

Sir,
The hereditary spastic paraplegias (HSPs) are a heterogeneous group of neurodegenerative disorders. Few epidemiological studies of HSPs have been done to date. The estimated prevalence is 3–10 cases/100,000 population in Europe.[1]
The diagnostic clinical findings are spasticity and pyramidal weakness in lower limbs, with hyperreflexia and extensor plantar responses. The genetics of HSPs is complex, and all modes of inheritance (autosomal dominant [AD], autosomal recessive [AR], and X-linked [XL] recessive) have been described.[2] AR HSPs are more frequent in consanguineous populations with a prevalence of 0.6/100,000 in Norway and up to 5.75/100,000 in Tunisia.[2,3] HSP type 35 is an AR form of HSPs caused by mutations in the fatty acid 2-hydroxylase (FA2H) gene at 1'q21-q23 chromosome.[2]
In the literature, a few cases have been reported with HSP type 35.[3,4,5,6] Here, we report HSP type 35 case of Turkish origin with a novel homozygous mutation in FA2H gene, presented with progressive gait disturbance and cognitive impairment.
A 16-years-old boy was admitted to our hospital because of progressive difficulty in walking, unsteady gait, cognitive impairment, and hand tremor. He had normal motor and intellectual development until the age of 10 years when gait disturbance and balance problems first appeared. Five years later, he began to show deterioration in academic skills. The patient was diagnosed with cerebral palsy because of these complaints in another hospital. The parents were consanguineous, and there was no family history of neurologic disease. He was born at term and had no neonatal problems. He had healthy two older sisters. Pedigree is shown in Figure 1.
Figure 1.
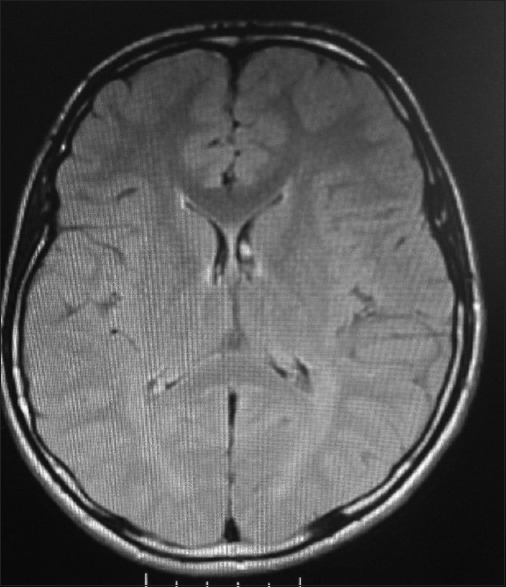
Pedigree of the patient
Neurological examination revealed mild cognitive impairment and spasticity. The deep tendon reflexes were increased in lower limbs, with ankle clonus and bilateral Babinski signs. He had muscle weakness of Grade 4/5 distally in both lower limbs and had mild bilateral foot drop, pes cavus deformities, and muscular atrophy. Ophthalmologic examination and the other physical examination were normal.
Complete blood count, serum biochemistry, lipid profile, thyroid function tests, and serum Vitamin E and B12 levels were all normal. Brain magnetic resonance imaging (MRI) showed bilateral symmetrical hyperintense lesions in the periventricular white matter in T-weighted images [Figure 2]. Nerve conduction studies revealed demyelinating form of polyneuropathy. Based on the clinical findings and nerve conduction studies, the patient was thought as HSP.
Figure 2.
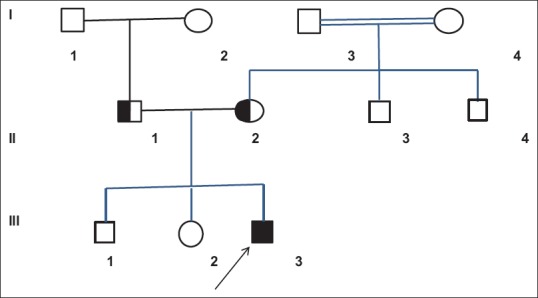
Cerebral magnetic resonance imaging showed bilateral symmetrical hyperintense lesions in the periventricular white matter
Clinical exome sequencing analysis was performed in the patient using TruSight One kits (Illumina Inc., San Diego, CA, USA). As a result of the clinical exome analysis, we identified a novel missense homozygous mutation at the FA2H gene (c.130C>T p. Pro44Ser p. P44S) which has not been reported previously. The mutation found was considered to be highly probable cause of disease according to in silico analysis (Sorting Tolerant From Intolerant, http://sift.jcvi.org and Mutation Taster, http://www.mutationtaster.org). Additional family screening revealed that both parents had heterozygous mutation. We confirmed that the patient was HSP type 35 due to clinical and genetically evaluation.
To date, 70 different gene loci associated with HSP were identified, involving XL, AR, AD, and maternal inheritance.[2]
AR spastic paraplegia-35 (SPG35) is a characterized by childhood onset of gait difficulties due to progressive spastic paraparesis, dysarthria, and mild cognitive decline associated with leukodystrophy on brain imaging. Other variable neurologic features, such as dystonia, optic atrophy, and seizures, may also occur.[3,4]
SPG35 is caused by mutations in the FA2H gene located on chromosome 16q23. FA2H was first described in 2008 as a rare leukodystrophy gene causing spasticity and dystonia.[7] In addition, FA2H gene has been shown to be associated with neurodegeneration with brain iron accumulation, thus expanding the phenotype. This phenotypic spectrum of disorders was then referred as fatty acid hydrolase-associated neurodegeneration (FAHN).[8]
FA2H gene encodes FA2H, a 372-amino-acid-long membrane-bound protein incorporated into the ceramide species which is necessary for the production of normal myelin. It contains two conserved domains, a cytochrome b5-like heme-binding domain, spanning residues 15–85 and responsible for the redox activity of FA2H, and a sterol desaturase domain at residues 210–367.[9]
To date, approximately 51 patients from 19 families have been reported in the literature.[5] Cao et al.[4] reported two siblings born to nonconsanguineous parents, who possess several typical clinical features of SPG35, a subtype of FAHN, owing to novel FA2H missense mutation. Two affected siblings had typical clinical features of SPG35. For the two siblings, brain MRI showed progressive leukoencephalopathy with cortical, cerebellar, and brainstem atrophy. Thinning of the corpus callosum was also noted. Soehn et al.[5] described four novel homozygous FA2H mutations in four nonconsanguineous families with SPG35. All four children presented with a complicated form of HSP with tetraspasticity and additional symptoms including limb ataxia (3/4), mild cognitive deficits (3/4), and extrapyramidal involvement (3/4). In another case, Liao et al.[3] reported three novel FA2H gene mutations in two unrelated Chinese families with SPG35. Bektaş et al.[6] described a 5-year-old boy presenting with spastic paraplegia without seizure, neuropathy, cognitive impairment, speech disturbance, and optic atrophy in Turkey. Their patient was rapid progressive spastic paraplegia, and he was early loss of ambulation. Our case had gait difficulties due to progressive spastic paraparesis, hand tremor, mild cognitive deficits, and additional findings including demyelinating form of polyneuropathy. He had no optic atrophy and cerebellar dysfunction.
Recently, identification of the neurodegeneration with brain iron accumulation expanded the phenotypic spectrum of the disorders associated with the FA2H gene mutation.[8] Brain iron accumulation was not shown in the patient.
Bektaş et al.[6] detected a novel homozygous mutation c. 160_169 dup (p. Asp57Glyfs*48) in the gene encoding FA2H in their case in Turkey. We detected novel homozygous mutation c.130C>T (p. Pro44Ser) (p. P44S) in the FA2H gene. Furthermore, the patient had demyelinating form of polyneuropathy which has not been reported in SPG35 previously. Table 1 presents the analysis of cases of SPG35 from the literature and the patient.
Table 1.
The analysis of cases of hereditary spastic paraplegia type 35 from the literature and our patient

In conclusion, HSPs are clinically and genetically heterogeneous disorders characterized by lower limb spasticity and weakness. SPG35 should be included in the differential diagnosis of lower limb spasticity and weakness when additional ataxia, mild cognitive deficits, and extrapyramidal involvement.
Informed consent
Informed consent was obtained from the parents of the child included in the study.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patient understands that name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.[18]
REFERENCES
- 1.McMonagle P, Webb S, Hutchinson M. The prevalence of “pure” autosomal dominant hereditary spastic paraparesis in the island of İreland. J Neurol Neurosurg Psychiatry. 2002;72:43–6. doi: 10.1136/jnnp.72.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finsterer J, Löscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G, et al. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci. 2012;318:1–8. doi: 10.1016/j.jns.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 3.Liao X, Luo Y, Zhan Z, Du J, Hu Z, Wang J, et al. SPG35 contributes to the second common subtype of AR-HSP in China: Frequency analysis and functional characterization of FA2H gene mutations. Clin Genet. 2015;87:85–9. doi: 10.1111/cge.12336. [DOI] [PubMed] [Google Scholar]
- 4.Cao L, Huang XJ, Chen CJ, Chen SD. A rare family with hereditary spastic paraplegia type 35 due to novel FA2H mutations: A case report with literature review. J Neurol Sci. 2013;329:1–5. doi: 10.1016/j.jns.2013.02.026. [DOI] [PubMed] [Google Scholar]
- 5.Soehn AS, Rattay TW, Beck-Wödl S, Schäferhoff K, Monk D, Döbler-Neumann M, et al. Uniparental disomy of chromosome 16 unmasks recessive mutations of FA2H/SPG35 in 4 families. Neurology. 2016;87:186–91. doi: 10.1212/WNL.0000000000002843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bektaş G, Yeşil G, Yıldız EP, Aydınlı N, Çalışkan M, Özmen M, et al. Hereditary spastic paraplegia type 35 caused by a novel FA2H mutation. Turk J Pediatr. 2017;59:329–34. doi: 10.24953/turkjped.2017.03.016. [DOI] [PubMed] [Google Scholar]
- 7.Dick KJ, Al-Mjeni R, Baskir W, Koul R, Simpson MA, Patton MA, et al. A novel locus for an autosomal recessive hereditary spastic paraplegia (SPG35) maps to 16q21-q23. Neurology. 2008;71:248–52. doi: 10.1212/01.wnl.0000319610.29522.8a. [DOI] [PubMed] [Google Scholar]
- 8.Kruer MC, Paisán-Ruiz C, Boddaert N, Yoon MY, Hama H, Gregory A, et al. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA) Ann Neurol. 2010;68:611–8. doi: 10.1002/ana.22122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eckhardt M, Yaghootfam A, Fewou SN, Zöller I, Gieselmann V. A mammalian fatty acid hydroxylase responsible for the formation of alpha-hydroxylated galactosylceramide in myelin. Biochem J. 2005;388:245–54. doi: 10.1042/BJ20041451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pedroso JL, Handfas BW, Abrahão A, Kok F, Barsottini OG, Oliveira AS, et al. Fatty acid 2-hydroxylase deficiency: Clinical features and brain iron accumulation. Neurology. 2015;84:960–1. doi: 10.1212/WNL.0000000000001316. [DOI] [PubMed] [Google Scholar]
- 11.Garone C, Pippucci T, Cordelli DM, Zuntini R, Castegnaro G, Marconi C, et al. FA2H-related disorders: A novel c.270+3A>T splice-site mutation leads to a complex neurodegenerative phenotype. Dev Med Child Neurol. 2011;53:958–61. doi: 10.1111/j.1469-8749.2011.03993.x. [DOI] [PubMed] [Google Scholar]
- 12.Edvardson S, Hama H, Shaag A, Gomori JM, Berger I, Soffer D, et al. Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am J Hum Genet. 2008;83:643–8. doi: 10.1016/j.ajhg.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rupps R, Hukin J, Balicki M, Mercimek-Mahmutoglu S, Rolfs A, Dias C, et al. Novel mutations in FA2H-associated neurodegeneration: An underrecognized condition? J Child Neurol. 2013;28:1500–4. doi: 10.1177/0883073812458538. [DOI] [PubMed] [Google Scholar]
- 14.Zaki MS, Selim L, Mansour L, Mahmoud IG, Fenstermaker AG, Gabriel SB, et al. Mutations in FA2H in three Arab families with a clinical spectrum of neurodegeneration and hereditary spastic paraparesis. Clin Genet. 2015;88:95–7. doi: 10.1111/cge.12516. [DOI] [PubMed] [Google Scholar]
- 15.Tonelli A, D'Angelo MG, Arrigoni F, Brighina E, Arnoldi A, Citterio A, et al. Atypical adult onset complicated spastic paraparesis with thin corpus callosum in two patients carrying a novel FA2H mutation. Eur J Neurol. 2012;19:e127–9. doi: 10.1111/j.1468-1331.2012.03838.x. [DOI] [PubMed] [Google Scholar]
- 16.Aguirre-Rodríguez FJ, Lucenilla MI, Alvarez-Cubero MJ, Mata C, Entrala-Bernal C, Fernandez-Rosado F, et al. Novel FA2H mutation in a girl with familial spastic paraplegia. J Neurol Sci. 2015;357:332–4. doi: 10.1016/j.jns.2015.07.042. [DOI] [PubMed] [Google Scholar]
- 17.Pierson TM, Simeonov DR, Sincan M, Adams DA, Markello T, Golas G, et al. Exome sequencing and SNP analysis detect novel compound heterozygosity in fatty acid hydroxylase-associated neurodegeneration. Eur J Hum Genet. 2012;20:476–9. doi: 10.1038/ejhg.2011.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donkervoort S, Dastgir J, Hu Y, Zein WM, Marks H, Blackstone C, et al. Phenotypic variability of a likely FA2H founder mutation in a family with complicated hereditary spastic paraplegia. Clin Genet. 2014;85:393–5. doi: 10.1111/cge.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]