

Abstract
AS is a common rheumatic condition characterized by inflammation and new bone formation. The pathogenesis of AS is likely multifactorial and has not been fully elucidated to date. A major genetic role has been demonstrated. The strongest genetic association is with HLA B27. Numerous other associated genetic polymorphisms have been identified, including those affecting the type 17 immune pathway, although the precise link between genetics and pathogenesis remains unexplained. Several immunological alterations, together with recent therapeutic advances, support a central role for IL-23- and IL-17-producing immune cells in disease pathogenesis. Recently, perturbations of gut microbiota of AS patients have further catalysed research and offer potential for future therapeutic intervention. In this review we outline the genetic basis of AS and describe the current hypotheses for disease pathogenesis. We synthesize recent experimental research data and clinical studies to support a central role for the type 17/23 immune axis in AS.
Keywords: ankylosing spondylitis, genetic predisposition, immunology, pathogenesis
Rheumatology key messages
AS is a complex immune-mediated condition whose pathogenesis is not fully understood.
Genetic factors influencing antigen presentation and the IL-23/17 A pathway confer predisposition to AS.
Therapeutic interventions in AS target inflammatory cytokines produced by innate and adaptive immune cells.
Introduction
AS is a common chronic immune-mediated disease characterized by inflammation of the axial skeleton, with early involvement of the SI joints. In AS inflammation likely originates at the tendon–bone interface (an example of enthesitis), leading to erosion and bone proliferation through mechanisms that are poorly understood. Extra-articular manifestations are common, with uveitis among the most frequent, as well as co-occurrence of psoriasis and IBD, which indicates a shared genetic predisposition for these conditions.
This review delineates our current understanding of the pathogenesis of AS. We summarize recent genetic and immunologic literature and argue that SpA constitutes a type 17 immune-driven disease. SpA lies, together with psoriasis and IBD, on the spectrum of immune-mediated diseases between autoimmunity and autoinflammation [1]. Here, we argue that these diseases likely share many pathogenic features.
Genetic predisposition to AS
HLA-B27
AS has long been associated with inheritance of the HLA allele B27 [2], representing one of the strongest genetic associations of any common human disease. HLA-B27 carriage results in an increased odds ratio for AS of ∼100 in many populations. The concordance rate in monozygotic twins is 63%, and the risk in first-degree relatives is 8.2% [3]. Molecular typing has shown that many closely related forms of HLA-B27 exist, differing at a few amino acid residues only. The likely ancestral form of HLA-B27 is B*2705, which is the most common form in white European populations. Of the 136 reported subtypes of HLA-B27, B*2702, B*2703, B*2704, B*2705 and B*2710 are reported to significantly increase risk in various populations and ethnicities, whereas B*2706 and B*2709 are not associated with disease. All causative variants produce modifications in the biochemical structure of the protein, leading to altered conformation of the HLA heavy chain or to alternative peptide presentation. Although the majority of patients with AS are HLA-B27 positive, this only accounts for ∼30% of the total heritability of the disease, which is estimated at ∼90% [4]. Thus, while HLA-B27 is by far the most important single gene, it contributes a minority of the total genetic risk. Many of the other genetic variants are shared with other immune-mediated conditions, some of which often overlap with AS. The most recent genome-wide association study implicated over 40 genetic regions, significant numbers of which are thought to have putative regulatory functions and thus may contribute to AS susceptibility [5].
Non-HLA genes
Genetic variations in endoplasmic reticulum (ER) aminopeptidase 1 (ERAP-1) are also associated with AS. ERAP-1 is involved in trimming peptides to optimal length for binding to HLA class 1 molecules (including HLA-B27). Disease-associated ERAP-1 variants increase the risk of AS only in the context of HLA-B27 [6], suggesting that ERAP-1 polymorphisms alter the interaction of HLA-B27 with peptides.
Other important associated genes are involved in the so-called IL-17 A/IL-23 immune inflammatory axis. These two cytokines are physiologically linked, because IL-23 signalling through the IL-23 receptor (IL-23 R) on CD4+ Th cells is required for the differentiation and expansion of Th17 cells, which are in turn major producers of the pro-inflammatory cytokine IL-17 A [7]. Polymorphisms of the IL23R gene, and also of the surrounding regulatory region, have been strongly associated with the risk of developing AS [8], IBD [9] and psoriasis [10]. This finding reinforces the concept of a common pathogenic mechanism involving the IL-17 A/IL-23 axis in these diseases. It has also been shown that the protective R381Q allele codes for a loss-of-function mutation, resulting in defective signalling and decreased Th17 cells [11]. IL-23 R signals via downstream cascades that include the Signal transducer and activator of transcription 3 (STAT3) and Tyrosine kinase 2 (TYK2) [12]. Polymorphisms in these molecules are also associated with disease susceptibility in AS [5, 13]. Other polymorphisms associated with AS are found in genes with effects on the IL-6 pathway (upstream of IL-17 A), those involved in T cell proliferation and survival (EOMES, RUNX3 and TBX21) [5], and those involved in the innate immune response (CARD9) [14].
Immunopathogenesis of AS
Recent genetic and immunological research has highlighted a key role for IL-17 A/IL-23 cytokine dysregulation in the aetiology of AS. Together with genetic data and the results of clinical trials of IL-17 A–neutralizing antibodies, this strongly supports the concept of SpA as a type 17-driven inflammatory disease.
HLA-B27 and AS
Despite intensive research, the pathogenic role of HLA-B27 remains unclear. Three major theories explaining the role of HLA-B27 in disease—the arthritogenic peptide theory, the misfolded HLA-B27 hypothesis and the cell-surface homodimers hypothesis—have been put forward with varying degrees of evidence (Fig. 1).
Fig. 1.

Proposed pathogenic mechanisms in AS
(1) Autoreactive CD8+ T cells may recognize the arthritogenic peptides displayed by HLA-B27 on the APC surface; (2) HLA-B27 misfolding in APCs leads to ER stress and consequent overproduction of IL-23; (3) abnormal cell-surface expression of HLA-B27 leads to interaction with innate immune receptors such as KIR3DL2 on CD4+ T cells and promotes type 17 immune responses; (4) HLA-B27 causes intestinal dysbiosis, resulting in overexpression of the IL-17A/IL-23 axis with activation of Th17 or other CD4+ cells, γδ T cells, mast cells, neutrophils and other innate immune cells. This leads to production of IL-17A, IL-22, TNF-α, IFN-γ and other cytokines that target organs and tissues directly or through tissue-resident effector cells. APC, antigen-presenting cell; ER, endoplasmic reticulum; IL23R, IL-23 receptor; KIR3DL2, killer immunoglobulin-like receptor; UPR, unfolded protein response.
The arthritogenic peptide theory proposes that HLA-B27 plays a central pathogenic role in the presentation of joint-specific peptides to CD8+ cytotoxic T cells. Specific self or environmental peptides are proposed to bind to and be presented by HLA-B27, to activate CD8+ cells. Although Chlamydia-specific CD8+ cells can be detected in joints (using HLA-B27 tetramers) in Chlamydia-triggered reactive arthritis [15], no convincing arthritogenic peptides have so far been identified in AS. Furthermore, disease occurs in the HLA-B27 transgenic rat model even in the absence of CD8+ cells [16].
A second theory for the pathogenesis of HLA-B27 in AS stems from the observation that HLA-B27 can misfold in the ER [17]. This misfolding leads to ER stress, which activates an unfolded protein response (UPR), leading to upregulation of IL-23 in dendritic cells [18]. This has been observed in the transgenic HLA-B27 rat model, in which HLA-B27 misfolding in macrophages led to upregulation of IL-23 [19]. However, in AS patients, studies of gut biopsies showed no evidence of upregulated UPR gene transcription, but instead showed autophagy (the intracellular response seen in macrophages that degrades improperly folded proteins) [20]. Hence, the role of the UPR in HLA-B27–driven inflammation remains unproven.
The third major theory for the pathogenesis of HLA-B27 in AS revolves around the ability of HLA-B27 to aberrantly fold to form homodimers [21] or beta-2-microglobulin-free heavy chains on the cell surface [22]. Aberrant forms of HLA-B27 can be recognized in vitro by killer-immunoglobulin-like receptors [23]. These receptors are primarily expressed on NK cells, but are also found on circulating CD4+ T cells [24]. HLA-B27 individuals with AS, and HLA-B27 healthy donors, have a higher frequency of T cells expressing this receptor, and these cells are polarized towards a Th17 phenotype (discussed below), with high levels of IL-17 A being produced [25]. It has also been shown that this receptor is expressed on CD4+ cells after activation, inducing transcription of the Th17 transcription factor, retinoic-acid–related orphan receptor-γt (ROR-γt) [26]. This body of work links cell-surface expression of HLA-B27 with the Th17 immune pathway for the first time.
Finally, HLA-B27 may have an indirect effect in disease causation by altering the individual’s microbiome (discussed below). It is possible that all these theoretical mechanisms of HLA-B27–driven pathogenesis may contribute to disease in an individual.
Interestingly, recent genetic evidence comparing many thousands of AS patients and controls suggested that the nature of the amino acid residue at position 97 in HLA-B molecules, including but not limited to HLA-B27, may be critical for disease predisposition [27]. Alterations at this residue may alter free heavy-chain expression [28].
The role of type 17 immunity in AS: different effector cells may be involved
Th17 cells
After selection in the thymus, CD4+ T cells are released as naïve CD4+ cells into the periphery, where they differentiate into different types of effector T cell (shown in simplified form in Fig. 2). The first T cell subtypes described were Th1 and Th2, each with a specific pattern of cytokine production and key master transcription factors. These were followed by the Tregs. Th17 cells, first described in around 2005, are a relatively new addition to the Th subset paradigm [29]. The key physiological effector function of these cells is thought to be immunity against extracellular bacterial and fungal infections. The importance of Th17 in immune-mediated inflammatory disease was first demonstrated in mouse models, where it was shown that knocking out IL-23 but not IL-12 (with which IL-23 shares the subunit p40) made mice resistant to experimental autoimmune encephalomyelitis, a model of human demyelinating diseases [30]. The effector functions of Th17 are mediated through the release of cytokines such as IL-17 A (the signature cytokine of the lineage), IL-17 F and IL-22. The differentiation of Th17 is driven by a combination of cytokines, including IL-1β, IL-6, TGF-β, and IL-23. However, IL-23 is thought to be the key driver of the pathogenic potential of Th17, promoting the expression of the key transcription factor ROR-γt.
Fig. 2.
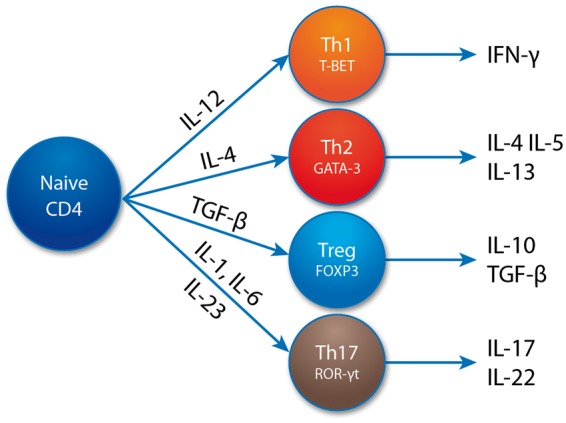
Cartoon showing induction, cytokine production and transcription factor expression of Th17 and other CD4+ T cell subsets
Recent studies have shown that the development of Th17 from naïve T cells can be regulated by hypoxia [31] and by dietary and environmental factors [32]. In addition, it is now apparent that Th17 cells show considerable plasticity and heterogeneity and that some are more pathogenic than others, in part due to production of additional inflammatory cytokines. Thus, IL-17 A cytokine production is often coupled with production of IL-17 F, IL-22 IFN-γ [33] and GM-CSF [34] in the same cell. Additional genes have been found that confer pathogenicity to murine Th17 cells [35]. However, little is known about the existence of such subtypes in humans.
Other type 17 immune cells may be important in AS pathogenesis
The cytokine repertoire and transcriptional profile of Th17 cells are also present in other lymphocyte subsets now being studied in AS pathogenesis. CD161-expressing CD8 T cells, including mucosal-associated invariant T cells [36], γδ T cells [37] and innate lymphoid cells [38] have also been shown to produce IL-17 A. The possible contribution of these cell subsets to the pathogenesis of AS, other than their production of IL-17 A, has only been explored very recently.
A role for tissue resident IL-23–responsive cells in AS pathogenesis?
One of the most specific pathologic features of SpA is inflammation at the enthesis, the site of attachment of bone into tendon or ligament [39]. This specificity of disease localization remains poorly understood, but it has been suggested that the enthesis is a site of high stress, and that local inflammation can be associated with dysregulation of normal homeostatic repair mechanisms at this site. Moreover, recent murine studies have identified resident entheseal cells that can respond to IL-23 and mediate spinal and peripheral inflammation [40]. The existence of these cell populations in humans has not yet been established, but if shown would be hugely significant, providing a pathological basis of inflammation at the enthesis.
Environmental factors in AS pathogenesis: the potential role of gut inflammation and the microbiome
Environmental factors, which may include the microbiome, mechanical stress, infections, and drug or toxin exposure, probably also play a significant role in AS pathogenesis. Bowel inflammation and AS are frequently clinically associated: up to 60% of patients with AS are estimated to show some degree of microscopic inflammatory involvement of the bowel [41], and up to 30% of patients with primary IBD will develop peripheral joint or axial symptoms [42]. The importance of the gut microenvironment in the pathogenesis of AS has been reinforced by mouse studies. The central role of the gut microbiome in SpA has long been suspected, following the observation that rats from the HLA-B27–transgenic model of AS did not develop the disease in germ-free conditions [43]. An influence of HLA-B27 in shaping the microbiome has also been proposed, and the caecal microbiota of Lewis rats transgenic for HLA-B27 and human β2-microglobulin showed differences in specific bacterial species in comparison with wild-type rats [44].
A recent study used 16 S microbial sequencing to compare the microbiomes in terminal ileal biopsies from nine patients with AS with those of healthy volunteers. The authors found significant increases in several bacterial families in AS (Lachnospiraceae, Ruminococcaceae, Rikenellaceae, Porphyromonadaceae, Bacteroidaceae) and a decrease in the abundance of two families (Veillonellaceae and Prevotellaceae) [45].
It seems reasonable to propose that there is a gut–joint immune axis. Notably, upregulation of mRNA for IL-23p19 has been found in the terminal ileum of patients with AS and those with Crohn’s disease [46]. Local alterations in the gut, such as dysbiosis, may alter type 17 immune responses directly or indirectly through altered barrier function or microbial metabolites. These factors might induce loss of tolerance and/or an increase in proinflammatory cytokines, such as IL-23, triggering SpA in susceptible individuals.
Inflammatory cytokines as drivers of inflammation in AS: a rationale for therapeutic inhibition
TNF-α blockade is a well-established and effective therapy in AS. Its introduction followed the successful treatment of other inflammatory conditions with TNF inhibitors and demonstration of overexpression of this cytokine in the SI joints of AS patients [47].
Overactivation of the type 17 immune pathway has been demonstrated more recently, with increased numbers of circulating Th 17 CD4+ cells in the peripheral blood of patients with AS [48]. Furthermore, a general increase in IL-17 A–producing cells across different lymphocyte lineages (CD8+ T cells, NK cells) is seen in AS compared with RA or healthy controls (Al Mossawi et al. unpublished data).
The downstream pathogenic role of IL-17 A in AS is probably a result of its effect on the recruitment of neutrophils, macrophages and epithelial cells, and subsequent release of other pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α. Other type 17–related cytokines, including IL-17 F, IL-22, GM-CSF and IL-9, might also be relevant in driving inflammation in AS, and may thus constitute novel therapeutic targets worthy of further investigation. We propose that, in addition to IL-17 A, a positive feedback loop may result, involving additional inflammatory cytokine production, perhaps as a result of predisposing genetic factors.
New strategies for treating AS
The validity of targeting IL-17 A is confirmed by evidence emerging from clinical trials, in which patients with AS (including those unresponsive to TNF-α blockers) responded to IL-17 A inhibition [49]. The emerging immunological and genetic evidence confirming the key role of the IL-17 A/IL-23 axis has led to the ongoing development of new compounds targeting different molecules, such as drugs inhibiting the p40 subunit of IL-12/IL-23 or the p19 subunit of IL-23. Preclinical studies targeting the Th17 transcription factor RORγt have also shown in vitro inhibition of IL-17 A production [50].
We predict that further advances in our understanding of the mechanisms driving AS pathogenesis will result in the development of additional new treatments for AS. These are exciting times in the AS field!
Acknowledgements
Editorial assistance was provided by Succinct Medical Communications, and funded by Novartis Pharmaceuticals UK Ltd.
Supplement: Novartis has fully funded the production and printing of this supplement. Novartis suggested the topic and authors and reviewed the content to ensure compliance with appropriate regulations. Content was peer reviewed and final editorial control remained with the authors.
Funding: No specific funding was received from any bodies in the public, commercial or not-for-profit sectors to carry out the work on this manuscript.
Disclosure statement: P.B. has received research funding from Merck Research Laboratories, GSK and Celgene and is a local PI for an investigative trial funded by Novartis. D.S. has received research grant support from Celgene. The other author has declared no conflicts of interest. M.H.A.-M. has received unrestricted research funding from UCB Pharma and honoraria from UCB Pharma, Novartis and Pfizer.
References
- 1. McGonagle D, McDermott MF.. A proposed classification of the immunological diseases. PLoS Med 2006;3:e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schlosstein L, Terasaki PI, Bluestone R, Pearson CM.. High association of an HL-A antigen, W27, with ankylosing spondylitis. N Engl J Med 1973;288:704–6. [DOI] [PubMed] [Google Scholar]
- 3. Brown MA, Laval SH, Brophy S, Calin A.. Recurrence risk modelling of the genetic susceptibility to ankylosing spondylitis. Ann Rheum Dis 2000;59:883–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bowness P. Hla-B27. Annu Rev Immunol 2015;33:29–48. [DOI] [PubMed] [Google Scholar]
- 5. Cortes A, Hadler J, Pointon JP. et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet 2013;45:730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Evans DM, Spencer CCA, Pointon JJ. et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet 2011;43:761–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McGeachy MJ, Chen Y, Tato CM. et al. Interleukin 23 receptor is essential for terminal differentiation of effector T helper type 17 cells in vivo. Nat Immunol 2009;10:314–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burton PR, Clayton DG, Cardon LR.. Association scan of 14,500 nsSNPs in four common diseases identifies variants involved in autoimmunity. Nat Genet 2007;39:1329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Duerr RH, Taylor KD, Brant SR. et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006;314:1461–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nair RP, Duffin KC, Helms C. et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat Genet 2009;41:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pidasheva S, Trifari S, Phillips A. et al. Functional studies on the IBD susceptibility gene IL23R implicate reduced receptor function in the protective genetic variant R381Q. PLoS One 2011;6:e25038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cho M-L, Kang J-W, Moon Y-M. et al. STAT3 and NF-κB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J Immunol 2006;176:5652–61. [DOI] [PubMed] [Google Scholar]
- 13. Davidson SI, Liu Y, Danoy PA. et al. Association of STAT3 and TNFRSF1A with ankylosing spondylitis in Han Chinese. Ann Rheum Dis 2011;70:289–92. [DOI] [PubMed] [Google Scholar]
- 14. Pointon JJ, Harvey D, Karaderi T. et al. Elucidating the chromosome 9 association with AS; CARD9 is a candidate gene. Genes Immun 2010;11:490–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Appel H, Kuon W, Kuhne M. et al. Use of HLA-B27 tetramers to identify low-frequency antigen-specific T cells in Chlamydia-triggered reactive arthritis. Arthritis Res Ther 2004;6:R521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Taurog JD, Dorris ML, Satumtira N. et al. Spondylarthritis in HLA–B27/human β2-microglobulin–transgenic rats is not prevented by lack of CD8. Arthritis Rheum 2009;60:1977–84. [DOI] [PubMed] [Google Scholar]
- 17. Mear JP, Schreiber KL, Münz C. et al. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunol 1999;163:6665–70. [PubMed] [Google Scholar]
- 18. Goodall JC, Wu C, Zhang Y. et al. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci U S A 2010;107:17698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. DeLay ML, Turner MJ, Klenk EI. et al. HLA-B27 misfolding and the unfolded protein response augment IL-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheum 2009;60:2633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ciccia F, Accardo-Palumbo A, Rizzo A. et al. Evidence that autophagy, but not the unfolded protein response, regulates the expression of IL-23 in the gut of patients with Ankylosing Spondylitis and subclinical gut inflammation. Ann Rheum Dis 2014;73:1566–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Allen RL, O’Callaghan CA, McMichael AJ, Bowness P.. Cutting edge: HLA-B27 can form a novel β2-microglobulin-free heavy chain homodimer structure. J Immunol 1999;162:5045–8. [PubMed] [Google Scholar]
- 22. Bird LA, Peh CA, Kollnberger S. et al. Lymphoblastoid cells express HLA-B27 homodimers both intracellularly and at the cell surface following endosomal recycling. Eur J Immunol 2003;33:748–59. [DOI] [PubMed] [Google Scholar]
- 23. Kollnberger S, Bird L, Sun M-Y. et al. Cell-surface expression and immune receptor recognition of HLA–B27 homodimers. Arthritis Rheum 2002;46:2972–82. [DOI] [PubMed] [Google Scholar]
- 24. Remtoula N, Bensussan A, Marie-Cardine A.. Cutting edge: selective expression of inhibitory or activating killer cell Ig-like receptors in circulating CD4+ T lymphocytes. J Immunol 2008;180:2767–71. [DOI] [PubMed] [Google Scholar]
- 25. Bowness P, Ridley A, Shaw J. et al. Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. J Immunol 2011;186:2672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ridley A, Hatano H, Wong-Baeza I. et al. Activation-induced killer cell immunoglobulin-like receptor 3DL2 binding to HLA–B27 licenses pathogenic T cell differentiation in spondyloarthritis. Arthritis Rheumatol 2016;68:901–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cortes A, Pulit SL, Leo PJ. et al. Major histocompatibility complex associations of ankylosing spondylitis are complex and involve further epistasis with ERAP1. Nat Commun 2015;6:7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen L, Shi H, Yuan J, Bowness P.. Position 97 of HLA-B, a residue implicated in pathogenesis of ankylosing spondylitis, plays a key role in cell surface free heavy chain expression. Ann Rheum Dis 2017;76:593–601. [DOI] [PubMed] [Google Scholar]
- 29. Harrington LE, Hatton RD, Mangan PR. et al. Interleukin 17–producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 2005;6:1123–32. [DOI] [PubMed] [Google Scholar]
- 30. Cua DJ, Sherlock J, Chen Y. et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003;421:744–8. [DOI] [PubMed] [Google Scholar]
- 31. Dang EV, Barbi J, Yang H-Y. et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell 2011;146:772–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Veldhoen M, Hirota K, Westendorf AM. et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008;453:106–9. [DOI] [PubMed] [Google Scholar]
- 33. Bluestone JA, Mackay CR, O’Shea JJ, Stockinger B.. The functional plasticity of T cell subsets. Nat Rev Immunol 2009;9:811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. El-Behi M, Ciric B, Dai H. et al. The encephalitogenicity of TH17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol 2011;12:568–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gaublomme JT, Yosef N, Lee Y. et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 2015;163:1400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gracey E, Qaiyum Z, Almaghlouth I. et al. IL-7 primes IL-17 in mucosal-associated invariant T (MAIT) cells, which contribute to the Th17-axis in ankylosing spondylitis. Ann Rheum Dis 2016;75:2124–32. [DOI] [PubMed] [Google Scholar]
- 37. Kenna TJ, Davidson SI, Duan R. et al. Enrichment of circulating interleukin-17–secreting interleukin-23 receptor–positive γ/δ T cells in patients with active ankylosing spondylitis. Arthritis Rheum 2012;64:1420–9. [DOI] [PubMed] [Google Scholar]
- 38. Cua DJ, Tato CM.. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol 2010;10:479–89. [DOI] [PubMed] [Google Scholar]
- 39. McGonagle D, Gibbon W, Emery P.. Classification of inflammatory arthritis by enthesitis. Lancet 1998;352:1137–40. [DOI] [PubMed] [Google Scholar]
- 40. Sherlock JP, Joyce-Shaikh B, Turner SP. et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4-CD8- entheseal resident T cells. Nat Med 2012;18:1069–76. [DOI] [PubMed] [Google Scholar]
- 41. De Vos M, Cuvelier C, Mielants H. et al. Ileocolonoscopy in seronegative spondylarthropathy. Gastroenterology 1989;96:339–44. [DOI] [PubMed] [Google Scholar]
- 42. Orchard TR, Thiyagaraja S, Welsh KI. et al. Clinical phenotype is related to HLA genotype in the peripheral arthropathies of inflammatory bowel disease. Gastroenterology 2000;118:274–8. [DOI] [PubMed] [Google Scholar]
- 43. Rath HC, Herfarth HH, Ikeda JS. et al. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J Clin Invest 1996;98:945–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin P, Bach M, Asquith M. et al. HLA-B27 and human β2-microglobulin affect the gut microbiota of transgenic rats. PLoS One 2014;9:e105684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Costello M-E, Ciccia F, Willner D. et al. Brief Report: intestinal dysbiosis in ankylosing spondylitis. Arthritis Rheumatol 2015;67:686–91. [DOI] [PubMed] [Google Scholar]
- 46. Ciccia F, Bombardieri M, Principato A. et al. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum 2009;60:955–65. [DOI] [PubMed] [Google Scholar]
- 47. Braun J, Bollow M, Neure L. et al. Use of immunohistologic and in situ hybridization techniques in the examination of sacroiliac joint biopsy specimens from patients with ankylosing spondylitis. Arthritis Rheum 1995;38:499–505. [DOI] [PubMed] [Google Scholar]
- 48. Shen H, Goodall JC, Hill Gaston JS.. Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis Rheum 2009;60:1647–56. [DOI] [PubMed] [Google Scholar]
- 49. Baeten D, Sieper J, Braun J. et al. Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med 2015;373:2534–48. [DOI] [PubMed] [Google Scholar]
- 50. Wit J. d, Al-Mossawi MH, Hühn MH. et al. RORγt inhibitors suppress TH17 responses in inflammatory arthritis and inflammatory bowel disease. J Allergy Clin Immunol 2016;137:960–3. [DOI] [PubMed] [Google Scholar]