

Abstract
The repair of chromosomal double-strand breaks (DSBs) by homologous recombination is essential to maintain genome integrity. The key step in DSB repair is the RecA/Rad51-mediated process to match sequences at the broken end to homologous donor sequences that can be used as a template to repair the lesion. Here, in reviewing research about DSB repair, I consider the many factors that appear to play important roles in the successful search for homology by several homologous recombination mechanisms.
Keywords: double-strand break repair, homologous recombination, search for homology, Rad51/RecA, DNA strand invasion
INTRODUCTION
Double-strand chromosome breaks (DSBs) threaten genome integrity and viability. In vertebrate cells, every round of DNA replication is accompanied by the appearance of several spontaneous DSBs, as judged by the frequency of sister chromatid exchanges or the number of chromatid breaks when the central Rad51 recombination protein is depleted. Indeed, when homologous recombination (HR) is impaired, vertebrate cells die. The consequences of lacking efficient homologous recombination mechanisms are particularly evident in the highly rearranged genomes of tumor cells. Repair appears to reflect a mixture of HR and nonhomologous end-joining (NHEJ) pathways, but the fact that the karyotype of normal cells remain so stable is a testament to the importance of HR mechanisms.
Here I wish to consider the rate-limiting homology search step in HR: How do the ends of a broken chromosome find a template with which to repair the DSB? Most of what we understand about this question has come from a detailed study of DSB repair in the budding yeast, Saccharomyces cerevisiae. Spontaneous DSBs in budding yeast are rare - about 1 in 8 cell divisions has a DSB that requires HR for repair, although DSBs arise in yeast at roughly the same rate per megabase pairs as in mammals. Most insights concerning DSB repair have come from synchronous induction of DSBs by site-specific endonucleases and the monitoring of repair events by a combination genetic, molecular biological and cytological approaches.
MECHANISMS OF HR
Recombination without strand invasion
Single-strand annealing (SSA).
The basis of HR is the ability of single stranded DNA to form Watson-Crick base pairs with another segment of DNA. In the simplest form of homologous recombination - single-strand annealing - DSB ends are resected to yield 3’-ended single-strand DNA (ssDNA) that can anneal if they share complementary sequences (Figure 1A). If homologous regions flank a single DSB, SSA produces a deletion; but cells efficiently create interchromosomal translocations via SSA between different DSBs [1, 2] (Figure 1B). When a strain can produce either a pair of intrachromosomal deletions or two interchromosomal translocations, there were as many translocations as deletions, suggesting that there is little “territoriality:” DSB ends initially a few kb away from each other are not strongly favored to promote a local deletion event. This result is interestingly different from the strong influence of chromosome architecture when there is a single DSB that must search for an ectopic homologous partner, as discussed below. In the case of SSA repair, the extensive 5’ to 3’ resection required to allow strand annealing (≥ 5 kb in each direction) may disrupt the normal association between DSB ends.
Figure 1.
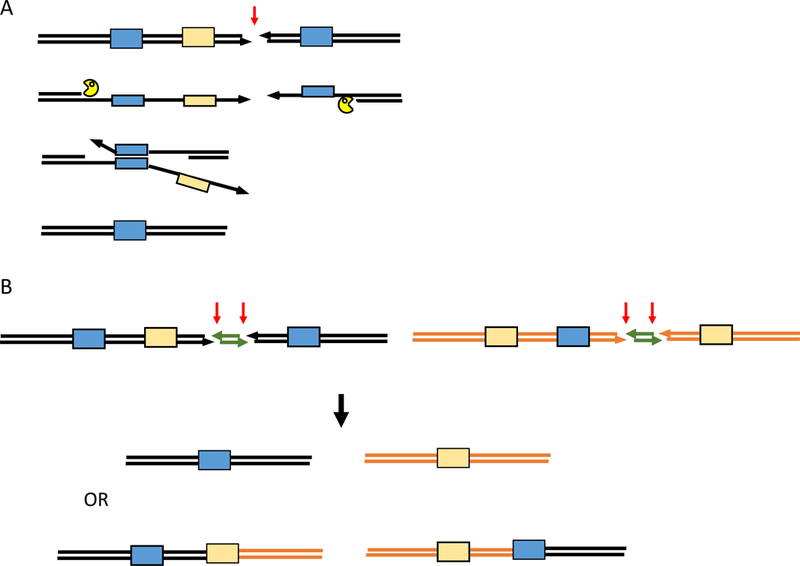
Single Strand Annealing (SSA). A. A DSB created between flanking repeated sequences (blue boxes) in direct orientation is resected by 5’ to 3’ exonucleases until complementary sequences are exposed, so that they can anneal with the aid of proteins such as Rad52. Nonhomologous 3’ tails are clipped off by the Rad1XPF-Rad10Ercc10 endonuclease complex and gaps are filled in to create a deletion. In the process, single stranded sequences that might provoke strand invasion and recombination elsewhere in the genome (light orange box) and interfere with the completion of SSA. B. Competition between alternative SSA events [1]. After induction of pairs of DSBs separated by about 1 kb, the ends are resected, allowing two viable alternative outcomes: either a pair of deletions or a pair of reciprocal translocations. Apparently because the sequences needed for the reciprocal translocation are rendered single-stranded more rapidly, translocations predominate over deletions.
Recombination requiring Rad51
Most types of homologous recombination, from bacteria to humans, rely on the RecA/Rad51/Dmc1 family of recombinase proteins to facilitate strand pairing and strand invasion (reviewed by [3-5]). These proteins assemble on single-stranded DNA (ssDNA) that is produced by 5’ to 3’ resection of the DSB end. The recombinase forms a nucleoprotein filament, with each recombinase subunit binding 3 nt of the ssDNA [6]. This structure has the capacity to search an entire genome, to locate a region of homology within dsDNA and then to promote an exchange of base pairs, leading to the formation of a displacement loop (D-loop). Within the D-loop, a DNA polymerase is recruited to extend the 3’ OH end of the invading strand, copying the template and eventually repairing the DSB. Three different outcomes from what appears to be a common initiating D-loop are illustrated in Figure 2. Studies of each of these processes have illuminated our understanding of the way Rad51 is able to locate a short region of homology in a sea of unrelated sequences.1
Figure 2.
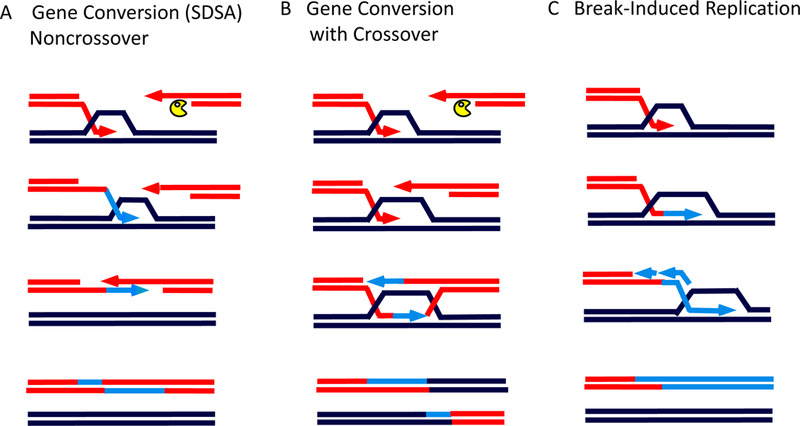
Modes of homologous recombination (HR). A. DSB repair by synthesis-dependent strand annealing (SDSA) creates a small patch of newly copied DNA to repair the DSB without an accompanying crossover. Here, strand invasion leads to the copying of the template, but the new strand is displaced from the donor and is captured by the second end of the DSB. This leads to another round of DNA synthesis and repair of the break. B. DSB repair through a double-Holliday junction (dHJ) intermediate engages both ends of the DSB with the donor sequences, creating a pair of Holliday junctions. After new DNA synthesis to fill in the gaps, the dHJ can either be “dissolved” by Sgs1BLM-Rmi1-Top3Top3α to produce an outcome similar to that in (A) or can be cleaved by structure-specific nucleases to create a crossover accompanying DSB repair. C. Break-induced replication (BIR) occurs when only one end of the DSB shares sufficient homology with other sequences in the genome. Repair occurs through a migrating “D-loop” in which the second strand of new synthesis is added only after some delay.
Both in vitro and in vivo studies have shed light on what occurs inside the RecA/Rad51 filament, to promote strand exchange. The X-ray crystallographic study by Pavletich’s lab [6] explained the long-standing observation that RecA (or Rad51) filaments bound to ssDNA elongate the DNA by 1.5 fold. Although the 3 bases bound by one subunit are still in roughly a B-DNA configuration, the adjacent phosphodiester bond is greatly stretched. This observation suggests that searching and strand exchange may occur in groups of 3 nucleotides (Box 1) [10].
BOX51‘s homology requirements.
In vitro experiments have suggested that the minimum length of homology that will form a stable structure could be as little as 6 bases [57]. Greene and Sung found that RecA or Rad51 can mediate strand exchange only if there are at least 8 consecutive bases of homology. Prentiss finds strand exchange can occur if there is a 9-base region of homology, even when one of these 9 is mismatched [10, 106, 107]. Our in vivo study of yeast Rad51’s tolerance for mismatches examined repair of a site-specific DSB by BIR, where the 108-bp donor template was altered to have various levels of heterology [97]. There was still tangible recombination (~5%, compared to the fully matched sequences) when every 6th base was mismatched (i.e. with only 5 consecutive bases of homology). The apparent lower stringency in this assay compared to in vitro studies may reflect the increased stability through imperfect base pairing between the template and the donor over 100 bp.
Although one can measure in vitro some aspect of strand exchange with very small substrates, efficient and complete RecA/Rad51-mediated recombination in vivo requires about 70 bases homology [11-13]. Linear DNA fragments with 50 bp homology on either side of a selectable marker (“ends-out” transformation [14]) are sufficient for gene replacement in yeast [15]. These findings are consistent with in vitro estimates [16]. However, when homology on either side of a DSB is reduced to 33 bases on each side of a DSB, repair occurs by a Rad51-independent BIR process and deleting Rad51 markedly improves repair, suggesting that a Rad51 filament is inhibitory when homology length is limited [11]. Rad51 also inhibits SSA [17].
The barrier to recombination by short homology can be significantly overcome if donor sequences can be brought into proximity with the DSB. For example, the yeast Recombination Enhancer (RE) binds multiple copies of the Fkh1 protein whose FHA phosphothreonine binding domain enables it to strongly associate with (as yet unknown) proteins near a DSB [18]. Thus, RE brings an adjacent, normally distant donor sequence close to the DSB and favors its usage. RE-mediated tethering a donor with only 50 bp homology near the DSB increases repair from <40% to nearly 80% [19]. Thus the time a donor spends in the vicinity of the DSB strongly influences its chances of success.
Break-induced recombination (BIR).
When only one end of a DSB shares adequate homology with a donor region, or when there is only one end (at eroding telomeres or at stalled and broken replication forks), repair can occur by BIR (Figure 1C). Most BIR events are Rad51-dependent [20-22]. Unlike normal DNA replication, BIR does not have coordinated leading- and lagging-strand synthesis; rather, a migrating D-loop, driven by copying the template, results in a long leading-strand intermediate that is later completed by lagging-strand synthesis [23]. Consequently, all the newly copied DNA is conservatively inherited, leaving the donor sequence unaltered [23, 24]. The copying process in BIR is 1000-fold more mutagenic than normal DNA replication, with frequent template jumps [25-27].
Gene conversions (with or without crossovers).
When both ends of a DSB share sufficient homology with a donor template, repair occurs by one of two gene conversion mechanisms. A synthesis-dependent strand invasion process (Figure 2A) leads to the repair of the break by a patch of new DNA synthesis, without an associated crossover. DNA density labeling revealed that all the newly copied DNA in gene conversion is found at the recipient, leaving the donor unchanged [28]. Alternatively, repair can be mediated through a double Holliday junction (dHJ) intermediate, and can yield either crossovers or noncrossovers through the agency of Holliday junction resolvases (Figure 2B). However, most dHJ-mediated events in mitotic cells result in noncrossovers because of an active dHJ “dissolvase” involving the BLM helicase and Topoisomerase 3 that collapses and removes the dHJ [29, 30].
How does chromosome architecture affect DSB repair?
In budding yeast, chromosome conformation capture has been used describe the 3D arrangement of chromosomes [31]. Chromosomes display a so-called Rabl architecture, with all the centromeres clustered at the spindle pole body throughout the cell cycle, with the chromosome arms of different lengths extending toward the nuclear periphery. Kupiec and Fabre [32] demonstrated that interchromosomal recombination between homologous sequences was more efficient when the donor and the recipient were either both close to the centromere or both near telomeres than when one was centromere-adjacent and the other telomere-proximal. We placed donor sequences at 20 sites on different chromosomes, each able to recombine with a recipient sharing 1 kb homology on either side of an HO endonuclease-induced DSB [33]. There was a nearly 10-fold difference between the best and worst donors and their relative efficiencies strongly correlated with the probability of each region surrounding the inserted donor coming into physical contact with the designated recipient. When the recipient was integrated at a different location, some “bad” donors became “good”. Hence, the restriction on using a “bad” donor was not some intrinsic lack of accessibility imposed by chromatin structure, but reflected how often two sites came into contact. In this regard, it should be noted that heterochromatin does not seem to be a severe impediment to efficient repair; the HML and HMR donor sequences used in MAT switching have highly positioned nucleosomes, resulting in transcriptional silencing and inaccessibility to HO endonuclease cleavage, but they are very efficient donors. In fact, unsilencing one of the two silent donors does not make it better [34]. Presumably, yeast have chromatin remodelers that are able to cope with this modest form of heterochromatin when the Rad51 filament encounters these transcriptionally silent donors. At least five such complexes have been implicated in promoting DSB repair: RSC [35-38], SWI/SNF[39-41], INO80 [41, 42], and Fun30 [43-46] all appear to affect the rate of 5’ to 3’ resection and perhaps later steps. Deleting the SWI/SNF-related chromatin remodeler, Rad54, does not prevent the association of the Rad51:::ssDNA filament with its heterochromatic donor, implying that there is still some form of heteroduplex DNA formation, but rad54Δ blocks the next step, the priming of new DNA synthesis from the 3’ end [47].
The constraints on using distant donors can be overcome; that is, a poor donor can be made more effective by increasing the level of shared homology from 1 kb on each side of the DSB to 2 kb [33]. Placing the Recombination Enhancer near a donor also improves the outcome. Finally, slowing down the rate of 5’ to 3’ resection or increasing the abundance of RPA can extend the time that a poor donor has to engage in repair and thus helps make a poor donor better [33].
Intrachromosomal donors obey the rules expected for a chromosome anchored at its centromere: donors close to the centromere were strongly constrained in interacting with a DSB-containing recipient 400 kb away on one chromosome arm [48]. A donor 100 kb from the centromere on the opposite arm from the DSB site was it insulated by the centromere, as it was as efficient in repair as a donor 100 kb from the centromere on the same arm as the DSB. In a diploid, intrachromosomal events are also favored over allelic events, even though the shared homology between two homologous chromosomes is vastly greater than between 2-kb ectopic sites [48]. Similarly, spontaneous gene conversions between intrachromosomal leu2 heteroalleles situated 17 kb apart were also strongly favored over interchromosomal locations [49].
How long does the search for homology take?
The time needed for two sites in the nucleus to find each other can be estimated by physical monitoring of synchronously induced recombination. Using Southern blots, PCR and chromatin immunoprecipitation (ChIP), one can identify the formation of specific intermediates as well as the association of certain proteins with both the broken chromosome ends and the donor locus. This has been accomplished in budding yeast where induced site-specific cleavage by the HO endonuclease takes about 20-30 min [50]. 5’ to 3’ exonuclease digestion of ends occurs at a rate of about 1 nt/sec. Binding of the RPA complex to ssDNA can be detected within minutes after the induction of the DSB, soon followed by the assembly of Rad51. There is a significant delay before Rad51 can be detected bound to the donor locus (15 - 30 min for intrachromosomal events and even longer for interchromosomal events) [47]. It is not clear if this delay represents the first encounter between Rad51 and its target, or if Rad51 can be detected at the donor only after there is a synaptic step, where the invading strand becomes interwound (plectonemic) with the donor locus. There could be - and probably are - several collisions on average before a stable synaptic intermediate is formed.
To estimate how frequently nonproductive collisions happen, we created a MAT switching system with competition between a favored donor (HMR) and a second, infrequently used donor (HML). If collisions between the recipient and the donor were 100% productive and the recipient interacted with the efficient donor 9 times more often than with the poor donor, adding more homology to the disfavored donor would have little effect. But if collisions were not always productive, then introducing extra homology between the recipient and the “wrong” donor should improve its probability of converting a collision to a more stable intermediate. By increasing homology from 300 bp to several kb, shared only by the “wrong” donor and the MAT locus, we increased its usage from < 10% to > 30% [34]. We concluded that the probability of any single encounter progressing to successful repair event is in fact about 25%.
One very useful concept that has come from in vitro single molecule experiments using bacterial RecA, is that the recombinase filament is long enough to interact with more than one site at a time. RecA bound to ssDNA was tested with for its ability to recombine with a 1 kb target embedded within 50 kb of DNA [51]. The efficiency was much greater when the 50 kb of DNA could fold up, compared to when the target DNA was extended. Forget and Kowalczykowski concluded that an efficient homology search is three-dimensional and that the RecA filament could “brachiate” - the way apes transfer from branch to branch - holding on to one place while reaching out to find another nearby contact. Multiple contacts of a Rad51 filament have also been inferred from in vivo experiments with budding yeast, by studying complex rearrangements caused by a single DSB [52].
There has been some confusion in the literature as to how long it should take for a DSB to find a partner anywhere in the nucleus. The process seems daunting for ectopic recombination even in a haploid yeast nucleus, where segments of 1-2 kb must locate homology in a sea of more than 105 kb. But for DSBs that arise during replication (i.e. most DSBs) the problem is much simpler, since sister chromatids are held in close proximity by cohesins. Moreover, additional cohesins are recruited around the DSB site through a process that responds to the formation of a domain of phosphorylated histone H2A (γ-H2AX) around the break2 [54, 55]. So, sister chromatid repair is likely to be rapid and efficient since the allelic donor and the recipient are firmly tethered. Unexpectedly, whereas essentially all ectopic DSB repair events require Rad51, a large proportion of spontaneous sister chromatid exchanges (SCE) appear to be Rad51-independent [56]. How repair occurs in this instance is not well-understood.
But the search for homology at an ectopic donor is much more difficult to comprehend. The timing itself is not in dispute: chromatin immunoprecipitation with an anti-Rad51 antibody reveals the time it takes to see the interaction of Rad51 with its donor. In asynchronous cells, without the facilitation by a Recombination Enhancer, intrachromosomal strand invasion after induction of a DSB takes about an hour [19]. A somewhat later, but much easier, measure of initial repair comes from quantitative PCR using primers that can only amplify after the invading end of the DSB elongates ~50 nt by copying the donor. The kinetics of intrachromosomal repair are about twice as rapid as interchromosomal events [33].
The duration of the search can be understood as the time of first passage So how does a DSB end find its partner? A DSB end sharing 1 kb with another site located somewhere among 105 different 1-kb sequences would seem to require on average 105 sequential interactions, likely many more if each possible alignment between a colliding Rad51 filament and donor sequences had to be tested individually. The search could be somewhat simplified if Rad51 slides along a segment of DNA to test each possible base-pair alignment over a certain distance rather than to engage in a separate collision [57]. Indeed, Rad51 ChIP evidence from Jentsch’s group supports the notion of extensive sliding of the filament along a chromosome [58], although the result might also be obtained by the Rad51 filament making many independent contacts with sequences in its proximity [59]. Still, one is left with the impression that each sequence alignment test would have to be impossibly brief to test all possible locations in the genome. Since it takes about 1 hour to find a donor, searching half of the 2 × 105 kilobase pairs would take 0.36 seconds. To search each of 107 unique alignments would leave only 0.004 sec per inquiry. To explain and overcome what seems like an impossible barrier to successful repair, several possible solutions have been proposed, including that homologous sequences are pre-aligned before a DSB occurs or that some diffusible copies of the template are used [60, 61]. But in fact, the search problem is not as formidable as it first seems.
A key to our understanding of the search problem can be found in biophysics, in the problem of first passage time, the “waiting time for the first encounter,” as discussed in an excellent introduction to these ideas by Zhang and Dudko [62]. Two regions on the same DNA molecule are linked such that their relative motion is still random, but not independent of each other. Their relative motion is “subdiffusive.” Yeast chromosomes exhibit this subdiffusive behavior [63-66]. Both simulations and direct observations of fluorescently tagged immunoglobulin loci in mouse B cells lead to the same conclusion [67]: for sites that are within a radius of 1 micron (the size of the yeast nucleus), the mean free passage time (MFPT) is about 2100 sec (i.e. about 30 min). If sites lie within a radius of 0.5 microns, the time shortens to less than 180 sec.3 These measurements and the simulated estimates are based on the apparent subdiffusion of mammalian chromatin, whose motion is substantially more constrained than for much smaller, less condensed yeast chromosomes. Simply restated, the time needed for two sites ≤ 1 micron apart to encounter each other is well within the observed limits derived from direct observation of repair, even without invoking any change in the mobility of broken ends.
Changes in chromosome mobility may facilitate homology searching
Repair can be made more efficient by increasing the probability that two loci will come into contact, by increasing rate of diffusion (or subdiffusion) of the DSB ends to more rapidly explore the volume of the nucleus, or by enlarging their “radius of confinement” (Rc). Several labs have observed that DNA damage causes at least two kinds of alterations of chromosome mobility [40, 68-72]. First, there is a “local” change. A fluorescent marker adjacent to a site-specific DSB exhibits an increased Rc when measured by mean square displacement (MSD), suggesting that the sequences are able to explore a much greater volume in a given time after DNA damage. This increase may reflect a change in the anomalous diffusion constant and as well as some alteration in the “stiffness” of chromatin around the DSB. Second, there are “global” changes: when yeast suffer several DSBs per cell in random positions created by drugs such as phleomycin or zeocin, there is a general, but more modest, change in the Rc (Figure 3).
Figure 3.
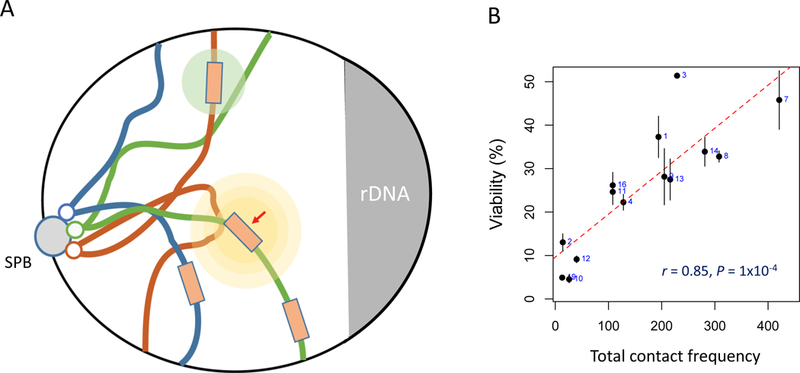
Ectopic repair of a DSB is influenced by proximity and possibly by DNA damage-stimulated changes in chromatin structure and chromosome movement. A. A cartoon of a budding yeast nucleus, with chromosomes tethered by their centromeres at the spindle pole body and displaying a so-called Rabl orientation. A DSB (red) causes both local (yellow circles) and a weaker global (light green circle) changes in chromatin structure and chromosome mobility. Image based on data and illustrations by Zimmer and Fabre [104]. B. The efficiency of intrachromosomal and interchromosomal repair, using donors of a few kb, is strongly influenced by the proximity - as measured by contact frequency [31] - when the efficiency of DSB repair, measured by viability, was compared for 20 budding yeast strains, each with a donor in a different interchromosomal location [33].
The mechanisms that underlie these changes remain only partially characterized. There are at least seven aspects to the change in Rc, which together lead to the observed response. Together these changes “stir” the chromosomes to facilitate the pairing of a homologous partner with the break site.
A single DSB in budding yeast provokes an extensive DNA damage checkpoint response by activating the ATM and ATR kinases, resulting in the phosphorylation of many proteins [73]. One modification is the phosphorylation of histone H2A (called γ-H2AX) that spreads for a distance of ~50 kb on either side of the DSB [74]. In cells lacking ATRMec1 or carrying a non-phosphorylatable histone H2A allele, Rc for a region near a DSB or the distance between two fluorescently marked sites around the DSB does not show the increases seen in wildtype cells [63, 68].
Checkpoint activation due to DNA damage also promotes the loss of telomere-associated proteins from the nuclear periphery [75-77], causing a general untethering of telomeric regions that likely affects all chromosomes and not just the one with a DSB.
There are profound changes in the pericentric chromatin (around centromeres), such as loosening of the association between kinetochores, although most evidence suggests that the kinetochore of the broken chromosome does not become dissociated from the spindle pole body [78, 79]. One kinetochore protein, Cep3, is phosphorylated by checkpoint kinases after DNA damage, but other observers have found that introducing a non-phosphorylatable Cep3 mutant does not seem to affect an increase in the distance between interchromosomal sites or to change in Rc of a broken chromosome [63, 78, 79].
Microtubule (MT) dynamics also enhance chromosome motion [80]. Disrupting microtubules with nocodazole or creating MT-related mutations impairs the normal increase in local Rc after damage and blocks the motion of DSB ends to which the Ddc2-GFP protein binds [81].
There are also changes in nuclear organization that depend on changes in the actin cytoskeleton [77]. The changes in motion are not dependent on the linking of nucleus to cytoplasm (LINC) proteins and may be related to the association of actin-related Arp4 and Arp8 proteins to the INO80 chromatin remodeler. DSB-induced recombination between subtelomeric sites was less efficient when actin polymerization was inhibited by latrunculin-A. Actin-dependent motion of chromosomes in budding yeast has an important role in homologous chromosome recombination in meiosis [82, 83] but also appears to affect mitotic DSB repair [77]. Actin-mediated changes have been documented in metazoans in the movement of damaged DNA from heterochromatin [84] and in the clustering of γ-H2AX foci [85].
DNA damage also provokes a global depletion of histones - astonishingly, as much as 20 to 40% [71]. This depletion requires ATRMec1-mediated damage signaling and the INO80 chromatin remodeler. The loss is not simply around the site of damage but global, and appears to cause a general decondensation of yeast chromatin (though yeast don’t have much higher-order chromatin beyond an 11 nm fiber) [86, 87]. Damage-provoked histone loss (or artificial histone depletion) was suggested to create increased fiber flexibility, which seems to be the opposite of the conclusion reached by Fabre and Zimmer [63] that damage leads to chromatin stiffening. However, another study of nucleosome depletion, but without DNA damage, also concluded that chromatin fibers became more stiff, with a reduced Rc [79].
Surprisingly, loss of Rad51, but not its necessary filament-forming mediator, Rad52, also blocks increased mobility [68]. This remains a fascinating, but poorly understood aspect of homology searching.
All of these studies imply that the damage-enhanced changes in exploration of the nuclear volume is an important feature of efficient ectopic recombination. But experiments designed to test the importance of mobility on repair have failed to provide the expected evidence. For example, several mutations that fail to increase Rc did not substantially change the use of a distant ectopic donor versus an intrachromosomal donor [33, 78]. Our expectation was that constraining Rc should improve the use of the intrachromosomal donor. Similarly, in an assay involving competition between two sets of Rad51-independent SSA events - either a pair of cis-annealings or a pair of trans events (Figure 1b) - were unaffected by deleting Rad51 (Q. Wu and JEH, unpublished). So, the changes in Rc seem still to be in search of a function.
DSBs appear to be moved to the nuclear periphery when repair is slow or when there is no donor [88, 89]. This seems to be a pathological response, perhaps bringing DSBs into a nuclear compartment enriched for Ku70/80 (and possibly other end-joining proteins) so that ends might be repaired by end-fusion. Many of the measurements of mobility change have been performed as long as 6 h after damage induction, whereas a lot of repair takes place much sooner.
Do DSB ends act independently?
By marking both sides of a DSB with fluorescent markers, it is possible to see how end of a DSB behaves after damage. Surprisingly, they appear to be tethered to each other, even though the ends are extensively resected and presumably loaded with Rad51 [90, 91]. The Mre11-Rad50 complex plays some role in restraining the ends, but even after arresting cells with nocodazole, only about 20% dissociate in the absence of Rad50; so, there must be other players. These results suggest that the two ends of a DSB somehow travel together during homology searching.
To examine whether ends move together, we devised a cis/trans test [2] (Figure 4A). In the cis arrangement, a DSB in the middle of a LEU2 gene on chromosome 5 can be repaired using a LEU2 template on another chromosome (Chr3); but at the same time a second DSB, on yet another chromosome (Chr11) was repaired by SSA of URA3 sequences flanking that DSB. In trans, one DSB (Chr5) was flanked by LE and URA3 sequences while Chr11 had URA3 and U2. In both arrangements the LE and U2 ends must invade the interchromosomal donor (effectively the same event except where the ends are derived). The other ends have URA3 sequences that undergo SSA, again in cis or in trans. Repair is more rapid when both ends of the same DSB are used for recombination simultaneously, suggesting there is an advantage in coordinating the search for homology and in subsequent steps. Deleting Rad50 did not change this outcome. However, the delay in trans repair could be eliminated by removing URA3 sequences on Chr11, preventing SSA. Thus, the two ends are somehow kept together but can be pulled away from completing one task by a competing task - despite the extensive resection that is needed for SSA.
Figure 4.
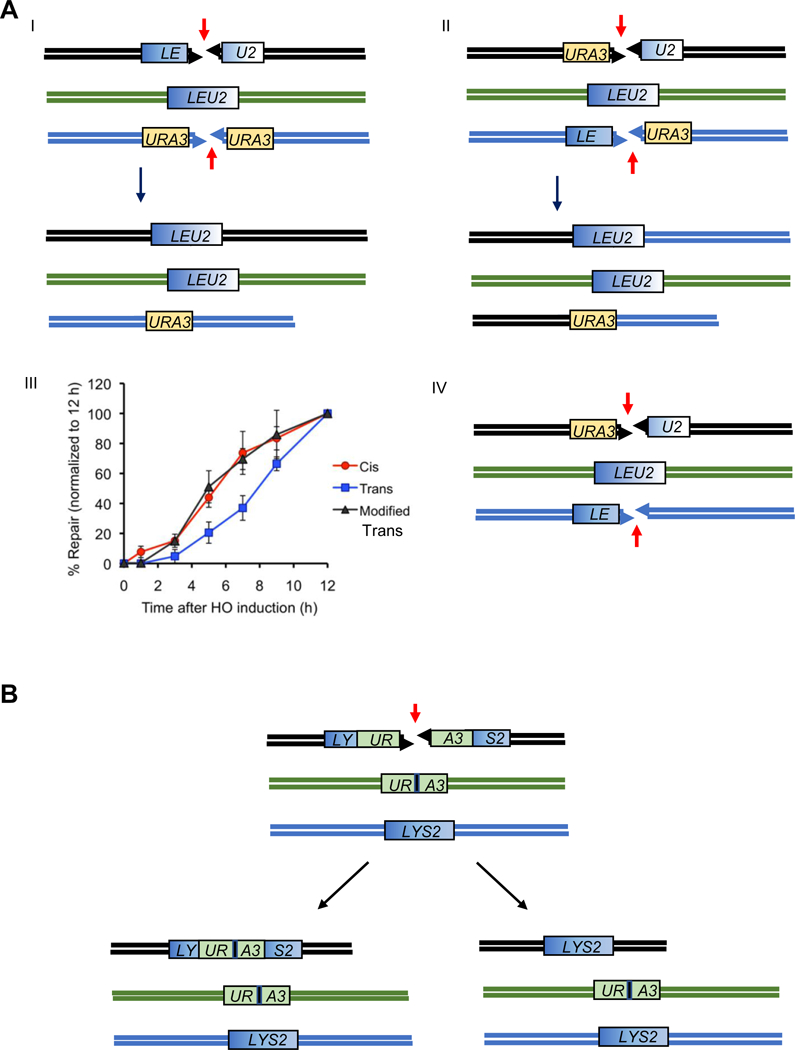
A. A Cis/Trans test of how DSB ends are linked together. Experiments by Jain et al. [2]. I. A DSB induced between two halves of a LEU2 gene (LE and U2) can be repaired by gene conversion with an unbroken LEU2 sequence on a different chromosome. Here, the two ends participating in repair are in Cis. A second DSB is repaired by single-strand annealing between flanking URA3 sequences. II. In Trans, the same two types of repair must occur as in (A), but now the LE and U2 ends are derived from two DSBs. The opposite DSB ends are repaired by SSA between URA3 sequences. III. Repair of LEU2 in trans is markedly slower than in Cis, but this delay can be suppressed by deleting one of the URA3 segments, so that there is no competition between the two ends of one DSB to participate in two different repair events (IV). B. Sequences close to the end of a DSB are not used preferentially. A DSB can either be repaired by gene conversion with an ectopic URA3 or LYS2 donor [92]. The DSB ends were perfectly matched, except for a single base pair substitution, to a mutated HO cleavage site (black vertical bar). Both outcomes are frequent.
The competition among sequences exposed by resection can also be seen in the assay illustrated in Figure 1A. A DSB that can be repaired by SSA between flanking homologous regions of ~ 1 kb is successful about 85% of the time, but the efficiency increased to ~100% when a retrotransposon LTR δ sequence (shown in light yellow) was removed [2]. These results imply that the δ sequences might engage in alternative recombination events that ultimately prevent recovery of the SSA deletion.
A point that needs to be stressed is that the search of homology is not strongly driven by the sequences closest to the 3’ end of a resected DSB. Kupiec [92] elegantly showed this by creating a DSB in the center of a URA3 gene that was flanked by “LY” and “S2” sequences (Figure 4B). Repair by gene conversion could occur either with an ectopic URA3 donor or with a LYS2 donor. More than half the events resulted in the loss of the UR and A3 sequences closest to the break. Thus, even when the sequences closest to the 3’ end were almost perfectly matched to a donor, sequences more distant from the DSB, requiring more extensive resection to be exposed, could efficiently recombine. The ratio of donor usage might also reflect their relative contact probabilities, as discussed above.
Are there “Repair Centers” when there are multiple DSBs?
Another aspect of homology searching and DSB repair is the possible existence of “repair centers,” where several DSBs appear to cluster [93]. This is especially evident when the reporter of multiple breaks is a fluorescent version of Rad52; however, when immunofluorescence was used to probe spread chromatin from lysed cells, three HO-induced DSBs yielded three foci of both Rad51 and Rad52 [94]. Recently, we monitored multiple, separate DSBs in live cells using two different GFP-marked probes: the checkpoint protein, Ddc2-GFP or a Rad51-GFP fusion protein that forms foci and is able to complete DSB repair when wildtype Rad51 is also present [81]. In cells with 3 HO-induced DSBs on different chromosomes, about 30% of cells have a single Rad51 or Ddc2 fluorescent focus with the intensity expected for all three individual foci grouped in one location. However, when Ddc2-GFP is used as the DSB probe, the co-localization of GFP spots is Rad52-independent, arguing that whereas broken ends can be transiently brought together, this clustering is not mediated by the core homologous recombination repair machinery. Rad52 fluorescent proteins themselves may aggregate; this may explain the many instances in which there are two Rad51-GFP spots but only one of them has a visible Rad52-RFP focus.
How does the search for homology differ from a search for homeology?
There is one other fundamental aspect of homology searching not yet addressed, namely the tolerance of the homology searching process for sequence divergence. For example, Rad51-independent SSA between a pair of ~200-bp sequences with 3% divergence is only about 20% as efficient as fully matched sequences [95]. The rejection of mismatched heteroduplex DNA depends on the Msh2-Msh6 (MutSα) mismatch repair proteins, but not on the Mlh1-Pms1 (MutLα) dimer, but correction of mismatches in the annealed heteroduplex still requires MutLα. Rejection is also promoted by the BLM helicase, Sgs1, and its partners, Rmi1 and Top3, apparently unwinding the mismatched duplex, leaving the sequences to anneal with other partners [95, 96].
As noted above, a BIR assay using 108-bp substrates showed that Rad51 has tolerance for a substantial degree of mismatching [97]. This study of BIR and sequence divergence revealed another unexpected finding: the presence of a nonhomologous sequence at the 3’ end of the invading strand (which is readily clipped off by Rad1-Rad10 [98]) dramatically changed the sensitivity of the system to mismatches; now, even a single mismatch was sufficient to reduce repair by a factor of two [97]. A similar finding has been made concerning SSA (E. Sapede, N. Sugawara and JEH, unpublished). When the same 3% divergent sequences that reduced repair to 20% were re-tested by using Cas9 to induce SSA without nonhomologous ends, there was no heteroduplex rejection. These data suggest that an annealed or strand-invaded intermediate with a 3’ protrusion recruits Msh2-Msh6 and provokes Sgs1-Top3-Rmi1 to dismantle the structure. The importance of a nonhomologous 3’-ended tail in heteroduplex rejection can be understood if such tails are a way of indicating to the cell’s surveillance machinery that the sequences being invaded or annealed are likely to be ectopic rather than on a sister chromatid or a homologous chromosome, where such tails would not be expected to be present.
Homologous recombination involving microhomology
Some complex chromosome rearrangements in cancer cells, called chromothripsis [99], appear to involve extensive DNA replication rather than simply reshuffling broken segments of a chromosome. These events, sometimes called chromoangenesis [100], have been proposed to arise from microhomology-mediated break induced replication (MMBIR), in which a DNA polymerase can jump even megabase distances to copy DNA segments into complex rearrangements whose junctions have only a few base pairs of homology [101]. These events might begin as a normal homologous recombination event that then leads to template switching between highly divergent substrates, as has been documented in the case of interchromosomal template switching (ICTS) in budding yeast [102] (Figure 5). It should be noted that such ICTS events are quite rare (~10−6) when the second donor is only 72% identical but they are found at > 10−3 when the second donor is identical. The jumps are made by DNA polymerase δ and are suppressed when the proofreading domain of the polymerase is mutated, apparently creating a more processive enzyme, less likely to dissociate from its template [103]. Whether these jumps are mediated solely by DNA polymerase or require the agency of Rad51 is a critical unanswered question.
Figure 5.
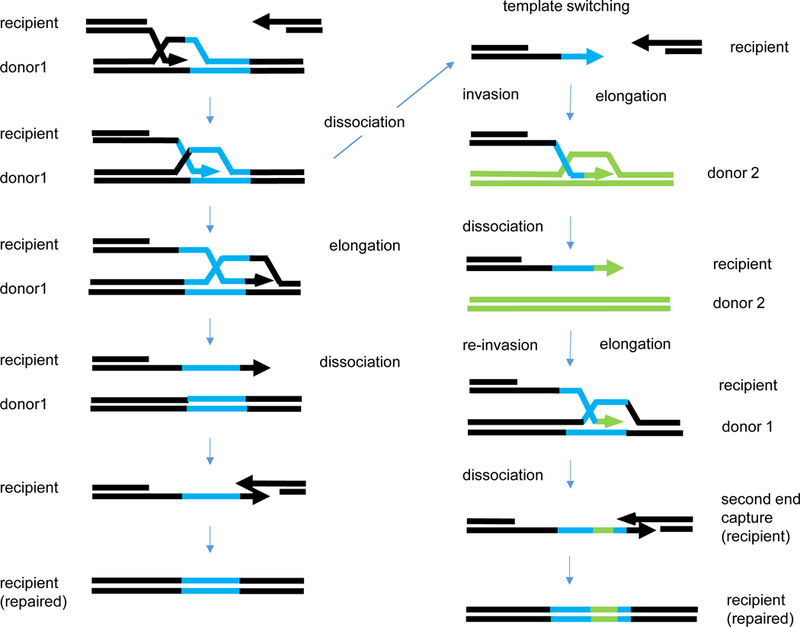
Template switching between divergent, unlinked donors during gap repair initiated by a DSB. Based on the observations of Hicks et al. [105] and Tasponina and Haber [102]. The sequence of events is that expected for gene conversion by SDSA. Black sequences are homologous and repair involves copying donor sequences. Left: simple SDSA results in the copying of the blue sequences from a single donor. Right: Template switching creates a blue/green/blue chimeric sequence. Note that the assay requires that there be two jumps to allow the completion of the DSB repair event.
Conclusions and outlook
Our understanding of how homologous and homeologous sequences recombine has grown dramatically in the past decade, not only with the development of new insights into Rad51’s strand invasion but especially by adding genomic approaches to genetic and molecular biological strategies. Homologous recombinational repair of a DSB is strongly influenced by contact probability, but poorly recombining partners can be improved by prolonging the time that repair is possible and by increasing homology length, thus increasing the probability of a stable initial contact. Intrachromosomal repair is general favored over interchromosomal repair, but proximity is the governing factor. DSBs provoke global and local changes in chromatin structure and mobility that may be critical to “agitate” chromosomes and facilitate the finding of a homologous donor, though evidence for this remains scanty.
The refinements in live-cell microscopy as well as correlative light and EM (CLEM) microscopy should make it possible to explore in greater detail the early steps in homologous recombination. Increases in both the efficiency and synchrony of DSB formation should also make it possible to extend the “in vivo biochemistry” experiments to mammalian cells. Moreover, the wealth of information coming from whole-genome studies comparing normal and cancer cells will likely reveal underlying principles in various kinds of DSB repair processes. New insights are undoubtedly just around the corner, where we cannot yet see them.
Acknowledgments
I am grateful to Kerry Bloom, Emmanuelle Fabre, Jane Kondev, Michael Lichten, Anna Malkova, Gonen Memisoglu, David Waterman and Christophe Zimmer for their invaluable comments on a draft of this paper, and to Gabriel Bronk, Kevin Li and Jane Kondev for long conversations. Work in my lab has been generously supported for more than 45 years by grants from the Institute of General Medicine of the NIH. Of course many of the results discussed here were the fruits of both imagination and insightful experiments from the talented postdocs, grad students, undergraduates and technicians with whom I have had the pleasure of working.
Footnotes
Most of the studies of recombination discussed here involve donors and recipients that share homology over a few kb of DNA, located at ectopic locations and - in budding yeast, where much of the work has been done - in haploids. Most DSBs arising during replication will be repaired using a nearby and already-aligned sister chromatid. This process has not been as thoroughly studied as ectopic repair because there is no discernable genetic signal of repair: donor and recipient are identical. One way to study these events is to use single-strand nicking enzymes that can cleave in G1, so that after replication, there will be a broken chromatid and an intact partner 7. Mayle, R., I.M. Campbell, C.R. Beck, Y. Yu, M. Wilson, C.A. Shaw, L. Bjergbaek, J.R. Lupski, and G. Ira, DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science, 2015. 349(6249): p. 742-7. Repair can also take place between homologous chromosomes that carry single nucleotide variants that can be scored. A great deal of work has been done on these processes in meiotic cells, which is beyond the scope of this review. Readers are encouraged to consult several excellent reviews on the complexities of meiotic homology search - why, for example, the search is directed away from a sister chromatid and toward a homolog 8. Brown, M.S. and D.K. Bishop, DNA strand exchange and RecA homologs in meiosis. Cold Spring Harb Perspect Biol, 2014. 7(1): p. a016659, 9. Lam, I. and S. Keeney, Mechanism and regulation of meiotic recombination initiation. Ibid.2015: p. a016634..
Evidence for recruitment of cohesin at DSB sites and the role of both γ-H2AX phosphorylation and the MRX complex is very strong in budding yeast. There is some supporting evidence for this same recruitment in mammals (53. Caron, P., F. Aymard, J.S. Iacovoni, S. Briois, Y. Canitrot, B. Bugler, L. Massip, A. Losada, and G. Legube, Cohesin protects genes against gammaH2AX Induced by DNA double-strand breaks. PLoS Genet, 2012. 8(1): p. e1002460., but the relation to γ-H2AX formation or involvement of MRN has yet to be examined.
The MDPT varies as a function of the radius, raised to the 4th power.
Literature cited
- 1.Haber JE and Leung WY, Lack of chromosome territoriality in yeast: promiscuous rejoining of broken chromosome ends. Proc. Natl. Acad. Sci. USA, 1996. 93(24): p. 13949–13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jain S, Sugawara N, and Haber JE, Role of Double-Strand Break End-Tethering during Gene Conversion in Saccharomyces cerevisiae. PLoS Genet, 2016. 12(4): p. e1005976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Symington LS, Rothstein R, and Lisby M, Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics, 2014. 198(3): p. 795–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell JC and Kowalczykowski SC, RecA: Regulation and Mechanism of a Molecular Search Engine. Trends Biochem Sci, 2016. 41(6): p. 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daley JM, Gaines WA, Kwon Y, and Sung P, Regulation of DNA pairing in homologous recombination. Cold Spring Harb Perspect Biol, 2014. 6(11): p. a017954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Z, Yang H, and Pavletich NP, Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature, 2008. 453(7194): p. 489–4. [DOI] [PubMed] [Google Scholar]
- 7.Mayle R, Campbell IM, Beck CR, Yu Y, Wilson M, Shaw CA, Bjergbaek L, Lupski JR, and Ira G, DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science, 2015. 349(6249): p. 742–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown MS and Bishop DK, DNA strand exchange and RecA homologs in meiosis. Cold Spring Harb Perspect Biol, 2014. 7(1): p. a016659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam I and Keeney S, Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb Perspect Biol, 2015. 7(1): p. a016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JY, Terakawa T, Qi Z, Steinfeld JB, Redding S, Kwon Y, Gaines WA, Zhao W, Sung P, and Greene EC, DNA RECOMBINATION. Base triplet stepping by the Rad51/RecA family of recombinases. Science, 2015. 349(6251): p. 977–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ira G and Haber JE, Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol Cell Biol, 2002. 22(18): p. 6384–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lovett ST, Hurley RL, Sutera VA Jr., Aubuchon RH, and Lebedeva MA, Crossing over between regions of limited homology in Escherichia coli. RecA-dependent and RecA-independent pathways. Genetics, 2002. 160(3): p. 851–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watt VM, Ingles CJ, Urdea MS, and Rutter WJ, Homology requirements for recombination in Escherichia coli. Proc Natl Acad Sci U S A, 1985. 82(14): p. 4768–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rothstein R, Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods in enzymology, 1991. 194: p. 281–301. [DOI] [PubMed] [Google Scholar]
- 15.Wach A, Brachat A, Pohlmann R, and Philippsen P, New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast, 1994. 10(13): p. 1793–1808. [DOI] [PubMed] [Google Scholar]
- 16.van der Heijden T, Modesti M, Hage S, Kanaar R, Wyman C, and Dekker C, Homologous recombination in real time: DNA strand exchange by RecA. Molecular cell, 2008. 30(4): p. 530–8. [DOI] [PubMed] [Google Scholar]
- 17.Ivanov EL, Sugawara N, Fishman-Lobell J, and Haber JE, Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics, 1996. 142(3): p. 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Coic E, Lee K, Lee CS, Kim JA, Wu Q, and Haber JE, Regulation of Budding Yeast Mating-Type Switching Donor Preference by the FHA Domain of Fkh1. PLoS genetics, 2012. 8(4): p. e1002630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mehta A, Beach A, and Haber JE, Homology Requirements and Competition between Gene Conversion and Break-Induced Replication during Double-Strand Break Repair. Mol Cell, 2017. 65(3): p. 515–526 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anand RP, Lovett SL, and Haber JE, Break-induced DNA replication, in DNA Replication, Bell SD, Méchali M, and DePamphilis ML, Editors. 2013, Cold Spring Harbor Press: Cold Spring Harbor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Llorente B, Smith CE, and Symington LS, Break-induced replication: what is it and what is it for? Cell Cycle, 2008. 7(7): p. 859–64. [DOI] [PubMed] [Google Scholar]
- 22.Signon L, Malkova A, Naylor M, and Haber JE, Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol. Cell. Biol, 2001. 21: p. 2048–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, and Malkova A, Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature, 2013. 502(7471): p. 389–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donnianni RA and Symington LS, Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci U S A, 2013. 110(33): p. 13475–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, and Malkova A, Break-induced replication is highly inaccurate. PLoS biology, 2011. 9(2): p. e1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malkova A and Haber JE, Mutations arising during repair of chromosome breaks. Annu Rev Genet, 2012. 46: p. 455–73. [DOI] [PubMed] [Google Scholar]
- 27.Smith CE, Llorente B, and Symington LS, Template switching during break-induced replication. Nature, 2007. 447: p. 102–105 [DOI] [PubMed] [Google Scholar]
- 28.Ira G, Satory D, and Haber JE, Conservative inheritance of newly synthesized DNA in double-strand break-induced gene conversion. Mol Cell Biol, 2006. 26(24): p. 9424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ira G, Malkova A, Liberi G, Foiani M, and Haber JE, Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell, 2003. 115(4): p. 401–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu L and Hickson ID, The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature, 2003. 426(6968): p. 870–4. [DOI] [PubMed] [Google Scholar]
- 31.Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA, and Noble WS, A three-dimensional model of the yeast genome. Nature, 2010. 465(7296): p. 363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agmon N, Liefshitz B, Zimmer C, Fabre E, and Kupiec M, Effect of nuclear architecture on the efficiency of double-strand break repair. Nat Cell Biol, 2013. 15(6): p. 694–9. [DOI] [PubMed] [Google Scholar]
- 33.Lee CS, Wang RW, Chang HH, Capurso D, Segal MR, and Haber JE, Chromosome position determines the success of double-strand break repair. Proc Natl Acad Sci U S A, 2016. 113(2): p. E146–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coic E, Martin J, Ryu T, Tay SY, Kondev J, and Haber JE, Dynamics of homology searching during gene conversion in Saccharomyces cerevisiae revealed by donor competition. Genetics, 2011. 189(4): p. 1225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chambers AL, Brownlee PM, Durley SC, Beacham T, Kent NA, and Downs JA, The two different isoforms of the RSC chromatin remodeling complex play distinct roles in DNA damage responses. PLoS One, 2012. 7(2): p. e32016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kent NA, Chambers AL, and Downs JA, Dual chromatin remodeling roles for RSC during DNA double strand break induction and repair at the yeast MAT locus. J Biol Chem, 2007. 282(38): p. 27693–701. [DOI] [PubMed] [Google Scholar]
- 37.Oum JH, Seong C, Kwon Y, Ji JH, Sid A, Ramakrishnan S, Ira G, Malkova A, Sung P, Lee SE, and Shim EY, RSC facilitates Rad59-dependent homologous recombination between sister chromatids by promoting cohesin loading at DNA double-strand breaks. Molecular and cellular biology, 2011. 31(19): p. 3924–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shim EY, Hong SJ, Oum JH, Yanez Y, Zhang Y, and Lee SE, RSC mobilizes nucleosomes to improve accessibility of repair machinery to the damaged chromatin. Mol Cell Biol, 2007. 27(5): p. 1602–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiest NE, Houghtaling S, Sanchez JC, Tomkinson AE, and Osley MA, The SWI/SNF ATP-dependent nucleosome remodeler promotes resection initiation at a DNA double-strand break in yeast. Nucleic Acids Res, 2017. 45(10): p. 5887–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seeber A, Dion V, and Gasser SM, Checkpoint kinases and the INO80 nucleosome remodeling complex enhance global chromatin mobility in response to DNA damage. Genes Dev, 2013. 27(18): p. 1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsukuda T, Lo YC, Krishna S, Sterk R, Osley MA, and Nickoloff JA, INO80-dependent chromatin remodeling regulates early and late stages of mitotic homologous recombination. DNA repair, 2009. 8(3): p. 360–9. [DOI] [PubMed] [Google Scholar]
- 42.van Attikum H, Fritsch O, and Gasser SM, Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. The EMBO journal, 2007. 26(18): p. 4113–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X, Cui D, Papusha A, Zhang X, Chu CD, Tang J, Chen K, Pan X, and Ira G, The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature, 2012. 489(7417): p. 576–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen X, Niu H, Yu Y, Wang J, Zhu S, Zhou J, Papusha A, Cui D, Pan X, Kwon Y, Sung P, and Ira G, Enrichment of Cdk1-cyclins at DNA double-strand breaks stimulates Fun30 phosphorylation and DNA end resection. Nucleic Acids Res, 2016. 44(6): p. 2742–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, Dubois K, Wiegant WW, Thierry A, Burma S, van Attikum H, and Llorente B, The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature, 2012. 489(7417): p. 581–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eapen VV, Sugawara N, Tsabar M, Wu WH, and Haber JE, The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol Cell Biol, 2012. 32(22): p. 4727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hicks WM, Yamaguchi M, and Haber JE, Real-time analysis of double-strand DNA break repair by homologous recombination. Proceedings of the National Academy of Sciences of the United States of America, 2011. 108(8): p. 3108–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang RW, Lee CS, and Haber JE, Position effects influencing intrachromosomal repair of a double-strand break in budding yeast. PLoS One, 2017. 12(7): p. e0180994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lichten M and Haber JE, Position effects in ectopic and allelic mitotic recombination in Saccharomyces cerevisiae. Genetics, 1989. 123(2): p. 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee CS and Haber JE, Mating-type Gene Switching in Saccharomyces cerevisiae. Microbiol Spectr, 2015. 3(2): p. MDNA3–0013-2014. [DOI] [PubMed] [Google Scholar]
- 51.Forget AL and Kowalczykowski SC, Single-molecule imaging brings Rad51 nucleoprotein filaments into focus. Trends in cell biology, 2010. 20(5): p. 269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piazza A, Wright WD, and Heyer WD, Multi-invasions Are Recombination Byproducts that Induce Chromosomal Rearrangements. Cell, 2017. 170(4): p. 760–773 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Caron P, Aymard F, Iacovoni JS, Briois S, Canitrot Y, Bugler B, Massip L, Losada A, and Legube G, Cohesin protects genes against gammaH2AX Induced by DNA double-strand breaks. PLoS Genet, 2012. 8(1): p. e1002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sjogren C and Nasmyth K, Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr Biol, 2001. 11(12): p. 991–5. [DOI] [PubMed] [Google Scholar]
- 55.Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, and Koshland D, DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell, 2004. 16(6): p. 991–1002. [DOI] [PubMed] [Google Scholar]
- 56.Dong Z and Fasullo M, Multiple recombination pathways for sister chromatid exchange in Saccharomyces cerevisiae: role of RAD1 and the RAD52 epistasis group genes. Nucleic Acids Res, 2003. 31(10): p. 2576–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ragunathan K, Liu C, and Ha T, RecA filament sliding on DNA facilitates homology search. Elife, 2012. 1: p. e00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Renkawitz J, Lademann CA, Kalocsay M, and Jentsch S, Monitoring homology search during DNA double-strand break repair in vivo. Mol Cell, 2013. 50(2): p. 261–72. [DOI] [PubMed] [Google Scholar]
- 59.Lee CS, Lee K, Legube G, and Haber JE, Dynamics of yeast histone H2A and H2B phosphorylation in response to a double-strand break. Nat Struct Mol Biol, 2014. 21(1): p. 103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barzel A and Kupiec M, Finding a match: how do homologous sequences get together for recombination? Nature reviews. Genetics, 2008. 9(1): p. 27–37. [DOI] [PubMed] [Google Scholar]
- 61.Elf J, Hypothesis: Homologous Recombination Depends on Parallel Search. Cell Syst, 2016. 3(4): p. 325–327. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y and Dudko OK, First-Passage Processes in the Genome. Annu Rev Biophys, 2016. 45: p. 117–34. [DOI] [PubMed] [Google Scholar]
- 63.Herbert S, Brion A, Arbona JM, Lelek M, Veillet A, Lelandais B, Parmar J, Fernandez FG, Almayrac E, Khalil Y, Birgy E, Fabre E, and Zimmer C, Chromatin stiffening underlies enhanced locus mobility after DNA damage in budding yeast. EMBO J, 2017. 36(17): p. 2595–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weber SC, Spakowitz AJ, and Theriot JA, Nonthermal ATP-dependent fluctuations contribute to the in vivo motion of chromosomal loci. Proc Natl Acad Sci U S A, 2012. 109(19): p. 7338–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bystricky K, Laroche T, van Houwe G, Blaszczyk M, and Gasser SM, Chromosome looping in yeast: telomere pairing and coordinated movement reflect anchoring efficiency and territorial organization. J Cell Biol, 2005. 168(3): p. 375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hajjoul H, Mathon J, Ranchon H, Goiffon I, Mozziconacci J, Albert B, Carrivain P, Victor JM, Gadal O, Bystricky K, and Bancaud A, High-throughput chromatin motion tracking in living yeast reveals the flexibility of the fiber throughout the genome. Genome Res, 2013. 23(11): p. 1829–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lucas JS, Zhang Y, Dudko OK, and Murre C, 3D trajectories adopted by coding and regulatory DNA elements: first-passage times for genomic interactions. Cell, 2014. 158(2): p. 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dion V, Kalck V, Horigome C, T. B.D., and S.M. Gasser, Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat Cell Biol, 2012. 14: p. 502–509. [DOI] [PubMed] [Google Scholar]
- 69.Mine-Hattab J and Rothstein R, Increased chromosome mobility facilitates homology search during recombination. Nat Cell Biol, 2012. 14(5): p. 510–7. [DOI] [PubMed] [Google Scholar]
- 70.Dion V, Kalck V, Seeber A, Schleker T, and Gasser SM, Cohesin and the nucleolus constrain the mobility of spontaneous repair foci. EMBO Rep, 2013. 14(11): p. 984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hauer MH, Seeber A, Singh V, Thierry R, Sack R, Amitai A, Kryzhanovska M, Eglinger J, Holcman D, Owen-Hughes T, and Gasser SM, Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat Struct Mol Biol, 2017. 24(2): p. 99–107. [DOI] [PubMed] [Google Scholar]
- 72.Mine-Hattab J, Recamier V, Izeddin I, Rothstein R, and Darzacq X, Multi-scale tracking reveals scale-dependent chromatin dynamics after DNA damage. Mol Biol Cell, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harrison JC and Haber JE, Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet, 2006. 40: p. 209–35. [DOI] [PubMed] [Google Scholar]
- 74.Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, and Lichten M, Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol, 2004. 14(19): p. 1703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martin S, Laroche T, Suka N, Grunstein M, and Gasser SM, Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell, 1999. 97: p. 621–633. [DOI] [PubMed] [Google Scholar]
- 76.Mills K, Sinclair D, and Guarente L, MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell, 1999. 97: p. 609–620. [DOI] [PubMed] [Google Scholar]
- 77.Spichal M, Brion A, Herbert S, Cournac A, Marbouty M, Zimmer C, Koszul R, and Fabre E, Evidence for a dual role of actin in regulating chromosome organization and dynamics in yeast. J Cell Sci, 2016. 129(4): p. 681–92. [DOI] [PubMed] [Google Scholar]
- 78.Strecker J, Gupta GD, Zhang W, Bashkurov M, Landry MC, Pelletier L, and Durocher D, DNA damage signalling targets the kinetochore to promote chromatin mobility. Nat Cell Biol, 2016. 18(3): p. 281–90. [DOI] [PubMed] [Google Scholar]
- 79.Verdaasdonk JS, Vasquez PA, Barry RM, Barry T, Goodwin S, Forest MG, and Bloom K, Centromere tethering confines chromosome domains. Mol Cell, 2013. 52(6): p. 819–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lawrimore J, Barry TM, Barry RM, York AC, Friedman B, Cook DM, Akialis K, Tyler J, Vasquez P, Yeh E, and Bloom K, Microtubule dynamics drive enhanced chromatin motion and mobilize telomeres in response to DNA damage. Mol Biol Cell, 2017. 28(12): p. 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Waterman DP, Lee C-S, Tsabar M, Zhou F, Eapen VV, and Massella AJ, Live cell monitoring of double strand breaks in S. cerevisiae. bioRxiv, 2018: p. BIORXIV/2018/265611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Scherthan H, Wang H, Adelfalk C, White EJ, Cowan C, Cande WZ, and Kaback DB, Chromosome mobility during meiotic prophase in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A, 2007. 104(43): p. 16934–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Koszul R, Kim KP, Prentiss M, Kleckner N, and Kameoka S, Meiotic chromosomes move by linkage to dynamic actin cables with transduction of force through the nuclear envelope. Cell, 2008. 133(7): p. 1188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Amaral N, Ryu T, Li X, and Chiolo I, Nuclear Dynamics of Heterochromatin Repair. Trends Genet, 2017. 33(2): p. 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aymard F, Aguirrebengoa M, Guillou E, Javierre BM, Bugler B, Arnould C, Rocher V, Iacovoni JS, Biernacka A, Skrzypczak M, Ginalski K, Rowicka M, Fraser P, and Legube G, Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nat Struct Mol Biol, 2017. 24(4): p. 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Arbona JM, Herbert S, Fabre E, and Zimmer C, Inferring the physical properties of yeast chromatin through Bayesian analysis of whole nucleus simulations. Genome Biol, 2017. 18(1): p. 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hsieh TH, Weiner A, Lajoie B, Dekker J, Friedman N, and Rando OJ, Mapping Nucleosome Resolution Chromosome Folding in Yeast by Micro-C. Cell, 2015. 162(1): p. 108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Horigome C, Bustard DE, Marcomini I, Delgoshaie N, Tsai-Pflugfelder M, Cobb JA, and Gasser SM, PolySUMOylation by Siz2 and Mms21 triggers relocation of DNA breaks to nuclear pores through the Slx5/Slx8 STUbL. Genes Dev, 2016. 30(8): p. 931–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Horigome C, Oma Y, Konishi T, Schmid R, Marcomini I, Hauer MH, Dion V, Harata M, and Gasser SM, SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Mol Cell, 2014. 55(4): p. 626–39. [DOI] [PubMed] [Google Scholar]
- 90.Kaye JA, Melo JA, Cheung SK, Vaze MB, Haber JE, and Toczyski DP, DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr Biol, 2004. 14(23): p. 2096–106. [DOI] [PubMed] [Google Scholar]
- 91.Lobachev K, Vitriol E, Stemple J, Resnick MA, and Bloom K, Chromosome fragmentation after induction of a double-strand break is an active process prevented by the RMX repair complex. Curr Biol, 2004. 14(23): p. 2107–12. [DOI] [PubMed] [Google Scholar]
- 92.Inbar O and Kupiec M, Homology search and choice of homologous partner during mitotic recombination. Mol Cell Biol, 1999. 19(6): p. 4134–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lisby M and Rothstein R, DNA damage checkpoint and repair centers. Curr Opin Cell Biol, 2004. 16(3): p. 328–34. [DOI] [PubMed] [Google Scholar]
- 94.Miyazaki T, Bressan DA, Shinohara M, Haber JE, and Shinohara A, In vivo assembly and disassembly of Rad51 and Rad52 complexes during double-strand break repair. Embo J, 2004. 23(4): p. 939–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sugawara N, Goldfarb T, Studamire B, Alani E, and Haber JE, Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc Natl Acad Sci U S A, 2004. 101(25): p. 9315–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chakraborty U, George CM, Lyndaker AM, and Alani E, A Delicate Balance Between Repair and Replication Factors Regulates Recombination Between Divergent DNA Sequences in Saccharomyces cerevisiae. Genetics, 2016. 202(2): p. 525–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Anand R, Beach A, Li K, and Haber J, Rad51-mediated double-strand break repair and mismatch correction of divergent substrates. Nature, 2017. 544(7650): p. 377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fishman-Lobell J and Haber JE, Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science, 1992. 258(5081): p. 480–484. [DOI] [PubMed] [Google Scholar]
- 99.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, Futreal PA, and Campbell PJ, Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell, 2011. 144(1): p. 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Holland AJ and Cleveland DW, Chromoanagenesis and cancer: mechanisms and consequences of localized, complex chromosomal rearrangements. Nat Med, 2012. 18(11): p. 1630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hastings PJ, Ira G, and Lupski JR, A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet, 2009. 5(1): p. e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tsaponina O and Haber JE, Frequent Interchromosomal Template Switches during Gene Conversion in S. cerevisiae. Mol Cell, 2014. 55(4): p. 615–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stith CM, Sterling J, Resnick MA, Gordenin DA, and Burgers PM, Flexibility of eukaryotic Okazaki fragment maturation through regulated strand displacement synthesis. J Biol Chem, 2008. 283(49): p. 34129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zimmer C and Fabre E, Principles of chromosomal organization: lessons from yeast. J Cell Biol, 2011. 192(5): p. 723–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hicks WM, Kim M, and Haber JE, Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science, 2010. 329(5987): p. 82–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Danilowicz C, Yang D, Kelley C, Prevost C, and Prentiss M, The poor homology stringency in the heteroduplex allows strand exchange to incorporate desirable mismatches without sacrificing recognition in vivo. Nucleic Acids Res, 2015. 43(13): p. 6473–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Qi Z, Redding S, Lee JY, Gibb B, Kwon Y, Niu H, Gaines WA, Sung P, and Greene EC, DNA sequence alignment by microhomology sampling during homologous recombination. Cell, 2015. 160(5): p. 856–69. [DOI] [PMC free article] [PubMed] [Google Scholar]