anasti K; Blinn JH ; et al. Recognition of Synthetic Glycopeptides by Hiv-1 Broadly Neutralizing Antibodies and Their Unmutated Ancestors. Proc. Natl. Acad. Sci. U. S.A. 2013, 110, 18214–18219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Hojo H; Tanaka H; Hagiwara M; Asahina Y; Ueki A; Katayama H; Nakahara Y; Yoneshige A; Matsuda J; Ito Y; et al. Chemoenzymatic Synthesis of Hydrophobic Glycoprotein: Synthesis of Saposin C Carrying Complex-Type Carbohydrate. J. Org. Chem. 2012, 77, 9437–9446. [DOI] [PubMed] [Google Scholar]
- (285).Klein AD; Futerman AH Lysosomal Storage Disorders: Old Diseases. Present and Future Challenges. PediatrEndocrinol Rev. 2013,11 (Suppl 1), 59–63. [PubMed] [Google Scholar]
- (286).Couture-Tosi E; Delacroix H; Mignot T; Mesnage S; Chami M; Fouet A; Mosser G Structural Analysis and Evidence for Dynamic Emergence of Bacillus Anthracis S-Layer Networks. J. Bacteriol. 2002, 184, 6448–6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Lachmann RH Enzyme Replacement Therapy for Lysosomal Storage Diseases. Curr. Opin. Pediatr. 2011, 23, 588–593. [DOI] [PubMed] [Google Scholar]
- (288).Sly WS Enzyme Replacement Therapy for Lysosomal Storage Disorders: Successful Transition from Concept to Clinical Practice. Mo Med. 2004, 101, 100–104. [PubMed] [Google Scholar]
- (289).Ghosh P; Dahms NM; Kornfeld S Mannose 6-Phosphate Receptors: New Twists in the Tale. Nat. Rev. Mol Cell Biol. 2003, 4, 202–212. [DOI] [PubMed] [Google Scholar]
- (290).Dahms NM; Olson LJ; Kim JJ Strategies for Carbohydrate Recognition by the Mannose 6-Phosphate Receptors. Glycobiology 2008, 18, 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Zhu Y; Li X; Kyazike J; Zhou Q; Thurberg BL; Raben N; Mattaliano RJ; Cheng SH Conjugation of Mannose 6-Phosphate-Containing Oligosaccharides to Acid Alpha-Glucosidase Improves the Clearance of Glycogen in Pompe Mice. J. Biol. Chem. 2004, 279, 50336–50341. [DOI] [PubMed] [Google Scholar]
- (292).Zhu Y; Li X; McVie-Wylie A; Jiang C; Thurberg BL; Raben N; Mattaliano RJ; Cheng SH Carbohydrate-Remodelled Acid Alpha-Glucosidase with Higher Affinity for the Cation Independent Mannose 6-Phosphate Receptor Demonstrates Improved Delivery to Muscles of Pompe Mice. Biochem. J. 2005, 389, 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Zhu Y; Jiang JL; Gumlaw NK; Zhang J; Bercury SD; Ziegler RJ; Lee K; Kudo M; Canfield WM; Edmunds T; et al. Glycoengineered Acid Alpha-Glucosidase with Improved Efficacy at Correcting the Metabolic Aberrations and Motor Function Deficits in a Mouse Model of Pompe Disease. Mol. Ther. 2009, 17, 954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Zhou Q; Stefano JE; Harrahy J; Finn P; Avila L; Kyazike J; Wei R; Van Patten SM; Gotschall R; Zheng X; et al. Strategies for Neoglycan Conjugation to Human Acid Alpha-Glucosidase. Bioconjugate Chem. 2011, 22, 741–751. [DOI] [PubMed] [Google Scholar]
- (295).Zhou Q; Avila LZ; Konowicz PA; Harrahy J; Finn P; Kim J; Reardon MR; Kyazike J; Brunyak E; Zheng X; et al. Glycan Structure Determinants for Cation-Independent Mannose 6-Phosphate Receptor Binding and Cellular Uptake of a Recombinant Protein. Bioconjugate Chem. 2013, 24, 2025–2035. [DOI] [PubMed] [Google Scholar]
- (296).Liu Y; Chan YM; Wu J; Chen C; Benesi A; Hu J; Wang Y; Chen G Chemical Synthesis of a Bisphosphorylated Mannose-6-Phosphate N-Glycan and Its Facile Monoconjugation with Human Carbonic Anhydrase Ii for in Vivo Fluorescence Imaging. Chem-BioChem 2011, 12, 685–690. [DOI] [PubMed] [Google Scholar]
- (297).McVie-Wylie AJ; Lee KL; Qiu H; Jin X; Do H; Gotschall R; Thurberg BL; Rogers C; Raben N; O’Callaghan M; et al. Biochemical and Pharmacological Characterization of Different Recombinant Acid Alpha-Glucosidase Preparations Evaluated for the Treatment of Pompe Disease. Mol. Genet. Metab. 2008, 94, 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (298).Song X; Lasanajak Y; Olson LJ; Boonen M; Dahms NM; Kornfeld S; Cummings RD; Smith DF Glycan Microarray Analysis of P-Type Lectins Reveals Distinct Phosphomannose Glycan Recognition. J. Biol. Chem. 2009, 284, 35201–35214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Priyanka P; Parsons TB; Miller A; Platt FM; Fairbanks AJ Chemoenzymatic Synthesis of a Phosphorylated Glycoprotein. Angew. Chem., Int. Ed. 2016, 55, 5058–5061. [DOI] [PubMed] [Google Scholar]
- (300).Yamaguchi T; Amin MN; Toonstra C; Wang LX Chemoenzymatic Synthesis and Receptor Binding of Mannose-6-Phosphate (M6p)-Containing Glycoprotein Ligands Reveal Unusual Structural Requirements for M6p Receptor Recognition. J. Am. Chem. Soc. 2016, 138, 12472–12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (301).Huang W; Yang Q; Umekawa M; Yamamoto K; Wang LX Arthrobacter Endo-Beta-N-Acetylglucosaminidase Shows Trans-glycosylation Activity on Complex-Type N-Glycan Oxazolines: One-Pot Conversion of Ribonuclease B to Sialylated Ribonuclease C. ChemBioChem 2010, 11, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Amin MN; Huang W; Mizanur RM; Wang LX Convergent Synthesis of Homogeneous Glc(l)Man(9)Glcnac(2)-Protein and Derivatives as Ligands ofMolecular Chaperones in Protein Quality Control. J. Am. Chem. Soc. 2011, 133, 14404–14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (303).Yang Q; Wang LX Mammalian Alpha-1,6-Fucosyltransferase (Fut8) Is the Sole Enzyme Responsible for the N-Acetylglucosaminyl-transferase I-Independent Core Fucosylation of High-Mannose N-Glycans. J. Biol. Chem. 2016, 291, 11064–11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (304).Yang Q; An Y; Zhu S; Zhang R; Loke CM; Cipollo JF; Wang LX Glycan Remodeling of Human Erythropoietin (Epo) through Combined Mammalian Cell Engineering and Chemo-enzymatic Transglycosylation. ACS Chem. Biol. 2017, 12, 1665–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Nimmerjahn F; Ravetch JV Fcgamma Receptors as Regulators of Immune Responses. Nat. Rev. Immunol. 2008, 8, 34–47. [DOI] [PubMed] [Google Scholar]
- (306).Wei Y; Li C; Huang W; Li B; Strome S; Wang LX Glycoengineering of Human Igg1-Fc through Combined Yeast Expression and in Vitro Chemoenzymatic Glycosylation. Biochemistry 2008, 47, 10294–10304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).Zou G; Ochiai H; Huang W; Yang Q; Li C; Wang LX Chemoenzymatic Synthesis and Fcgamma Receptor Binding of Homogeneous Glycoforms of Antibody Fc Domain. Presence of a Bisecting Sugar Moiety Enhances the Affinity of Fc to Fcgammaiiia Receptor. J.Am. Chem. Soc. 2011, 133, 18975–18991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (308).Lin CW; Tsai MH; Li ST; Tsai TI; Chu KC; Liu YC; Lai MY; Wu CY; Tseng YC; Shivatare SS; et al. A Common Glycan Structure on Immunoglobulin G for Enhancement of Effector Functions. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10611–10616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (309).Kurogochi M; Mori M; Osumi K; Tojino M; Sugawara S; Takashima S; Hirose Y; Tsukimura W; Mizuno M; Amano J; et al. Glycoengineered Monoclonal Antibodies with Homogeneous Glycan (M3, G0, G2, and A2) Using a Chemoenzymatic Approach Have Different Affinities for Fcgammariiia and Variable Antibody-Dependent Cellular Cytotoxicity Activities. PLoS One 2015, 10, e0132848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (310).Quast I; Keller CW; Maurer MA; Giddens JP; Tackenberg B; Wang L-X; Munz C; Nimmerjahn F; Dalakas MC; Lunemann JD Sialylation of Igg Fc Domain Impairs Complement-Dependent Cytotoxicity. J. Clin. Invest. 2015, 125, 4160–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (311).Parsons TB; Struwe WB; Gault J; Yamamoto K; Taylor TA; Raj R; Wals K; Mohammed S; Robinson CV; Benesch JL; et al. Optimal Synthetic Glycosylation of a Therapeutic Antibody. Angew. Chem., Int. Ed. 2016, 55, 2361–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (312).Ahmed AA; Giddens J; Pincetic A; Lomino JV; Ravetch JV; Wang LX; Bjorkman PJ Structural Characterization of AntiInflammatory Immunoglobulin G Fc Proteins. J. Mol. Biol. 2014, 426, 3166–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (313).Smith EL; Giddens JP; Iavarone AT; Godula K; Wang LX; Bertozzi CR Chemoenzymatic Fc Glycosylation Via Engineered Aldehyde Tags. Bioconjugate Chem. 2014, 25, 788–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (314).Liu R; Giddens J; McClung CM; Magnelli PE; Wang LX; Guthrie EP Evaluation of a Glycoengineered Monoclonal Antibody Via Lc-Ms Analysis in Combination with Multiple Enzymatic Digestion. MAbs 2016, 8, 340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (315).Sjogren J; Struwe WB; Cosgrave EF; Rudd PM; Stervander M; Allhorn M; Hollands A; Nizet V; Collin M Endos2 Is a Unique and Conserved Enzyme of Serotype M49 Group a Streptococcus That Hydrolyses N-Linked Glycans on Igg and Alpha1-Acid Glycoprotein. Biochem. J. 2013, 455, 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (316).Li T; DiLillo DJ; Bournazos S; Giddens JP; Ravetch JV; Wang LX Modulating Igg Effector Function by Fc Glycan Engineering. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 3485–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (317).Li T; Li C; Quan DN; Bentley WE; Wang LX Site-Specific Immobilization of Endoglycosidases for Streamlined Chemo-enzymatic Glycan Remodeling of Antibodies. Carbohydr. Res. 2018, 458–459, 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (318).Suda M; Sumiyoshi W; Kinoshita T; Ohno S Reaction of Sugar Oxazolines with Primary Amines. Tetrahedron Lett. 2016, 57, 5446–5448. [Google Scholar]
- (319).Wang N; Seko A; Daikoku S; Kanie O; Takeda Y; Ito Y Non-Enzymatic Reaction of Glycosyl Oxazoline with Peptides. Carbohydr. Res. 2016, 436, 31–35. [DOI] [PubMed] [Google Scholar]
- (320).Liu CP; Tsai TI; Cheng T; Shivatare VS; Wu CY; Wu CY; Wong CH Glycoengineering of Antibody (Herceptin) through Yeast Expression and in Vitro Enzymatic Glycosylation. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 720–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (321).Iwamoto M; Sekiguchi Y; Nakamura K; Kawaguchi Y; Honda T; Hasegawa J Generation of Efficient Mutants of Endoglycosidase from Streptococcus Pyogenes and Their Application in a Novel One-Pot Transglycosylation Reaction for Antibody Modification. PLoS One 2018, 13, e0193534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Fairbanks AJ Synthetic and Semi-Synthetic Approaches to Unprotected N-Glycan Oxazolines. Beilstein J. Org. Chem. 2018, 14, 416–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (323).Kobayashi S; Uyama H; Kimura S Enzymatic Polymerization. Chem. Rev. 2001, 101, 3793–3818. [DOI] [PubMed] [Google Scholar]
- (324).Kobayashi S New Developments of Polysaccharide Synthesis Via Enzymatic Polymerization. Proc. Jpn. Acad. Ser. B 2007, 83, 215–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (325).Nakabayashi S; Warren CD; Jeanloz RW A New Procedure for the Preparation of Oligosaccharide Oxazolines. Carbohydr. Res. 1986, 150, C7–C10. [DOI] [PubMed] [Google Scholar]
- (326).Colon M; Staveski MM; Davis JT Mild Conditions for the Preparation of High-Mannose Oligosaccharide Oxazolines: EntryPoint for Beta -Glycoside and Neoglycoprotein Syntheses. Tetrahedron Lett. 1991, 32, 4447–4450. [Google Scholar]
- (327).Noguchi M; Tanaka T; Gyakushi H; Kobayashi A; Shoda SI Efficient Synthesis of Sugar Oxazolines from Unprotected N-Acetyl-2-Amino Sugars by Using Chloroformamidinium Reagent in Water. J. Org. Chem. 2009, 74, 2210–2212. [DOI] [PubMed] [Google Scholar]
- (328).Kajihara Y; Suzuki Y; Yamamoto N; Sasaki K; Sakakibara T; Juneja L R Prompt Chemoenzymatic Synthesis of Diverse Complex-Type Oligosaccharides and Its Application to the Solid-Phase Synthesis of a Glycopeptide with Asn-Linked Sialyl-Undeca- and Asialo-Nonasaccharides. Chem. - Eur. J 2004, 10, 971–985. [DOI] [PubMed] [Google Scholar]
- (329).Seko A; Koketsu M; Nishizono M; Enoki Y; Ibrahim HR; Juneja LR; Kim M; Yamamoto T Occurence of a Sialylglycopeptide and Free Sialylglycans in Hen’s Egg Yolk. Biochim. Biophys. Acta, Gen. Subj. 1997, 1335, 23–32. [DOI] [PubMed] [Google Scholar]
- (330).Sun B; Bao W; Tian X; Li M; Liu H; Dong J; Huang W A Simplified Procedure for Gram-Scale Production of Sialylglycopeptide (Sgp) from Egg Yolks and Subsequent Semi-Synthesis of Man3glcnac Oxazoline. Carbohydr. Res. 2014, 396, 62–69. [DOI] [PubMed] [Google Scholar]
- (331).Liu L; Prudden AR; Bosman GP; Boons GJ Improved Isolation and Characterization Procedure of Sialylglycopeptide from Egg Yolk Powder. Carbohydr. Res. 2017, 452, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Wu Z; Liu Y; Ma C; Li L; Bai J; Byrd-Leotis L; Lasanajak Y; Guo Y; Wen L; Zhu H; et al. Identification of the Binding Roles of Terminal and Internal Glycan Epitopes Using Enzymatically Synthesized N-Glycans Containing Tandem Epitopes. Org. Biomol. Chem. 2016, 14, 11106–11116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (333).Wang LX; Ni J; Singh S; Li H Binding of High-Mannose-Type Oligosaccharides and Synthetic Oligomannose Clusters to Human Antibody 2g12: Implications for Hiv-1 Vaccine Design. Chem. Biol. 2004, 11, 127–134. [DOI] [PubMed] [Google Scholar]
- (334).Wang N; Seko A; Takeda Y; Ito Y Preparation of Asparagine-Linked Monoglucosylated High-Mannose-Type Oligosaccharide from Egg Yolk. Carbohydr. Res. 2015, 411, 37–41. [DOI] [PubMed] [Google Scholar]
- (335).Nakakita S; Sumiyoshi W; Miyanishi N; Hirabayashi J A Practical Approach to N-Glycan Production by Hydrazinolysis Using Hydrazine Monohydrate. Biochem. Biophys. Res. Commun. 2007, 362, 639–645. [DOI] [PubMed] [Google Scholar]
- (336).Evers DL; Hung RL; Thomas VH; Rice KG Preparative Purification of a High-Mannose Type N-Glycan from Soy Bean Agglutinin by Hydrazinolysis and Tyrosinamide Derivatization. Anal. Biochem. 1998, 265, 313–316. [DOI] [PubMed] [Google Scholar]
- (337).Makimura Y; Kiuchi T; Izumi M; Dedola S; Ito Y; Kajihara Y Efficient Synthesis of Glycopeptide-Alpha-Thioesters with a High-Mannose Type Oligosaccharide by Means of Tert-Boc-Solid Phase Peptide Synthesis. Carbohydr. Res. 2012, 364, 41–48. [DOI] [PubMed] [Google Scholar]
- (338).Li T; Huang M; Liu L; Wang S; Moremen KW; Boons GJ Divergent Chemoenzymatic Synthesis of Asymmetrical-Core-Fucosylated and Core-Unmodified N-Glycans. Chem. - Eur. J 2016, 22, 18742–18746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (339).Gagarinov IA; Li T; Torano JS; Caval T; Srivastava AD; Kruijtzer JA; Heck AJ; Boons GJ Chemoenzymatic Approach for the Preparation of Asymmetric Bi-, Tri-, and Tetra-Antennary N-Glycans from a Common Precursor. J. Am. Chem. Soc. 2017, 139, 1011–101S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (340).Wang Z; Chinoy ZS; Ambre SG; Peng W; McBride R; de Vries RP; Glushka J; Paulson JC; Boons GJ A General Strategy for the Chemoenzymatic Synthesis of Asymmetrically Branched N-Glycans. Science 2013, 341, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (341).Li L; Liu Y; Ma C; Qu J; Calderon AD; Wu B; Wei N; Wang X; Guo Y; Xiao Z; et al. Efficient Chemoenzymatic Synthesis of an N-Glycan Isomer Library. Chem. Sci. 2015, 6, 5652–5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (342).Helenius A; Aebi M Roles of N-Linked Glycans in the Endoplasmic Reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [DOI] [PubMed] [Google Scholar]
- (343).Imperiali B; Hendrickson TL Asparagine-Linked Glyco-sylation: Specificity and Function of Oligosaccharyl Transferase. Bioorg. Med. Chem. 1995, 3, 1565–1578. [DOI] [PubMed] [Google Scholar]
- (344).Dempski RE Jr.; Imperiali B Oligosaccharyl Transferase: Gatekeeper to the Secretory Pathway. Curr. Opin. Chem. Biol. 2002, 6, S44–S50. [DOI] [PubMed] [Google Scholar]
- (345).Ruiz-Canada C; Kelleher DJ; Gilmore R Cotranslational and Posttranslational N-Glycosylation of Polypeptides by Distinct Mammalian Ost Isoforms. Cell 2009, 136, 272–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (346).Srinivasan A; Coward JK A Biotin Capture Assay for Oligosaccharyltransferase. Anal. Biochem. 2002, 306, 328–335. [DOI] [PubMed] [Google Scholar]
- (347).Reddy A; Gibbs BS; Liu YL; Coward JK; Changchien LM; Maley F Glycosylation of the Overlapping Sequons in Yeast External Invertase: Effect of Amino Acid Variation on Site Selectivity in Vivo and in Vitro. Glycobiology 1999, 9, 547–555. [DOI] [PubMed] [Google Scholar]
- (348).Gibbs BS; Coward JK Dolichylpyrophosphate Oligosaccharides: Large-Scale Isolation and Evaluation as Oligosac-charyltransferase Substrates. Bioorg. Med. Chem. 1999, 7, 441–447. [DOI] [PubMed] [Google Scholar]
- (349).Xu T; Werner RM; Lee KC; Fettinger JC; Davis JT; Coward JK Synthesis and Evaluation of Tripeptides Containing Asparagine Analogues as Potential Substrates or Inhibitors of Oligosaccharyltransferase. J. Org. Chem. 1998, 63, 4767–477S. [Google Scholar]
- (350).Liggins J; Bluck LJ; Coward WA; Bingham SA Extraction and Quantification ofDaidzein and Genistein in Food. Anal. Biochem. 1998, 264, 1–7. [DOI] [PubMed] [Google Scholar]
- (351).Xu T; Coward JK C13- and N15-Labeled Peptide Substrates as Mechanistic Probes of Oligosaccharyltransferase. Biochemistry 1997, 36, 14683–14689. [DOI] [PubMed] [Google Scholar]
- (352).Fang X; Gibbs BS; Coward JK Synthesis and Evaluation of Synthetic Analogues of Dolichyl-P-P-Chitobiose as Oligosaccharyl-transferase Substrates. Bioorg. Med. Chem. Lett. 1995, 5, 2701–2706. [Google Scholar]
- (353).Lee J; Coward JK Oligosaccharyltransferase: Synthesis and Use of Deuterium-Labeled Peptide Substrates as Mechanistic Probes. Biochemistry 1993, 32, 6794–6801. [DOI] [PubMed] [Google Scholar]
- (354).Tai VW; Imperiali B Substrate Specificity of the Glycosyl Donor for Oligosaccharyl Transferase. J. Org. Chem. 2001, 66, 6217— 622S. [DOI] [PubMed] [Google Scholar]
- (355).Peluso S; de LUM; O’Reilly MK; Imperiali B Neoglycopeptides as Inhibitors of Oligosaccharyl Transferase: Insight into Negotiating Product Inhibition. Chem. Biol. 2002, 9, 1323–1328. [DOI] [PubMed] [Google Scholar]
- (356).Aebi M N-Linked Protein Glycosylation in the Er. Biochim. Biophys. Acta, Mol. Cell Res 2013, 1833, 2430–2437. [DOI] [PubMed] [Google Scholar]
- (357).Bieberich E Synthesis, Processing, and Function of N-Glycans in N-Glycoproteins. Adv. Neurobiol 2014, 9, 47–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (358).Szymanski CM; Yao R; Ewing CP; Trust TJ; Guerry P Evidence for a System of General Protein Glycosylation in Campylobacter Jejuni. Mol. Microbiol. 1999, 32, 1022–1030. [DOI] [PubMed] [Google Scholar]
- (359).Szymanski CM; Logan SM; Linton D; Wren BW Campylobacter-a Tale of Two Protein Glycosylation Systems. Trends Microbiol. 2003, 11, 233–238. [DOI] [PubMed] [Google Scholar]
- (360).Larsen JC; Szymanski C; Guerry P N-Linked Protein Glycosylation Is Required for Full Competence in Campylobacter Jejuni 81—176. J. Bacteriol 2004, 186, 6508–6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (361).Szymanski CM; Wren BW Protein Glycosylation in Bacterial Mucosal Pathogens. Nat. Rev. Microbiol. 2005, 3, 225–237. [DOI] [PubMed] [Google Scholar]
- (362).Kelly J; Jarrell H; Millar L; Tessier L; Fiori LM; Lau PC ; Allan B; Szymanski CM Biosynthesis of the N-Linked Glycan in Campylobacter Jejuni and Addition onto Protein through Block Transfer. J. Bacteriol. 2006, 188, 2427–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (363).Dell A; Galadari A; Sastre F; Hitchen P Similarities and Differences in the Glycosylation Mechanisms in Prokaryotes and Eukaryotes. Int. J. Microbiol. 2010, 2010, 148178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (364).Glover KJ; Weerapana E; Imperiali B In Vitro Assembly of the Undecaprenylpyrophosphate-Linked Heptasaccharide for Prokaryotic N-Linked Glycosylation. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 14255–14259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).Weerapana E; Glover KJ; Chen MM; Imperiali B Investigating Bacterial N-Linked Glycosylation: Synthesis and Glycosyl Acceptor Activity ofthe Undecaprenyl Pyrophosphate-Linked Bacillos-amine. J. Am. Chem. Soc. 2005, 127, 13766–13767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (366).Lukose V; Whitworth G; Guan Z; Imperiali B Chemo-enzymatic Assembly of Bacterial Glycoconjugates for Site-Specific Orthogonal Labeling. J. Am. Chem. Soc. 2015, 137, 12446–12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (367).Liu F; Vijayakrishnan B; Faridmoayer A; Taylor TA; Parsons TB; Bernardes GJ; Kowarik M; Davis BG Rationally Designed Short Polyisoprenol-Linked Pglb Substrates for Engineered Polypeptide and Protein N-Glycosylation. J. Am. Chem. Soc. 2014, 136, 566–569. [DOI] [PubMed] [Google Scholar]
- (368).Chen MM; Weerapana E; Ciepichal E; Stupak J; Reid CW; Swiezewska E; Imperiali B Polyisoprenol Specificity in the Campylobacter Jejuni N-Linked Glycosylation Pathway. Biochemistry 2007, 46, 14342–14348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (369).Li L; Woodward R; Ding Y; Liu XW; Yi W; Bhatt VS; Chen M; Zhang LW; Wang PG Overexpression and Topology of Bacterial Oligosaccharyltransferase Pglb. Biochem. Biophys. Res. Commun. 2010, 394, 1069–1074. [DOI] [PubMed] [Google Scholar]
- (370).Nita-Lazar M; Wacker M; Schegg B; Amber S; Aebi M The N-X-S/T Consensus Sequence Is Required but Not Sufficient for Bacterial N-Linked Protein Glycosylation. Glycobiology 2005, 15, 361–367. [DOI] [PubMed] [Google Scholar]
- (371).Kowarik M; Numao S; Feldman MF; Schulz BL; Callewaert N; Kiermaier E; Catrein I; Aebi M N-Linked Glycosylation of Folded Proteins by the Bacterial Oligosaccharyl-transferase. Science 2006, 314, 1148–1150. [DOI] [PubMed] [Google Scholar]
- (372).Kowarik M; Young NM; Numao S; Schulz BL; Hug I; Callewaert N; Mills DC; Watson DC; Hernandez M; Kelly JF; et al. Definition of the Bacterial N-Glycosylation Site Consensus Sequence. EMBOJ 2006, 25, 1957–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (373).Silverman JM; Imperiali B Bacterial N-Glycosylation Efficiency Is Dependent on the Structural Context of Target Sequons. J. Biol. Chem. 2016, 291, 22001–22010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (374).Chen MM; Glover KJ; Imperiali B From Peptide to Protein: Comparative Analysis of the Substrate Specificity of N-Linked Glycosylation in C. Jejuni. Biochemistry 2007, 46, 5579–5585. [DOI] [PubMed] [Google Scholar]
- (375).Ollis AA; Chai Y; Natarajan A; Perregaux E; Jaroentomeechai T; Guarino C; Smith J; Zhang S; DeLisa MP Substitute Sweeteners: Diverse Bacterial Oligosaccharyltransferases with Unique N-Glycosylation Site Preferences. Sci. Rep. 2015, 5, 15237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (376).Schwarz F; Lizak C; Fan YY; Fleurkens S; Kowarik M; Aebi M Relaxed Acceptor Site Specificity of Bacterial Oligosacchar-yltransferase in Vivo. Glycobiology 2011, 21, 45–54. [DOI] [PubMed] [Google Scholar]
- (377).Matsumoto S; Shimada A; Nyirenda J; Igura M; Kawano Y; Kohda D Crystal Structures of an Archaeal Oligosaccharyltransferase Provide Insights into the Catalytic Cycle of N-Linked Protein Glycosylation. Proc. Natl. Acad. Sci. U. S.A. 2013, 110, 17868–17873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (378).Matsumoto S; Taguchi Y; Shimada A; Igura M; Kohda D Tethering an N-Glycosylation Sequon-Containing Peptide Creates a Catalytically Competent Oligosaccharyltransferase Complex. Biochemistry 2017, 56, 602–611. [DOI] [PubMed] [Google Scholar]
- (379).Gerber S; Lizak C; Michaud G; Bucher M; Darbre T; Aebi M; Reymond JL; Locher KP Mechanism of Bacterial Oligosaccharyltransferase: In Vitro Quantification of Sequon Binding and Catalysis. J. Biol. Chem. 2013, 288, 8849–8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (380).Lizak C; Gerber S; Michaud G; Schubert M; Fan YY; Bucher M; Darbre T; Aebi M; Reymond JL; Locher KP Unexpected Reactivity and Mechanism of Carboxamide Activation in Bacterial N-Linked Protein Glycosylation. Nat. Commun. 2013, 4, 2627. [DOI] [PubMed] [Google Scholar]
- (381).Napiorkowska M; Boilevin J; Sovdat T; Darbre T; Reymond JL; Aebi M; Locher KP Molecular Basis of Lipid-Linked Oligosaccharide Recognition and Processing by Bacterial Oligosaccharyltransferase. Nat. Struct. Mol Biol. 2017, 24, 1100–1106. [DOI] [PubMed] [Google Scholar]
- (382).Schwarz F; Huang W; Li C; Schulz BL; Lizak C; Palumbo A; Numao S; Neri D; Aebi M; Wang LX A Combined Method for Producing Homogeneous Glycoproteins with Eukaryotic N-Glycosylation. Nat. Chem. Biol. 2010, 6, 264–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (383).Valderrama-Rincon JD; Fisher AC; Merritt JH; Fan YY; Reading CA; Chhiba K; Heiss C; Azadi P; Aebi M; DeLisa MP An Engineered Eukaryotic Protein Glycosylation Pathway in Escherichia Coli. Nat. Chem. Biol. 2012, 8, 434–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (384).Wacker M; Feldman MF; Callewaert N; Kowarik M; Clarke BR; Pohl NL; Hernandez M; Vines ED; Valvano MA; Whitfield C; et al. Substrate Specificity of Bacterial Oligosaccharyl-transferase Suggests a Common Transfer Mechanism for the Bacterial and Eukaryotic Systems. Proc. Natl. Acad. Sci. U. S. A. 2006,103, 7088–7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (385).Cuccui J; Thomas RM, Moule MG, D’Elia RV, Laws TR, Mills DC, Williamson D, Atkins TP, Prior JL, Wren BW Exploitation of Bacterial N-Linked Glycosylation to Develop a Novel Recombinant Glycoconjugate Vaccine against Francisella Tularensis. Open Biol. 2013, 3, 130002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (386).Cuccui J; Wren B Hijacking Bacterial Glycosylation for the Production of Glycoconjugates, from Vaccines to Humanised Glycoproteins. J. Pharm. Pharmacol. 2015, 67, 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (387).Ihssen J; Haas J; Kowarik M; Wiesli L; Wacker M; Schwede T; Thony-Meyer L Increased Efficiency of Campylobacter Jejuni N-Oligosaccharyltransferase Pglb byStructure-Guided Engineerng. Open Biol 2015, 5, 140227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (388).Kampf MM; Braun M; Sirena D; Ihssen J; Thony-Meyer L; Ren Q In Vivo Production of a Novel Glycoconjugate Vaccine against Shigella Flexneri 2a in Recombinant Escherichia Coli: Identification of Stimulating Factors for in Vivo Glycosylation. Microb. Cel Fact 2015, 14, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (389).Ravenscroft N; Haeuptle MA; Kowarik M; Fernandez FS; Carranza P; Brunner A; Steffen M; Wetter M; Keller S; Ruch C ; et al. Purification and Characterization of a Shigella Conjugate Vaccine, Produced by Glycoengineering Escherichia Coli. Glycobiology 2016, 26,51–62. [DOI] [PubMed] [Google Scholar]
- (390).Ihssen J; Kowarik M; Dilettoso S; Tanner C; Wacker M; Thony-Meyer L Production of Glycoprotein Vaccines in Escherichia Coli. Microb. Cell Fact. 2010, 9, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (391).Terra VS; Mills DC; Yates LE; Abouelhadid S; Cuccui J; Wren BW Recent Developments in Bacterial Protein Glycan Coupling Technology and Glycoconjugate Vaccine Design. J. Med. Microbiol. 2012, 61, 919–926. [DOI] [PubMed] [Google Scholar]
- (392).Ohuchi T; Ikeda-Araki A; Watanabe-Sakamoto A; Kojiri K; Nagashima M; Okanishi M; Suda H Cloning and Expression of a Gene Encoding N-Glycosyltransferase (Ngt) from Saccarothrix Aerocolonigenes Atcc39243. J. Antibiot. 2000, 53, 393–403. [DOI] [PubMed] [Google Scholar]
- (393).Gross J; Grass S; Davis AE; Gilmore-Erdmann P; Townsend RR; St Geme JW 3rd. The Haemophilus Influenzae Hmwl Adhesin Is a Glycoprotein with an Unusual N-Linked Carbohydrate Modification. J. Biol. Chem. 2008, 283, 26010–26015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Grass S; Buscher AZ; Swords WE; Apicella MA; Barenkamp SJ; Ozchlewski N; St Geme JW 3rd. The Haemophilus Influenzae Hmwl Adhesin Is Glycosylated in a Process That Requires Hmwlc and Phosphoglucomutase, an Enzyme Involved in Lipooligosaccharide Biosynthesis. Mol. Microbiol. 2003, 48, 737–751. [DOI] [PubMed] [Google Scholar]
- (395).Grass S; Lichti CF; Townsend RR; Gross J; St Geme JW 3rd. The Haemophilus Influenzae Hmwlc Protein Is a Glycosyltransferase That Transfers Hexose Residues to Asparagine Sites in the Hmwl Adhesin. PLoS Pathog 2010, 6, e1000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (396).Choi KJ; Grass S; Paek S; St Geme JW 3rd; Yeo HJ The Actinobacillus Pleuropneumoniae Hmwlc-Like Glycosyltransferase Mediates N-Linked Glycosylation of the Haemophilus Influenzae Hmwl Adhesin. PLoS One 2010, 5, el5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (397).Kawai F; Grass S; Kim Y; Choi KJ; St Geme JW 3rd; Yeo HJ Structural Insights into the Glycosyltransferase Activity of the Actinobacillus Pleuropneumoniae Hmwlc-Like Protein. J. Biol. Chem. 2011, 286,38546–38557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (398).Naegeli A; Neupert C; Fan YY; Lin CW; Poljak K; Papini AM; Schwarz F; Aebi M Molecular Analysis of an Alternative N-Glycosylation Machinery by Functional Transfer from Actinobacillus Pleuropneumoniae to Escherichia Coli. J. Biol. Chem. 2014, 289, 2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (399).Lomino JV; Naegeli A; Orwenyo J; Amin MN; Aebi M; Wang LX A Two-Step Enzymatic Glycosylation of Polypeptides with Complex N-Glycans. Bioorg. Med. Chem. 2013, 21, 2262–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (400).Song Q; Wu Z; Fan Y; Song W; Zhang P; Wang L; Wang F; Xu Y; Wang PG; Cheng J Production of Homogeneous Glycoprotein with Multisite Modifications by an Engineered N-Glycosyltransferase Mutant. J. Biol. Chem. 2017, 292, 8856–8863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (401).Xu Y; Wu Z; Zhang P; Zhu H; Song Q; Wang L; Wang F ; Wang PG; Cheng J A Novel Enzymatic Method for Synthesis of Glycopeptides Carrying Natural Eukaryotic N-Glycans. Chem. Commun. (Cambridge, U. K.) 2017, 53, 9075–9077. [DOI] [PubMed] [Google Scholar]
- (402).Kong Y; Li J; Hu X; Wang Y; Meng Q; Gu G; Wang PG ; Chen M N-Glycosyltransferase from Aggregatibacter Aphrophilus Synthesizes Glycopeptides with Relaxed Nucleotide-Activated Sugar Donor Selectivity. Carbohydr. Res. 2018, 462, 7–12. [DOI] [PubMed] [Google Scholar]
- (403).Yang X; Qian K Protein O-Glcnacylation: Emerging Mechanisms and Functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (404).Wu Z; Jiang K; Zhu H; Ma C; Yu Z; Li L; Guan W; Liu Y; Zhu H; Chen Y; et al. Site-Directed Glycosylation of Peptide/ Protein with Homogeneous O-Linked Eukaryotic N-Glycans. Bioconjugate Chem. 2016, 27, 1972–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (405).Schjoldager KT; Clausen H Site-Specific Protein O-Glycosylation Modulates Proprotein Processing - Deciphering Specific Functions of the Large Polypeptide Galnac-Transferase Gene Family. Biochim. Biophys. Acta, Gen. Subj 2012, 1820, 2079–2094. [DOI] [PubMed] [Google Scholar]
- (406).Tran DT; Ten Hagen KG Mucin-Type O-Glycosylation During Development. J. Biol. Chem. 2013, 288, 6921–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (407).Hang HC; Bertozzi CR The Chemistry and Biology of Mucin-Type O-Linked Glycosylation. Bioorg. Med. Chem. 2005, 13, 5021–5034. [DOI] [PubMed] [Google Scholar]
- (408).Dziadek S; Kunz H Synthesis of Tumor-Associated Glycopeptide Antigens for the Development of Tumor-Selective Vaccines. Chem. Rec. 2004, 3, 308–321. [DOI] [PubMed] [Google Scholar]
- (409).Liakatos A; Kunz H Synthetic Glycopeptides for the Development of Cancer Vaccines. Curr. Opin Mol. Ther 2007, 9, 35–44. [PubMed] [Google Scholar]
- (410).Tarp MA; Clausen H Mucin-Type O-Glycosylation and Its Potential Use in Drug and Vaccine Development. Biochim. Biophys. Acta, Gen. Subj. 2008, 1780, 546–563. [DOI] [PubMed] [Google Scholar]
- (411).Wilson RM; Danishefsky SJ A Vision for Vaccines Built from Fully Synthetic Tumor-Associated Antigens: From the Laboratory to the Clinic. J. Am. Chem. Soc. 2013, 135, 14462–14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Marcaurelle LA; Bertozzi CR Recent Advances in the Chemical Synthesis of Mucin-Like Glycoproteins. Glycobiology 2002, 12, 69R–77R. [PubMed] [Google Scholar]
- (413).Sames D; Chen XT; Danishefsky SJ Convergent Total Synthesis of a Tumour-Associated Mucin Motif. Nature 1997, 389, 587–591. [DOI] [PubMed] [Google Scholar]
- (414).Shin Y; Winans KA; Backes BJ; Kent SBH; Ellman JA; Bertozzi CB Fmoc-Based Synthesis of Peptide-Athioesters: Application to the Total Chemical Synthesis of a Glycoprotein by Native Chemical Ligation. J.Am. Chem. Soc. 1999, 121, 11684–11689. [Google Scholar]
- (415).Marcaurelle LA; Mizoue LS; Wilken J; Oldham L; Kent SB; Handel TM; Bertozzi CR Chemical Synthesis of Lymphotactin: A Glycosylated Chemokine with a C-Terminal Mucin-Like Domain. Chem. - Eur. J 2001, 7, 1129–1132. [DOI] [PubMed] [Google Scholar]
- (416).Macmillan D; Bertozzi CR Modular Assembly of Glycoproteins: Towards the Synthesis of Glycam-1 by Using Expressed Protein Ligation. Angew. Chem., Int. Ed. 2004, 43, 1355–1359. [DOI] [PubMed] [Google Scholar]
- (417).Cai H; Huang ZH; Shi L; Sun ZY; Zhao YF; Kunz H; Li YM Variation of the Glycosylation Pattern in Mucl Glycopeptide Bsa Vaccines and Its Influence on the Immune Response. Angew. Chem., Int. Ed. 2012, 51, 1719–1723. [DOI] [PubMed] [Google Scholar]
- (418).Leppanen A; Mehta P; Ouyang YB; Ju T; Helin J; Moore KL; van Die I; Canfield WM; McEver RP; Cummings RD A Novel Glycosulfopeptide Binds to P-Selectin and Inhibits Leukocyte Adhesion to P-Selectin. J. Biol. Chem. 1999, 274, 24838–24848. [DOI] [PubMed] [Google Scholar]
- (419).Leppanen A; White SP; Helin J; McEver RP; Cummings RD Binding of Glycosulfopeptides to P-Selectin Requires Stereo-specific Contributions of Individual Tyrosine Sulfate and Sugar Residues. J. Biol. Chem. 2000, 275, 39569–39578. [DOI] [PubMed] [Google Scholar]
- (420).Leppanen A; Parviainen V; Ahola-Iivarinen E; Kalkkinen N; Cummings RD Human L-Selectin Preferentially Binds Synthetic Glycosulfopeptides Modeled after Endoglycan and Containing Tyrosine Sulfate Residues and Sialyl Lewis X in Core 2 O-Glycans. Glycobiology 2010, 20, 1170–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (421).Koeller KM; Smith ME; Wong CH Chemoenzymatic Synthesis of Psgl-1 Glycopeptides: Sulfation on Tyrosine Affects Glycosyltransferase-Catalyzed Synthesis of the O-Glycan. Bioorg. Med. Chem. 2000, 8, 1017–1025. [DOI] [PubMed] [Google Scholar]
- (422).Blixt O; Allin K; Pereira L; Datta A; Paulson JC Efficient Chemoenzymatic Synthesis of O-Linked Sialyl Oligosaccharides. J. Am. Chem. Soc. 2002, 124, 5739–5746. [DOI] [PubMed] [Google Scholar]
- (423).George SK; Schwientek T; Holm B; Reis CA; Clausen H ; Kihlberg J Chemoenzymatic Synthesis of Sialylated Glycopeptides Derived from Mucins and T-Cell Stimulating Peptides. J. Am. Chem. Soc. 2001, 123, 11117–11125. [DOI] [PubMed] [Google Scholar]
- (424).Zeng X; Nakaaki Y; Murata T; Usui T Chemoenzymatic Synthesis ofGlycopolypeptides Carrying Alpha-Neu5ac-(2-> 3)-Beta-D-Gal(l->3)Alpha-D-Galnac, Beta-D-Gal(l->3)Alpha-D-Galnac, and Related Compounds and Analysis of Their Specific Interactions with Lectins. Arch. Biochem. Biophys. 2000, 383, 28–37. [DOI] [PubMed] [Google Scholar]
- (425).Sorensen AL; Reis CA; Tarp MA; Mandel U; Ramachandran K; Sankaranarayanan V; Schwientek T; Graham R; Taylor-Papadimitriou J; Hollingsworth MA; et al. Chemoenzymatically Synthesized Multimeric Tn/Stn Mucl Glycopeptides Elicit Cancer-Specific Anti-Muc1 Antibody Responses and Override Tolerance. Glycobiology 2006, 16, 96–107. [DOI] [PubMed] [Google Scholar]
- (426).Bennett EP; Mandel U; Clausen H; Gerken TA; Fritz TA; Tabak LA Control of Mucin-Type O-Glycosylation: A Classification of the Polypeptide Galnac-Transferase Gene Family. Glycobiology 2012, 22, 736–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (427).Gerken TA; Revoredo L; Thome JJ; Tabak LA; Vester-Christensen MB; Clausen H; Gahlay GK; Jarvis DL; Johnson RW; Moniz HA; et al. The Lectin Domain of the Polypeptide Galnac Transferase Family of Glycosyltransferases (Ppgalnac Ts) Acts as a Switch Directing Glycopeptide Substrate Glycosylation in an N- or C-Terminal Direction, Further Controlling Mucin Type O-Glycosylation. J. Biol. Chem. 2013, 288, 19900–19914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (428).Gerken TA; Jamison O; Perrine CL; Collette JC; Moinova H; Ravi L; Markowitz SD; Shen W; Patel H; Tabak LA Emerging Paradigms for the Initiation of Mucin-Type Protein O-Glycosylation by the Polypeptide Galnac Transferase Family of Glycosyltransferases. J. Biol. Chem. 2011, 286, 14493–14507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (429).Hassan H; Reis CA; Bennett EP; Mirgorodskaya E; Roepstorff P; Hollingsworth MA; Burchell J; Taylor-Papadimitriou J; Clausen H The Lectin Domain of Udp-N-Acetyl-D-Galactosamine: Polypeptide N-Acetylgalactosaminyltransferase-T4 Directs Its Glycopeptide Specificities. J. Biol. Chem. 2000, 275, 38197–38205. [DOI] [PubMed] [Google Scholar]