

Abstract
Enzyme replacement therapy (ERT) is available for mucopolysaccharidosis (MPS) I, MPS II, MPS VI, and MPS IVA. The efficacy of ERT has been evaluated in clinical trials and in many post-marketing studies with a long-term follow-up for MPS I, MPS II, and MPS VI. While ERT is effective in reducing urinary glycosaminoglycans (GAGs) and liver and spleen volume, cartilaginous organs such as the trachea and bronchi, bones and eyes are poorly impacted by ERT probably due to limited penetration in the specific tissue. ERT in the present formulations also does not cross the blood–brain barrier, with the consequence that the central nervous system is not cured by ERT. This is particularly important for severe forms of MPS I and MPS II characterized by cognitive decline. For severe MPS I patients (Hurler), early haematopoietic stem cell transplantation is the gold standard, while still controversial is the role of stem cell transplantation in MPS II. The use of ERT in patients with severe cognitive decline is the subject of debate; the current position of the scientific community is that ERT must be started in all patients who do not have a more effective treatment. Neonatal screening is widely suggested for treatable MPS, and many pilot studies are ongoing. The rationale is that early, possibly pre-symptomatic treatment can improve prognosis. All patients develop anti-ERT antibodies but only a few have drug-related adverse reactions. It has not yet been definitely clarified if high-titre antibodies may, at least in some cases, reduce the efficacy of ERT.
Keywords: Enzyme replacement therapy, ERT, Mucopolysaccharidosis, MPS
Background
Enzyme replacement therapy (ERT), based on the periodic intravenous administration of specific enzymes produced with recombinant DNA technology, is at present the most appropriate available therapy for several lysosomal storage disorders.
The recombinant enzymes are produced in continuous human (fibroblasts) or animal cell lines (Chinese hamster ovary (CHO) cells) and plant cells [1] and are a purified form of the lysosomal enzymes. The resulting glycoproteins present mannose-6-phosphate (M6P) residues on the oligosaccharide chains. This allows specific binding of the enzyme to M6P receptors on the cell surface, thus enabling the enzymes to enter the cell and to be targeted to lysosomes, with subsequent catabolism of accumulated substrates [2] (Fig. 1).
Fig. 1.
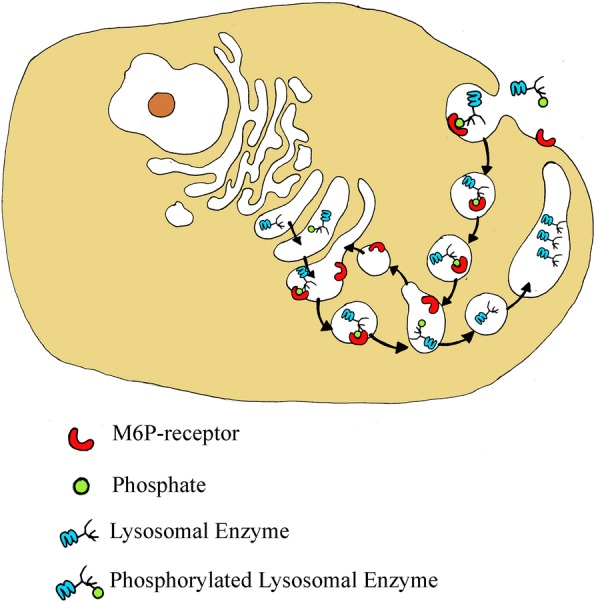
Mannose-6-phosphate (M6P) residues on the oligosaccharide chains of lysosomal enzymes are recognized by specific receptors present in the cell. Thanks to these receptors, the neo-synthesized enzymes are directed to the lysosomal compartment, where they perform their function. The M6P receptors are also expressed on the plasmatic membrane and this allows recombinant lysosomal enzymes to be “captured” by the cells and, following the pathway of the endocytic pathway, to be properly transported to the lysosome. Once lysosomes are reached, recombinant enzymes can replace the enzymatic deficit and degrade the accumulated substrate
The first effective treatment with ERT was performed in patients with Gaucher disease [3] and in the last 15 years ERT has become available for other lysosomal storage disorders including some types of mucopolysaccharidoses (MPS).
MPS I (Hurler, Hurler-Scheie, Scheie syndrome) was the first MPS type treated with ERT (available since 2003); subsequently the treatment became available for MPS VI (Maroteaux-Lamy syndrome; 2005), MPS II (Hunter syndrome; 2006), and MPS IVA (Morquio A syndrome; 2014) (Table 1). Recently, the recombinant enzyme β-glucuronidase has been tested for patients with MPS VII (Sly syndrome) [4, 5] and, to date, the treatment is available for commercial use in the United States where it was approved by the US Food and Drug Administration on 15 November 2017 (https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm585308.htm accessed on 27 June 2018) and is under review by the European Medicines Agency (EMA) (EMA/CHMP/181307/2018 Committee for medicinal products for human use (CHMP) Draft agenda for the meeting on 23–26 April 2018).
Table 1.
Enzyme replacement therapy (ERT) regimens for mucopolysaccharidoses (MPS)
| MPS I | MPS II | MPS IVA | MPS VI | |
|---|---|---|---|---|
| Enzyme deficiency | α-l Iduronidase (IDUA) | Iduronate-2-sulphatase (IDS) | N-acetylgalactosamine-6-sulphatase (GALNS) | N-acetylgalactosamine-4-sulphatase (arylsulphatase B; ARSB) |
| Glycosaminoglycan accumulation | Dermatan sulphate and heparan sulphate | Dermatan sulphate and heparan sulphate | Keratan sulphate and chondroitin-6-sulphate | Dermatan sulphate |
| Drug | Laronidase (Aldurazyme®; Genzyme Europe B.V., Gooimeer 10, NL-1411 DD Naarden, The Netherlands), available since 2003 | Recombinant human idursulphase (Elaprase®; Shire Human Genetic Therapies, Inc., Cambridge, MA, USA), available since 2006 | Elosulphase alpha (Vimizim™; Bio Marin Pharmaceutical, Inc., Novato, CA, USA), available since 2014 | Galsulphase (Naglazyme®; Bio Marin Pharmaceutical, Inc., Novato, CA, USA), available since 2005 |
| Dosage | 0.58 mg/kg body weight administered once every week as an intravenous infusion. The initial infusion rate of 10 μg/kg/h may be increased every 15 min, if tolerated, to a maximum of 200 μg/kg/h. The total volume of the administration should be delivered in approximately 3–4 h | 0.5 mg/kg body weight administered once a week as intravenous infusions over 3 h. The duration of infusion can be shortened gradually to 1 h if there are no infusion-associated reactions (IARs) | 2 mg/kg body weight administered once a week. The total volume of the infusion should be delivered over approximately 4 h | 1 mg/kg body weight administered once a week as an intravenous infusion over 4 h |
| Official suggested premedication | With initial administration of Aldurazyme or upon re-administration following interruption of treatment due to previous IARs, pre-treatment with antihistamines and/or antipyretics approximately 60 min prior to the start of the infusion is recommended | Antihistamines and/or corticosteroids can be considered for those patients who have experienced previous IARs during the infusions | Patients should receive antihistamines with or without antipyretics 30 to 60 min prior to start of infusion | Antihistamines with or without antipyretics approximately 30–60 min prior to the start of infusion |
| Home treatment | Available | Available | Not available | Not available |
Results from clinical trials and the real-world setting confirm the efficacy and safety of ERT in the treatment of these multisystem, progressive disorders [6]. The major proportion of the infused recombinant enzymes for MPS is delivered to the visceral organs such as the liver, kidney, and spleen [7, 8]. The infused enzymes have a short half-life in the circulation due to rapid binding to M6P receptors and uptake into visceral organs. It is known that only a small fraction of the recombinant enzyme can reach the bone cartilage and the eye, explaining why improvements of these organ/systems are limited even after long-term treatment [7, 9]. Moreover, due to the inefficacy of recombinant enzymes to cross the blood–brain barrier (BBB), there are no benefits of ERT for central nervous system (CNS) involvement [10, 11].
The ERT regimen for MPS requires weekly intravenous infusions of the recombinant enzyme. ERT is a life-long therapy, and each infusion takes 3 to 4 h depending on the enzyme and the dose (Table 1). There is the potential for severe infusion reactions; life-threatening anaphylaxis has rarely occurred in patients receiving ERT [12]. Most infusions are given in a hospital setting because of this risk, but home infusions are reported to be feasible and safe for some patients and thus home treatment is now available for selected patients with MPS I and MPS II [13, 14]. The feasibility of home therapy for any MPS patient should be based on a risk/benefit evaluation by the treating physician, the patient, and the patient’s caregiver.
A comprehensive search of journal articles regarding safety and effectiveness of ERT in MPS I, MPS II, MPS IV, and MPS VI from 2003 to July 2017 was carried out on PubMed. The subject headings were Mucopolysaccharidosis I, Mucopolysaccharidosis II, Mucopolysaccharidosis IV, and Mucopolysaccharidosis VI, MPS I, MPS II, MPS IV, MPS VI, enzyme replacement therapy, ERT, laronidase and Aldurazyme, idursulfase and Elaprase, elosulfase and Vimizim, galsulfase and Naglazyme. They were used alone and in combination. All the results of the clinical trials are reported and commented upon, while only the most relevant and/or interesting (in our judgement) clinical studies were considered in this review.
Objectives of ERT
The various types of MPS have differences and similarities in their clinical pictures (see Galimberti et al. [15] and Rigoldi et al. [16] in this supplement) but we can generally say that the ideal aims of ERT are the same for all of them: reducing glycosaminoglycan (GAG) accumulation and organomegaly, improving growth (by ameliorating bone structure) and reducing bone deformities, improving the range of motion (ROM) of joints, and improving respiratory function, heart function, hearing, visual acuity, and quality of life (QoL). The major drawback of ERT molecules is their inability to cross the BBB and cure CNS pathology [10, 11].
What are the major effects and limits of ERT in MPS?
GAG and organomegaly
The demonstration that ERT is biochemically effective is given by the impressive fast decline of urinary GAG concentration (uGAG) over the first 3–6 months of administration followed by a slow continuous decline during the following years [17–23]. From the clinical point of view, a prompt reduction in liver and spleen volumes is observed after few months of therapy, which is subsequently maintained [17–23]; this effect was somehow expected from the beginning considering that tissue distribution studies in animals [8, 24] had shown very high uptake of the recombinant enzyme in the liver and spleen. Reduction in liver size may be relevant for the outcome of patients because it can directly help in improving respiratory function through facilitating diaphragm excursions.
In summary, ERT is very effective in reducing urinary GAG to approximately normal values and improving liver and spleen size. This effect is sustained over time.
Joints
One of the major complaints of patients affected by MPS I, II, and VI is joint stiffness which hampers the easy execution of normal activities of daily life (combing, bathing, dressing, putting a hat on the head). MPS IVA patients instead have joint laxity and other different disturbances such as pectus carenatum, wrist subluxation, early presentation of genu valgum, and frequent osteoarthritis in adults [25]. Passive joint ROM improved in MPS I, II, and VI during clinical trials and improvement was maintained in the long term, although never reaching a normal extension/abduction of joints. Improvement is reported mainly for the shoulder, while the changes for the other joints have not been significant [12, 18, 19, 26–29]. The improvements in ROM were partial but allowed the accomplishment of many activities of daily living according to Sifuentes et al. [17] and Lampe et al. [19]. Although the majority of the authors agree that ERT has an effect, albeit limited, on joint stiffness, other papers report no effect of ERT on joint limitations [14, 30].
In summary, the effect of ERT on joint movement is probably variable from one individual to another, partial even after many years of ERT, is limited to the shoulder and does not significantly affect the other joints. Furthermore, the different responses to therapy may be explained by different joint conditions at the start of ERT [31].
Heart
Heart involvement is typical of MPS. GAG deposition in the myocardium and cardiac valves is the first step of a complex pathway starting with the release of pro-inflammatory cytokines and matrix metalloproteinases consequently activating the macrophages that ultimately damage the tissues [30]. While valve disease, when present at the start of ERT, is not reversible and progressively worsens, myocardial hypertrophy (or pseudohypertrophy) is responsive to ERT and the ejection fraction improves [20, 26, 27, 29, 31] (see also Boffi et al.in this Supplement [30]).
In summary, ERT improves the geometry and contraction of the cardiac muscle but has no clear effect on the valve structure.
Ear, nose, and throat, trachea, and pulmonary function
Ear, nose, and throat (ENT) disturbances are much frequent in MPS and consist of recurrent otitis and rhinosinusitis, tonsil and adenoid hypertrophy, sleep-related breathing disorders (oral breathing, snoring, obstructive sleep apnoea syndrome), and both conductive and sensorineural hearing loss [32–37]. Few data are reported about the effects of ERT on ENT signs and symptoms; ERT is acknowledged to reduce the number of upper airway infections and to improve sleep apnoea in the long term [12, 17, 19, 37, 38], mainly in patients with low-titre inhibitory antibodies [36]. Tomanin et al. [39], however, showed no effect of ERT on sleep apnoea in MPS II. Besides this, ERT does not seem to be very effective in reducing tonsil and adenoid hypertrophy or hearing deficit [20, 26, 40].
Spirometric tests evaluating forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC) (usually expressed as percent predicted FVC, or FVC%) have been used in clinical trials for all four MPS, showing improvements of 3–5% in the first year of treatment in MPS I and MPS IVA [23, 41]. For MPS II and MPS VI, FVC% did not significantly improve in double-blind trials [42–44]. In the long-term follow-up studies, results for FEV1 and FVC% range from stabilization to 11 ± 17% change from baseline [12, 18, 44, 45]. However, it seems that most patients probably reach a plateau after improvement at around 1–2 years of ERT and then stabilize or slowly progressively decline [12, 45]. The reason for this may be that ERT has effects only on one of the components responsible for airway insufficiency, which is multifactorial in MPS: GAG deposition in the soft tissues causes obstructive upper airway disease; tracheobronchial narrowing due to stenosis and malacia is responsible for obstructive lower airway disease; and chest deformities and poor mobility of the ribs lead to restrictive airway signs and symptoms. ERT is expected to be more effective on soft tissues and upper airway obstruction than on the other two factors. Chest deformities cannot be reverted, and the structure of the cartilaginous skeleton of the trachea and bronchi is likely to be marginally modified by the presently available ERTs [46]. Two recent papers address in detail the issue of tracheal and bronchial narrowing in MPS patients [47, 48]. In many of these individuals, a severe tracheal collapse during expiration is seen and, the longer they survive, the more frequent become the complications of bronchial and tracheal stenosis and malacia. These deformities are frequently the basis of the severe obstructive respiratory symptoms of adult MPS patients, mainly MPS I, II, and VI, and a satisfactory treatment still needs to be found [47, 48].
In summary, ERT partially improves the functional capacity of the lung with great variability in different individuals; probably the improvement is limited to the first years of treatment, reaching a plateau. ERT has no effect on the anatomic structure of the trachea and bronchi which are narrow and tend to collapse during expiration.
Endurance
Energy and endurance have been most commonly assessed in MPS-treated patients with the 6-min walk test (6MWT). Other tests of endurance are the 12MWT, used in the clinical trials for MPS VI, and the 3-min stair climb used in the trials for MPS VI and IV [23, 45, 49]. The 6MWT is a submaximal exercise tolerance test which includes evaluations of the responses and functional reserves of pulmonary, cardiovascular, and musculoskeletal systems [50]. An improvement in the 6MWT was seen after short-term treatment in clinical trials and in long-term studies for all four MPS, although in the very long term it seems they reach a plateau [12, 18, 31, 41, 42, 45, 49].
However, both spirometry and 6MWT can be performed only in patients who are not too young or cognitively impaired; there are therefore categories of patients for whom these parameters cannot be applied.
In summary, tests of endurance are much suitable for testing improvements attributable to ERT in clinical trials because their results are seen early, a few months after starting treatment. Their improvement is sustained over the subsequent years. However, only patients with no cognitive impairment and who are not too young are able to undergo these tests.
Bones and growth
Bio-distribution of ERT in bones, articular, and growth cartilage is modest, probably mainly due to their poor vascular supply [10, 12]. No effect on skeletal deformities was shown in clinical trials [23, 41, 42, 51]; it is generally agreed that bone disease cannot be reversed or even stabilized by ERT [52].
As for growth, there are reports showing improvement of growth after ERT in MPS I, MPS II, and MPS VI [53–55], but this effect is usually limited unless the patient is treated from the first weeks or months of life. This has been demonstrated in familial case reports of affected siblings where the earlier treated siblings had less skeletal deformities and better growth than the first sibling [19, 56–62].
These family cases show that ERT may have an effect on growth and bone development if started very early. An improvement in growth during ERT has also been demonstrated in Morquio A patients under 5 years of age who were included in the open-label ERT MOR-007 trial [63].
In summary, the effect of ERT on bones and cartilage is limited, probably partially due to scarce penetration. However, a very early ERT start seems to improve bone health and growth as demonstrated by studies on siblings [64].
Eyes
Eyes are frequently involved in the clinical picture of MPS. Corneal clouding is more often reported in MPS I, VI, and IVA, and optic disk swelling, optic atrophy, papilloedema, and retinal pigment degeneration in all [65]. Few data are available on the efficacy of ERT. Stabilization and improvement of photophobia and, in some cases, improvement of visual acuity and reversal of papilloedema have been reported. It seems that if there are improvements, they are partial and possibly variable among individuals [33, 66–70].
In summary, some patients had an improvement in photophobia and visual acuity and other eye problems after ERT, but this is not observed in most patients.
Health-related quality of life (HRQoL)
The demonstration of biochemical and clinical improvements after ERT with all the assessments described above does not clarify if these effects really mean an improvement in QoL for patients and their families. Is it relevant for patients having a mild improvement in pulmonary function tests or in metres walked at the 6MWT? For the patients it is probably more relevant and meaningful to be more autonomous in performing activities of daily living (ADL), having less pain and satisfactory relationships with schoolmates or in the working environment. With the purpose of exploring this area, many studies have included the assessment of ADL, HRQoL, and pain in the parameters evaluated to demonstrate the efficacy of ERT. A recent review reports a critical comment on all the published studies and the different tests used [44]. The most frequently used test was MPS-HAQ (CHAQ), an adaptation of the Health Assessment Questionnaire (HAQ)/Childhood Health Assessment Questionnaire (CHAQ) test used for rheumatoid arthritis [71]. ADL and HRQoL were reported to improve after long-term ERT in MPS I patients who underwent clinical trials [12, 17]. The MPS-HAQ disability index improved after long-term ERT in cognitively normal MPS II patients [18, 20]. MPS-HAQ/CHAQ and pain control also improved in MPS IVA and MPS VI patients [21, 23, 26, 29, 51, 72–75]. However, the vast majority of the patients in these studies did not have cognitive delay. Demonstration of improvement in these parameters in the patients with CNS involvement (i.e. the most severe forms of MPS I and II) is lacking [20].
In summary, ERT is effective in improving ADL, HRQoL, and pain in those patients with no cognitive delay. We do not have enough data on the more severe patients with MPS I and II.
CNS
It is generally accepted that all the intravenous ERTs developed for MPS and other lysosomal storage diseases do not reach the CNS in amounts sufficient to prevent deterioration of CNS and neurocognitive functions [7, 11, 76]. This is particularly true for MPS I and MPS II, the MPS types with CNS involvement in the majority of the patients. The role of ERT in MPS I and MPS II severe phenotypes has been subject of debate [38, 77]. For MPS I, the main point is “when you should not offer HSCT to an MPS I Hurler patient balancing risks of HSCT with forecasted results?”
On the basis of the progressively reduced harm and mortality of HSCT in recent years, it is at present performed even beyond 2.5 years of age and in MPS I Hurler-Scheie patients who have a slower decline of cognitive functions [77–80]. For MPS II, the option of HSCT is not recommended at present, although a recent paper shows better results compared to ERT in a considerable number of patients [81]. Most MPS II severe patients thus receive ERT from the diagnosis. The treatment is usually decided together with the family taking into consideration advantages for somatic organs and disadvantages related to possible infusion reactions and worsening of behavioural disturbances due to veni-puncture every week followed by 4-h infusion treatment. In our personal experience, only 2 of 19 families refused starting ERT in their children with a severe form of MPS II. This is consistent with the results of a survey conducted in MPS families where 77% of respondents were in favour of starting ERT in a patient with a severe phenotype, even knowing that treatment cannot alter the intellectual deterioration associated with the disease [82]. At present, the opinion of the experts is that “withholding a therapeutic that has the potential to improve some of the somatic manifestations of the disease because of an eventual cognitive decline”, or even “if the cognitive decline is manifest”, “is not justifiable” [38]. The response to ERT would be periodically assessed after starting treatment and, in case of apparent lack of clinical benefit, the decision to withdraw will then be discussed with the family [38].
In summary, ERTs developed for MPS and other lysosomal storage diseases do not reach the CNS in amounts sufficient to prevent deterioration of the CNS and neurocognitive function.
Safety and immunogenicity
Safety
Based on clinical trials, ERT for MPS is considered well tolerated and has an acceptable safety profile. Infusion adverse reactions (IAR), such as rash, urticaria, angioedema, bronchoconstriction, rhinitis, and anaphylaxis, have been reported in approximately 50% of MPS I patients treated with laronidase [12], approximately 30% of MPS II patients treated with idursulphase [83], approximately 90% of MPS IVA patients treated with elosulphase [23], and approximately 50% of MPS VI patients treated with galsulphase [84]. The majority of IARs are usually mild and/or successfully treated by interrupting or slowing the rate of infusion and/or by the administration of anti-histamines, antipyretics and/or corticosteroids. Most patients who experience an IAR receive and tolerate subsequent infusions. Serious adverse reactions have been rarely reported such as anaphylaxis requiring emergency tracheotomy for an associated airway obstruction in a 16-year-old patient with MPS I Hurler-Scheie after 44 laronidase infusions [12]. The reactions experienced during ERT can be caused by either IgE-mediated or non-immunological mechanisms. In the case of recurrent IARs, with failure of pre-medication to prevent hypersensitivity reactions, desensitization is indicated. Effective desensitization has been reported in patients affected by MPS I, MPS II, and MPS VI [85, 86].
Immunogenicity
Most ERTs used for treating lysosomal storage disorders produce an anti-drug antibody (ADA) response which can potentially reduce efficacy or lead to hypersensitivity reactions. The enzymes are taken up by antigen-presenting cells which process and present them to helper T cells specific for the generated peptide. Helper T-cell signals activate antigen-specific B cells to proliferate and differentiate into memory B cells, and into antibodies secreting plasma cells. ADAs may impair the desired biological effects of the therapeutic enzyme through several mechanisms, including altered enzyme targeting, increased enzyme turnover, and/or inhibition of the catalytic site. They can bind to segments of the therapeutic enzyme that are not associated with particular functional activities (non-neutralizing antibodies) or bind to the uptake or catalytic domains (neutralizing antibodies). The level and nature of the residual endogenous enzyme affects the propensity of the patient to generate ADAs. More than 90% of MPS I patients developed antibodies to laronidase during the first few months of treatment [12], about 50% of MPS II patients produced antibodies against idursulphase [22], and almost all patients treated with elosulphase [87] and galsulphase [88] produced ADAs. A clear correlation between ADA titre and clinical outcome has been shown in infantile-onset Pompe disease [89]; less is known about the role of immunogenicity in MPS, and the possible interference of antibodies with the efficacy of ERT is still unclear. A relationship between exposure to ADAs and a pharmacodynamic biomarker, uGAG, has been demonstrated in MPS I and MPS II. Some authors analysed the role of inhibitory antibodies on metabolic biomarkers and sleep disorders in ERT-treated MPS I patients. They showed that increasing inhibition of enzyme activity by antibodies correlated significantly with poorer substrate reduction [36].
A case of allo-immune membranous nephropathy has been reported in a patient with MPS VI treated with galsulphase. The finding of high titres of circulating ADA, which peaked at the onset of the nephrotic syndrome, indicates a mechanism of allo-immunization against the recombinant enzyme [90].
Undoubtedly, the effect of ADAs in MPS is more difficult to evaluate than in infantile Pompe disease due to the slowly progressive course of MPS and the fact that no consistent relationship between ADA titre and clinical outcome has been documented until now. A possible role of the time of uptake of the drug from the plasma into target cells via the M6P receptor, and therefore the mean plasma half-life of distinct ERT, has been recently hypothesized [86]. Laronidase has a mean plasma half-life ranging from 1.5 to 3.6 h, while idursulphase, galsulphase, and elosulphase exhibit a mean plasma half-life of 44, 26, and 36 min, respectively. This rapid uptake may limit the drug’s exposure to antibodies in the plasma and might reduce the formation of immune complexes and their downstream effects.
Some attempts at immune tolerance induction in MPS patients treated with ERT have been performed. An open-label, phase II trial was undertaken to determine the safety and effectiveness of a prophylactic immunosuppressive regimen (cyclosporine and azathioprine) in treatment-naive patients with severe MPS I caused by two nonsense mutations [91]. Unfortunately, the study was terminated early due to changing standards of care for this patient population with inconclusive results. An immune tolerance induction regimen similar to that used in infantile Pompe disease patients has been used in a 4-year-old MPS II patient with sustained high antibody titre and limited clinical efficacy of idursulphase treatment. Over 18 months, therapy with atumumab, bortezomib, methotrexate, short-term dexamethasone, and IVIG resulted in a significant reduction in neutralizing anti-idursulphase IgG titre and a moderate reduction in uGAG levels compared with baseline, while modest clinical improvements were observed [92].
Real-time access to ADA testing is not always easy in the clinical setting and the time to obtain assay results may reduce its clinical utility. However, the reader is reminded that ERT is a lifelong therapy for a devastating disease and that routine monitoring of ADAs is essential and should be part of the routine management of each patient on ERT.
Further prospective and more detailed investigations are needed to understand the real impact of the immune response to ERT and therefore the long-term safety and efficacy. Furthermore, more studies are needed to evaluate the type and the risk/benefit ratio of immunosuppressive therapy.
In summary, ERT for MPS is considered well tolerated and has an acceptable safety profile. However, the real impact of immune response on long-term efficacy remains to be elucidated.
Conclusions
Experience with ERT in MPS I, II, and VI is now reaching more than 10 years, while in MPS IVA the time of observation is shorter. As clearly reported in the literature, we could observe the benefits of this treatment in our patients in terms of reduction of organomegaly, improving pulmonary and heart function, and ameliorating HRQoL, but we also progressively realized that the patients had a big burden of “residual disease” accompanied by the need for several surgical treatments with increased risk of anaesthesia and bone and cartilage abnormalities which are not cured. This implies a risk of cord compression at any time and progressive tracheo-bronchomalacia with severe obstructive lower airway disease. Many patients develop antibodies to the recombinant enzyme and it is not clear yet if high-titre antibodies might influence the efficacy of the treatment with ERT [36].
The use of ERT in cognitively involved patients is a subject of debate and the most accepted position in the scientific community at present is that ERT should be started in any patient because it has the potential to improve some of the somatic manifestations of the disease [38].
Over the years we have also learned from many anecdotal familiar reports that ERT is much more effective, even on the bones, if administered very early. Since ERT is more powerful if it is started early, in a pre-symptomatic phase, new-born screening of treatable MPS has been proposed and pilot studies have been developed in many countries [93]. Whether starting ERT at a very precocious age would be able to halt the progression of all the signs of the disease (excluding as usual the CNS) is not known. For certain, in the case of treatment of a pre-symptomatic or oligo-symptomatic patient, other ethical questions would arise, such as how to distinguish between severe and attenuated forms which might deserve different treatments (HSCT vs ERT for example). The elevated costs of these treatments complicate the choice.
Other drugs are at present being developed: different kinds of more powerful ERT, specifically targeting tissues such as bones where the disease is prominent, drugs based on different principles to enzyme replacement such as substrate deprivation, chaperone therapy, exon skipping, and gene therapy [94, 95]. We hope that all these research lines will develop good treatments for MPS to be used alone or associated with ERT.
Acknowledgements
The authors wish to thank the Italian MPS family Association ONLUS (AIMPS) and patients with their families. RP wishes to thank Fondazione Pierfranco and Luisa Mariani, Milano, for providing financial support for clinical assistance to metabolic patients and Mrs Vera Marchetti for her smart secretarial assistance to the clinical work at the Metabolic Unit in Monza.
Funding
The publication costs for this paper in the IJP supplement were made possible with unconditional financial support from BioMarin, Sanofi Genzyme, and Shire. The sponsors had no input into the content of articles, which were independently prepared by the authors and have undergone the journal’s standard peer-review process.
Availability of data and materials
Not applicable.
About this supplement
This article has been published as part of Italian Journal of Pediatrics, Volume 44 Supplement 2, 2018: Mucopolysaccharidoses: state of the art. The full contents of the supplement are available online at https://ijponline.biomedcentral.com/articles/supplements/volume-44-supplement-2 .
Abbreviations
- ADA
Anti-drug antibody
- ADL
Activities of daily living
- BBB
Blood–brain barrier
- CHAQ
Childhood Health Assessment Questionnaire
- CHO
Chinese hamster ovary
- CNS
Central nervous system
- ENT
Ear, nose, and throat
- ERT
Enzyme replacement therapy
- FDA
Food and Drug Administration
- FEV1
Forced expiratory volume in 1 s
- FVC
Forced vital capacity
- GAG
Glycosaminoglycan
- HAQ
Health Assessment Questionnaire
- HRQoL
Health-related quality of life
- HSCT
Haematopoietic stem cell transplantation
- IAR
Infusion adverse reactions
- M6P
Mannose-6-phosphate
- MPS
Mucopolysaccharidosi(e)s
- QoL
Quality of life
- ROM
Range of motion
- uGAG
Urinary glycosaminoglycan
Authors’ contributions
Each author wrote a part of the manuscript. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
RP participated as principal investigator in the phase III trial for ERT in MPS IVA (BioMarin) and has recently participated as subinvestigator to the phase I/II trial for intrathecal ERT in MPS IIIA (Shire). RP has received honoraria for consulting or speaking engagement from BioMarin, Sanofi Genzyme, SOBI, and Shire. DC has received honoraria for speaking engagements from Shire and Sanofi Genzyme. FD has received honoraria for speaking engagements from Sanofi Genzyme and Shire.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Daniela Concolino, Email: dconcolino@unicz.it.
Federica Deodato, Email: federica.deodato@opbg.net.
Rossella Parini, Phone: 0392333286, Email: rossella.parini@unimib.it.
References
- 1.Ries M. Enzyme replacement therapy and beyond—in memoriam roscoe O. Brady, M.D. (1923–2016) J Inherit Metab Dis. 2017;40:343–356. doi: 10.1007/s10545-017-0032-8. [DOI] [PubMed] [Google Scholar]
- 2.Sly WS. Receptor-mediated transport of acid hydrolases to lysosomes. Curr Top Cell Regul. 1985;26:27–38. doi: 10.1016/B978-0-12-152826-3.50010-3. [DOI] [PubMed] [Google Scholar]
- 3.Barton NW, Furbish FS, Murray GJ, Garfield M, Brady RO. Therapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher disease. Proc Nat AcadSci U S A. 1990;87:1913–1916. doi: 10.1073/pnas.87.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fox JE, Volpe L, Bullaro J, Kakkis ED, Sly WS. First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol Genet Metab. 2015;114:203–208. doi: 10.1016/j.ymgme.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montaño AM, Lock-Hock N, Steiner RD, Graham BH, Szlago M, Greenstein R, et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII) J Med Genet. 2016;53:403–418. doi: 10.1136/jmedgenet-2015-103322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valayannopoulos V, Wijburg FA. Therapy for the mucopolysaccharidoses. Rheumatology (Oxford) 2011;50(Suppl 5):v49–v59. doi: 10.1093/rheumatology/ker396. [DOI] [PubMed] [Google Scholar]
- 7.Kakkis ED, McEntee MF, Schmidtchen A, Neufeld EF, Ward DA, Gompf RE, et al. Long-term and high-dose trials of enzyme replacement therapy in the canine model of mucopolysaccharidosis I. BiochemMol Med. 1996;58:156–167. doi: 10.1006/bmme.1996.0044. [DOI] [PubMed] [Google Scholar]
- 8.Turner CT, Hopwood JJ, Brooks DA. Enzyme replacement therapy in mucopolysaccharidosis I: altered distribution and targeting of alpha-L-iduronidase in immunized rats. Mol Genet Metab. 2000;69:277–285. doi: 10.1006/mgme.2000.2979. [DOI] [PubMed] [Google Scholar]
- 9.Shull RM, Kakkis ED, McEntee MF, Kania SA, Jonas AJ, Neufeld EF. Enzyme replacement in a canine model of hurler syndrome. Proc Natl Acad Sci U S A. 1994;91:12937–12941. doi: 10.1073/pnas.91.26.12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sly WS. Enzyme replacement therapy: from concept to clinical practice. Acta Paediatr. 2002;439(Suppl 91):71–78. doi: 10.1111/j.1651-2227.2002.tb03115.x. [DOI] [PubMed] [Google Scholar]
- 11.Anson DS, McIntyre C, Byers S. Therapies for neurological disease in the mucopolysaccharidoses. Curr Gene Ther. 2011;11:132–143. doi: 10.2174/156652311794940791. [DOI] [PubMed] [Google Scholar]
- 12.Clarke LA, Wraith JE, Beck M, Kolodny EH, Pastores GM, Muenzer J, et al. Long-term efficacy and safety of laronidase in the treatment of mucopolysaccharidosis I. Pediatrics. 2009;123:229–240. doi: 10.1542/peds.2007-3847. [DOI] [PubMed] [Google Scholar]
- 13.Ceravolo F, Mascaro I, Sestito S, Pascale E, Lauricella A, Dizione E, et al. Home treatment in paediatric patients with Hunter syndrome: the first Italian experience. Ital J Pediatr. 2013;39:53. doi: 10.1186/1824-7288-39-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox-Brinkman J, Timmermans RG, Wijburg FA, Donker WE, van der Ploeg AT, Aerts JM, et al. Home treatment with enzyme replacement therapy for mucopolysaccharidosis type I is feasible and safe. J Inherit Metab Dis. 2007;30:984. doi: 10.1007/s10545-007-0686-8. [DOI] [PubMed] [Google Scholar]
- 15.Galimberti C, Madeo A, Di Rocco M, Fiumara A. Mucopolysaccharidoses: early diagnostic signs in infants and children. Ital J Pediatr. 2018; 10.1186/s13052-018-0550-5. [DOI] [PMC free article] [PubMed]
- 16.Rigoldi M, Verrecchia E, Manna R, Mascia MT. Clinical hints to diagnosis of attenuated forms of mucopolysaccharidosis. Ital J Pediatr. 2018; 10.1186/s13052-018-0551-4. [DOI] [PMC free article] [PubMed]
- 17.Sifuentes M, Doroshow R, Hoft R, Mason G, Walot I, Diament M, et al. A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol Genet Metab. 2007;90:171–180. doi: 10.1016/j.ymgme.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 18.Muenzer J, Beck M, Eng CM, Giugliani R, Harmatz P, Martin R, et al. Long term, open-labeled extension study of idursulfase in the treatment of hunter syndrome. Genet Med. 2011;13:95–101. doi: 10.1097/GIM.0b013e3181fea459. [DOI] [PubMed] [Google Scholar]
- 19.Lampe C, Bosserhoff AK, Burton BK, Giugliani R, de Souza CF, Bittar C, et al. Long-term experience with enzyme replacement therapy (ERT) in MPS II patients with a severe phenotype: an international case series. J Inherit Metab Dis. 2014;37:823–829. doi: 10.1007/s10545-014-9686-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parini R, Rigoldi M, Tedesco L, Boffi L, Brambilla A, Bertoletti S, et al. Enzymatic replacement therapy for hunter disease: up to 9 years experience with 17 patients. Mol Genet Metab Rep. 2015;3:65–74. doi: 10.1016/j.ymgmr.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giugliani R, Lampe C, Guffon N, Ketteridge D, Leão-Teles E, Wraith JE, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)—ten year follow-up of patients who previously participated in an MPS VI survey study. Am J Med Genet A. 2014;164A:1953–1964. doi: 10.1002/ajmg.a.36584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hendriksz CJ, Burton B, Fleming TR, Harmatz P, Hughes D, Jones SA, et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomized placebo-controlled study. J Inherit Metab Dis. 2014;37:979–990. doi: 10.1007/s10545-014-9715-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kılıç M, Dursun A, Coşkun T, Tokatlı A, Özgül RK, Yücel-Yılmaz D, et al. Genotypic-phenotypic features and enzyme replacement therapy outcome in patients with mucopolysaccharidosis VI from Turkey. Am J Med Genet A. 2017;173(11):2954–2967. doi: 10.1002/ajmg.a.38459. [DOI] [PubMed] [Google Scholar]
- 24.Kakkis ED, Schuchman E, He X, Wan Q, Kania S, Wiemelt S, et al. Enzyme replacement therapy in feline mucopolysaccharidosis I. Mol Genet Metab. 2001;72:199–208. doi: 10.1006/mgme.2000.3140. [DOI] [PubMed] [Google Scholar]
- 25.Hendriksz CJ, Harmatz P, Beck M, Jones S, Wood T, Lachman R, et al. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol Genet Metab. 2013;110:54–64. doi: 10.1016/j.ymgme.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brands MMG, Oussoren E, Ruijter GJG, Vollebregt AAM, van den Hout HMP, Joosten KFM, et al. Up to five years experience with 11 mucopolysaccharidosis type VI patients. Mol Genet Metab. 2013;9:70–76. doi: 10.1016/j.ymgme.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 27.Okuyama T, Tanaka A, Suzuki Y, Ida H, Tanaka T, Cox GF, et al. Japan Elaprase treatment (JET) study: idursulfase enzyme replacement therapy in adult patients with attenuated hunter syndrome (mucopolysaccharidosis II, MPS II) Mol Genet Metab. 2010;99:18–25. doi: 10.1016/j.ymgme.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 28.Tylki-Szymanska A, Marucha J, Jurecka A, Syczewska M, Czartoryska B. Efficacy of recombinant human alpha-L-iduronidase (laronidase) on restricted range of motion of upper extremities in mucopolysaccharidosis type I patients. J Inherit Metab Dis. 2010;33:151–157. doi: 10.1007/s10545-010-9059-9. [DOI] [PubMed] [Google Scholar]
- 29.Lin HY, Chuang CK, Wang CH, Chien YH, Wang YM, Tsai FJ, et al. Long-term galsulfase enzyme replacement therapy in Taiwanese mucopolysaccharidosis VI patients: a case series. Mol Genet Metab Rep. 2016;7:63–69. doi: 10.1016/j.ymgmr.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boffi L, Russo P, Limongelli G. Early diagnosis and management of cardiac manifestations in mucopolysaccharidoses: a practical guide for paediatric and adult cardiologists. Ital J Pediatr. 2018; 10.1186/s13052-018-0560-3. [DOI] [PMC free article] [PubMed]
- 31.Guarany NR, Schwartz IV, Guarany FC, Giugliani R. Functional capacity evaluation of patients with mucopolysaccharidosis. J Pediatr Rehabil Med. 2012;5:37–46. doi: 10.3233/PRM-2012-0194. [DOI] [PubMed] [Google Scholar]
- 32.Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Blaudet AL, Sly WS, Valle D, editors. The metabolic bases of inherited disease. 8. New York: McGraw Hill; 2001. pp. 3421–3452. [Google Scholar]
- 33.Wraith JE. The first 5 years of clinical experience with laronidase enzyme replacement therapy for mucopolysaccharidosis I. Expert Opin Pharmacother. 2005;6:489–506. doi: 10.1517/14656566.6.3.489. [DOI] [PubMed] [Google Scholar]
- 34.Santamaria F, Andreucci MV, Parenti G, Polverino M, Viggiano D, Montella S, et al. Upper airway obstructive disease in mucopolysaccharidoses: polysomnography, computed tomography and nasal endoscopy findings. J Inherit Metab Dis. 2007;30:743–749. doi: 10.1007/s10545-007-0555-5. [DOI] [PubMed] [Google Scholar]
- 35.Berger KI, Fagondes SC, Giugliani R, Hardy KA, Lee KS, McArdle C, et al. Respiratory and sleep disorders in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36:201–210. doi: 10.1007/s10545-012-9555-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pal AR, Langereis EJ, Saif MA, Mercer J, Church HJ, Tylee KL, et al. Sleep disordered breathing in mucopolysaccharidosis I: a multivariate analysis of patient, therapeutic and metabolic correlators modifying long term clinical outcome. Orphanet J Rare Dis. 2015;10:42. doi: 10.1186/s13023-015-0255-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dualibi Ana Paula Fiuza Funicello, Martins Ana Maria, Moreira Gustavo Antônio, de Azevedo Marisa Frasson, Fujita Reginaldo Raimundo, Pignatari Shirley Shizue Nagata. The impact of laronidase treatment in otolaryngological manifestations of patients with mucopolysaccharidosis. Brazilian Journal of Otorhinolaryngology. 2016;82(5):522–528. doi: 10.1016/j.bjorl.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muenzer J, Bodamer O, Burton B, Clarke L, Schulze-Frenking G, Giugliani R, et al. The role of enzyme replacement therapy in severe hunter syndrome—an expert panel consensus. Eur J Pediatr. 2012;171:181–188. doi: 10.1007/s00431-011-1606-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomanin R, Zanetti A, D’Avanzo F, Rampazzo A, Gasparotto N, Parini R, et al. Clinical efficacy of enzyme replacement therapy in paediatric hunter patients, an independent study of 3.5 years. Orphanet J Rare Dis. 2014;9:129. doi: 10.1186/s13023-014-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horovitz DD, Magalhães TS, Acosta A, Ribeiro EM, Giuliani LR, Palhares DB, et al. Enzyme replacement therapy with galsulfase in 34 children younger than five years of age with MPS VI. Mol Genet Metab. 2013;109:62–69. doi: 10.1016/j.ymgme.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 41.Wraith JE, Clarke L, Beck M, Kolodny EH, Pastores GM, Muenzer J, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human a-l-iduronidase (laronidase) J Pediatr. 2004;144:581–588. doi: 10.1016/j.jpeds.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 42.Muenzer J, Wraith JE, Beck M, Giugliani R, Harmatz P, Eng C, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (hunter syndrome) Genet Med. 2006;8:465–473. doi: 10.1097/01.gim.0000232477.37660.fb. [DOI] [PubMed] [Google Scholar]
- 43.Beck M, Muenzer J, Scarpa M. Evaluation of disease severity in mucopolysaccharidoses. J Pediatr Rehabil Med. 2010;3:39–46. doi: 10.3233/PRM-2010-0100. [DOI] [PubMed] [Google Scholar]
- 44.Harmatz P, Yu ZF, Giugliani R, Schwartz IV, Guffon N, Teles EL, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: evaluation of long-term pulmonary function in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase. J Inherit Metab Dis. 2010;33:51–60. doi: 10.1007/s10545-009-9007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hendriksz CJ, Berger KI, Parini R, AlSayed MD, Raiman J, Giugliani R, et al. Impact of long-term elosulfase alfa treatment on respiratory function in patients with Morquio A syndrome. J Inherit Metab Dis. 2016;39:839–847. doi: 10.1007/s10545-016-9973-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khan S, Alméciga-Díaz CJ, Sawamoto K, Mackenzie WG, Theroux MC, Pizarro C, et al. Mucopolysaccharidosis IVA and glycosaminoglycans. Mol Genet Metab. 2017;120:78–95. doi: 10.1016/j.ymgme.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rutten M, Ciet P, van den Biggelaar R, Oussoren E, Langendonk JG, van der Ploegand AT, et al. Severe tracheal and bronchial collapse in adults with type II mucopolysaccharidosis. Orphanet J Rare Dis. 2016;11:50. doi: 10.1186/s13023-016-0425-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kampmann C, Wiethoff CM, Huth RG, Staatz G, Mengel E, Beck M, et al. Management of life-threatening tracheal stenosis and tracheomalacia in patients with mucopolysaccharidoses. J Inherit Metab Dis Rep. 2017;33:33–39. doi: 10.1007/8904_2016_578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harmatz P, Whitley CB, Waber L, Pais R, Steiner R, Plecko B, et al. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) J Pediatr. 2004;144:574–580. doi: 10.1016/j.jpeds.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 50.Enright PL. The six-minute walk test. Respir Care. 2003;48:783–785. [PubMed] [Google Scholar]
- 51.Harmatz P, Giugliani R, Schwartz I, Guffon N, Teles EL, Sá Miranda MC, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J Pediatr. 2006;148:533–539. doi: 10.1016/j.jpeds.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 52.Opoka-Winiarska V, Jurecka A, Emeryky A, Tylki-Szymanska A. Osteoimmunology in mucopolysaccharidoses type I, II, VI and VII. Immunological regulation of the osteoarticular system in the course of metabolic inflammation. Osteoarthr Cartil. 2013;21:1813–1823. doi: 10.1016/j.joca.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 53.Wraith JE, Beck M, Lane R, van der Ploeg A, Shapiro E, Xue Y, et al. Enzyme replacement therapy in patients who have mucopolysaccharidosis I and are younger than 5 years: results of a multinational study of recombinant human alpha-L-iduronidase (laronidase) Pediatrics. 2007;120:e37–e46. doi: 10.1542/peds.2006-2156. [DOI] [PubMed] [Google Scholar]
- 54.Giugliani R, Federhen A, Rojas MV, Vieira T, Artigalas O, Pinto LL, et al. Mucopolysaccharidosis I, II, and VI: brief review and guidelines for treatment. Genet Mol Biol. 2010;33:589e604. doi: 10.1590/S1415-47572010005000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harmatz P. Enzyme replacement therapy with galsulfase for mucopolysaccharidosis VI: clinical facts and figures. Turkish J Pediatr. 2010;52:443e9. [PubMed] [Google Scholar]
- 56.Laraway S, Breen C, Mercer J, Jones S, Wraith EJ. Does early use of enzyme replacement therapy alter the natural history of mucopolysaccharidosis I? Experience in three siblings. Mol Genet Metab. 2013;109:315–316. doi: 10.1016/j.ymgme.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 57.Al-Sannaa NA, Bay L, Barbouth DS, Benhayoun Y, Goizet C, Guelbert N, et al. Early treatment with laronidase improves clinical outcomes in patients with attenuated MPS I: a retrospective case series analysis of nine sibships. Orphanet J Rare Dis. 2015;10:131. doi: 10.1186/s13023-015-0344-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tajima G, Sakura N, Kosuga M, Okuyama T, Kobayashi M. Effects of idursulfase enzyme replacement therapy for mucopolysaccharidosis type II when started in early infancy: comparison in two siblings. Mol Genet Metab. 2013;108:172–177. doi: 10.1016/j.ymgme.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 59.Gabrielli O, Clarke LA, Bruni S, Coppa GV. Enzyme-replacement therapy in a 5-month-old boy with attenuated presymptomatic MPS I: 5-year follow-up. Pediatrics. 2010;125:e183–e187. doi: 10.1542/peds.2009-1728. [DOI] [PubMed] [Google Scholar]
- 60.Gabrielli O, Clarke LA, Ficcadenti A, Santoro L, Zampini L, Volpi N, et al. 12 year follow-up of enzyme-replacement therapy in two siblings with attenuated mucopolysaccharidosis I: the important role of early treatment. BMC Med Genet. 2016;17:19. doi: 10.1186/s12881-016-0284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tylki-Szymanska A, Jurecka A, Zuber Z, Rozdzynska A, Marucha J, Czartoryska B. Enzyme replacement therapy for mucopolysaccharidosis II from 3 months of age: a 3-year follow-up. Acta Paediatr. 2012;101:e42–e47. doi: 10.1111/j.1651-2227.2011.02385.x. [DOI] [PubMed] [Google Scholar]
- 62.McGill JJ, Inwood AC, Coman DJ, Lipke ML, de Lore D, Swiedler SJ, et al. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age—a sibling control study. Clin Genet. 2010;77:492–498. doi: 10.1111/j.1399-0004.2009.01324.x. [DOI] [PubMed] [Google Scholar]
- 63.Jones SA, Bialer M, Parini R, Martin K, Wang H, Yang K, et al. Safety and clinical activity of elosulfase alfa in pediatric patients with Morquio A syndrome (mucopolysaccharidosis IVA) less than 5 y. Pediatr Res. 2015;78:717–722. doi: 10.1038/pr.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111:63–72. doi: 10.1016/j.ymgme.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 65.Ashworth JL, Kruse FE, Bachmann B, Tormene AP, Suppiej A, Parini R, et al. Ocular manifestations in the mucopolysaccharidoses—a review. Clin Exp Ophthalmol. 2010;28:12–22. doi: 10.1111/j.1442-9071.2010.02364.x. [DOI] [Google Scholar]
- 66.Pitz S, Ogun O, Bajbouj M, Arash L, Schulze-Frenking G, Beck M. Ocular changes in patients with mucopolysaccharidosis I receiving enzyme replacement therapy: a 4-year experience. Arch Ophthalmol. 2007;125:1353–1356. doi: 10.1001/archopht.125.10.1353. [DOI] [PubMed] [Google Scholar]
- 67.Pitz S, Ogun O, Arash L, Miebach E, Beck M. Does enzyme replacement therapy influence the ocular changes in type VI mucopolysaccharidosis? Graefes Arch Clin Exp Ophthalmol. 2009;247:975–980. doi: 10.1007/s00417-008-1030-1. [DOI] [PubMed] [Google Scholar]
- 68.Fenzl CR, Teramoto K, Moshirfar M. Ocular manifestations and management recommendations of lysosomal storage disorders I: mucopolysaccharidoses. Clin Ophthalmol. 2015;9:1633–1644. doi: 10.2147/OPTH.S78368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fahnehjelm KT, Ashworth JL, Pitz S, Olsson M, Törnquist AL, Lindahl P, et al. Clinical guidelines for diagnosing and managing ocular manifestations in children with mucopolysaccharidosis. Acta Ophthalmol. 2012;90:595–602. doi: 10.1111/j.1755-3768.2011.02280.x. [DOI] [PubMed] [Google Scholar]
- 70.Laraway S, Mercer J, Jameson E, Ashworth J, Hensman P, Jones SA. Outcomes of long-term treatment with laronidase in patients with mucopolysaccharidosis type I. J Pediatr. 2016;178:219–226. doi: 10.1016/j.jpeds.2016.08.033. [DOI] [PubMed] [Google Scholar]
- 71.Singh G, Athreya BH, Fries JF, Goldsmith DP. Measurement of health status in children with juvenile rheumatoid arthritis. Arthritis Rheum. 1994;37:1761–1769. doi: 10.1002/art.1780371209. [DOI] [PubMed] [Google Scholar]
- 72.Harmatz P, Treadwell M, Burton BK, Mitchell J, Muschol N, Jones S, et al. Impact of elosulfase alfa on pain in patients with Morquio syndrome type A. Mol Genet Metab. 2015;114:S51–S52. doi: 10.1016/j.ymgme.2014.12.104. [DOI] [Google Scholar]
- 73.Hendriksz Christian J., Giugliani Roberto, Harmatz Paul, Mengel Eugen, Guffon Nathalie, Valayannopoulos Vassili, Parini Rossella, Hughes Derralynn, Pastores Gregory M., Lau Heather A., Al-Sayed Moeenaldeen D., Raiman Julian, Yang Ke, Mealiffe Matthew, Haller Christine. Multi-domain impact of elosulfase alfa in Morquio A syndrome in the pivotal phase III trial. Molecular Genetics and Metabolism. 2015;114(2):178–185. doi: 10.1016/j.ymgme.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 74.Hendriksz Christian J, Lavery Christine, Coker Mahmut, Ucar Sema, Jain Mohit, Bell Lisa, Lampe Christina. Burden of disease in patients with Morquio A syndrome: results from an international patient-reported outcomes survey. Orphanet Journal of Rare Diseases. 2014;9(1):32. doi: 10.1186/1750-1172-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harmatz P, Ketteridge D, Giugliani R, Guffon N, Teles EL, Miranda MC, et al. Direct comparison of measures of endurance, mobility, and joint function during enzyme-replacement therapy of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): results after 48 weeks in a phase 2 open-label clinical study of recombinant human N-acetylgalactosamine 4-sulfatase. Pediatrics. 2005;115:e681–e689. doi: 10.1542/peds.2004-1023. [DOI] [PubMed] [Google Scholar]
- 76.Boado RJ, Hui EK, Lu JZ, Sumbria RK, Pardridge WM. Blood–brain barrier molecular trojan horse enables imaging of brain uptake of radioiodinated recombinant protein in the rhesus monkey. Bioconjug Chem. 2013;24:1741–1749. doi: 10.1021/bc400319d. [DOI] [PubMed] [Google Scholar]
- 77.de Ru MH, Boelens JJ, Das AM, Jones SA, van der Lee JH, Mahlaoui N, et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet J Rare Dis. 2011;6:55. doi: 10.1186/1750-1172-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boelens JJ, Orchard PJ, Wynn RF. Transplantation in inborn errors of metabolism: current considerations and future perspectives. Br J Haematol. 2014;167:293–303. doi: 10.1111/bjh.13059. [DOI] [PubMed] [Google Scholar]
- 79.Aldenhoven M, Wynn RF, Orchard PJ, O'Meara A, Veys P, Fischer A, et al. Long-term outcome of hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125:2164–2172. doi: 10.1182/blood-2014-11-608075. [DOI] [PubMed] [Google Scholar]
- 80.Poe MD, Chagnon SL, Escolar ML. Early treatment is associated with improved cognition in hurler syndrome. Ann Neurol. 2014;76:747–753. doi: 10.1002/ana.24246. [DOI] [PubMed] [Google Scholar]
- 81.Kubaski F, Yabe H, Suzuki Y, Seto T, Hamazaki T, Mason RW, et al. Hematopoietic stem cell transplantation for patients with mucopolysaccharidosis II. Biol Blood Marrow Transplant. 2017;30551–7:S1083–S8791. doi: 10.1016/j.bbmt.2017.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Coman DJ, Hayes IM, Collins V, Sahhar M, Wraith JE, Delatycki MB. Enzyme replacement therapy for mucopolysaccharidoses: opinions of patients and families. J Pediatr. 2008;152:723–727. doi: 10.1016/j.jpeds.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 83.Burton BK, Whiteman DA, Hunter Outcome Survey Investigators Incidence and timing of infusion-related reactions in patients with mucopolysaccharidosis type II (Hunter syndrome) on idursulfase therapy in the real-world setting: a perspective from the Hunter Outcome Survey (HOS) Mol Genet Metab. 2011;103:113–120. doi: 10.1016/j.ymgme.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 84.Harmatz P, Giugliani R, Schwartz IV, Guffon N, Teles EL, Miranda MC, Wraith JE, et al. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol Genet Metab. 2008;94:469–475. doi: 10.1016/j.ymgme.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 85.Serrano CD, Gomez JF. Successful desensitization to idursulfase in a patient with type II mucopolysaccharidosis (hunter syndrome) J Investig Allergol Clin Immunol. 2011;21:571–572. [PubMed] [Google Scholar]
- 86.Guvenir H, Dibek Misirlioglu E, Capanoglu M, Buyuktiryaki B, Unal O, Toyran M, Kocabas CN. Successful desensitization of elosulfase alfa-induced anaphylaxis in a pediatric patient with Morquio syndrome. J Allergy Clin Immunol Pract. 2017;5:1156–1157. doi: 10.1016/j.jaip.2017.02.020. [DOI] [PubMed] [Google Scholar]
- 87.Long B, Tompkins T, Decker C, Jesaitis L, Khan S, Slasor P, et al. Long-term immunogenicity of elosulfase alfa in the treatment of Morquio A syndrome: results from MOR-005, a phase III extension study. Clin Thera. 2017;39:118–129.e3. doi: 10.1016/j.clinthera.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 88.Brands MM, Hoogeveen-Westerveld M, Kroos MA, Nobel W, Ruijter GJ, Özkan L, et al. Mucopolysaccharidosis type VI phenotypes-genotypes and antibody response to galsulfase. Orphanet J Rare Dis. 2013;8:51. doi: 10.1186/1750-1172-8-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Banugaria SG, Prater SN, Ng JK, Kobori JA, Finkel RS, Ladda RL, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med. 2011;13:729–736. doi: 10.1097/GIM.0b013e3182174703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Debiec H, Valayannopoulos V, Boyer O, Nöel LH, Callard P, Sarda H, et al. Allo-immune membranous nephropathy and recombinant arylsulfatase replacement therapy: a need for tolerance induction therapy. J Am Soc Nephrol. 2014;25(4):675–680. doi: 10.1681/ASN.2013030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Giugliani R, Vieira TA, Carvalho CG, Muñoz-Rojas MV, Semyachkina AN, Voinova VY, et al. Immune tolerance induction for laronidase treatment in mucopolysaccharidosis I. Mol Genet Metab Rep. 2017;10:61–66. doi: 10.1016/j.ymgmr.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim KH, Messinger YH, Burton BK. Successful reduction of high-sustained anti-idursulfase antibody titers by immune modulation therapy in a patient with severe mucopolysaccharidosis type II. Mol Genet Metab Rep. 2015;2:20–24. doi: 10.1016/j.ymgmr.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Clarke LA, Atherton AM, Burton BK, Day-Salvatore DL, Kaplan P, Leslie ND, Scott CR, et al. Mucopolysaccharidosis type I newborn screening: best practices for diagnosis and management. J Pediatr. 2017;182:363–370. doi: 10.1016/j.jpeds.2016.11.036. [DOI] [PubMed] [Google Scholar]
- 94.Nestrasil I, Shapiro E, Svatkova A, Dickson P, Chen A, Wakumoto A, et al. Intrathecal enzyme replacement therapy reverses cognitive decline in mucopolysaccharidosis type I. Am J Med Genet A. 2017;173:780–783. doi: 10.1002/ajmg.a.38073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Giugliani R, Federhen A, Vairo F, Vanzella C, Pasqualim G, da Silva LM, et al. Emerging drugs for the treatment of mucopolysaccharidoses. Expert Opin Emerg Drugs. 2016;21:9–26. doi: 10.1517/14728214.2016.1123690. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.