

Abstract
Although Interleukin‐22 (IL‐22) is produced by various leukocytes, it preferentially targets cells with epithelial origins. IL‐22 exerts essential roles in modulating various tissue epithelial functions, such as innate host defense against extracellular pathogens, barrier integrity, regeneration, and wound healing. Therefore, IL‐22 is thought to have therapeutic potential in treating diseases associated with infection, tissue injury or chronic tissue damage. A number of in vitro and in vivo nonclinical studies were conducted to characterize the pharmacological activity and safety parameters of UTTR1147A, an IL‐22 recombinant fusion protein that links the human cytokine IL‐22 with the Fc portion of a human immunoglobulin. To assess the pharmacological activity of UTTR1147A, STAT3 activation was evaluated in primary hepatocytes isolated from human, cynomolgus monkey, minipig, rat, and mouse after incubation with UTTR1147A. UTTR1147A activated STAT3 in all species evaluated, demonstrating that all were appropriate nonclinical species for toxicology studies.
The nonclinical safety profile of UTTR1147A was evaluated in rats, minipigs, and cynomolgus monkeys to establish a safe clinical starting dose for humans in Phase I trials and to support clinical intravenous, subcutaneous and/or topical administration treatment regimen. Results demonstrate the cross‐species translatability of the biological response in activating the IL‐22 pathway as well as the translatability of findings from in vitro to in vivo systems. UTTR1147A was well tolerated in all species tested and induced the expected pharmacologic effects of epidermal hyperplasia and a transient increase in on‐target acute phase proteins. These effects were all considered to be clinically predictable, manageable, monitorable, and reversible.
Keywords: Epithelial tissues, Fc fusion protein, IL‐22, pharmacology, preclinical translation, toxicology
Abbreviations
- ADA
Anti‐drug antibodies
- CRP
C‐reactive protein
- ELISA
Enzyme‐linked immune‐absorbent assay
- GLP
Good laboratory practice
- H&E
Hematoxylin and eosin
- IL‐22R
IL‐22 receptor
- LLOQ
Lowest limit of quantification
- NOAEL
No observable adverse effect level
- PD
Pharmocodynamic
- PK
Pharmacokinetic
- REG3A
Regenerating islet‐derived protein 3 alpha
- STAT3
Signal transducer and activator of transcription 3
- TK
Toxicokinetic
1. INTRODUCTION
Interleukin 22 (IL‐22) belongs to the IL‐10 cytokine family1 and binds specifically to the IL‐22 receptor (IL‐22R), a heterodimeric complex consisting of an IL‐22R and an IL‐10R2 chain.2 While IL‐10R2 chain is ubiquitously expressed, the IL‐22RA1 chain is preferentially expressed on a variety of cells with epithelial origins tissues, including epidermal keratinocytes, the gastrointestinal (GI) tract epithelium, liver hepatocytes, pancreatic epithelium, and renal tubular epithelium.3 IL‐22 binding on epithelial tissues results in activation of the STAT3 (Signal transducer and activator of transcription 3), which further induces expression of genes involved in innate immunity, regeneration, barrier function, tissue remodeling and wound healing. While the IL‐22 pathway is implicated in the regulation of responses involved in host defense,4, 5 inflammation,6, 7 and repair,8 there is also evidence to suggest that an activation of the IL‐22 pathway may induce or exacerbate inflammatory processes in psoriasis and in arthritis.7, 9, 10, 11
IL‐22 induces the expression of mucus‐associated molecules and the restitution of mucus‐producing goblet cells.12 In addition, IL‐22 increases the production of antimicrobial peptides,5, 12 which is essential for host defense against various pathogens and leads to modulation of the colonic microflora.5, 13 IL‐22 also increases epithelial proliferation and repair of the epithelial barrier 8, 14, 15 and elevated systemic levels of IL‐22 appear to have a protective effect against mucosal damage. Evidence for IL‐22′s role in wound healing comes from its ability to stimulate keratinocyte proliferation to accelerate re‐epithelialization, induce local vascular endothelial growth factor (VEGF) production to increase neovascularization, enhance chemokine production to accelerate inflammatory responses, boost antimicrobial responses to reduce infection, and facilitate tissue remodeling.15, 16 All these functions are considered critical to normal cutaneous wound repair. IL‐22 induces the hepatic production of lipopolysaccharide (LPS)‐binding protein (LBP), which inhibits systemic inflammation induced by LPS.6 Through all of these protective mechanisms, IL‐22 has demonstrated efficacy in various murine models of colitis,7, 8, 12, 17, 19 hepatitis,20 and pneumonitis.21
As a result of its potential beneficial effects on innate immunity and regenerative and protective mechanisms in epithelial tissues‐expressing IL‐22R,2, 5, 14, 16 an IL‐22 fusion protein is being evaluated as a potential therapeutic in treating epithelial tissue injury. We developed UTTR1147A, a human interleukin‐22 (IL‐22) fusion protein that links the cytokine IL‐22 with the Fc portion of human immunoglobulin G4 (hIgG4) to improve the cytokine's pharmacokinetic (PK) characteristics. The Fc portion of the fusion protein incorporates a mutation (N227G) to minimize the potential for Fc effector function. UTTR1147A is currently being produced by Chinese hamster ovary cells and has a deglycosylated molecular weight of approximately 85 kD.
To ensure that we had a clear understanding of safety risks as a result of expected pharmacologic effects of activating the IL‐22 pathway, a series of nonclinical studies were conducted in in vitro and in vivo systems. The toxicology program for UTTR1147A was designed to support the intravenous (IV), subcutaneous (SC), and topical administration of UTTR1147A in the clinic where dosing is performed once every 2 or 3 weeks for various indications. Based on STAT3 activation by UTTR1147A in primary hepatocytes of various species, the rat, minipig, and cynomolgus monkey were selected as appropriate toxicity species for safety assessment, with the cynomolgus monkey considered the most sensitive species. Data presented in this paper suggest a conservation of the IL‐22 pathway and its biological function across species and demonstrate that the safety risks identified nonclinically translate very well to the clinic.20 And based on the postulated mechanism of action of IL‐22 in the repair of epithelial barrier, UTTR1147A may be beneficial in the treatment of infectious or inflammatory diseases.
2. MATERIALS AND METHODS
2.1. In vitro
Cryopreserved human and cynomolgus monkey primary hepatocytes (Bioreclamation IVT, Baltimore, MD) were thawed in prewarmed invitro GRO CP Plating and plated at 106 cells/well into 6‐well collagen‐coated tissue culture plates (Life Technologies, Carlsbad, CA) and allowed to adhere for 4 hours. Media was then changed to serum‐free Maintenance medium (Life Technologies) and incubated overnight (18 hours) at 37°C (5% CO2). Freshly isolated and plated primary hepatocytes from Yucatan minipig (Triangle Research Labs, Charlottesville, VA), CD1 mouse (Bioreclamation IVT; Life Technologies), and Sprague Dawley rats (Bioreclamation IVT; Triangle Research Labs) were replenished with fresh serum‐free Maintenance medium upon receiving from the vendor and incubated overnight (18 hours) at 37°C (5% CO2). After overnight incubation, media was removed and replaced for cell stimulation with serum‐free Maintenance media either with vehicle, UTTR1147A (test material), positive control (rhIL22 or rhIL6 for the rat, due to decreased sensitivity of rat to rhIL‐22) for STAT3 activation. After 5, 30 minutes and/or 24 hours incubation at 37°C (5% CO2), cells were put immediately on ice, media removed, rinsed in cold PBS. PBS was removed and cells lysed in 100 μL/well of 1X RIPA buffer, including protease and phosphatase inhibitors. Cell lysates were clarified by centrifugation at 18,078 g for 10 minutes at 4°C. An aliquot was removed to determine protein concentration, and the remainder frozen at −80°C until Western blot analysis. Protein concentration was determined using the BCA Assay (Calbiochem, Darmstadt, Germany).
2.1.1. Western blot analysis
Protein lysates were thawed and 50 μg of protein per sample was loaded onto 4%‐20% Tris‐Gly gels (Novex by Life Technologies). Proteins were separated by standard SDS‐PAGE, transferred to nitrocellulose membranes, and detected, using a STAT3 or pSTAT3‐Tyr705 primary antibody (Cell Signaling, Danvers, MA) and peroxidase conjugated goat anti‐rabbit F(ab’)2 secondary antibody (Jackson Immuno Research, West Grove, PA). Blots were imaged, using Gene Gnome (SynGene Bio imaging, Cambridge, UK) and quantified with the Adobe Photoshop CS4. Quantitative analyses of phosphorylation were performed by determining the ratio between relative density of the total protein and the phosphorylation band. Using the data from three separate experiments, values are expressed as mean relative density ± SD (n = 3).
2.1.2. Statistical analysis
Student's two‐tailed t‐test with Welch's correction was used to compare data between two groups; P < 0.05 was considered statistically significant.
2.2. In vivo
All animal studies were conducted at contract research organization laboratories (details for each study are given below) according to their Standard Operating Procedures and in compliance with applicable regulations concerning the use of laboratory animals. All protocol and procedures employed were reviewed and approved by the respective institutional review committees.
Assessment of toxicity was based on clinical signs, postdosing observations, dose site reactions, activity monitoring, bodyweights, food consumptions, ophthalmoscopy, hematology, clinical chemistry, urinalysis, macroscopic findings at necropsy, organ weights, and microscopic histopathology. Blood samples were also analyzed for toxicokinetic (TK) evaluation, pharmacodynamic (PD) parameters, and anti‐therapeutic antibodies. Safety pharmacology assessment was also conducted as a part of the 11‐week GLP study in monkeys and included cardiovascular (electrocardiogram and hemodynamic endpoints in surgically implanted telemetry animals), respiratory, and neurobehavioral endpoints at predefined intervals throughout the study duration.
Specifically, acute phase proteins serum C‐reactive protein (CRP) was measured as part of the standard clinical chemistry panel and plasma fibrinogen was measured as part of the coagulation panel, conducted at the contract research organization laboratories. Circulating levels of REG3A were quantified in serum using a commercially available enzyme‐linked immunoabsorbent assay (ELISA) kit developed for detection of REG3A (Dynabio, Marseille, France). Cynomolgus monkey serum samples were analyzed for detection of antibody responses to UTTR1147A, using an ELISA for detecting anti‐therapeutic antibodies (Genentech, South San Francisco, USA). The concentration of UTTR1147A in rat, minipig, and cynomolgus monkey sera was determined using a validated ELISA that used a mouse MAb against human IL‐22 to capture UTTR1147A and another anti−IL‐22 for detection (Genentech, South San Francisco, USA). The assay lower limit of quantitation (LLOQ) is 4‐5.0 ng/mL.
Tissues obtained from various organs were preserved in 10% neutral‐buffered formalin, embedded in paraffin, sectioned, mounted on glass slides, and stained with hematoxylin and eosin (H&E) for histology. Microscopic evaluation of tissue was subsequently performed by a veterinary pathologist.
2.3. Rats
2.3.1. 11‐Week study
A GLP toxicity study was conducted at Covance Laboratories in Harrogate, UK. In this study, UTTR1147A was administered to each dose group (50, 150, or 1500 μg/kg, twice weekly via IV administration, for a total of 22 doses over 11 weeks at dose levels of (15 rats/sex/group for toxicity group, 9 rats/sex/group for toxicokinetics group and, 3 rats/sex/group for pharmacodynamics group). Five male and five female rats per treatment group were assessed for reversibility of any drug‐related effects after an 8‐week recovery period. These doses were chosen based on initial tolerability data from a pilot toxicity study (data not shown) and were expected to cover the efficacious dose range in a mouse DSS colitis model,19 where UTTR1147A demonstrated efficacy by inducing both a significant decrease in colon histologic scores and protection from weight loss. The trough serum concentration that was associated with the lowest efficacious dose was 10.4 ng/mL.19
Crl:CD(SD) strain from Charles River Laboratories, Margate, UK was used in the study. Animal care, use, and handling are in full accordance with SOP HAR‐GEN‐609 (Covance Harrogate, UK). The animals were assigned to treatment groups on arrival using a total randomization procedure. Animals were approximately 6 weeks old at the start of dosing. Males weighed between 114.7 and 192.2 g and females weighed between 93.6 and 174.3 g. The animals were housed in groups of up to five in cages that conform to the ‘Code of practice for the housing and care of animals used in scientific procedures’ (Home Office, London, 1989). Animals were housed singly during the functional observation battery (FOB) assessment to facilitate clear observation of the animal in the home cage and to effect use of the ‘blinded’ test procedure. Animals were transferred to single housing 24 hours (nominal) prior to the procedure. The scheduled necropsies were performed on Day 78 (Week 12) of the dosing phase and Day 53 (Week 8) of the recovery phase.
2.4. Minipigs
2.4.1. Local subcutaneous study
A non‐GLP local subcutaneous (SC) toxicity study was conducted at Charles River Laboratories in Spencerville, OH. Female melanotic Yucatan minipigs (3 animals per dose group) were administered a single SC injection of UTTR1147A at 0, 0.930, and 4660 μg to evaluate skin toxicity, as well as to evaluate the TK characteristics of IL‐22Fc. These doses were chosen to be within the range of the projected efficacious dose in humans.
Melanotic Yucatan minipigs from Sinclair Research Center, Inc., Auxvasse, MO were used in the study. Animal care, use, and handling are in full accordance with the Animal Welfare and the Humane Treatment of Animals Policy of Charles River Laboratories. The animals were 18 weeks old and weighted between 16.1 and 21.7 kg at initiation of dosing. Animals were randomly assigned to groups by a stratified randomization scheme designed to achieve similar group mean body weights. Animals were group‐housed (2‐3 animals/swine pen or cage) for 3‐5 days and then individually housed in swine pens/cages equipped with an automatic watering valve. Housing and care were as specified in the USDA animal welfare Act (9 CFR, Parts 1, 2, and 3) and as described in the Guide for the Care and Use of Laboratory Animals from the National Research Council.
Following sample collection on Day 16, the surviving animals were observed for 7 days and then transferred to the testing facility's training colony.
2.4.2. Wounded minipig model study
A non‐GLP study was conducted at the CiTox Laboratory in Lille Skensved, Denmark. A wounded minipig model was used to assess the impact of topically administered UTTR1147A on PK, PD, and toxicity parameters. Minipigs were anesthetized and wounds were established on the dorsolateral area of both sides of the back of the animal. Eight circular full thickness wounds were made on each animal and hemostasis was achieved with a sterile gauze. Gel formulations were applied on each wound with 0, 64, 318, or 1059 μg/cm2 when dressings were changed on Days 3, 5, 7, 11, 13, 15, 17, 19, and 21 at a dose volume of 1 mL per wound. Each wound was evaluated macroscopically at each change in dressing and at necropsy on Day 25. The doses of 64‐1059 μg/cm2 were chosen based on results from a wound model in db/db mice demonstrating 100 μg/cm2 UTTR1147A to be efficacious in wound closure (data not shown;.14
Gottingen SPF minipigs from Ellengard Gottingen (Dalmose, Denmark) were used in the study. Animal care, use, and handling are in full accordance with SOPs 1.1.3/01, 1.1.2/12, 1.1.2/13, 1.1/26 (Lille Skensved, DK). The animals weighed between 28.8 and 35 kg at initiation of dosing. Animals were randomly assigned to groups by a stratified randomization scheme designed to achieve similar group mean body weights. Animals were individually housed in floor pens of at least 2 m2 with sawdust as bedding and equipped with an automatic watering valve.
2.5. Monkeys
Two GLP toxicity studies were conducted in cynomolgus monkeys.
2.5.1. 11‐week study
This study was conducted at Charles River Laboratories in Reno, NV. UTTR1147A was administered to 5 male and 5 female cynomolgus monkeys once every 2 weeks, for a total of 6 doses, by intravenous IV injection for 11 weeks at 0, 15, 75, or 300 μg/kg and by SC injection at 0 or 75 μg/kg. However, 2 males and 2 females from each group were assessed for reversibility of any drug‐related effects over an 8‐week recovery period. The doses selected for this study were based on preliminary tolerability data from a pilot 2‐week toxicity study (data not shown) as well as the projected human efficacious dose of 25 μg/kg (based on the 0.03 mg/kg efficacious dose in the mouse DSS colitis model;19 In addition, a subset of animals (2M/2F) from all dose groups were observed for an additional period of 8 weeks following the final dose (i.e., treatment‐free recovery period) to assess the reversibility of toxicity and PD biomarker effects.
Naïve Cynomolgus monkeys (Macaca fascicularis) of Chinese origin were transferred from the colony (originally acquired from a Charles River approved vendor) for the study. Animal care, use, and handling are in full accordance with SOP 1798 (Charles River Laboratories, Reno, NV). The animals were between 2.5 and 5.5 years old and weighed between 2.3 and 3.4 kg prior to initiation of dosing. Animals were randomized and assigned to groups using a computer‐based randomization procedure. Animals were socially housed (up to 3 animals of same sex and same dosing group together) in stainless steel cages equipped with a stainless steel mesh floor and an automatic watering valve. Primary enclosures were as specified in the USDA Animal Welfare Act (9 CFR, Parts 1, 2 and 3) and as described in the Guide for the Care and Use of Laboratory Animals. However, 3 animals/sex/group were necropsied (terminal necropsy on Study Day 74) and 2 animals/sex/group were monitored for an additional 8‐week recovery period (recovery necropsy on Study Day 130).
2.5.2. Chronic 6‐month study
This study conducted at Covance Laboratories in Muenster, Germany. UTTR1147A was administered to 5 sexually mature male and 5 sexually mature female cynomolgus monkeys by IV injection Q2W for a total of 14 doses over 26 weeks at 0, 30, 100, and 300 μg/kg. As in the 11‐week study, 2 males and 2 females per group were assessed for reversibility of any drug‐related effects over a 12‐week recovery period. These doses were selected based on the results of the previous 11‐week study where we observed minimal findings starting at 75 μg/kg.
Naïve Cynomolgus monkeys were purpose bred for use in this study. Animal care, use, and handling are in full accordance with SOP 07.05.11, 07.05.14, 07.05.27, and 07.05.31 (Covance Muenster, DE). The monkeys ranged in age from 2.5 to 5.5 years and weighed between 2.3 and 3.4 kg in the 11‐week study and 4 years of age or older for females and 5 years of age or older for males and weighed between 2.8 and 7.4 kg in the 26‐week study group. Animals were assigned to treatment groups, where possible, based on existing social groups and body weight. After group assignment, the mean body weight for each group/sex was not statistically different at the 5.0% probability level, as indicated by analysis of variance F probability. As appropriate, animals of the same gender and dose group were group‐housed following successful compatibility evaluation to provide for psychological enrichment unless precluded for scientific or health/behavioral reasons, and except during study‐related procedures, if necessary. The 3 animals/sex/group were necropsied (terminal necropsy on Study day 186/187) and 2 animals/sex/group were monitored for an additional 12‐week recovery period (recovery necropsy on Study Day 274).
2.6. Compliance with design and statistical analysis requirements
Animal experiments were designed with groups with an equal N value. In cases where N values were <5 per group, statistical analyses were not performed and data were qualitatively interpreted. In vitro hepatocyte data was derived from hepatocytes isolated from 3 different animals and experiments ran in triplicate and was used only to test precision of n = 1 , but not analyzed as independent experiments.
3. RESULTS
3.1. In vitro STAT3 activation in primary hepatocytes
Sequence homology in IL‐22 and IL‐22Ra1 protein across species is shown in Figure A1. The percent identity of the nonclinical species to human IL22 and IL22Ra1 ranges from 75% to 95% and 71% to 88%, respectively (Table A1, A2). IL‐22 has been shown to signal via JAK kinases, leading to phosphorylation of STAT3 and the generation of activated phospho STAT3 (pSTAT3) in vivo.2, 8 Therefore, we utilized the phosphorylation of STAT3 as a marker of IL‐22 pathway activation by UTTR1147A with the objective of characterizing the biological species sensitivity by evaluating in vitro potency of UTTR1147A in primary hepatocytes from human, cynomolgus monkey, minipig, rat, and mouse; and to confirm the use of appropriate species for nonclinical assessment.
A representative Western blot image is shown in Figure 1. UTTR1147A‐induced STAT3 phosphorylation in primary hepatocytes from human, cynomolgus monkey, minipig, and mouse was observed after incubation with UTTR1147A at 0.5 μg/mL for 30 minutes. To achieve similar level of STAT3 phosphorylation in rat hepatocytes, higher concentrations of UTTR1147A (5 and 50 μg/mL) and longer incubations (30 minutes and 24 hours) were required.
Figure 1.
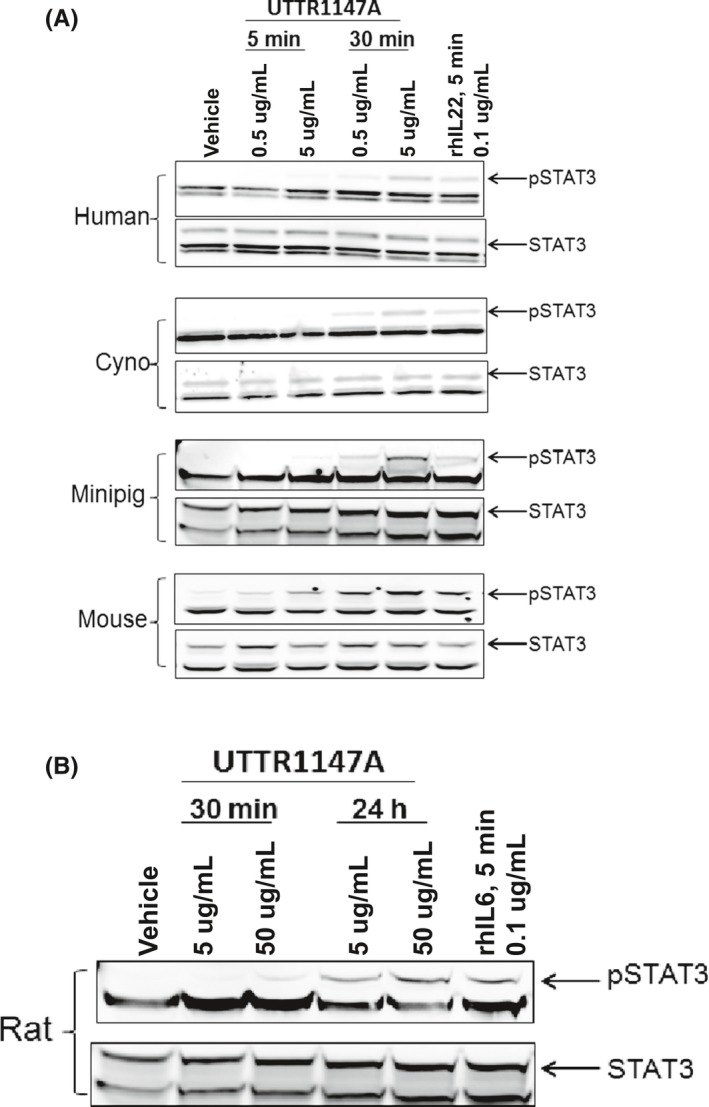
Cultured primary hepatocytes across species were treated with vehicle, UTTR1147A, or positive control. Representative Western blot from 3 separate independent experiments is shown here. Top panel: Concentration‐ and time‐dependent elevations in STAT3 phosphorylation were observed in hepatocytes from human, cynomolgus monkey, minipig, and mouse upon treatment with 0.5 and 5 μg/mL UTTR1147A (0.1 μg/mL rhIL‐22 was used as a positive control) for 5 or 30 minutes. Bottom panel: Higher concentrations (up to 50 μg/mL) and longer duration treatment (up to 24 hours) with UTTR1147A was needed to demonstrate STAT3 phosphorylation in rat hepatocytes. (0.1 μg/mL rhIL‐6 was used as a positive control). Quantification of these blots is presented in Figure A2. STAT3, Signal transducer and activator of transcription 3; pSTAT3, phosphorylated STAT3; rh, recombinant human protein
Densitometric quantification of these blots is presented in Figure A2. The results illustrate the ability of UTTR1147A in activating STAT3 phosphorylation after engaging IL‐22 receptors on hepatocytes from all tested species, albeit with rat needing higher concentrations. Based on these data, studies proceeded with rat, minipig, and cynomolgus monkeys as appropriate nonclinical species for the in vivo translational safety assessment of UTTR1147A.
3.2. In vivo
3.2.1. Repeat dose toxicity study in rats
While the in vitro response of rat hepatocytes indicated that rats were less sensitivity to IL‐22 pathway activation than other species, we chose the rat as the main rodent species for safety assessment for several reasons. We demonstrated that rat hepatocytes did respond to UTTR1147A, rats are larger than mice and may be better suited for multiple blood draws (thereby reducing the number of rats used), and rats are very commonly used in nonclinical safety assessment studies and as such and have a large and well‐established reference database.
Rats tolerated the repeated administration in this study with no significant weight loss and no neurobehavioral and ophthalmic abnormalities over the period of study (data not shown). The only UTTR1147A‐related finding was reversible, minimal to mild epidermal hyperplasia in the highest dose group (1500 μg/kg; Figure 2).
Figure 2.
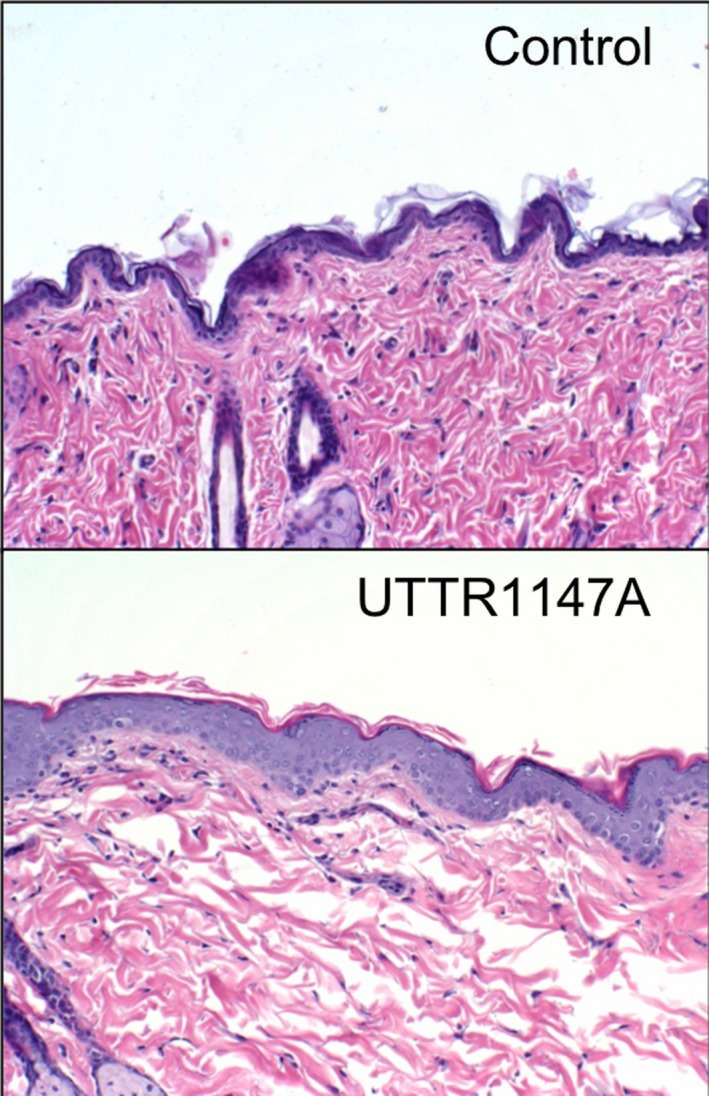
H&E stained sections of skin obtained from 1500 μg/kg UTTR1147A‐treated rats on Study Day 78 show a thickening of the epidermal layer, compared to skin from control rats. The observed epidermal hyperplasia was reversed by the end of the 8‐week recovery period (data not shown)
While fibrinogen and CRP elevations were considered a direct downstream pharmacological effect of IL‐22R activation, the rats did not exhibit increases in these acute phase proteins as rats typically use different acute phase response pathways, which were not tested23 (Table 1).
Table 1.
A summary of toxicologic, toxicokinetic, and pharmacodynamic parameters from the various toxicity studies is presented here
| Species (Study) | Epidermal hyperplasia (dose observed) | Biomarkers of target engagement | NOAEL (μg/kg) | % ATA | Doses μg/kg (IV) | C max (ng/mL) | AUC0‐t (ng · day/mL) |
|---|---|---|---|---|---|---|---|
| Rat (11‐week) | Yes (1500 μg/kg) |
|
150 | 24 | 50 | 667 (136) | 547 |
| 150 | 2780 (499) | 2370 | |||||
| 1500 | 32 000 (9680) | 38,000 | |||||
| Miniplg (SC) | Yes (930, 4660 μg) | Not measured | ‐ | ‐ | 930 μg (SC) | 109 (21.4) | 434 (279) |
| 4660 μg (SC) | 929 (123) | 3790 (2650) | |||||
| Miniplg (Wound) | Not measured | ≥318 μg/cm2 :
|
1059 μg/cm2 | 64 μg/cm2 | – | – | |
| 318 μg/cm2 | – | – | |||||
| 1059 μg/cm2 | 7.6 | – | |||||
| Monkey (11‐week) | Yes (≥75 μg/kg) | ≥75 μg/kg:
|
15 | 13 | 15 | 188 (2S.4) | 333 (163) |
| 75 | 1020 (132) | 1890 (581) | |||||
| 300 | 4300 (452) | 7010 (2650) | |||||
| 75 (SC) | 246 | 1009 | |||||
| Monkey (26‐week) | Yes (300 μg/kg) | >100 μg/kg:
|
100 | 20 | 30 | 547 (51.4) | 2350 (343) |
| 100 | 1900 (263) | 8760 (1840) | |||||
| 300 | 5900 (1690) | 36 000 (12 220) |
NOAEL, No observable adverse effect level; ATA, anti‐therapeutic antibody; C max, maximum serum concentration; AUC, are under the curve indicating exposure of drug over time; standard deviation is added in parentheses for mean AUC and C max values (male and female animals combined). “‐” indicates parameter was not determined.
Overall, based on the toxicity finding of epidermal hyperplasia in the high‐dose group given 1500 μg/kg, the no‐observable adverse effect level (NOAEL) was determined to be 150 μg/kg. At this dose level, the steady‐state C max and cumulative exposure [AUC from Study Day 0‐Study Day 133 {AUC0‐133}] of 2780 ng/mL and 2370 ng·day/mL, respectively, were observed (Table 1).
3.3. Repeat dose toxicity studies in cynomolgus monkeys
Cynomolgus monkeys are considered an appropriate nonrodent species for the safety assessment of biotherapeutics, based on their relevance to antigen‐binding and pharmacologic response.18 Additionally, the in vitro hepatocyte data demonstrating STAT3 activation in response to UTTR1147A (Figure 1) provided a strong rationale that the cynomolgus monkeys would provide relevant translatable information on the impact of systemic IL‐22 pathway activation. To be able to support First‐in‐Human (FIH) entry and a clinical dosing regimen of 6‐12 weeks and beyond, UTTR1147A was administered to cynomolgus monkeys in 2 separate studies.
In the 11‐week study, there were no UTTR1147A‐related changes in body weights or other endpoints evaluated. Additionally, there were no toxicologically relevant changes in cardiovascular, respiratory, or neurologic safety pharmacology parameters observed with either IV or SC administration of UTTR1147A (data not shown).
However, UTTR1147A‐related findings were limited to cutaneous erythema at 300 μg/kg, which correlated with the microscopic finding of epidermal hyperplasia at 75 μg/kg (SC) and 300 μg/kg (IV) (Figure 3A), these were the expected pharmacologic effects of UTTR1147A. These observations were trending toward recovery (clinical signs) or had fully recovered (microscopic findings) by the end of the 12‐week dose‐free period (data not shown).
Figure 3.
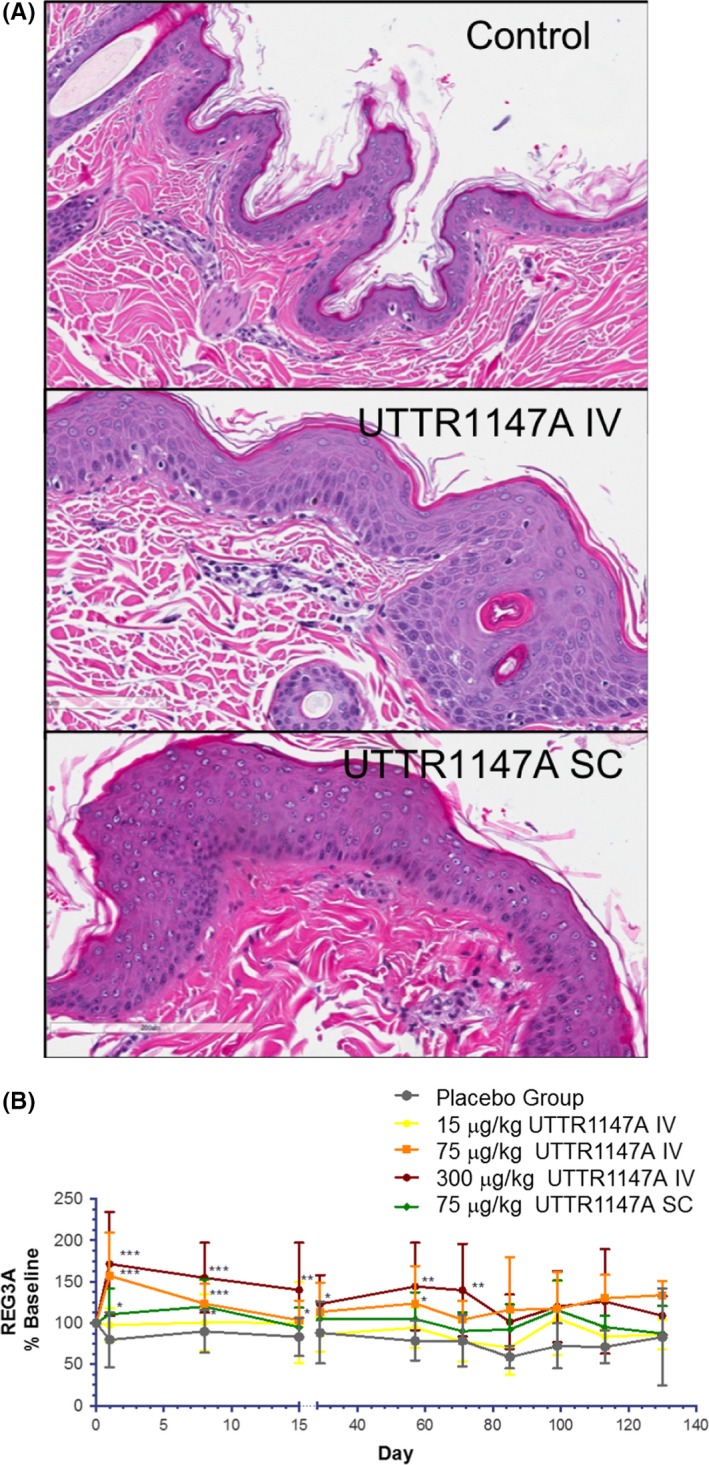
(A) H&E stained sections of skin obtained on Study Day 79, from UTTR1147A‐treated monkeys of the 11‐week study, show substantial thickening of the epidermal layer at injection sites in animals administered 300 μg/kg UTTR1147A by intravenous injections (labeled UTTR1147A IV) as well as by local subcutaneous injection administering 75 μg/kg UTTR1147A (labeled UTTR1147A SC), compared to skin from vehicle‐treated monkeys (labeled Control). The observed epidermal hyperplasia was reversed by the end of the 12‐week recovery period (data not shown). (B) Serum collected from monkeys at various time points of the 11‐week study were measured for REG3A levels, which were elevated and sustained post UTTR1147A administration at IV doses ≥ 75 μg/kg (from pre‐dose to Study Day 130). REG3A levels were only significantly elevated after the first SC dose cycle. Data are plotted as individual REG3A concentrations as a percentage of baseline over time. The data shown are represented as group mean ± SD normalized to baseline. ***P < 0.001; **P < 0.01; *P < 0.05. Significance was assessed using Student's t‐test, comparing individual normalized animals in the treated group to the placebo group at each time point
While five of 40 animals (13%) given UTTR1147A developed anti‐drug antibodies (ADAs) after dosing (Table 1), there was no obvious impact of ADAs on the exposure of UTTR1147A. The exposure of the males was slightly higher than for the females at all dose levels, but did not differ substantially. There was less than 2fold accumulation noted following repeat IV or SC dosing every 2 weeks and TK was roughly linear and dose proportional at these doses (Figure A3).
Ex vivo treatment of mouse colon with IL‐22 upregulates the murine REG3 gene isoforms, REG3β, and REG3γ.5 We evaluated REG3A, the closest‐related NHP/human isoform of rodent REG3β, as an exploratory PD biomarker in cynomolgus monkeys. REG3A is an antimicrobial protein produced by the intestinal epithelial cells and pancreatic acinar cells.25, 26 Increases in REG3A levels were sustained through the dosing period after each dose administration of UTTR1147A ≥ 75 μg/kg and returned to levels similar to control within 2 weeks after the last dose at all dose levels for IV dose groups (Figure 3B). Alterations in REG3A did not persist past the first dose cycle for the SC dose group, compared to the IV dose group at 75 μg/kg, presumably due to lower exposure (C max and AUC; Table 1) in the SC dose group. The dose of 300 μg/kg IV, where we observed sustained elevation in REG3A, was the only dose to produce a C max above the EC50 for pSTAT in human hepatocytes (~3 μg/mL).
At doses ≥75 μg/kg, there were also UTTR1147A‐related increases in the levels of the acute phase proteins fibrinogen and CRP with complete reversibility (Table 1). There were no increases in systemic levels of proinflammatory cytokines or chemokines (data not shown), demonstrating that UTTR1147A was not activating inflammatory pathways that might indirectly induce CRP such as IL‐6 but rather the observed elevations were on‐target dose‐dependent increases as a result of UTTR1147A binding to the IL‐22R.
Based on a lack of the adverse effects of skin toxicity, the NOAEL following UTTR1147A administration was considered to be the lowest dose of 15 μg/kg IV (C max/AUC0t of 188 ng/mL/333 ng·day/mL; Table 1).
In a chronic toxicity study in cynomolgus monkeys designed to support longer‐term dosing in humans, the only UTTR1147A‐related effect in this study was cutaneous erythema and the correlating microscopic finding of epidermal hyperplasia at the dose level of 300 μg/kg at terminal necropsy (data not shown). Affected animals at this dose level also had increased serum fibrinogen levels, consistent with an acute phase response (Table 1). Both the skin findings and the elevated fibrinogen levels were fully reversed following a 12‐week recovery period.
Dose‐dependent elevations in the serum PD biomarker REG3A followed the first dose administration at doses ≥100 μg/kg (Table 1), confirming the pharmacologic activity of UTTR1147A and suggested that IL‐22R engagement was achieved at these dose levels. Levels of REG3A appeared to correlate with serum levels of UTTR1147A, and returned to baseline levels by the end of the 12‐week recovery period.
We used sexually mature monkeys in the chronic toxicity study, and thus were also able to assess the impact of UTTR1147A on sexual reproduction endpoints. There were no effects of UTTR1147A observed on male and female reproductive endpoints (testicular and semen evaluation; menstrual cyclicity, and ovarian and uterus maturation stage) and there were no UTTR1147A‐related macroscopic and microscopic findings in the male and female reproductive organs (data not shown).
In contrast to the 11‐week toxicity study, there was a higher rate of ADA formation in in the 26‐week study, and the formation of ADA had a substantial impact on exposure, particularly in the high‐dose group administered 300 μg/kg, where lower exposure and rapid elimination corresponded with higher ADA titer values (1.88‐2.18 on Study Day 29 and 2.90‐4.58 on Study Day 85). Additionally, the ADA‐positive animals that had markedly reduced exposure starting on Study Day 85 had a correlated with loss of the clinical observation of cutaneous erythema and/or loss of the clinical pathology finding of elevated serum fibrinogen.
Based on these observations and under these study conditions, the NOAEL was determined to be the mid‐dose level of 100 μg/kg every 2 weeks (steadystate C max and steady‐state AUCtau of 1900 ng/mL and 8760 ng·day/mL, respectively; Table 1) for 26 weeks in this study.
3.4. Toxicity studies in minipigs
Due to the anatomic similarity in human and pig skin, the minipig was selected as an appropriate nonclinical species to assess the local toxicity of either a subcutaneous injection or topical administration of UTTR1147A.
In the local subcutaneous study, UTTR1147A‐related, dose‐dependent microscopic findings in the skin at the injection site were observed at 930 and 4660 μg and were characterized by epidermal hyperplasia in both dose groups with the additional findings of epidermal melanosis and increased vascular profiles and a perivascular lymphoplasmacytic infiltrate in the superficial dermis in the 4660 μg dose group,27 similar to the skin findings observed in the cynomolgus monkeys (Figure 3A).
UTTR1147A was well tolerated locally and systemically up to an absolute dose of 4660 μg. The C max and exposure of UTTR1147A were roughly dose proportional across the dose levels tested [930 μg (109 ng/mL and 434 ng/mL) and 4660 μg (929 day·ng/mL and 3790 day·ng/mL); Table 1] and correlated with the dose‐dependent severity and incidence in microscopic skin findings.
Based on the expected pharmacologic findings of IL‐22 on the skin, consistent with the other nonclinical species tested, we were also interested in assessing the local tolerability of UTTR1147A in a minipig wound model, which would enable another route of administration in humans.
In the wound‐healing minipig model, UTTR1147A did not impair the normal wound healing process (Figure 4A shows Study Day 11 clinical observations), whereby all wounds were closed and healed by the end of the study (data not shown). UTTR1147A was only detected in the serum of 2 animals in the high dose group (Table 1). This increase in systemic exposure corresponded with slight elevations in the levels of the systemic biomarker serum amyloid protein A (SAA) in a few individual animals (Figure 4B), suggesting target engagement of the IL‐22 pathway in this model.
Figure 4.
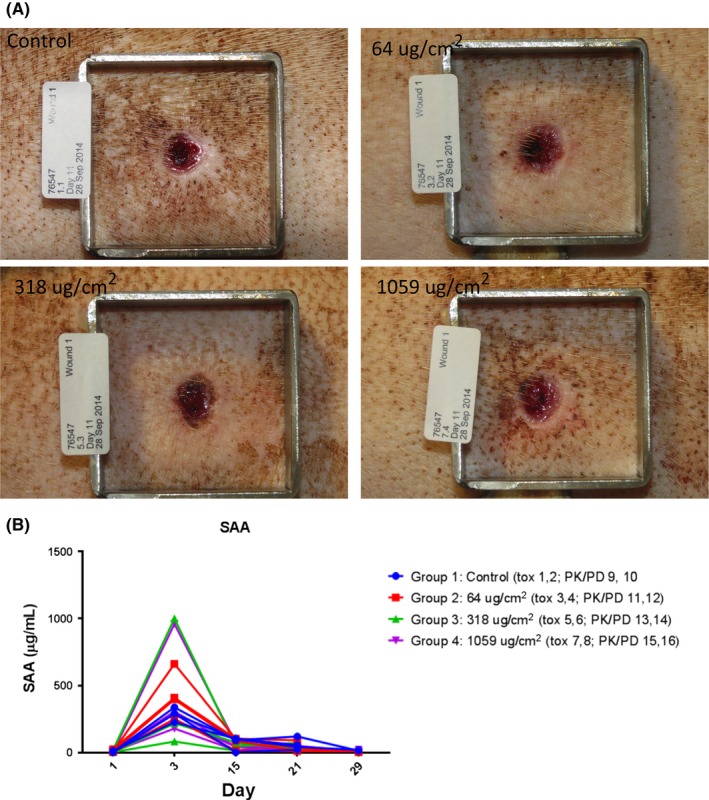
(A) Wounds of minipigs were topically administered up to 1059 μg/cm2 UTTR1147A every other day up to Study Day 21. Wounds were macroscopically observed on days of dosing, which also corresponded with days of dressing change. Clinical observations of all wounds in minipigs on Study Day 11 is presented here and do not indicate a treatment‐related impairment of the wound healing process. (B) Blood was collected from minipigs on Study Days ‐1 (pre‐dosing on Day 1), 3, and 21. Serum amyloid protein A was measured and demonstrated to be slightly elevated 2 days post‐ UTTR1147A administration in wounded minipigs topically administered UTTR1147A. Individual animal responses are shown here
Overall, topical administration of UTTR1147A to full‐thickness dermal wounds was well tolerated and caused no adverse effects.
4. DISCUSSION
Nonclinical studies with UTTR1147A have demonstrated a positive benefit to risk ratio in the use of this molecule for the treatment of inflammatory and infectious diseases. Key safety risks are related to expected pharmacologic activity in vivo, including acute phase protein responses and skin toxicity. All UTTR1147A‐related findings were consistent with the expected pharmacologic effect of activating the IL‐22 pathway, given that IL‐22Ra1 is expressed on a variety of epithelial tissues, including epidermal keratinocytes, gastrointestinal tract epithelium, liver hepatocytes, pancreatic epithelium, and renal tubular epithelium.3
In support of species selection for toxicology studies, in vitro results demonstrated similarities in UTTR1147A‐induced response of STAT3 and provided additional justification in the use of rat, minipig and cynomolgus monkeys as relevant species for translating pharmacologic activation of the IL‐22 pathway to humans. Despite a high degree of conservation of the IL‐22 pathway across species, different response levels, both in vitro and in vivo, were seen as a result of differential IL‐22 pathway activation. Rats were less sensitive to the effects of UTTR1147A than cynomolgus monkeys, both in vitro, as evidenced by lower sensitivity of rat hepatocytes to pSTAT3 phosphorylation, and in vivo, as evidenced by the lack of clinical signs; clinical pathology findings or skin reddening; and lessened severity of the microscopic findings of epidermal hyperplasia in rats. We postulate that this reduced sensitivity could be due to lesser protein sequence homology and/or differential expression in IL‐22/IL‐22Ra1 levels in the rats.
UTTR1147A was well tolerated in both cynomolgus monkeys and rats up to the highest dose tested, 300 μg/kg IV and 75 μg/kg SC in cynomolgus monkeys; and 1500 μg/kg in rats. Results from all systemic and local injection toxicity studies were similar, with UTTR1147A causing expected pharmacologic effects in the skin, consistent with activating the IL22 pathway in epidermal keratinocytes,16, 24 including skin reddening (in cynomolgus monkeys, only) with microscopic findings limited to reversible, dose‐dependent, epidermal hyperplasia (all species tested), sometimes accompanied by variably increased vascularity in the superficial dermis (primarily in the local toxicity study in minipigs). These skin findings have also been seen in human clinical trials,20 demonstrating the appropriate nonclinical species selection in evaluating the pharmacologic effects of UTTR1147A.
While ADA formation was observed in both rat and monkey repeat‐dose toxicity studies, ADA had no obvious impact on UTTR1147A exposure, except for in the 26‐week chronic study where animals that tested positive for ADA had lower exposure. The markedly reduced exposure is likely to have contributed to the loss of target engagement starting on Study Day 85.
Elevated CRP and fibrinogen are consistent with activating the IL22 pathway in liver hepatocytes,6, 24 and elevated REG3A is consistent with activating the IL22 pathway in intestinal and pancreatic acinar epithelial cells.5, 25, 26 In contrast to the cynomolgus monkeys, rats did not exhibit increases in CRP and fibrinogen levels as rats utilize a different acute‐phase response than monkeys.23 Results from the monkey studies demonstrated that serum REG3A levels were increased, and correlated with UTTR1147A drug levels. These data demonstrate pharmacological activity of UTTR1147A and suggest that IL22R engagement was achieved. While the functional aspects of the increase in REG3A in cynomolgus monkeys given UTTR1147A were not explored in the toxicity studies reported here, systemic administration of recombinant REG3γ, a murine homologue of REG3A, partially protected against weight loss and mortality in IL22−null mice infected with Citrobacter rodentium.5 As with the skin findings, these transient dose‐dependent elevations in acutephase proteins as well as in REG3A were observed in human clinical trials.22
Doses of 75 μg/kg and above in monkey studies presented here produced C max at or above the in vitro and in vivo EC50 level estimated from human hepatocytes (~3 μg/mL) and cynomolgus monkeys (~1 μg/mL), respectively.19 In general, the doses of IL‐22Fc required to elicit measurable serum biomarker elevations in healthy mice and monkeys (muIL‐22Fc or UTTR1147A, respectively) were associated with a serum C max above 1 μg/mL and these biomarkers returned to baseline when serum levels were below the 10‐100 ng/mL range.19 However, diseased animals may have different levels and/or turnover of IL‐22 such that different concentrations of IL‐22Fc are required in order to induce pharmacological activity compared to healthy animals.
The low systemic exposure of UTTR1147A in the minipig wound model indicated that either the systemic absorption or bioavailability of UTTR1147A through topical administration to the wound was minimal or that the total dose administered was too low for systemic detection. This could be beneficial to indications where local IL‐22 is deficient at sites of injury and is needed for tissue repair.
There were no increases in systemic levels of proinflammatory cytokines or chemokines in nonclinical studies conducted in cynomolgus monkeys. These findings are consistent with IL‐22Ra1 expression being limited to epithelial tissues, and no expression of IL‐22Ra1 on leukocytes, including T lymphocytes. These findings have also translated clinically, in that UTTR1147A administration in humans has not elicited a proinflammatory cytokine response at the given doses.20
In summary, results from these nonclinical studies demonstrate the cross‐species translatability of the biological response in activating the IL‐22 pathway as well as the translatability of findings from in vitro to in vivo systems. Thus, the toxicology program has appropriately identified relevant toxicities associated with UTTR1147A as reversible, minimal to mild epidermal hyperplasia with minimal inflammation, and a reversible acute‐phase inflammatory response. All toxicities are considered monitorable, manageable, and reversible; and have reliably translated to the toxicities observed in humans in ongoing clinical trials.22
Lastly, it is also important to note, that depending on the tissue environment in which IL‐22 is being upregulated, the functional consequence of activating the IL‐22 pathway can lead to either further inflammation or tissue repair.11 More recently, contributions of IL‐22 from mast cells 28 and innate lymphoid cells (ILCs;29 have been postulated to contribute to the pathophysiology of diseases such as psoriasis and colon cancer. Understanding how different cellular mechanisms, such as IL‐18 stimulating IL‐22 production in ILCs29 and IL‐37 neutralizing proinflammatory activities,30, 31 may be essential to further explore interactions of IL‐22 underlying inflammatory diseases. Therefore, it is important for drug developers to take into account disease pathophysiology, tissue microenvironments in which IL‐22 will be targeting, as well as gaining a deeper understanding of the translatability of animal models to human disease.
AUTHOR CONTRIBUTION
Lee and Danilenko wrote and contributed to the writing of the manuscript:; Pai, Doudement, and Rae designed and conducted in vitro experiments; Lee, Danilenko, and Zhong designed and conducted in vivo toxicology experiments; Wang and Ouyang contributed to pharmacology experiments and editorial review of manuscript; and Lee, Danilenko, Lutman, Stefanich, Sukumaran, Harder, Herman, Lekkerkerker, Wang, and Ouyang performed sample and data analysis .
DISCLOSURE
None declared.
ACKNOWLEDGEMENTS
Naruhisa “Neko” Ota, Dept of Immunology (Genentech) for preparation of manuscript.
APPENDIX 1.
1.1.
Figure A1.

Comparison of amino‐acid sequences of IL22 across species. The human IL22 sequence is shown in full‐length. Aligned with human IL22 sequence, identical amino acid sequences are marked with blank for cynomolgus monkey (cyno), mouse, rat and minipig, and only the different amino acids are displayed. The absence of amino acids are indicated by “‐“
Figure A2.
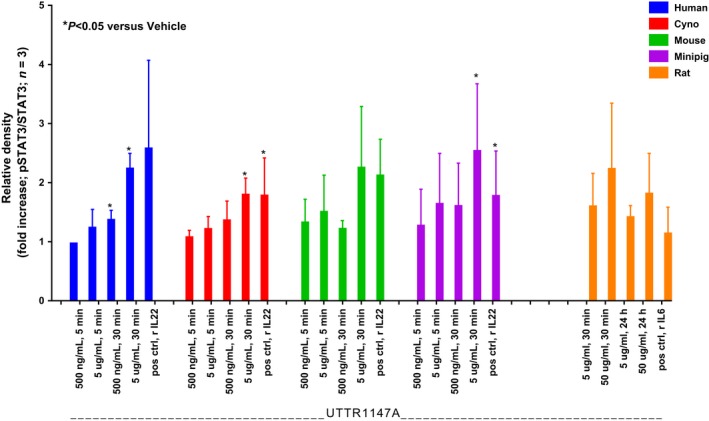
Treatment of isolated primary hepatocytes with UTTR1147A substantially induced STAT3 phosphorylation indicating IL‐22 pathway activation, a biological response across species. Hepatocytes from human, cynomolgus monkey, minipig, and mouse were treated with 0.5 and 5 μg/mL UTTR1147A (or 0.1 μg/mL rhIL‐22 as a positive control) for 5 or 30 minutes. Rat hepatocytes were treated with 5 and 50 μg/mL UTTR1147A (or 0.1 μg/mL rhIL‐6 as a positive control) for 30 minutes or 24 h. Quantitative analyses of STAT3 phosphorylation, presented in Figure 1, were performed by determining the ratio between relative density of the total protein and the phosphorylation band of western blots. Values are expressed as fold change in mean relative density ± SD relative to vehicle control (n = 3). Student's two‐tailed t‐test with Welch's correction was used to compare data between vehicle control and each treatment, within each species. P < 0.05 were considered statistically significant (vs. vehicle)
Figure A3.
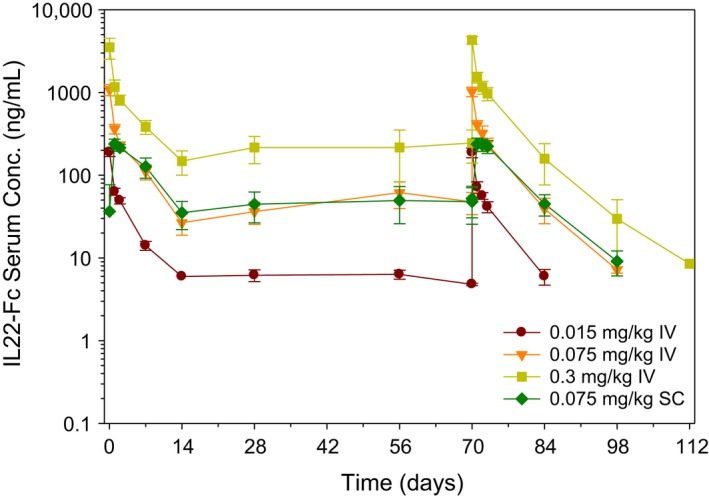
Cynomolgus monkeys were given IV or SC dose of UTTR1147A every 2 weeks for 11 weeks, for a total of 6 doses. Serum from monkeys was collected at various time points throughout the study and UTTR1147A was measured using ELISA. Serum concentrations of combined male and female monkeys are plotted over time (Study Day) as mean ± SD
Table A1.
Sequence homology of IL‐22 protein across species
| Percent similarity | |||||
| Human | Cyno | Mouse | Rat | Minipig | |
| Human | 100 | 96 | 88 | 87 | 82 |
| Cyno | — | 100 | 80 | 88 | 82 |
| Mouse | — | — | 100 | 97 | 79 |
| Rat | — | — | — | 100 | 78 |
| Minipig | — | — | — | — | 100 |
| Percent identity | |||||
| Human | Cyno | Mouse | Rat | Minipig | |
| Human | 100 | 95 | 78 | 75 | 75 |
| Cyno | — | 100 | 89 | 76 | 74 |
| Mouse | — | 100 | 90 | 68 | |
| Rat | — | — | 100 | 68 | |
| Minipig | — | — | — | — | 100 |
—, no entry; cyno, cynomolgus monkey; IL, interleukin.
Table A2.
Sequence homology of IL‐22RA1 receptor across species
| Percent similarity | |||||
| Human | Cyno | Mouse | Rat | Minipig | |
| Human | 100 | 90 | 80 | 79 | 82 |
| Cyno | — | 100 | 74 | 74 | 76 |
| Mouse | — | — | 100 | 95 | 75 |
| Rat | — | — | — | 100 | 74 |
| Minipig | — | — | — | — | 100 |
| Percent identity | |||||
| Human | Cyno | Mouse | Rat | Minipig | |
| Human | 100 | 88 | 71 | 71 | 75 |
| Cyno | — | 100 | 66 | 65 | 69 |
| Mouse | — | — | 100 | 91 | 66 |
| Rat | — | — | — | 100 | 64 |
| Minipig | — | — | — | — | 100 |
—, no entry; cyno, cynomolgus monkey; IL, interleukin.
Lee DW, Zhong S, Pai R, et al. Nonclinical safety assessment of a human interleukin‐22FC IG fusion protein demonstrates in vitro to in vivo and cross‐species translatability. Pharmacol Res Perspect. 2018;e434 10.1002/prp2.434
Funding information
This work was supported by Genentech, Inc.
REFERENCES
- 1. Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL‐10 family of cytokines in inflammation and disease. Annu Rev Immunol. 2011;29:71‐109. [DOI] [PubMed] [Google Scholar]
- 2. Xie MH, Aggarwal S, Ho WH, et al. Interleukin (IL)‐22, a novel human cytokine that signals through the interferon receptor‐related proteins CRF2‐4 and IL‐22R. J Biol Chem. 2000;6(275):31335‐31339. [DOI] [PubMed] [Google Scholar]
- 3. Gurney AL. IL‐22, a Th1 cytokine that targets the pancreas and select other peripheral tissues. Int Immunopharmacol. 2004;4:669‐677. [DOI] [PubMed] [Google Scholar]
- 4. Aujla SJ, Chan YR, Zheng M, et al. IL‐22 mediates mucosal host defense against Gram‐negative bacterial pneumonia. Nat Med. 2008;14:275‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zheng Y, Valdez PA, Danilenko DM, et al. Interleukin‐22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282‐289. [DOI] [PubMed] [Google Scholar]
- 6. Wolk K, Witte E, Hoffman U, et al. IL‐22 induces lipopolysaccharide‐binding protein in hepatocytes: a potential systemic role of IL‐22 in Crohn's disease. J Immunol. 2007;178:5973‐5981. [DOI] [PubMed] [Google Scholar]
- 7. Zheng Y, Danilenko DM, Valdez P, et al. Interleukin‐22, a T(H)17 cytokine, mediates IL‐23‐induced dermal inflammation and acanthosis. Nature. 2007;8(445):648‐651. [DOI] [PubMed] [Google Scholar]
- 8. Pickert G, Neufert C, Leppkes M, et al. STAT3 links IL‐22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465‐1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Geboes L, Dumoutier L, Kelchtermans H, et al. Proinflammatory role of the Th17 cytokine interleukin‐22 in collagen‐induced arthritis in C57BL/6 mice. Arthritis Rheum. 2009;60:390‐395. [DOI] [PubMed] [Google Scholar]
- 10. Ma HL, Liang S, Li J, et al. IL‐22 is required for Th17 cell‐mediated pathology in a mouse model of psoriasis‐like skin inflammation. J Clin Invest. 2008;118:597‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL‐22. Nat Immunol. 2011;12:383‐390. [DOI] [PubMed] [Google Scholar]
- 12. Sugimoto K, Ogawa A, Mizoguchi E, et al. IL‐22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin‐22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947‐957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kolumam G, Wu X, Lee WP, et al. IL‐22R Ligands IL‐20, IL‐22, and IL‐24 Promote Wound Healing in Diabetic db/db Mice. PLoS ONE. 2017;26:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL‐22 increases the innate immunity of tissues. Immunity. 2004;21(2):241‐254. [DOI] [PubMed] [Google Scholar]
- 16. Sa SM, Valdez PA, Wu J, et al. The effects of IL‐20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J Immunol. 2007;178:2229‐2240. [DOI] [PubMed] [Google Scholar]
- 17. Ota N, Wong K, Valdez PA, et al. IL‐22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium. Nat Immunol. 2011;12:941‐948. [DOI] [PubMed] [Google Scholar]
- 18. Deng R, Iyer S, Theil FP, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? MAbs; 3:61‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stefanich EG, Rae J, Sukumaran S, et al. Pre‐clinical and translational pharmacology of a human interleukin‐22 IgG fusion protein for potential treatment of infectious or inflammatory diseases. Biochem Pharmacol. 2018;152:224‐235. [DOI] [PubMed] [Google Scholar]
- 20. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin‐22 but not interleukin‐17 provides protection to hepatocytes during acute liver inflammation. Immunity. 2007;27:647‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simonian PL, Wehrmann F, Roark CL, Born WK, O'Brien RL, Fontenot AP. γδ T cells protect against lung fibrosisvia IL‐22. J Exp Med. 2010;207:2239‐2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rothernberg M, Wang Y, Lekkerkerker A, et al. Randomized phase 1 healthy volunteer study of UTTR1147A (IL‐22Fc), a potential therapy for epithelial injury. Clin Pharm & Ther. 2018; 10.1002/cpt.1164. [DOI] [PubMed] [Google Scholar]
- 23. Watterson C, Lanevschi A, Horner J, Louden C. A comparative analysis of acute‐phase proteins as inflammatory biomarkers in preclinical toxicology studies: implications for preclinical to clinical translation. Toxicol Pathol. 2009;37:28‐33. [DOI] [PubMed] [Google Scholar]
- 24. Park O, Wang H, Weng H, Feigenbaum L, Li H, Yin S, Ki SH, Yoo SH, Dooley S, Wang FS, Young HA, Gao B. In vivo consequences of liver‐specific interleukin‐22 expression: implications for human liver disease progression. Hepatology;54:252‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choi JH, Lee MY, Kim Y, et al. Isolation of genes involved in pancreas regeneration by subtractive hybridization. Biol Chem. 2010;391:1019‐1029. [DOI] [PubMed] [Google Scholar]
- 26. Ogawa H, Fukushima K, Naito H, et al. Increased expression of HIP/PAP and regenerating gene III in human inflammatory bowel disease and a murine bacterial reconstitution model. Inflamm Bowel Dis. 2003;9:162‐170. [DOI] [PubMed] [Google Scholar]
- 27. Danilenko, DM , Phillips, GD , Diaz, D . In vitro skin models and their predictability in defining normal and disease biology, pharmacology, and toxicity. Toxicol Pathol. 2016;44:555‐563. [DOI] [PubMed] [Google Scholar]
- 28. Mashiko S, Bouguermouh S, Rubio M, Baba N, Bissonnette R, Sarfati M. Human mast cells are major IL‐22 producers in patients with psoriasis and atopic dermatitis. J Allergy Clin Immunol. 2015;136:351‐359. [DOI] [PubMed] [Google Scholar]
- 29. Victor AR, Nalin AP, Dong W, et al. IL‐18 Drives ILC3 Proliferation and Promotes IL‐22 Production via NF‐κB. J Immunol. 2017;199:2333‐2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Caraffa A, Conti CD, Ovidio C, et al. New concepts in neuroinflammation: mast cells pro‐inflammatory and anti‐inflammatory cytokinemediators. J Biol Regul Homeost Agents. 2018;32:449‐454. [PubMed] [Google Scholar]
- 31. Robuffo I, Toniato E, Tettamanti L, et al. Mast cell in innate immunity mediated by proinflammatory and antiinflammatory IL‐1 family members. J Biol Regul Homeost Agents. 2017;31:837‐842. [PubMed] [Google Scholar]