

Abstract
Background:
Cerebrospinal fluid (CSF) levels of T-cell activation marker soluble CD27 (sCD27) are associated with subsequent disease activity after a first attack of suspected MS in adults. The predictive value for disease course in children with acquired demyelinating syndromes (ADS) is unknown.
Objectives:
To assess the predictive value of sCD27 levels for clinically definite multiple sclerosis (CDMS) diagnosis in childhood ADS.
Methods:
Children <18 years with a first demyelinating event were prospectively included and followed. Soluble CD27 was determined in CSF using an enzyme-linked immunosorbent assay (ELISA). Cox regression analyses were used to calculate hazard ratios (HRs) for CDMS.
Results:
A total of 94 ADS children were included (ADS with encephalopathy (ADS+) n = 33 and ADS without encephalopathy (ADS–) n = 61). Of the 61 ADS– children, 21 (48%) were diagnosed with CDMS during follow-up. At baseline, sCD27 levels were higher in patients with a future CDMS diagnosis (n = 29) than in monophasic ADS+ (n = 30), monophasic ADS– (n = 28) and relapsing non-MS patients (n = 7; p < 0.001). In ADS– patients, sCD27 was associated with CDMS (HR = 1.8 per 100 U/mL increase in sCD27 levels, p = 0.031), after adjustments for age, oligoclonal bands and the presence of dissemination in space on baseline magnetic resonance imaging (MRI).
Conclusion:
CSF sCD27 levels at first attack of demyelination were associated with CDMS diagnosis in children. This makes sCD27 a potential clinically relevant quantitative marker when performing routine CSF diagnostics.
Keywords: Acquired demyelinating syndromes, multiple sclerosis, biomarker, soluble CD27, children
Introduction
Clinical manifestations of acute onset inflammatory demyelinating disease of the central nervous system (CNS) in children are termed acquired demyelinating syndromes (ADS).1,2 ADS encompasses, for example, optic neuritis (ON), transverse myelitis (TM) as well as other presentations that localise to monofocal or polyfocal locations in the CNS, such as acute disseminated encephalomyelitis (ADEM). Up to one-third of the children with ADS receive a later diagnosis of multiple sclerosis (MS).1,3–5 At the time of the first attack, it can be a challenge to determine the disease course of these patients. Early identification of children who will have an active disease course is important and can have therapeutic implications.6
Soluble CD27 (sCD27) is a soluble form of CD27 secreted by activated T-cells after activation via the T-cell receptor and is introduced as a potential biomarker for T-cell–mediated inflammation.7 CD27 and sCD27 have a role in maturation, activation and proliferation of T- and B-cells.8,9 High sCD27 levels are reported in autoimmune diseases like rheumatoid arthritis, systemic lupus erythematosus and MS.10–12 Komori et al.,13 validated cerebrospinal fluid (CSF) sCD27 as a biomarker for intrathecal T-cell activation in MS, using an extensive and validated battery of biomarkers for CNS inflammation. Serum sCD27 does not discriminate between healthy individuals and MS patients.14 In a recent study, high sCD27 levels in CSF associate with MS diagnosis and disease course in adult patients with clinically isolated syndromes (CISs).15
These observations in adults have not yet been validated in the paediatric population with CNS demyelination. It is shown that children with MS tend to have a more inflammatory disease course than adults.16,17 Therefore, we hypothesised that the predictive value of sCD27 levels for a second attack of MS will be equal or even higher in children than in adults. Furthermore, ADS with encephalopathy (ADEM) are known to have extensive intracerebral inflammation on magnetic resonance imaging (MRI) scans and may have severe clinical presentations.18 The levels of sCD27 might therefore differ between ADS subtypes.
Here, we examined whether sCD27 levels at first attack in children differ between ADS subtypes and assessed the predictive value of sCD27 for a second attack of MS in paediatric ADS patients.
Methods
Study participants
Patients <18 years were included in the Dutch prospective and multicentre study for children with ADS (PROUD-kids study).2 All patients with a lumbar puncture and baseline MRI, performed for routine diagnostics <6 months after onset of first symptoms, were included between June 2006 and February 2017. Patients with alternative diagnosis were excluded. Patients were assessed at baseline and reassessed regularly. Patients were instructed to contact the hospital in case of suspected exacerbation.
Definitions
ADS in children encompass the first attack of demyelination in the CNS, including patients presenting with encephalopathy (ADEM, defined as ADS+) and ADS without encephalopathy (defined as ADS–).2,3 Clinically definite multiple sclerosis (CDMS) was defined as two non-encephalopathic attacks based on the clinical criteria proposed by the International Paediatric multiple sclerosis (MS) study group for paediatric MS diagnosis.3 Relapsing patients who have a distinct clinical phenotype other than CDMS were also included in this study, such as ADEM followed by relapsing ON (ADEM-ON),19 anti-aquaporin 4 antibody (AQP4-ab)-positive and -negative relapsing disease.3,20,21
A relapse was defined as acute worsening of existing symptoms or new symptoms after 30 days of improvement or stable disease and no evidence of an alternative diagnosis. The symptoms should exist for more than 24 hours and not be preceded by fever.22 Exacerbations were confirmed by neurological examination.
Follow-up (FU) duration was calculated by subtracting the date of first symptoms from the last visit date. Disability was expressed by the Expanded Disability Status Scale (EDSS).23
CSF samples and sCD27 enzyme-linked immunosorbent assay
CSF samples were centrifuged for 10 minutes at 3000 r/min to separate the supernatant from cells and cellular components. After centrifugation, all samples were stored in −80 C until use.24 Routine diagnostics of CSF included oligoclonal band (OCB), IgG index, cell count and total protein. Soluble CD27 levels were measured in duplo using the available commercial enzyme-linked immunosorbent assay (ELISA) kit (Pelikine compact human sCD27 kit) manufactured by Sanquin in Amsterdam, the Netherlands.7 The manufacturer’s instructions were followed when performing the sCD27 ELISA. Levels of sCD27 were expressed by U/mL by reference to a standard curve supplied with the ELISA kit. The clinical diagnosis was blinded for the analysts who performed the ELISA. The detection limit of the ELISA was 6 U/mL.
Standard protocol approvals, registrations and patient consents
The PROUD-kids study was approved by the Erasmus MC ethical committee and by the ethical committees of the other participating centres. Written informed consent was obtained from patients and/or their families.
Statistical analysis
Statistical analyses were performed using SPSS 24.0. Kolmogorov–Smirnov test was used to assess the normality of the data. Figures are made in Graphpad Prism5. Soluble CD27 levels were not normally distributed and were therefore log transformed to attain normally distributed data. Due to log-transformation, geometric means were calculated. For group comparisons, Student’s t-test and Mann–Whitney U test were used for continuous variables when appropriate. Student’s t-test was performed to compare the sCD27 levels in different ADS subgroups. Chi-square and Fisher’s exact test were used for categorical data. Correlation analyses were done for two continuous variables.
Cox proportional hazard regression models were used to calculate univariate and multivariate hazard ratios (HRs) in the ADS– group, with CDMS set as endpoint. The Cox proportional hazard assumption was tested by including a time-dependent covariate in the model. Known predictors for MS diagnosis such as age of onset, OCB, and fulfilling dissemination in space (DIS) at baseline MRI are used for adjustments in the multivariate analysis for sCD27 levels.
Annualised relapse rate (ARR) for CDMS patients was compared between groups with high and low levels of sCD27 using a binomial regression model with the natural logarithm of number of FU years after a second clinical attack as offset. This offset corrects for the difference in FU duration between patients. The data were overdispersed and therefore the Poisson regression model was not suitable for our data set. P-value of <0.05 was considered significant.
Results
Patients
A total of 94 children with a first attack of ADS were included in this study. Of these children, 33 presented with ADS+ and 61 with ADS–. The median age for ADS+ was 4.5 years (interquartile range (IQR) = 2.6–6.3) and for ADS– patients 14.5 years (IQR = 11.3–16.0). During FU, 30/33 (91%) of the ADS+ patients remained monophasic. Three ADS+ patients (9%) had a relapsing disease and fulfilled the criteria for ADEM-ON. No patient presenting with ADS+ was diagnosed with CDMS. Within the ADS– patients, 33/61 (54%) had a second attack. Of these 33 relapsing patients, 29/33 (88%) children were diagnosed with a second attack, fulfilling the criteria for CDMS, and the other 4/33 (12%) were diagnosed with a relapsing demyelinating disorder other than MS (2 AQP4-ab–positive and 2 AQP4-ab–negative patients). The median time to CDMS was 10.3 months (IQR = 4.3–15.7). The median FU duration for all included patients was 2.5 years (IQR = 1.4–4.9).
The following flowchart (Figure 1) illustrates the presenting phenotypes (ADS+ and ADS–) and diagnoses during FU.
Figure 1.
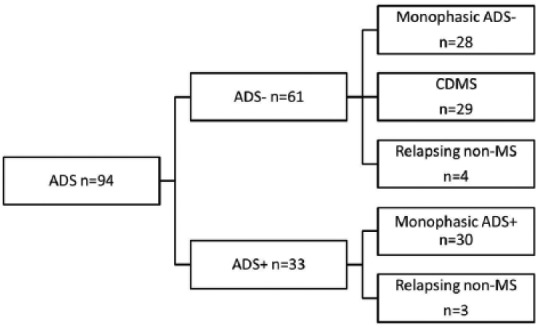
Flowchart of included ADS patients. Patients presented as ADS without encephalopathy (ADS–) and ADS with encephalopathy (ADS+; ADEM). The disease course during follow-up are shown dividing patients in monophasic ADS–, monophasic ADS+, CDMS and relapsing non-MS. The relapsing non-MS patients included three ADEM followed by optic neuritis, two AQP4-positive NMOSD and two patients with AQP4-negative NMOSD. ADS: acquired demyelinating syndromes; ADS–: ADS without encephalopathy; ADS+: ADS with encephalopathy; ADEM: acute disseminated encephalomyelitis; AQP4: anti-aquaporin 4 antibodies; CDMS: clinically definite multiple sclerosis.
The median time between onset of symptoms and CSF sampling was 2.2 weeks (IQR = 0.7–5.9). No correlation was found between the levels of sCD27 and time between first symptoms and CSF sampling in the total group and in all groups separately. Of the 94 patients, 24 (26%) received acute treatment (intravenous corticosteroids) before CSF sampling. No difference in sCD27 was found in patients who did or did not receive intravenous corticosteroids. No patients were on disease-modifying therapy (DMT) or oral steroids before CSF sampling. Patient characteristics are shown in Table 1.
Table 1.
Patient characteristics.
| Characteristic | Mono- ADS+ (n = 30) | Mono- ADS– (n = 28) | CDMS (n = 29) | Relapsing non-MS (n = 7) | All (n = 94) | P-valuea |
|---|---|---|---|---|---|---|
| Female, no. (%) | 20 (67) | 13 (46) | 19 (66) | 4 (57) | 56 (60) | 0.380 |
| Age, median (IQR), years | 4.1 (2.5–6.1) | 11.7 (6.2–16.0) | 15.1 (13.8–16.0) | 10.7 (6.0–16.3) | 11.3 (5.1–15.2) | <0.001 |
| Follow-up time, median (IQR), years | 3.7 (1.3–6.1) | 2.1 (1.1–3.9) | 2.6 (1.8–4.6) | 2.3 (1.2–4.3) | 2.6 (1.4–4.9) | 0.343 |
| Clinical presentation, no. (%) | ||||||
| Isolated optic neuritis, no. (%) | 0 | 7 (25) | 7 (24) | 0 | 14 (15) | 0.940b |
| Isolated bilateral ON, no. (%) | N/A | 2/7 (22) | 0 | N/A | 2/14 (14) | |
| Isolated transverse myelitis, no.(%) | 0 | 8 (29) | 7 (24) | 1 (14) | 16 (17) | 0.704b |
| Isolated LETM, no. (%) | N/A | 4/8 (50) | 0 | 1/1 (100) | 5/16 (31) | |
| Other CIS, no. (%) | 0 | 6 (21) | 3 (10) | 1 (14) | 10 (11) | 0.251b |
| Polyfocal CIS, no. (%) | 0 | 7 (25) | 12 (41) | 2 (29) | 21 (22) | 0.190b |
| LETM and ON, no. (%) | 3/7 (60) | 0 | 2/2 (100) | 5 (5) | ||
| Polyfocal CIS with encephalopathy, no. (%) | 30 (100) | 0 | 0 | 3 (43) | 26 (37) | N/Ab |
| CSF OCB, (⩾2 bands), no. (%) (n = 78) | 0/21 | 14/24 (58) | 25/27 (93) | 0/6 | 39/78 (48) | <0.001 |
| IgG index, median (IQR) (n = 67) | 0.57 (0.53–0.74) | 0.57 (0.53–0.77) | 1.11 (0.88–1.84) | 0.63 (0.52–0.66) | 0.72 (0.56–1.02) | <0.001 |
| CSF WBC count, median (IQR) (n = 90) | 22 (7–48) | 5 (3–10) | 14 (7–28) | 43 (9–77) | 10 (5–35) | 0.491 |
| % of CSF mononuclear WBC, median (IQR) (46/90) | 77 (55–95) | 75 (45–90) | 100 (90–100) | 90 (70–95) | 90 (65–100) | 0.004 |
| Time from symptom onset to CSF sampling, median (IQR), weeks | 1.6 (0.6–2.8) | 1.3 (0.4–5.9) | 5.0 (2.4–12.9) | 2.6 (1.3–10.6) | 2.3 (0.7–5.9) | <0.001 |
| CSF sampling prior to acute treatment, no. (%) | 23 (77) | 21 (75) | 23 (79) | 3 (43) | 70 (75) | 0.249 |
ADS: acquired demyelinating syndrome; mono-ADS+: monophasic ADS with encephalopathy; mono-ADS-: monophasic ADS without encephalopathy; CDMS: clinically definite multiple sclerosis; relapsing non-MS: relapsing ADS not diagnosed as MS; ON: optic neuritis; CIS: clinically isolated syndrome; LETM: longitudinally extended transverse myelitis; OCB: oligoclonal band; IQR: interquartile range; WBC: white blood cell; NA: not applicable.
Comparison between all subgroups.
Comparison between monophasic ADS– and CDMS.
Soluble CD27 levels in subgroups of ADS
Soluble CD27 levels at first attack of ADS were higher in patients with a future second attack of MS (n = 29) than in ADS– patients who remained monophasic (n = 28) (geometric mean = 65 U/mL; 95% confidence interval (CI) = 47–89 vs 13 U/mL; 95% CI = 9–18, p < 0.001). Patients with monophasic ADS+ (n = 30) did not differ in sCD27 levels from monophasic ADS– patients (n = 28) (geometric mean = 18 U/mL; 95% CI = 11–30 vs 13 U/mL; 95% CI = 9–18) but did differ from ADS– patients with future CDMS diagnosis (geometric mean = 13 U/mL; 95% CI = 9–18 vs 65 U/mL; 95% CI = 48–89; p < 0.001). Patients with a relapsing non-MS disease course (n = 7; four patients with ADS– onset and three with ADS+ onset) had lower sCD27 levels at onset than patients with a future CDMS diagnosis (geometric mean = 18 U/mL; 95% CI = 8–42 vs 65 U/mL; 95% CI = 48–89; p = 0.001) but did not differ from monophasic ADS+ and monophasic ADS– patients. These results are displayed in Figure 2. No differences were found in sCD27 levels between anti-myelin oligodendrocyte glycoprotein (MOG)-positive (n = 13) and anti-MOG–negative (n = 81) patients (data not shown).
Figure 2.
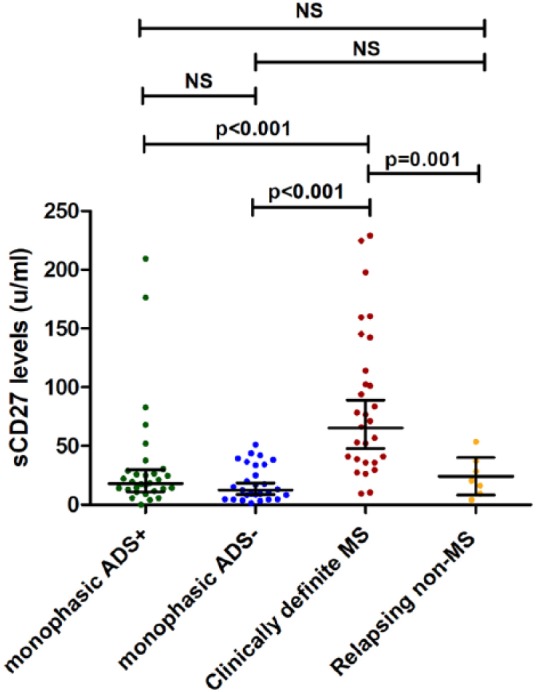
Comparison of CSF sCD27 levels in ADS at time of first demyelinating event. Comparison of CSF soluble CD27 levels between patients with ADS+ (ADEM), monophasic ADS–, CDMS and relapsing non-MS patients. The relapsing non-MS patients included three ADEM followed by optic neuritis, two AQP4-positive NMOSD and two patients with AQP4-negative NMOSD. Horizontal lines with error bars indicate geometric means with 95% CI. ADS: acquired demyelinating syndromes; ADS–: ADS without encephalopathy; ADS+: ADS with encephalopathy; ADEM: acute disseminated encephalomyelitis; CDMS: clinically definite multiple sclerosis.
Of the 59 ADS– patients, 14 (24%; after excluding the two AQP4-positive patients) fulfilled MS diagnosis at baseline by fulfilling the IPMSSG 2012 criteria for MS on first MRI.3 The sCD27 levels of these 14 children are significantly higher than ADS– patients who did not fulfil the criteria at baseline (geometric mean = 78 U/mL; 95% CI = 53–115 vs 20 U/mL; 95% CI = 15–30, p < 0.001).
Of the 28 monophasic ADS– patients, 10 patients showed sCD27 levels that exceed the upper bound of the 95% CI of the geometric mean (13 U/mL; 95% CI = 9–18), as shown in Figure 2. Of these patients, 8/10 fulfilled the diagnosis of MS by FU MRI scans but did not experience a second attack during FU. The sCD27 geometric mean of these eight patients was higher than the other 20 monophasic ADS– patients (geometric mean = 35 U/mL; 95% CI = 27–45 vs 9 U/mL; 95% CI = 6–12; p < 0.001) but lower than the patients with future CDMS diagnosis (geometric mean = 35 U/mL; 95% CI = 27–45 vs 65 U/mL; 95% CI = 48–89; p = 0.04).
A subgroup analysis (n = 59) was performed after excluding patients with <2 years of FU (patients with CDMS n = 20). This did not change our observation that sCD27 levels are elevated in patients with a future second attack of MS compared to the three other subgroups (geometric mean = 61 U/mL, 95% CI = 42–89 vs 15 U/mL, 95% CI = 10–22; p < 0.001).
Soluble CD27 correlates with CSF and MRI parameters
Patients with OCB (n = 39) had higher sCD27 levels compared to patients without OCB (geometric means = 42 U/mL; 95% CI = 30–60 vs 17 U/mL; 95% CI = 12–23, p < 0.001). Soluble CD27 levels were positively correlated with IgG index (Spearman rho = 0.695, p < 0.001) as well as white blood cell count (Spearman rho = 0.444, p < 0.001).
Patients fulfilling DIS on baseline MRI (n = 50) showed higher sCD27 levels than patients without DIS (geometric mean 35 U/mL; 95% CI = 26–46 vs 16 U/mL; 95% CI = 11–24, p = 0.003).
A total of 72 patients (77%) received gadolinium when the first MRI was performed. The sCD27 levels were significantly higher in children who showed enhancement (n = 31, 58%) than patients without enhancement (geometric mean = 49 U/mL; 95% CI = 34–71 vs 19 U/mL; 95% CI = 12–28, p = 0.001).
For the analyses depicted above, all patients were analysed. Exclusion of patients with ADS+ (ADEM) and relapsing non-MS from the analysis (thus only including patients with monophasic ADS– and CDMS), did not alter the results. Results are shown in Figure 3.
Figure 3.

Comparison of sCD27 levels with CSF and MRI parameters. Comparing CSF and MRI parameters with the levels of sCD27 in ADS patients. Horizontal lines with error bars indicate geometric means with 95% CI. ADS: acquired demyelinating syndromes; OCB: oligoclonal bands; DIS: dissemination in space.
High levels of sCD27 at first attack in ADS– patients are independently associated with a shorter time to CDMS
As described above, all patients with a second attack of MS had an ADS– presentation. No patient with ADS+ was diagnosed with CDMS. Therefore, in the following analyses ADS+ patients were excluded as well as the relapsing non-MS patients with AQP4-ab.
A Kaplan–Meier curve was obtained after dichotomising the sCD27 levels, using the median of the included ADS– patients (n = 59); median sCD27: 36.0 U/mL. Six out of 29 patients in the low sCD27 group were diagnosed with CDMS (21%) versus 23/30 (79%) in the high-level group. Figure 4 shows the Kaplan–Meier curve for time to CDMS diagnosis (Log-rank test, p = 0.006).
Figure 4.

Kaplan–Meier curve for time to CDMS diagnosis in ADS without encephalopathy. Kaplan–Meier curve for time to CDMS diagnosis. All ADS– patients were stratified into two groups by the median CSF sCD27 level of all CIS patients; median = 36 U/mL. ADS: acquired demyelinating syndromes; ADS–: ADS without encephalopathy; CDMS: clinically definite multiple sclerosis.
The univariate HR for CDMS was 2.8 (95% CI = 1.7–4.6) per 100 U/mL increase in sCD27 levels (p < 0.001). In the multivariate COX regression analyses, we corrected for age of onset, OCB and DIS. After these corrections, sCD27 was independently associated with time to CDMS diagnosis with an HR of 1.8 (95% CI = 1.0–3.3) per 100 U/mL increase in sCD27 levels (p = 0.031).
Eight out of 29 patients (28%) who received MS diagnosis based on MRI received DMT before CDMS, and this might have postponed the second attack. We performed a sub-analysis using the COX regression analysis, where we excluded these patients, resulting in the same HR for the univariate (2.8, 95% CI = 1.7–4.9; p < 0.001) and multivariate analyses after adjusting for age of onset, OCB and DIS (1.8, 95% CI = 0.96–3.4; p = 0.061).
ARR and disability
In patients who were diagnosed with CDMS, we used a negative binomial regression model for analysing the ARR. The median of sCD27 levels in CDMS patients (n = 29; 71 U/mL) was used to stratify patients into two groups with either high or low sCD27 levels. There was no difference found in ARR between high- and low-level groups after correcting for FU duration. No correlation was found between sCD27 levels and EDSS score during FU.
Discussion
Here, we show that the T-cell activation marker sCD27 in CSF at first attack in paediatric ADS differs among ADS subtypes. Soluble CD27 levels in patients with a future diagnosis of CDMS were higher than in monophasic ADS+, monophasic ADS– and relapsing non-MS patients. There was no difference between the latter three groups. We analysed ADS+ patients separately from ADS– patients as MS diagnoses after ADS+ is extremely low and also in our cohort no patients with ADS+ were diagnosed with CDMS during FU. Soluble CD27 was associated with a shorter time to CDMS diagnosis independently of known relevant clinical parameters such as MRI and CSF characteristics.
An interesting observation was that 80% of the monophasic ADS– patients, whose sCD27 levels exceeded the 95% CI intervals of the whole monophasic ADS– group, fulfilled the IPMSSG diagnostic criteria for MS based on FU MRI. The sCD27 levels of this group were higher than monophasic ADS– and somewhat lower than patients with CDMS diagnosis. This may be related to the fact that these patients (who are diagnosed with MS by MRI alone) have a less-active clinical disease course, compared to CDMS patients with higher levels of sCD27.
In line with Van der Vuurst de Vries et al.,15 we observed higher sCD27 levels in patients with a future CDMS diagnosis than in monophasic ADS patients without encephalopathy (including CIS). The levels were even higher in children than in adults with MS (geometric mean = 65 U/mL; 95% CI = 48–89 vs 42 U/mL; 95% CI = 29–51, respectively).15 Our data not only validate the conclusion of Van der Vuurst et al but are also congruent with previous observations that paediatric MS patients have a more inflammatory disease course compared to adults. 16,25–30
The higher levels in patients with a future CDMS diagnosis most likely correspond to a higher intrathecal T-cell activation and higher inflammatory activity. The correlation we found between sCD27, OCB and IgG index is in line with previous adult studies. 13–15 In vitro, a functional role of sCD27 on stimulation and differentiation of B-cells is described earlier.31,32 However, the exact role for sCD27 on IgG production remains to be investigated.
In adult MS patients, a higher ARR was found in patients with high sCD27 levels at the time of CIS.15 One would expect also a higher relapse rate in paediatric MS patients with high sCD27 levels at time of the first attack; however, no association with ARR was found. This finding might be explained by a ceiling effect, as the overall relapse rate in our paediatric MS cohort is high.16
No association was found with EDSS, which is little surprising given the slower disease progression in paediatric MS.27,33 Longer FU duration will be needed to investigate a possible relationship between sCD27 levels and chronic disease progression.
There were a few limitations of this study. First, the FU duration varied between patients. We addressed this problem by correcting for FU duration in the survival analyses. We also performed a sub-analysis in which we excluded patients with less than 2 years of FU. This did not alter the conclusions. Second, we did not perform an FU MRI on a regular basis. However, we aimed to assess the value of sCD27 levels on the clinical disease course and therefore chose CDMS as a study endpoint instead of the McDonald criteria. In addition, the use of DMTs may have delayed a second attack and could have influenced the results of the survival analyses. Therefore, we performed sub-analyses after excluding patients with ADS– who used DMT before CDMS diagnosis. After excluding these patients, the multivariate analysis was not significant (p = 0.061); however, there was still a clear trend, and the univariate analysis remained significant. Finally, patients who remained monophasic had a shorter time to CSF sampling compared to patients who were diagnosed with CDMS during FU. This may have been caused by the difference in severity of the presenting symptoms and the differential diagnosis at onset, for example, in ADEM, where acute non-demyelinating pathology needs to be ruled out. Yet, we found no correlation between the time to CSF sampling and the levels of sCD27, making it unlikely that this influenced our results.
In summary, we show that CSF sCD27 in children with ADS at time of the first attack is associated with a future CDMS diagnosis, independently of MRI and CSF parameters. This result is in line with the earlier finding in adult CIS patients that higher sCD27 was associated with subsequent MS diagnosis.14 Therefore, we can conclude that sCD27 is a potential clinically relevant quantitative marker when performing routine CSF diagnostics not only in adults but also in children with ADS. The next step will be validation of these findings in international cohorts.
Acknowledgments
The authors thank all the children and their families who participated in the Dutch Study for Paediatric MS. We thank the members of the Dutch Study Group for Paediatric MS and Acute. Disseminated Encephalomyelitis: D.P. Bakker, Departments of Paediatric Neurology, VU Medical Centre, Amsterdam; M. Boon, Department of Paediatric Neurology, UMCG, Groningen; K.P.J. Braun, Department of Paediatric Neurology, University Medical Centre Utrecht, Utrecht; K.G.J. van Dijk, Department of Paediatrics, Rijnstate Hospital, Arnhem; M.J. Eikelenboom, Department of Neurology, Westfriesgasthuis, Hoorn; M. Engelen, Department of Paediatric Neurology, Academic Medical Centre Amsterdam, Amsterdam; K. Geleijns, Department of Paediatric Neurology, University Medical Centre Utrecht, Utrecht; C.A. Haaxma, Department of Paediatric Neurology, Radboud UMC, Nijmegen; J.M.F. Niermeijer, Department of Neurology, Elisabeth-Tweesteden hospital, Tilburg; E.H. Niks, Department of Neurology, Leiden University Medical Centre, Leiden; C.M.P.C.D. Peeters-Scholte, Department of Neurology, Leiden University Medical Centre, Leiden; E.A.J. Peeters, Department of Neuropediatrics, Haga Hospital, The Hague; B.T. Poll, The Department of Paediatric Neurology, Emma Children’s Hospital/AMC, Amsterdam; R.P. Portier, Department of Neurology, Medisch Spectrum Twente, Enschede; J.F. de Rijk-van Andel, Department of Neurology, Amphia Hospital, Breda; J.P.A. Samijn, Department of Neurology, Maasstad Hospital, Rotterdam; H.M. Schippers, Department of Neurology, St. Antonius Hospital, Nieuwegein; I.N. Snoeck, Department of Neuropediatrics, Haga Hospital, The Hague; H. Stroink, Department of Neurology, Canisius-Wilhelmina Hospital, Nijmegen; R.J. Vermeulen, Department of Paediatric Neurology, Maastricht UMC, Maastricht; A Verrips, Department of Neurology, Canisius-Wilhelmina Hospital, Nijmegen; F Visscher, Department of Paediatric Neurology, Admiraal de Ruyter Hospital, Goes; J.H.S. Vles, Department of Paediatric Neurology, Maastricht UMC, Maastricht; M.A.A.P. Willemsen, Department of Paediatric Neurology, Radboud UMC, Nijmegen. Y.Y.M.W contributed to study design, acquisition of data, statistical analysis, interpretation of data and drafting of the manuscript. R.M.V.D.V.D.V. contributed to the interpretation of data and revision of manuscript for content. E.D.V.P., I.A.K., M.J.M., A.F.W., C.E.C., R.F.N. contributed to acquisition of data, revision of manuscript for content. R.Q.H contributed to study design, study supervision, interpretation of data and revision of manuscript for content.
Footnotes
Author Disclosures: R.F.N. participates in paediatric MS trials sponsored by Sanofi and Novartis. R.Q.H received honoraria for serving on advisory boards for Biogen Idec, Roche and Sanofi. He participated in trials with Biogen Idec, Merck-Serono, Roche, Genzyme and Novartis. The remaining authors have no reports to disclose.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: The study was supported by the Dutch MS research Foundation. This study was not industry sponsored.
Contributor Information
Yu Yi M Wong, Department of Neurology, Erasmus MC, Rotterdam, The Netherlands.
Roos M van der Vuurst de Vries, Department of Neurology, Erasmus MC, Rotterdam, The Netherlands.
E Daniëlle van Pelt, Department of Neurology, Erasmus MC, Rotterdam, The Netherlands.
Immy A Ketelslegers, Department of Neurology, Erasmus MC, Rotterdam, The Netherlands.
Marie-José Melief, Department of Immunology, Erasmus MC, Rotterdam, The Netherlands.
Annet F Wierenga, Department of Immunology, Erasmus MC, Rotterdam, The Netherlands.
Coriene E Catsman-Berrevoets, Department of Paediatric Neurology, Erasmus MC-Sophia, Rotterdam, The Netherlands.
Rinze F Neuteboom, Department of Paediatric Neurology, Erasmus MC-Sophia, Rotterdam, The Netherlands.
Rogier Q Hintzen, Department of Neurology, Erasmus MC, Rotterdam, The Netherlands/Department of Immunology, Erasmus MC, Rotterdam, The Netherlands.
References
- 1. Banwell B, Kennedy J, Sadovnick D, et al. Incidence of acquired demyelination of the CNS in Canadian children. Neurology 2009; 72: 232–239. [DOI] [PubMed] [Google Scholar]
- 2. Hintzen RQ, Dale RC, Neuteboom RF, et al. Pediatric acquired CNS demyelinating syndromes: features associated with multiple sclerosis. Neurology 2016; 87: S67–S73. [DOI] [PubMed] [Google Scholar]
- 3. Krupp LB, Tardieu M, Amato MP, et al. International pediatric multiple sclerosis study group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler 2013; 19: 1261–1267. [DOI] [PubMed] [Google Scholar]
- 4. Tantsis EM, Prelog K, Brilot F, et al. Risk of multiple sclerosis after a first demyelinating syndrome in an Australian paediatric cohort: clinical, radiological features and application of the McDonald 2010 MRI criteria. Mult Scler 2013; 19: 1749–1759. [DOI] [PubMed] [Google Scholar]
- 5. Ketelslegers IA, Catsman-Berrevoets CE, Neuteboom RF, et al. Incidence of acquired demyelinating syndromes of the CNS in Dutch children: a nationwide study. J Neurol 2012; 259: 1929–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ghezzi A, Amato MP, Makhani N, et al. Pediatric multiple sclerosis: conventional first-line treatment and general management. Neurology 2016; 87: S97–S102. [DOI] [PubMed] [Google Scholar]
- 7. Hintzen RQ, De Jong R, Hack CE, et al. A soluble form of the human T cell differentiation antigen CD27 is released after triggering of the TCR/CD3 complex. J Immunol 1991; 147: 29–35. [PubMed] [Google Scholar]
- 8. Hendriks J, Gravestein LA, Tesselaar K, et al. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol 2000; 1: 433–440. [DOI] [PubMed] [Google Scholar]
- 9. Han BK, Olsen NJ, Bottaro A. The CD27-CD70 pathway and pathogenesis of autoimmune disease. Semin Arthritis Rheum 2016; 45: 496–501. [DOI] [PubMed] [Google Scholar]
- 10. Font J, Pallares L, Martorell J, et al. Elevated soluble CD27 levels in serum of patients with systemic lupus erythematosus. Clin Immunol Immunopathol 1996; 81: 239–243. [DOI] [PubMed] [Google Scholar]
- 11. Tak PP, Hintzen RQ, Teunissen JJ, et al. Expression of the activation antigen CD27 in rheumatoid arthritis. Clin Immunol Immunopathol 1996; 80: 129–138. [DOI] [PubMed] [Google Scholar]
- 12. Gattorno M, Prigione I, Vignola S, et al. Levels of soluble CD27 in sera and synovial fluid and its expression on memory T cells in patients with juvenile idiopathic arthritides. Clin Exp Rheumatol 2002; 20: 863–866. [PubMed] [Google Scholar]
- 13. Komori M, Blake A, Greenwood M, et al. Cerebrospinal fluid markers reveal intrathecal inflammation in progressive multiple sclerosis. Ann Neurol 2015; 78: 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hintzen RQ, Van Lier RA, Kuijpers KC, et al. Elevated levels of a soluble form of the T cell activation antigen CD27 in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol 1991; 35: 211–217. [DOI] [PubMed] [Google Scholar]
- 15. Van Der Vuurst, De Vries RM, Mescheriakova JY, Runia TF, et al. Soluble CD27 levels in cerebrospinal fluid as a prognostic biomarker in clinically isolated syndrome. JAMA Neurol 2017; 74: 286–292. [DOI] [PubMed] [Google Scholar]
- 16. Van Der Vuurst, De Vries RM, Van Pelt ED, Mescheriakova JY, et al. Disease course after clinically isolated syndrome in children versus adults: a prospective cohort study. Eur J Neurol 2016. [DOI] [PubMed] [Google Scholar]
- 17. Waldman A, Ness J, Pohl D, et al. Pediatric multiple sclerosis: clinical features and outcome. Neurology 2016; 87: S74–S81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pohl D, Alper G, Van Haren K, et al. Acute disseminated encephalomyelitis: updates on an inflammatory CNS syndrome. Neurology 2016; 87: S38–S45. [DOI] [PubMed] [Google Scholar]
- 19. Huppke P, Rostasy K, Karenfort M, et al. Acute disseminated encephalomyelitis followed by recurrent or monophasic optic neuritis in pediatric patients. Mult Scler 2013; 19: 941–946. [DOI] [PubMed] [Google Scholar]
- 20. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015; 85: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chitnis T, Ness J, Krupp L, et al. Clinical features of neuromyelitis optica in children: US Network of Pediatric MS Centers report. Neurology 2016; 86: 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the ‘McDonald Criteria’. Ann Neurol 2005; 58: 840–846. [DOI] [PubMed] [Google Scholar]
- 23. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983; 33: 1444–1452. [DOI] [PubMed] [Google Scholar]
- 24. Teunissen CE, Petzold A, Bennett JL, et al. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology 2009; 73: 1914–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boiko A, Vorobeychik G, Paty D, et al. Early onset multiple sclerosis: a longitudinal study. Neurology 2002; 59: 1006–1010. [DOI] [PubMed] [Google Scholar]
- 26. Benson LA, Healy BC, Gorman MP, et al. Elevated relapse rates in pediatric compared to adult MS persist for at least 6 years. Mult Scler Relat Disord 2014; 3: 186–193. [DOI] [PubMed] [Google Scholar]
- 27. Renoux C, Vukusic S, Mikaeloff Y, et al. Natural history of multiple sclerosis with childhood onset. N Engl J Med 2007; 356: 2603–2613. [DOI] [PubMed] [Google Scholar]
- 28. Yeh EA, Weinstock-Guttman B, Ramanathan M, et al. Magnetic resonance imaging characteristics of children and adults with paediatric-onset multiple sclerosis. Brain 2009; 132: 3392–3400. [DOI] [PubMed] [Google Scholar]
- 29. Waubant E, Chabas D, Okuda DT, et al. Difference in disease burden and activity in pediatric patients on brain magnetic resonance imaging at time of multiple sclerosis onset vs adults. Arch Neurol 2009; 66: 967–971. [DOI] [PubMed] [Google Scholar]
- 30. Gorman MP, Healy BC, Polgar-Turcsanyi M, et al. Increased relapse rate in pediatric-onset compared with adult-onset multiple sclerosis. Arch Neurol 2009; 66: 54–59. [DOI] [PubMed] [Google Scholar]
- 31. Bohnhorst JO, Bjorgan MB, Thoen JE, et al. Abnormal B cell differentiation in primary Sjogren’s syndrome results in a depressed percentage of circulating memory B cells and elevated levels of soluble CD27 that correlate with serum IgG concentration. Clin Immunol 2002; 103: 79–88. [DOI] [PubMed] [Google Scholar]
- 32. Dang LV, Nilsson A, Ingelman-Sundberg H, et al. Soluble CD27 induces IgG production through activation of antigen-primed B cells. J Intern Med 2012; 271: 282–293. [DOI] [PubMed] [Google Scholar]
- 33. Simone IL, Carrara D, Tortorella C, et al. Course and prognosis in early-onset MS: comparison with adult-onset forms. Neurology 2002; 59: 1922–1928. [DOI] [PubMed] [Google Scholar]