

Abstract
Protein-based therapeutics have been revolutionizing the oncology space since they first appeared in the clinic two decades ago. Unlike traditional small-molecule chemotherapeutics, protein biologics promote active targeting of cancer cells by binding to cell surface receptors and other markers specifically associated with or overexpressed on tumors versus healthy tissue. While the first approved cancer biologics were monoclonal antibodies, the burgeoning field of protein engineering is spawning research on an expanded range of protein formats and modifications that allow tuning of properties such as target binding affinity, serum half-life, stability, and immunogenicity. In this review, we highlight some of these strategies and provide examples of modified and engineered proteins under development as preclinical and clinicalstage drug candidates for treating cancer.
Keywords: Protein engineering, biologics, biotherapeutics, targeted therapies, oncology drugs
Recombinant Protein Therapeutics
Starting with the FDA approval of Humulin (recombinant human insulin; Eli Lilly, Indianapolis) in 1982, protein therapeutics have dominated the worldwide pharmaceutical market [1]. Since then, the ability to customize the biochemical and biophysical properties of proteins has offered a powerful and fundamentally new direction for the pharmaceutical industry to augment the clinical potential of proteins. As the enabling field of protein engineering evolved throughout the 1980s, modified proteins soon joined recombinant versions of natural proteins as a major class of new therapeutics. The steadily rising market value of biopharmaceuticals is now over $140 billion, and for scale, thus exceeds the reported gross domestic product (GDP) of three-quarters of the economies listed in the World Bank GDP rankings [1]. The enormous size of this market underscores both the clinical and economic importance of protein engineering in the translational medicine space.
Protein therapeutics have maintained their research and development spotlight over the past several decades (Box 1), and can mainly be grouped into four categories: (1) proteins with enzymatic or regulatory activity, (2) proteins that block, stimulate, or tag their molecular targets, (3) vaccines, and (4) diagnostics [2]. Proteins from each of these categories have had major clinical success, particularly in oncology. However, despite the success of early protein drugs, numerous challenges have reduced their efficacy in the clinical setting, such as limited therapeutic index, acquired resistance, inefficient delivery, and individual patient variation [3]. In parallel, the persistent need to develop cancer therapeutics with improved safety and efficacy provides constant fuel to drive the optimization of protein biologics [4,5].
In this review, we provide a survey of principles underlying the development of protein-based cancer therapeutics, focusing on research efforts underway to generate next-generation drugs. We first briefly describe antibodies, antibody fragments, and alternative proteins under development as preclinical and clinical drug candidates. Next, to provide context for these efforts, we discuss common mechanisms of action of cancer therapeutics, which rely on tumor targeting or modulation of tumor-specific ligand/receptor interactions. We then define passive and active tumor targeting, describing principles of affinity and selectivity that underlie efficient targeting, and detail methods to achieve these properties using multivalent and multispecific ligands. Finally, we describe protein modifications and strategies that are being used to improve pharmacological properties including serum half-life extension, protein stability, and immunogenicity.
Box 1. Advantages of Protein Therapeutics
Benefits of proteins over small molecule drugs have helped facilitate their widespread traction in the pharmaceutical market. First, protein therapeutics have significantly more surface area for binding compared to small molecule drugs, allowing access to a much wider range of protein targets. The increased contact surface also helps to create a more specific binding interaction, decreasing the potential for off-target effects [2,6]. Conversely, small molecule drugs are typically nearly completely buried within a hydrophobic pocket of their protein binding partner to maximize hydrophobic contact and create a more stable complex [7]. This effectively restricts targetable proteins to those with solvent-accessible pockets. Second, proteins can often be more readily adapted for therapeutic purposes by augmenting their existing properties or installing new activities [8]. The unique form and function of proteins can provide an advantage over small molecules in allowing researchers to obtain stronger patent protection, which incentivizes development [9]. Additionally, although proteins therapeutics are not as commonly administered orally as compared to small molecule drugs, their increased blood circulation time can allow for far less frequent dosing [3].
Monoclonal antibodies historically have been shown to have significantly faster USA FDA approval success times than small molecules [10] and nearly double the overall approval rates [11,12]. In 2015, small molecule drugs had a 20% success rate in Phase II trials versus 40% for large molecules. Further in Phase III, small molecule drugs had only 65% approval as compared to 79% for large molecules.Despite the clear advantages of protein therapeutics, small molecule drugs do have beneficial properties such as oral bioavailability, intracellular targeting, ease of manufacturing, and generally long shelf life. Nevertheless, the enormous potential of protein therapeutics in cancer treatment drives significant interest in further developing this class of drugs [13].
Cancer therapeutics based on antibodies and alternative protein scaffolds
Antibody Scaffolds
Monoclonal antibodies (mAbs) have had much success as targeted cancer therapeutics, beginning in November 1997 with the approval of Rituximab for cancer therapy [14]. In 2015, the top three grossing cancer treatments were all monoclonal antibodies: Rituximab (Rituxan®), bevacizumab (Avastin®), and trastuzumab (Herceptin®), all produced by Genentech / Roche, sold a combined revenue of over $20 billion [15]. Key advantages of antibodies include high antigen binding affinity and selectivity and long circulatory half-lives as a result of neonatal Fcreceptor (FcRn) recycling. Antibodies also benefit from established protein engineering and modification strategies and well-defined manufacturing and clinical development pathways. For these reasons, antibodies currently dominate the landscape of approved protein cancer therapies. Intact immunoglobulin G (IgG) molecules comprise 18 out of 21 approved therapeutic antibodies first indicated for oncology [16]. However, the large size of full-length antibodies can make extravasation, tissue penetration, and tumor access challenging [17,18]. Antibody fragments (e.g. scFvs, scFv-Fc fusions, minibodies, diabodies) are also being increasingly investigated in preclinical and clinical research (Figure 1). The development of these and other antibody-based cancer therapeutics has been extensively reviewed elsewhere [19].
Figure 1:
Summary of representative antibody-based and alternative scaffolds commonly used for engineering next-generation protein therapeutics. Molecules are drawn to scale, using anticalins as a reference point for two different size classifications.
Alternative Scaffolds
To address limitations of antibodies the field has turned to so-called ‘alternative protein scaffolds’, which are smaller and more modular than their antibody counterparts (Figure 1). These alternatives include affibodies and DARPins (which are primarily α-helical in structure), adnectins, avimers, fynomers, and affilins (which are primarily composed of β-sheets), and atrimers, knottins, Kunitz domains, and anticalins (which utilize both secondary structures) [20]. More than 20 non-IgG scaffolds along with their origin, structure, and biochemical properties have been recently reviewed [21]. Potential advantages of many of these scaffolds include protein folding under non-reducing conditions, production via bacterial expression or chemical synthesis, improved tissue penetrance due to smaller size, and the ability to access grooves or catalytic pockets that may be inaccessible to larger mAbs. Due to their small size and consequently rapid renal clearance, alternative scaffolds often require strategies to increase serum half-life; an array of modifications enables tunable control of this parameter (described in detail below). Ecallantide (Kalbitor®, Dyax), a rationally designed Kunitz domain, provided clinical proof-of-concept for non-IgG scaffold therapeutics with approval in 2009 for treatment of hereditary angioedema [22]. Twelve other alternative scaffold therapeutics (including 3 adnectins, 2 affibodies, and 2 DARPins) are currently in clinical trials, four of which target cancer [23]. As examples, MP0250, a bispecific DARPin blocking VEGF and HGF developed by Molecular Partners, is currently in Phase I clinical trials for treatment of solid tumors and hematological malignancies, with enrollment for a Phase II trial beginning in late 2016, and Angiocept® (Bristol-Myers Squibb), a VEGFR2-targeting adnectin, is currently in Phase II clinical trials for glioblastoma, colorectal cancer, and non-small cell lung cancer [24].
Native Ligand and Receptor Scaffolds
An elegant alternative to antibodies and alterative protein scaffolds involves using natural ligands and receptors as a starting point for drug discovery [25]. A critical advantage of this strategy is that the native ligand or receptor is known a priori to bind the target at a clinicallyrelevant epitope. Furthermore, engineered natural ligands and receptors are more likely to exhibit broad specificity profiles, a characteristic that may confer significant therapeutic benefit. As a prominent example, in 1998 etanercept (Enbrel®, Amgen/Pfizer) received FDA approval for rheumatoid arthritis. Enbrel, which comprises of two soluble human tumor necrosis factor (TNF) receptors fused to an antibody Fc domain [26], functions as a decoy receptor to bind to and neutralize the activity of TNFα. In 2011, Regeneron received FDA approval for the treatment of wet age-related macular degeneration using aflibercept (Eylea®), a VEGF ligand trap engineered by fusing fragments of the VEGFR1 and VEGFR2 receptors to an Fc domain. In 2012 the same molecule (Zaltrap®, Sanofi) was approved for the treatment of colorectal cancer in combination with chemotherapy [27]. A number of other engineered natural ligands and receptors are under development as potential cancer therapeutics including SIRPα [28], Axl [29], VEGF [30], and FGF (FP-1039, Five Prime Therapeutics) [31].
Targeting protein therapeutics to tumor cells
Cancer therapeutics based on antibodies and alternative scaffolds require some component of targeting a protein biologic to receptors present on the surface of a tumor cell, or otherwise interfering with a ligand/receptor interaction. Cancer therapeutics can be selectively localized to tumors via passive or active targeting mechanisms [32]. Passive targeting exploits the enhanced permeability and retention (EPR) effect, a unique phenomenon of solid tumors that arises from their physiological aberration [33]. The increased metabolic demand from solid tumors results in significantly increased vascular density and permeability, and more loose-cell junctions as compared to healthy tissue. This defective vascular architecture facilitates the selective extravasation and subsequent retention of macromolecules into tumor tissue [34]. Because the EPR effect does not occur in normal tissue, an improved therapeutic window can be achieved due to the increased effective drug concentration that accumulates within a tumor.
In contrast, active targeting utilizes proteins specific to the tumor cells to help guide localization of a therapeutic modality, with a goal of minimal collateral cell killing. A requirement of active targeting is the identification of molecular signatures that help distinguish diseased cells from healthy ones. Protein therapeutics, including monoclonal antibodies, antibody fragments, and alternative scaffolds (Figure 1), can be engineered to recognize these targets and induce tumor cell killing by one of the mechanisms described above. Active targeting results in reduced offtarget cell killing, potentially translating into a larger therapeutic window. In addition to the examples presented earlier, the FDA-approved monoclonal antibody rituximab (Rituxan®) targets CD20 on the surface of B cells for treatment of non-Hodgkin’s lymphoma or chronic lymphocyte leukemia [35], and cetuximab (Erbitux®, Bristol Myers Squibb, Merck KGaA) targets EGFR (epidermal growth factor receptor) for treatment of metastatic colorectal, nonsmall cell lung cancer and head and neck cancer [36]. Preclinical discovery and development of novel targeted protein therapeutics is an active area of research, and includes applications in immuno-oncology, blood-tumor-barrier transcytosis, and intracellular drug delivery.
Unlike cell-permeable small molecule drugs, protein biologics almost exclusively target extracellular proteins due to limitations in intracellular delivery of large biomacromolecules [6]. This leaves many promising untapped intracellular targets, catalyzing significant efforts to develop proteins capable of accessing and binding targets inside the cell. Major strategies for cellular internalization include peptide stapling [37], protein supercharging [38], toxin-based delivery [39,40], and fusion of proteins to cell-penetrating peptides [41,42]. Despite these efforts, the field of intracellular targeting remains inchoate, imposing restrictions on the targets currently tractable to protein therapeutics.
In the next section we describe common mechanisms that targeted cancer therapeutics employ to carry out their therapeutic effects.
Mechanisms of action of targeted cancer therapeutics
Most approaches for the development of protein-based cancer therapeutics exploit ubiquitous features of tumors and tumor-associated cells. Accordingly, cancer therapeutics generally utilize one or more of three common mechanisms for tumor cell killing [19]. These mechanisms are direct killing, vascular and stromal cell ablation, and immune-mediated killing. An understanding of these mechanisms is helpful in guiding the development of therapeutics with improved potency.
A wide variety of cell-surface receptors can deliver pro-apoptotic signals upon binding of their cognate ligands. Thus, protein therapeutics have been developed that are capable of directly inducing cell death by blocking cellular signals important for survival or by inducing apoptosis. Some of the most ubiquitous protein therapeutics in this class target cell surface receptors to induce apoptosis. A prominent example includes anti-TRAIL antibodies [43] from companies including Human Genome Sciences (HGSETR1, HGS-ETR2 and HGS-TR2J), Genentech (Apomab), Amgen (AMG 655), Novartis (LBY135), and Daiichi Sankyo (CS-1008). Other prominent therapeutics act by localizing cytotoxic agents directly to tumor cells. An important strategy leveraged in oncology has been the creation of drug conjugates, which use proteins to target cytotoxic payloads to tumor cells. In most cases, these compounds consist of antibodies attached to small molecule chemotherapeutics by a chemical linker. Two antibody-drug conjugates are currently approved for clinical use: Adcetris® (Seattle Genetics), a CD30targeting antibody, brentuximab, linked to the potent antimitotic monomethyl auristatin E; and Kadcyla® (Genentech/Roche), comprised of the HER2-targeting antibody trastuzumab, linked to the cytotoxic agent emtansine. The design, optimization, and application of highly potent drug conjugates with improved safety profiles is an active area of oncology research, and has been summarized elsewhere in the literature [44,45].
Protein therapeutics may also target tumor vasculature and stroma as a means of inducing tumor cell death. The stromal microenvironment is an intricate structure consisting of the extracellular matrix, fibroblasts, myofibrils, inflammatory cells, and blood and lymphatic vessels. Interactions between cancer cells and this microenvironment are critical for tumor growth, progression, and metastasis [46]. In some cases, tumors contain a surfeit of stromal cells with poor vascular perfusion [47], which hinders effective delivery of many conventionally-targeted therapeutics. Targeting tumor-associated stroma and vasculature provides a strategy to destroy the tumor microenvironment and prevent tumor survival and proliferation [48]. The most notable clinical example in this category is bevacizumab, a monoclonal antibody that inhibits angiogenesis through the binding of vascular endothelial growth factor (VEGF), blocking its ability to activate VEGF receptor [49,50]. Alternatively, more recent approaches aim to promote angiogenesis or vasculature normalization to enhance cancer drug delivery, and hence therapeutic effectiveness [51].
Other protein therapeutics opsonize cells and elicit immune-mediated killing via complement activation [52], induction of phagocytosis [53], antibody-dependent cellular cytotoxicity [54], or modulation of T cell function [55]. Immune checkpoint inhibitors, in particular monoclonal antibodies against PD-1, PDL-1, and CTLA-4, block signaling axes between cancer cells, Tcells, and antigen presenting cells, to unleash a T-cell response against the tumor. By stimulating the immune system, these therapies have shown significant clinical impact by conferring longterm survival in a subset of patients [56]. To date, four checkpoint blockade inhibitors have received FDA approval: Yervoy® (ipilimumab, targets CTLA-4, Bristol-Myers Squibb), Opdivo® (nivolumab, a PD-1 inhibitor, Bristol-Myers Squibb), Keytruda® (pembrolizumab, a PD-1 inhibitor, Merck), and Tecentriq® (atezolizumab, a PD-L1 inhibitor, Genentech/Roche). Based on these successes, immuno-oncology is one of the fastest growing fields in the pharmaceutical industry. These transformative cancer treatments, and others under development, have been aptly described in recent reviews and thus will not be discussed here [57].
In the next two sections we describe the principles of target affinity and specificity, followed by approaches to achieve them using multivalent and multispecific protein therapeutics.
Target affinity and selectivity
The therapeutic efficacy of protein-based therapeutics is mediated by properties such as binding affinity, tumor penetration, and tissue retention [58]. A protein’s affinity for its molecular target is most commonly described by its dissociation constant, Kd. Accordingly, one primary goal in optimizing a protein therapeutic is to increase target binding affinity using directed evolution. With this technology, diverse libraries of millions of protein variants are generated and screened to identify proteins with the highest binding affinity. Directed evolution has been utilized in protein engineering for decades, and as a result these methods now enable the production of novel proteins with improved or previously inaccessible properties. The scope of this review does not allow for a recapitulation of these technological developments, but advances have been recently reviewed elsewhere in the literature [59,60], with highlights including new screening strategies, the inclusion of unnatural amino acids, and workflows that combine rational design with traditional random mutagenesis. While increased target binding affinity often correlates with increased potency and therapeutic efficacy, there are notable exceptions, especially when targeting solid tumors [61]. For example, ultra-high affinity therapeutics can suffer from limited tumor penetration because they are unable to diffuse into the tumor core without saturating all antigen presented on the periphery [62]. Moreover, for large proteins that have long serum halflives, such as antibodies, affinity is less critical than for smaller proteins, which have only minutes to localize to their targets before they are cleared from the body [63,64].
Selectivity is the ability of a protein to distinguish between its target and all other biomolecules, and can be expressed as the ratio of ligand bound to target versus ligand bound to non-targets. Improved target selectivity often directly correlates to the therapeutic window, with some exceptions in cases of therapies requiring high systemic exposure [65]. Towards this end, an ideally selective therapeutic would have an orthogonal target and an inability to bind healthy tissue, such as a mutated receptor that appears only on tumor-associated cells. In practice, such scenarios are rare; instead the same target is typically present at much higher levels on tumor cells compared to healthy cells. Nevertheless, protein therapeutics only need to be functionally orthogonal; that is, the target expression on healthy tissue is well below the Kd of the protein biologic or too low to cause significant accumulation. In cases where the therapeutic target is highly expressed by non-cancerous cells, masking strategies can be used to occlude binding domains until the protein therapeutic reaches the tumor microenvironment. As an example, the potential to create tumor-activated antibodies was demonstrated using single-chain Fv (scFv) fragments fused to a masking domain that blocks antibody-antigen binding. The masking domain is tethered to the antibody using a protease-cleavable linker that can be degraded in the tumor microenvironment [66]. Cytomx Therapeutics has recently developed this approach as a technology called Probodies, with demonstrated efficacy in animal models [67].
A complementary approach for increasing the affinity or selectivity of a binding interaction is through avidity effects promoted by multivalent and multispecific protein-protein interactions, discussed below.
Multivalent and multispecific targeting
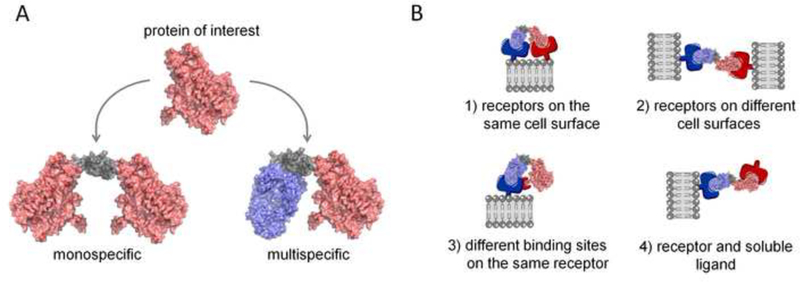
Multivalent and multispecific proteins provide a way to substantially augment the affinity and selectivity of a therapeutic. Multivalent proteins have more than one binding site that can either have affinity for the same target (monospecific) or multiple targets (multispecific) (Figure 2). As an example, natural antibodies (IgGs) are bivalent, monospecific proteins that require engineering to achieve multispecificity. In contrast, alternative scaffolds commonly require engineering to achieve both multivalency and multispecificity. Multivalent proteins benefit from avidity effects, where, after the first binding interaction, the effective local concentration of the next binding site is significantly increased, facilitating further ligand-receptor association. Avidity effects are only realized where there is a sufficiently high density of receptors on the cell surface [68]. Thus, because target protein expression is typically much higher in tumor versus normal cells, the apparent affinity of the therapeutic is augmented for tumor cells. In the case of multispecific proteins, selectivity is greatly enhanced on cells that express all of the targets of interest. For example, an engineered tandem bs-scFv antibody against ErbB3 and HER2 was shown to bind exclusively to ErbBr+HER2+ cells and did not have significant binding to cells that expressed a single target [69].
Figure 2:

A) Avidity effects and enhanced selectivity to diseased versus healthy tissue can be achieved by multivalent therapeutic proteins that to bind the same target (monospecific) or different targets (multispecific). A flexible linker (grey) connects two protein subunits in this example, however, antibody domains, which are naturally bivalent, are often used to create multivalent and multispecific proteins. B) Multispecific proteins are particularly useful for: (1) binding multiple receptors on the same cell surface, (2) binding receptors on separate cell surfaces, (3) binding multiple epitopes on the same receptor, or (4) binding a cell surface receptor and one or more soluble ligands.
Researchers have developed a number of elegant strategies for creating multispecific proteins. Many of these constructs are created by fusing protein domains to the N- or C-terminus of the heavy or light chain of a full-length antibody. Others are created by simple fusion of protein domains through a flexible linker (Figure 2). To date, more than 30 bispecific and multispecific molecules are actively under clinical development [70]. Multispecific protein therapeutics have generated great interest as drugs as they offer opportunities for: 1) improved therapeutic efficacy, 2) lower/less frequent dosing, and 3) lower risk of systemic exposure and off-target effects. Additional benefits include development of one protein compared to co-administration of a drug combination, and as a result lower costs of goods [71]. However, there are also significant challenges with multispecific proteins, including difficulties with manufacturing (low yields and misfolded products), stability, and aggregation [72]. In addition, pre-clinical development is more complex in that each component of the multispecific protein needs to cross-react with the appropriate pre-clinical species (typically rodent and cynomologous monkey) for toxicology and pharmacokinetic studies. Furthermore, while doses can be tailored in a cocktail strategy, they are tied together in a multispecific protein, which has led to concerns about the unpredictable effects of modulating multiple drug targets with a single protein construct. While Phase III clinical trials with multispecifics have been reassuring [70], more studies need to be done to compare combination monotherapies with their multispecific counterparts.
Multispecific proteins enable a variety of targeting strategies (Figure 2): (1) binding multiple receptors on the same cell surface; (2) binding multiple epitopes on the same receptor; (3) binding receptors on separate cell surfaces; or (4) binding a cell surface receptor and a soluble ligand. These molecular mechanisms have enabled several new clinical applications otherwise inaccessible to monospecific proteins, including T-cell recruitment in immunotherapy [73,74], dual blockade of two disease mediators [75–77], bi- or triepitotic targeting of a single protein target [78,79], improving access to the brain [80,81] and delivery of cytokines and other payloads [82,83].
The advantage of binding to multiple receptors on a single cell is especially powerful in cancer, where redundant signaling pathways and receptor crosstalk have traditionally limited the efficacy of single-target monospecific antibodies [84]. For example, an anti-EGFR/HER2 bispecific antibody was shown to outperform anti-EGFR and anti-HER2 monospecific antibodies, as well as the two in combination, in both in vitro and in vivo preclinical cancer models, demonstrating the synergistic effect of down-regulating both receptor tyrosine kinases simultaneously [75]. More recently, the tetraspecific, tetravalent antibody CRTB6 (which targets EGFR, HER2, HER3, and VEGF simultaneously) was shown to disrupt HER/MET crosstalk and more effectively inhibit the growth of anti-HER-resistant cancer cells when compared to the EGFR/HER2 bispecific, further validating the principle that multispecifics may be promising agents for avoiding or delaying acquired resistance as well as boosting efficacy [85]. Biparatopic receptor targeting agents have also been shown to be more effective than monospecific agents, for example, in the case of HER2 inhibition via a biparatopic DARPin, which traps HER2 in a dimerization-independent state [86], and is also effective as an antibody drug conjugate [87]. In another example, COVA420 (FynomAb), a bispecific antibody created through genetic fusion of fynomer domains that target CD3 and HER2, provides an improved therapeutic window by selectively inducing T-cell-mediated cytotoxicity in tumor cells, but not in cells with lower HER2 expression [88].
Multispecific proteins have also been successfully leveraged to target the immune system against cancer. In this case, the simultaneous engagement of two receptors allows bispecific proteins to bind both tumor cells (at tumor-associated receptor targets) and T cells (at the CD3 receptor), resulting in T-cell activation and secretion of cytokines. The first multispecific to receive approval, catumaxomab (Removab®, Trion Pharmaceuticals), is a trifunctional antiEpCAM/anti-CD3 bsAb that targets the tumor, uses binding to the CD3 domain to recruit T effector cells, and uses Fcγ-receptor binding through the Fc domain to activate monocytes, macrophages, dendritic cells, and NK cells [89]. Alternatively, Amgen (formerly Micromet) developed a bi-specific T-cell engager (BiTE) format consisting of two scFvs fused via a flexible linker, with constructs engineered to bind to a plethora of target receptors, including CD19, EpCAM, HER2, CEA, and PSMA [90]. Blinatumomab (Amgen), an FDA-approved BiTE [91], utilizes precisely this mechanism to elicit T-cell effector functions against malignant B cells in acute lymphoblastic leukemia via an immunologic synapse. While immune system modulators represent a significant portion of the multispecific protein pipeline, emerging research trends for multispecific proteins include engineered antibodies and alternative scaffolds that coordinate inhibition of multiple oncology targets or facilitate transcytosis for improved or novel drug delivery applications [70,90].
Protein Modifications and Strategies for Enhancing Pharmacological Properties
Advances in protein engineering over the past decade have enabled unprecedented control of pharmacological attributes of would-be therapeutics. Specifically, these strategies have been utilized to tune the serum half-life, stability, and immunogenicity of cancer-targeting proteins, and have been leveraged to develop therapeutic candidates with enhanced efficacy and reduced off-target effects.
Extending Serum Half-Life
The pharmacokinetic profile of a protein molecule in the body is determined in part by size, shape, hydrodynamic radius, and charge. In particular, proteins and peptides below the size cutoff for glomerular filtration (generally reported in the literature to be ~70 kDa) are more likely to be eliminated by kidney filtration than larger proteins [92], and negatively charged proteins may be eliminated more slowly due to repulsion by the charged basement membrane of the kidney [93]. The growing number of protein therapeutics being pursued, as well as the development of antibody fragments and alternative scaffolds beyond native IgG molecules, has necessitated innovative methods of tuning protein residence time in the blood. A favorable pharmacokinetic profile has the potential to increase efficacy of a molecule by extending the window of exposure to the target and in many cases decreasing the number or frequency of doses, thus offering economic as well as therapeutic benefits.
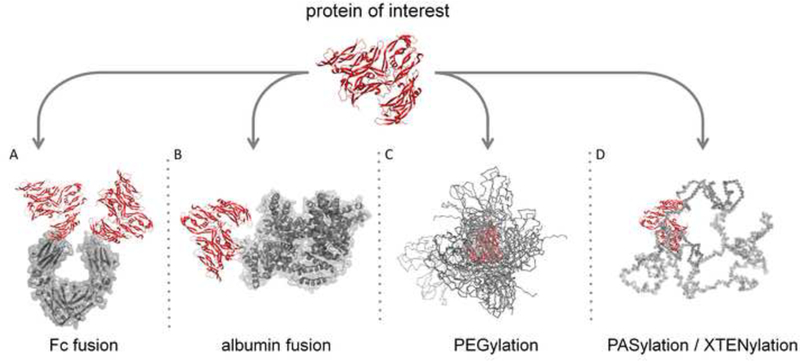
As an example of the importance of half-life extension, 20 of the 43 fusion proteins recently entering Phase II or Phase III clinical trials were constructed strictly to extend half-life [94]. Alternatively, proteins that have not been modified to extend half-life require frequent or continuous administration, such as BiTEs, which are administered over 28 days of continuous infusion [95]. As shown in Figure 3, researchers have devised a number of strategies for half-life extension [96]. These approaches and their respective advantages and disadvantages have been recently reviewed [42,94,97,98]. The range of options available for half-life extension allows protein engineers to increasingly use rational design to select the approach most appropriate for their desired molecule and clinical application.
Figure 3:
Major protein modification strategies for serum half-life extension. (A, B) Fc and albumin recombinant protein fusions that leverage FcRn-mediated recycling. (C) PEG, a nonbiologic (synthetic) polymer. (D) PAS and XTEN, recombinant polypeptide based polymers.
The most commonly utilized strategy to extend the serum half-life of cancer therapeutics is to engineer the protein to increase receptor recycling [99]. This method prevents degradation of the molecule by altering cellular trafficking via fusion to a natural protein or protein domain known to be recycled such as albumin or an antibody Fc domain. Within the endocytic pathway, vesicle acidification enables sorting of proteins bound to certain receptors. Once internalized, proteins not bound to recycled receptors are trafficked for lysosomal degradation, while bound proteins are transported back to the cell surface where they may be subsequently released back into circulation [100]. The neonatal Fc receptor (FcRn) is among the transmembrane proteins that shuttles internalized protein ligands back to the cell surface and is known to contribute to the long half-lives of antibodies and albumin [101].
Fusing a protein therapeutic to the Fc domain of an antibody has been one of the most commonly used strategies to boost half-life by taking advantage of FcRn-mediated recycling, as human Fc IgG isotypes have been shown to have a 2- to 3-week half-life in human serum [102]. A proteinFc fusion also results in a bivalent construct, which can confer beneficial avidity effects as described above. An early example comprised the fusion of an IgG Fc fragment to the ligandbinding domains of CD4 [103]; since then, 11 Fc fusion proteins have been approved by the FDA [82], including Enbrel® and Eylea®/Zaltrap®, which are described earlier in this review. Moreover, engineering an Fc domain to possess increased binding to FcRn can also extend halflife by several fold, a property that has been exploited to further enhance receptor recycling [104,105].
FcRn binding may also facilitate epithelial transcytosis to increase access to more protected targets such as intracranial tumors [106]. Another application of FcRn recycling has been the development of “sweeping” antibodies. Sweeping antibodies bind antigen at extracellular pH but unlike conventional antibodies that form a stable antigen-antibody complex, they dissociate upon internalization. This allows for degradation of the antigen and recycling of the antibody, which can continue to bind antigen. Sweeping antibodies have been shown to reduce antigen concentration 50–1000 fold compared to conventional antibodies [107]. A similar format of antigen-clearing antibodies has been developed that uses IGF-IR as a membrane anchor instead of FcRn [108].
Fusion or conjugation of a protein therapeutic to albumin, the most abundant serum protein, can also extend half-life. Albumin is highly soluble, very stable, and has an extremely long circulatory half-life as a result of its size and interaction with the FcRn recycling pathway, where albumin binds to FcRn in a pH-dependent manner at a site that does not overlap with the IgG binding site [109]. Recent efforts have focused on further extending the half-life of albumin fusions by modulating the affinity of albumin to FcRn [110]. Two albumin-fusion proteins have garnered FDA approval in recent years: albiglutide (Tanzeum®, Glaxo-Smith Kline) and Idelvion® (CSL Behring), although neither is indicated for cancer. Albuleukin (Human Genome Sciences, Inc.), a genetic fusion of human serum albumin to interleukin-2 (IL-2) which significantly increases the circulation time of IL-2 [111], is in Phase I clinical trials for patients with solid tumors. MMM-111 (Merrimack), a unique bispecific antibody fusion comprising two scFvs that bind HER2 and HER3 fused to human serum albumin, is currently in Phase II clinical development for treating solid tumors. MMM-111 increases the half-life of free scFvs from <1 h to a half-life of 86–90 hours [112]. To minimize the size of a protein therapeutic, fusions can also be made to peptides or alternative scaffolds engineered to bind to serum albumin [113,114]. In addition, modifying the protein of interest with fatty acids that bind to serum albumin has been shown to result in prolonged half-life [115].
Beyond increased recycling, methods have also been utilized to increase the size and negative charge of the protein as a way to increase serum half-life. PEGylation is one of the earliest and most widely used methods for extending serum half-life and involves conjugation of proteins to linear or branched-chain monomethoxy poly-ethylene glycol (PEG) to increase the mass and decrease the rate of glomerular filtration of the target molecule [116]. Several PEGylated compounds are now on the market for various indications, including Neulasta® (peg-granulocyte colony stimulating factor, Amgen), PegIntron® (peginterferon alfa-2b, Merck), and Pegasys® (peginterferon alfa-2a, Genentech/Roche), all of which received FDA approval in the early 2000s. For oncology applications, a clever modification of PEG was recently demonstrated by NKTR-214 (Nektar Therapeutics), a biologic prodrug consisting of six releasable PEG chains attached to IL-2. NKTR-214 had greatly improved tumor exposure and efficacy in a murine melanoma model compared to aldesleukin (recombinant IL-2) and was well tolerated in nonhuman primates [117]. PEG has also been used to reduce dosing frequency and improve safety of L-asparaginase, a chemotherapy agent approved for the treatment of acute lymphoblastic leukemia (ALL). Oncaspar® (PEG-L-asparaginase; Enzon Pharmaceuticals) is effective when administered every two weeks, as opposed to the non-PEGylated L-asparaginase, which must be administered two or three times per week to be effective [118].
Despite the clinical success of PEGylated molecules and the designation of PEG by the FDA as a “generally recognized as safe” molecule, this approach has been associated with vascularization of renal cortical tubular epithelium cells [119]. Furthermore, the cost of PEG and the necessity for additional manufacturing steps of chemical conjugation and purification have prompted research towards methods of serum half-life extension that could alleviate safety concerns while lowering production costs. To address these needs, protein-based polymers are being developed as homogeneous, biocompatible alternatives to PEG for serum half-life extension that do not require chemical synthesis for protein attachment. These include fusion or conjugation to inert genetically-encoded biomacromolecules such as a randomized polypeptide (XTEN) [120], a homo-amino acid polymer (HAP) [121], a proline-alanine-serine repeat polypeptide (PAS) [122], or an elastin-like peptide (ELP) biopolymer [123]. As an example, an XTEN fusion of exenatide (VRS-859; Amunix/Versartis) has successfully completed Phase I trials in type 2 diabetic patients [124] and a fusion of XTEN and human growth hormone (VRS-317; Versartis) has completed Phase IIa trials in growth hormone deficient patients [125], demonstrating the feasibility of this strategy in a clinical setting. Finally, attachment of mono- or poly-sialyl groups [126] or fusion to a highly sialyated, negatively charged peptide such as the human CG β-subunit [127] may also be a useful option to increase half-life, however, carbohydrate modification can result in protein products that are heterogeneous and difficult to characterize, so these approaches are less common.
Improving Protein Stability
Stability is a critically important feature of any protein therapeutic, as elegant protein engineering methodologies to improve targeting, efficacy, and pharmacokinetic parameters are irrelevant should the molecule physically or chemically degrade before carrying out the desired function. Moreover, stability is critical for cost effective large-scale production. An effective therapeutic must retain physical and chemical integrity during both production and storage both to retain potency and to ensure lack of degraded or aggregated product. Furthermore, an increasingly crowded landscape for protein therapies favors the design of more stable molecules that can be brought quickly to market and, once there, possess longer shelf lives.
Dominant processes leading to protein degradation during storage include formation of aggregates, oxidation, deamination, and isomerization [128–130]. Transient or partial unfolding of otherwise soluble proteins can provide a pathway for aggregate formation based on strong covalent or noncovalent (hydrophobic, hydrogen bonding, and van der Waals) protein interactions. These aggregates significantly elevate the risk of an adverse immune response in patients, even in small doses, and have thus become a particular focus both of pharmaceutical development companies and regulatory authorities.
In addition to changes and optimizations in formulation, which will not be discussed here, researchers can select from antibody formats and alternative scaffolds that have a range of properties including high thermal stability (for example, melting points of around 70–80° C for some alternative scaffolds and 70–90° C for some Fab fragments), chemical stability upon exposure to pH changes and solvents, and in some cases proteolytic stability. These properties have been recently reviewed [20].
Various engineering techniques have also been leveraged to stabilize protein therapies. One such method is identifying protein variants with higher cell surface expression under different temperature stresses, which has been shown to correlate with higher thermal stability and recombinant expression yield [131]. An alternative method involves determining antibody regions prone to aggregation using a spatial aggregation propensity (SAP) technology, then making targeted mutations in those regions to improve antibody stability [132]. Another more recent technology involves using affinity-capture self-interaction nanoparticle spectroscopy (ACSINS) to screen large panels of antibodies for their propensity to self-associate [133]. Other methods for engineering protein stability have recently been reviewed, including physical crosslinking, isotype switching, and screening for off-target binding which can lead to rapid clearance [134–137].
Immunogenicity
An inherent risk of introducing non-native proteins into the human body as therapeutics is the potential to provoke an unwanted immune response. Thus, managing the immunogenicity profile of a therapeutic candidate is a critically important facet of successful drug development. Factors influencing the immunogenicity of a protein therapeutic include both patient- and product-related factors, the latter has been extensively reviewed in recent literature [138,139].
The species origin of the protein therapeutic has been identified as a significant factor in determining immunogenic responses. Non-human proteins tend to elicit longer and more pronounced immune responses than therapies developed from human or humanized molecules due to differences in amino acid sequence and glycosylation influencing the categorization of the protein as self or non-self [140]. However, significant immunogenicity of some fully human proteins suggest that other factors such as formulation, impurities, and aggregation contribute to the formation of anti-drug antibodies [138]. Aggregation in particular has been implicated in triggering unfavorable immune responses [141–143], a challenging phenomenon to mitigate through rational engineering due to gaps in understanding of the molecular mechanisms that trigger aggregation and the absence of reliable aggregation prediction tools.
Pragmatic approaches to mitigating immunogenicity in protein drug development currently involve humanization and modification of the primary sequence to remove or mask potential T and B cell epitopes, and rigorous immunogenicity characterization. A panel of tools has been developed to predict CD4+ T-cell responses, including in silico (iTope™, TCED™, Epibase™, EpiMatrix™), and in vitro (EpiScreen™, Epibase™, REVEAL®) methods. In addition, conventional mouse models, immune tolerant transgenic mice, HLA-immune-tolerant transgenic mice, and non-human primates models have found use as in vivo tools [141].
Concluding Remarks
Protein-based therapeutics have enabled new, targeted approaches for treating cancer over the last two decades. The clinical and commercial impact of these strategies have been notable, and interest in developing protein-based cancer drugs continues to grow, fueled by both the particular advantages of proteins over small-molecule drugs (Box 1) and the ability to customize and optimize proteins with an ever expanding repertoire of protein engineering tools.
In this review, we have surveyed strategies applied to and under development for engineering tumor targeting proteins, highlighting some examples of biologics that have entered the clinic or gained FDA approval. In particular, the approaches discussed leverage the diversity of therapeutic candidates based on antibodies and alternative scaffolds, use multivalent and multispecific formats to increase target binding affinity and selectivity, and exploit various modification techniques to extend half-life, enhance stability, and reduce immunogenicity. These powerful advancements in our ability to optimize pharmacological properties are poised to improve the next generation of cancer therapeutics by increasing potency, reducing toxicity due to off-target effects, and decreasing the required frequency and amount of drug administered.
Several critical aspects of protein design and optimization for cancer treatment remain beyond the reach of current technologies (Figure 4, Key Figure)(see Outstanding Questions). The subject of ongoing and intensive research efforts, some immediate unmet needs include achieving effective therapeutic delivery to intracellular targets, developing methods to penetrate the bloodbrain-barrier to access intracranial tumors [131], and building better models to better predict immunogenicity in humans [129]. A less defined but important goal involves developing therapies that can escape or overcome cancer’s powerful resistance mechanisms, as the efficacy of many current therapeutics is limited by acquired resistance. A growing area of interest and promise also lies in our ability to better deliver therapeutic modalities to tumors, including chemotherapeutics and components of the immune system. The application of engineering and design strategies to these approaches has only just begun, but is sure to continue to yield exciting discoveries in the years to come.
Figure 4.
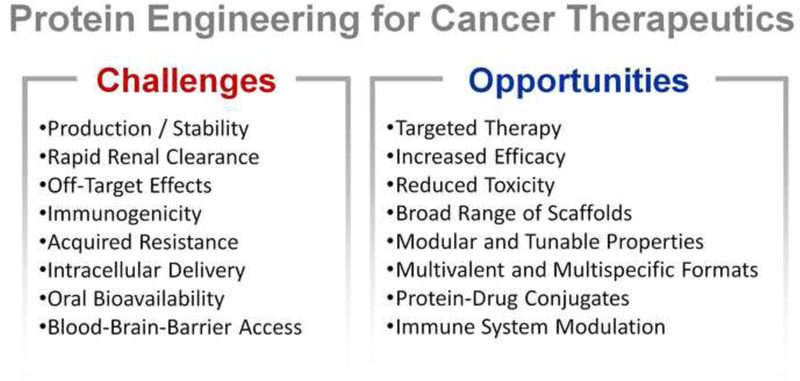
(Key Figure) The future of protein engineering for cancer therapeutics has both challenges and opportunities.
Outstanding Questions.
How can protein therapeutics be developed to work in concert with precision medicine, enabling specific and customized treatments for different patients and cancer types, while being mindful of cost and regulatory challenges?
What improvements can be made to protein production methods, regulatory oversight processes, and clinical trial design to reduce development time and costs?
How reliable are models for predicting therapeutic efficacy or immunogenicity in humans and what advancements can be made?
Does half-life extension increase the off-target effects of a protein therapeutics? If so, is there an ideal half-life that maximizes efficacy while mitigating side effects?
How can protein therapeutics be effectively delivered to intracellular targets?
How can protein therapeutics be engineered for increased delivery to intracranial tumors?
How can protein therapeutics be optimized to better escape or overcome cancer’s resistance methods?
How can proteins be exploited to better deliver therapeutic modalities to tumors, including chemotherapeutics and immune system modulators?
Trends.
Biotherapeutics have come to dominate the oncology space, with blockbuster monoclonal antibody drugs Rituxan®, Avastin®, and Herceptin® generating billions of dollars of revenue annually and comprising 3 of the top 5 highest-revenue cancer drugs.
There is a critical need to supersede moderately effective cancer treatments with next-generation protein therapeutics optimized for safety and efficacy.
Emerging protein therapeutics under development increasingly employ some form of combinatorial or rational engineering and commonly comprise novel protein architectures and tailored modifications.
Active areas of exploration in protein optimization and engineering include utilization of new protein formats/scaffolds, half-life extension, stability enhancement, and leveraging of multivalency and multispecificity to increase target affinity and selectivity.
Acknowledgements
JRK is supported by an NSF Graduate Research Fellowship. MVFI receive support from the Stanford University Medical Scientist Training Program (T32GM007365).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walsh G: Biopharmaceutical benchmarks 2014. Nat Biotech 2014, 32:992–1002. [DOI] [PubMed] [Google Scholar]
- 2.Leader B, Baca Q, Golan D: Protein therapeutics- summary and pharmacological classification. Nat Rev Drug Discov 2008, 7:21–39. [DOI] [PubMed] [Google Scholar]
- 3.Dimitrov D: Therapeutic proteins. In Therapeutic Proteins Edited by Voynov V, Carvella J: Humana Press; 2012:1–27. [Google Scholar]
- 4.Carter PJ: Introduction to current and future protein therapeutics: a protein engineering perspective. Exp Cell Res 2011, 317:1261–1269. [DOI] [PubMed] [Google Scholar]
- 5.Beck A, Wurch T, Bailly C, Corvaia N: Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 2010, 10:345–352. [DOI] [PubMed] [Google Scholar]
- 6.Verdine GL, Walensky LD: The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin Cancer Res 2007, 13:7264–7270. [DOI] [PubMed] [Google Scholar]
- 7.Hopkins A, Greoom C: The druggable genome. Nat Rev Drug Discov 2002, 1:727–730. [DOI] [PubMed] [Google Scholar]
- 8.Huang S, Armstron E, Benavente S, Chinnaiyan P, Harari P: Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res 2004, 64:5355–5363. [DOI] [PubMed] [Google Scholar]
- 9.Kresse GB: Biosimilars--science, status, and strategic perspective. Eur J Pharm Biopharm 2009, 72:479–486. [DOI] [PubMed] [Google Scholar]
- 10.Reichert JM: Trends in development and approval times for new therapeutics in the United States. Nat Rev Drug Discov 2003, 2:695–702. [DOI] [PubMed] [Google Scholar]
- 11.Hay M, Thomas D, Craighead J, Economides C, Rosenthal J: Clinical development success rates for investigational drugs Nat Biotech 2014, 32:40–51. [DOI] [PubMed] [Google Scholar]
- 12.DiMasi JA, Feldman L, Seckler A, Wilson A: Trends in risks associated with new drug development: success rates for investigational drugs. Clin Pharmacol Ther 2010, 87:272–277. [DOI] [PubMed] [Google Scholar]
- 13.Wen F, Rubin-Pitel S, Zhao H: Engineering of Therapeutic Proteins. In Protein Engineering and Design Edited by Park S, Cochran JR: CRC Press; 2009:153–177. [Google Scholar]
- 14.Dillman RO: Magic bullets at last! Finally--approval of a monoclonal antibody for the treatment of cancer!!! Cancer Biotherapy & Radiopharmaceuticals 1997, 12:223–225. [DOI] [PubMed] [Google Scholar]
- 15.pharmaceutical-technology.com: The world’s most sold cancer drugs in 2015 Edited by: Kable; 2016. [Google Scholar]
- 16.Therapeutic monoclonal antibodies approved or in review in the European Union or the United States. Edited by. Antibody Society; 2016:1–3. [Google Scholar]
- 17.Fujimori K, Covell D, Fletcher J, Weinstein J: Modeling analysis of the global and microscopic distribution of immunoglobulin G, F(ab’)2, and Fab in tumors. Cancer Res 1989, 49:5656–5663. [PubMed] [Google Scholar]
- 18.Thurber GM, Schmidt MM, Wittrup KD: Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Advanced drug delivery reviews 2008, 60:1421–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scott AM, Wolchok JD, Old LJ: Antibody therapy of cancer. Nat Rev Cancer 2012, 12:278–287. [DOI] [PubMed] [Google Scholar]
- 20.Weidle UH, Auer J, Brinkmann U, Georges G, Tiefenthaler G: The emerging role of new protein scaffold-based agents for treatment of cancer. Cancer Genomics and Proteomics 2013, 13:155–168. [PubMed] [Google Scholar]
- 21.Löfblom J, Frejd FY, Ståhl S: Non-immunoglobulin based protein scaffolds. Current Opinion in Biotechnology 2011, 22:843–848. [DOI] [PubMed] [Google Scholar]
- 22.Dennis MS, Herzka A, Lazarus RA: Potent and selective kunitz domain inhibitors of plasma kallikrein designed by phage display. Journal of Biological Chemistry 1995, 270:25411–25417. [DOI] [PubMed] [Google Scholar]
- 23.Vazquez-Lombardi R, Phan TG, Zimmermann C, Lowe D, Jermutus L, Christ D: Challenges and opportunities for non-antibody scaffold drugs. Drug Discovery Today 2015, 20:1271–1283. [DOI] [PubMed] [Google Scholar]
- 24.Rodon J, Omlin A, Herbschleb KH, Garcia-Corbacho J, Steiner J, Dolado I, Zitt C, Feurstein D, Turner D, Dawson KM, et al. : Abstract B25: First-in-human Phase I study to evaluate MP0250, a DARPin blocking HGF and VEGF, in patients with advanced solid tumors. Molecular Cancer Therapeutics 2016, 14:B25. [Google Scholar]
- 25.Kariolis MS, Kapur S, Cochran JR: Beyond antibodies: using biological principles to guide the development of next-generation protein therapeutics. Current Opinion in Biotechnology 2013, 24:1072–1077. [DOI] [PubMed] [Google Scholar]
- 26.Weinblatt ME, Kremer JM, Bankhurst AD, Bulpitt KJ, Fleischmann RM, Fox RI, Jackson CG, Lange M, Burge DJ: A Trial of Etanercept, a Recombinant Tumor Necrosis Factor Receptor:Fc Fusion Protein, in Patients with Rheumatoid Arthritis Receiving Methotrexate. New England Journal of Medicine 1999, 340:253–259. [DOI] [PubMed] [Google Scholar]
- 27.Tang PA, Moore MJ: Aflibercept in the treatment of patients with metastatic colorectal cancer: latest findings and interpretations. Therapeutic Advances in Gastroenterology 2013, 6:459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiskopf K, Ring AM, Ho CCM, Volkmer J- P, Levin AM, Volkmer AK, Özkan E, Fernhoff NB, van de Rijn M, Weissman IL, et al. : Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science 2013, 341:88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kariolis MS, Miao YR, Jones Ii DS, Kapur S, Mathews II, Giaccia AJ, Cochran JR: An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat Chem Biol 2014, 10:977983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papo N, Silverman AP, Lahti JL, Cochran JR: Antagonistic VEGF variants engineered to simultaneously bind to and inhibit VEGFR2 and alphavbeta3 integrin. Proc Natl Acad Sci U S A 2011, 108:14067–14072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tolcher AW, Papadopoulos KP, Patnaik A, Wilson K, Thayer S, Zanghi J, Gemo AT, Kavanaugh WM, Keer HN, LoRusso PM: A phase I, first in human study of FP-1039 (GSK3052230), a novel FGF ligand trap, in patients with advanced solid tumors. Annals of Oncology 2016, 27:526–532. [DOI] [PubMed] [Google Scholar]
- 32.Torchilin VP: Passive and active drug targeting: drug delivery to tumors as an example. Handb Exp Pharmacol 2010:3–53. [DOI] [PubMed] [Google Scholar]
- 33.Matsumura Y, Maeda H: A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Research 1986, 46:6387–6392. [PubMed] [Google Scholar]
- 34.Maeda H: Vascular permeability in cancer and infection as related to macromolecular drug delivery, with emphasis on the EPR effect for tumor-selective drug targeting. Proceedings of the Japan Academy. Series B, Physical and Biological Sciences 2012, 88:53–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McLaughlin P, Grillo-Lopez A, Link B, Levy R, Czuczman M, Williams M, Heyman M, Bence-Bruckler I, White C, Cabanillas F, et al. : Rituximab Chimeric Anti-CD20 Monoclonal Antibody Therapy for Relapsed Indolent Lymphoma: Half of Patients Respond to a Four-Dose Treatment Program. J Clin Oncol 1998, 16:2825–2833. [DOI] [PubMed] [Google Scholar]
- 36.Mendelsohn J: Epidermal growth factor receptor inhibition by a monoclonal antibody as anticancer therapy. Clinical Cancer Research 1997, 3:2703–2707. [PubMed] [Google Scholar]
- 37.Chu Q, Moellering RE, Hilinski GJ, Kim Y- W, Grossmann TN, Yeh JTH, Verdine GL: Towards understanding cell penetration by stapled peptides. Med. Chem. Commun 2015, 6:111–119. [Google Scholar]
- 38.Thompson DB, Cronican JJ, Liu DR: Engineering and identifying supercharged proteins for macromolecule delivery into mammalian cells. Methods Enzymol 2012, 503:293–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liao X, Rabideau AE, Pentelute BL: Delivery of antibody mimics into mammalian cells via anthrax toxin protective antigen. Chembiochem 2014, 15:2458–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verdurmen WP, Luginbuhl M, Honegger A, Pluckthun A: Efficient cell-specific uptake of binding proteins into the cytoplasm through engineered modular transport systems. J Control Release 2015, 200:13–22. [DOI] [PubMed] [Google Scholar]
- 41.Copolovici DM, Langel K, Eriste E, Langel U: Cell-penetrating peptides: design, synthesis, and applications. ACS Nano 2014, 8:1972–1994. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt SR: Fusion proteins for half-life extension. In Fusion Protein Technologies for Biopharmaceuticals Edited by Schmidt SR: John Wiley & Sons, Inc.; 2013:91–106. [Google Scholar]
- 43.Takeda K, Stagg J, Yagita H, Okumura K, Smyth MJ: Targeting death-inducing receptors in cancer therapy. Oncogene 2007, 26:3745–3757. [DOI] [PubMed] [Google Scholar]
- 44.Sievers EL, Senter PD: Antibody-drug conjugates in cancer therapy. Annu Rev Med 2013, 64:15–29. [DOI] [PubMed] [Google Scholar]
- 45.Chari RVJ, Miller ML, Widdison WC: Antibody-drug conjugates: an emerging concept in cancer therapy. Angewandte Chemie International Edition 2014, 53:3796–3827. [DOI] [PubMed] [Google Scholar]
- 46.Tchou J, Conejo-Garcia J: Targeting the tumor stroma as a novel treatment strategy for breast cancer: shifting from the neoplastic cell-centric to a stroma-centric paradigm. Adv Pharmacol 2012, 65:45–61. [DOI] [PubMed] [Google Scholar]
- 47.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. : Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324:1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frey K, Neri D: Antibody-based targeting of tumor vasculature and stroma 2011:419–450. [Google Scholar]
- 49.Presta LG, Chen H, O’Connor SJ, Chisholm V, Meng YG, Krummen L, Winkler M, Ferrara N: Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Research 1997, 57:4593–4599. [PubMed] [Google Scholar]
- 50.Keating GM: Bevacizumab: a review of its use in advanced cancer. Drugs 2014, 74:1891–1925. [DOI] [PubMed] [Google Scholar]
- 51.Huang D, Lan H, Liu F, Wang S, Chen X, Jin K, Mou X: Anti-angiogenesis or pro-angiogenesis for cancer treatment: focus on drug distribution. International Journal of Clinical and Experimental Medicine 2015, 8:8369–8376. [PMC free article] [PubMed] [Google Scholar]
- 52.Golay J, Cittera E, Gaetano ND, Manganini M, Mosca M, Nebuloni M, van Rooijen N, Vago L, Introna M: The role of complement in the therapeutic activity of rituximab in a murine B lymphoma model homing in lymph nodes. Haematologica 2006, 91:176–183. [PubMed] [Google Scholar]
- 53.Willingham SB, Volkmer J- P, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, et al. : The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proceedings of the National Academy of Sciences 2012, 109:6662–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weiner GJ: Rituximab: mechanism of action. Semin Hematol 2010, 47:115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. : Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. New England Journal of Medicine 2010, 363:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shin DS, Ribas A: The evolution of checkpoint blockade as a cancer therapy: what’s here, what’s next? Current Opinion in Immunology 2015, 33:23–35. [DOI] [PubMed] [Google Scholar]
- 57.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD: The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol 2016, 13:273–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou Y, Goenaga A- L, Harms BD, Zou H, Lou J, Conrad F, Adams GP, Schoeberl B, Nielsen UB, Marks JD: Impact of intrinsic affinity on functional binding and biological activity of EGFR antibodies. Molecular Cancer Therapeutics 2012, 11:1467–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Packer MS, Liu DR: Methods for the directed evolution of proteins. Nat Rev Genet 2015, 16:379–394. [DOI] [PubMed] [Google Scholar]
- 60.Lane MD, Seelig B: Advances in the directed evolution of proteins. Current Opinion in Chemical Biology 2014, 22:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adams GP, Schier R, McCall AM, Simmons HH, Horak EM, Alpaugh RK, Marks JD, Weiner LM: High affinity restricts the localization and tumor penetration of single-chain Fv antibody molecules. Cancer Research 2001, 61:4750–4755. [PubMed] [Google Scholar]
- 62.Cilliers C, Guo H, Liao J, Christodolu N, Thurber GM: Multiscale Modeling of Antibody-Drug Conjugates: Connecting Tissue and Cellular Distribution to Whole Animal Pharmacokinetics and Potential Implications for Efficacy. The AAPS Journal 2016, 18:1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmidt MM, Wittrup KD: A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Molecular Cancer Therapeutics 2009, 8:2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zahnd C, Kawe M, Stumpp MT, de Pasquale C, Tamaskovic R, Nagy-Davidescu G, Dreier B, Schibli R, Binz HK, Waibel R, et al. : Efficient Tumor Targeting with High-Affinity Designed Ankyrin Repeat Proteins: Effects of Affinity and Molecular Size. Cancer Research 2010, 70:1595. [DOI] [PubMed] [Google Scholar]
- 65.Muller PY, Milton MN: The determination and interpretation of the therapeutic index in drug development. Nat Rev Drug Discov 2012, 11:751–761. [DOI] [PubMed] [Google Scholar]
- 66.Donaldson JM CK, Fragoso R, Rodeck U, WIlliams JC: Design and development of masked therapeutic antibodies to limit off-target effects: Application to anti-EGFR antibodies. Cancer Biol Ther 2009, 8:2147–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EEM, Liang TW, Wong C, Bessette PH, Kamath K, Moore SJ, et al. : Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Science Translational Medicine 2013, 5:207ra144. [DOI] [PubMed] [Google Scholar]
- 68.Mammen M, Choi S- K, Whitesides GM: Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed Engl 1998, 37:2754–2794. [DOI] [PubMed] [Google Scholar]
- 69.Robinson MK, Hodge KM, Horak E, Sundberg AL, Russeva M, Shaller CC, von Mehren M, Shchaveleva I, Simmons HH, Marks JD, et al. : Targeting ErbB2 and ErbB3 with a bispecific single-chain Fv enhances targeting selectivity and induces a therapeutic effect in vitro. Br J Cancer 2008, 99:1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spiess C, Zhai Q, Carter PJ: Alternative molecular formats and therapeutic applications for bispecific antibodies. Molecular Immunology 2015, 67:95–106. [DOI] [PubMed] [Google Scholar]
- 71.Fitzgerald J, Lugovskoy A: Rational engineering of antibody therapeutics targeting multiple oncogene pathways. mAbs 2011, 3:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klein C, Sustmann C, Thomas M, Stubenrauch K, Croasdale R, Schanzer J, Brinkmann U, Kettenberger H, Regula JT, Schaefer W: Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. mAbs 2012, 4:653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chelius D, Ruf P, Plöscher M, Liedtke R, Gansberger E, Hess J, Wasiliu M, Lindhofer H: Structural and functional characterization of the trifunctional antibody catumaxomab. mAbs 2010, 2:309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bossi G, Buisson S, Oates J, Jakobsen BK, Hassan NJ: ImmTAC-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer immunology, immunotherapy: CII 2014, 63:437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang S, Chen C, Meng Y, Hu S, Zheng L, Song J, Zhang D, Li B, Guo Y: Effective suppression of breast tumor growth by an anti-EGFR/ErbB2 bispecific antibody. Cancer Letters 2012, 325:214–219. [DOI] [PubMed] [Google Scholar]
- 76.McDonagh CF, Huhalov A, Harms BD, Adams S, Paragas V, Oyama S, Zhang B, Luus L, Overland R, Nguyen S, et al. : Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Molecular Cancer Therapeutics 2012, 11:582–593. [DOI] [PubMed] [Google Scholar]
- 77.Castoldi R, Ecker V, Wiehle L, Majety M, Busl-Schuller R, Asmussen M, Nopora A, Jucknischke U, Osl F, Kobold S, et al. : A novel bispecific EGFR/Met antibody blocks tumor-promoting phenotypic effects induced by resistance to EGFR inhibition and has potent antitumor activity. Oncogene 2013, 32:5593–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, Guntas G, Leaver-Fay A, Smith EM, Ho C, et al. : Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nature Biotechnology 2014, 32:191–198. [DOI] [PubMed] [Google Scholar]
- 79.Spangler JB, Manzari MT, Rosalia EK, Chen TF, Wittrup KD: Triepitopic antibody fusions inhibit cetuximab-resistant BRAF and KRAS mutant tumors via EGFR signal repression. J Mol Biol 2012, 422:532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Farrington GK, Caram-Salas N, Haqqani AS, Brunette E, Eldredge J, Pepinsky B, Antognetti G, Baumann E, Ding W, Garber E, et al. : A novel platform for engineering blood-brain barriercrossing bispecific biologics. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 2014, 28:4764–4778. [DOI] [PubMed] [Google Scholar]
- 81.Watts RJ, Dennis MS: Bispecific antibodies for delivery into the brain. Current Opinion in Chemical Biology 2013, 17:393–399. [DOI] [PubMed] [Google Scholar]
- 82.Metz S, Haas AK, Daub K, Croasdale R, Stracke J, Lau W, Georges G, Josel H- P, Dziadek S, Hopfner K- P, et al. : Bispecific digoxigenin-binding antibodies for targeted payload delivery. Proceedings of the National Academy of Sciences of the United States of America 2011, 108:8194–8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rossi EA, Rossi DL, Stein R, Goldenberg DM, Chang C- H: A bispecific antibody-IFNalpha2b immunocytokine targeting CD20 and HLA-DR is highly toxic to human lymphoma and multiple myeloma cells. Cancer Research 2010, 70:7600–7609. [DOI] [PubMed] [Google Scholar]
- 84.Zhu Y, Choi SH, Shah K: Multifunctional receptor-targeting antibodies for cancer therapy. The Lancet Oncology 2015, 16:e543–e554. [DOI] [PubMed] [Google Scholar]
- 85.Hu S, Fu W, Xu W, Yang Y, Cruz M, Berezov SD, Jorissen D, Takeda H, Zhu W: Four-in-one antibodies have superior cancer inhibitory activity against EGFR, HER2, HER3, and VEGF through disruption of HER/MET crosstalk. Cancer Research 2015, 75:159–170. [DOI] [PubMed] [Google Scholar]
- 86.Tamaskovic R, Schwill M, Nagy-Davidescu G, Jost C, Schaefer DC, Verdurmen WPR, Schaefer JV, Honegger A, Plückthun A: Intermolecular biparatopic trapping of ErbB2 prevents compensatory activation of PI3K/AKT via RAS–p110 crosstalk. Nature Communications 2016, 7:11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li John Y, Perry Samuel R, Muniz-Medina V, Wang X, Wetzel Leslie K, Rebelatto Marlon C, Hinrichs Mary Jane M, Bezabeh Binyam Z, Fleming Ryan L, Dimasi N, et al. : A biparatopic HER2-targeting antibody-drug conjugate induces tumor regression in primary models refractory to or ineligible for HER2-targeted therapy. Cancer Cell 2016, 29:117–129. [DOI] [PubMed] [Google Scholar]
- 88.Wuellner U, Klupsch K, Buller F, Attinger-Toller I, Santimaria R, Zbinden I, Henne P, Grabulovski D, Bertschinger J, Brack S: Bispecific CD3/HER2 Targeting FynomAb Induces Redirected T CellMediated Cytolysis with High Potency and Enhanced Tumor Selectivity. Antibodies 2015, 4. [Google Scholar]
- 89.Zeidler R, Mysliwietz J, Csánady M, Walz A, Ziegler I, Schmitt B, Wollenberg B, Lindhofer H: The Fcregion of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. British Journal of Cancer 2000, 83:261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kontermann RE, Brinkmann U: Bispecific antibodies. Drug Discovery Today 2015, 20:838–847. [DOI] [PubMed] [Google Scholar]
- 91.Frankel SR, Baeuerle PA: Targeting T cells to tumor cells using bispecific antibodies. Current Opinion in Chemical Biology 2013, 17:385–392. [DOI] [PubMed] [Google Scholar]
- 92.Hamano Y, Grunkemeyer JA, Sudhakar A, Zeisberg M, Cosgrove D, Morello R, Lee B, Sugimoto H, Kalluri R: Determinants of vascular permeability in the kidney glomerulus. Journal of Biological Chemistry 2002, 277:31154–31162. [DOI] [PubMed] [Google Scholar]
- 93.Tang L, Persky A: Pharmacokinetic aspects of biotechnology products. J Pharm Sci 2004, 93:2184–2204. [DOI] [PubMed] [Google Scholar]
- 94.Strohl WR: Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs 2015, 29:215–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Topp MS, Kufer P, Gökbuget N, Goebeler M, Klinger M, Neumann S, Horst H- A, Raff T, Viardot A, Schmid M, et al. : Targeted Therapy With the T-Cell–Engaging Antibody Blinatumomab of Chemotherapy-Refractory Minimal Residual Disease in B-Lineage Acute Lymphoblastic Leukemia Patients Results in High Response Rate and Prolonged Leukemia-Free Survival. Journal of Clinical Oncology 2011, 29:2493–2498. [DOI] [PubMed] [Google Scholar]
- 96.Kontermann RE: Half-life extended biotherapeutics. Expert Opinion on Biological Therapy 2016, 16:903–915. [DOI] [PubMed] [Google Scholar]
- 97.Yousefpour P, Chilkoti A: Co-opting biology to deliver drugs. Biotechnology and Bioengineering 2014, 111:1699–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mannucci PM: Half-life extension technologies for haemostatic agents. Thrombosis and Haemostasis 2015, 113:165–176. [DOI] [PubMed] [Google Scholar]
- 99.Czajkowsky DM, Hu J, Shao Z, Pleass RJ: Fc-fusion proteins: new developments and future perspectives. EMBO molecular medicine 2012, 4:1015–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mitragotri S, Burke PA, Langer R: Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov 2014, 13:655–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim J, Bronson CL, Hayton WL, Radmacher MD, Roopenian DC, Robinson JM, Anderson CL: Albumin turnover: FcRn-mediated recycling saves as much albumin from degradation as the liver produces. American Journal of Physiology - Gastrointestinal and Liver Physiology 2006, 290:G352–G360. [DOI] [PubMed] [Google Scholar]
- 102.Vidarsson G, Dekkers G, Rispens T: IgG Subclasses and Allotypes: From Structure to Effector Functions. Frontiers in Immunology 2014, 5:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Byrn RA, Mordenti J, Lucas C, Smith D, Marsters SA, Johnson JS, Cossum P, Chamow SM, Wurm FM, Gregory T, et al. : Biological properties of a CD4 immunoadhesin. Nature 1990, 344:667–670. [DOI] [PubMed] [Google Scholar]
- 104.Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, Ober RJ, Ward ES: Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotech 1997, 15:637–640. [DOI] [PubMed] [Google Scholar]
- 105.Olafsen T: Fc engineering: serum half-life modulation through FcRn binding. In Antibody Engineering: Methods and Protocols, Second Edition Edited by Chames P: Humana Press; 2012:537–556. [DOI] [PubMed] [Google Scholar]
- 106.Sockolosky JT, Tiffany MR, Szoka FC: Engineering neonatal Fc receptor-mediated recycling and transcytosis in recombinant proteins by short terminal peptide extensions. Proc Natl Acad Sci U S A 2012, 109:16095–16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Igawa T, Maeda A, Haraya K, Tachibana T, Iwayanagi Y, Mimoto F, Higuchi Y, Ishii S, Tamba S, Hironiwa N, et al. : Engineered monoclonal antibody with novel antigen-sweeping activity in vivo. PLoS One 2013, 8:e63236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shen Y, Zeng L, Novosyadlyy R, Forest A, Zhu A, Korytko A, Zhang H, Eastman SW, Topper M, Hindi S, et al. : A bi-functional antibody-receptor domain fusion protein simultaneously targeting IGF-IR and VEGF for degradation. mAbs 2015, 7:931–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sleep D, Cameron J, Evans LR: Albumin as a versatile platform for drug half-life extension. Biochimica et Biophysica Acta (BBA) - General Subjects 2013, 1830:5526–5534. [DOI] [PubMed] [Google Scholar]
- 110.Andersen JT, Dalhus B, Viuff D, Ravn BT, Gunnarsen KS, Plumridge A, Bunting K, Antunes F, Williamson R, Athwal S, et al. : Extending serum half-life of albumin by engineering neonatal Fc receptor (FcRn) binding. Journal of Biological Chemistry 2014, 289:13492–13502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Osborn BL, Gu M, Grzegorzewski KJ, Logan TF, Crowder K, Weiss GR, Syed S, Rowensky E, Tolcher A, Agarwala SS, et al. : Preliminary pharmacokinetic evaluation of Albuleukin; an interleukin-2 human serum albumin fusion protein, in solid tumor patients. Cancer Research 2004, 64:1099–1099. [Google Scholar]
- 112.Richards D, Braiteh F, Garcia A, Denlinger C, Conkling P, Edenfield W, Anthony S, Hellerstedt B, Raju R, Becerra C, et al. : A phase 1 study of MM-111, a bispecific HER2/HER3 antibody fusion protein, combined with multiple treatment regimens in patients with advanced HER2-positive solid tumors. J Clin Oncol (Meeting Abstracts) 2014, 32:651. [Google Scholar]
- 113.Tijink BM, Laeremans T, Budde M, Walsum MS-v, Dreier T, de Haard HJ, Leemans CR, van Dongen GAMS: Improved tumor targeting of anti–epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology. Molecular Cancer Therapeutics 2008, 7:2288. [DOI] [PubMed] [Google Scholar]
- 114.Langenheim JF, Chen WY: Improving the pharmacokinetics/pharmacodynamics of prolactin, GH, and their antagonists by fusion to a synthetic albumin-binding peptide. Journal of Endocrinology 2009, 203:375–387. [DOI] [PubMed] [Google Scholar]
- 115.Havelund S, Plum A, Ribel U, Jonassen I, Vølund A, Markussen J, Kurtzhals P: The mechanism of protraction of insulin detemir, a long-acting, acylated analog of human insulin. Pharm Res 2004, 21:1498–1454. [DOI] [PubMed] [Google Scholar]
- 116.Caliceti P: Pharmacokinetic and biodistribution properties of poly(ethylene glycol)–protein conjugates. Advanced Drug Delivery Reviews 2003, 55:1261–1277. [DOI] [PubMed] [Google Scholar]
- 117.Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, Sheng D, Liu X, Sims PW, VanderVeen LA, et al. : NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clinical Cancer Research 2016, 22:680. [DOI] [PubMed] [Google Scholar]
- 118.Graham ML: Pegaspargase: a review of clinical studies. Advanced Drug Delivery Reviews 2003, 55:1293–1302. [DOI] [PubMed] [Google Scholar]
- 119.Bendele A, Seely J: Short Communication: renal tubular vacuolation in animals treated with polyethylene-glycol-conjugated proteins. Toxicological sciences : an official journal of the Society of Toxicology 1998, 42:152–157. [DOI] [PubMed] [Google Scholar]
- 120.Schellenberger V, Wang C-w, Geething NC, Spink BJ, Campbell A, To W, Scholle MD, Yin Y, Yao Y, Bogin O, et al. : A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nature Biotechnology 2009, 27:1186–1190. [DOI] [PubMed] [Google Scholar]
- 121.Schlapschy M, Theobald I, Mack H, Schottelius M, Wester H- J, Skerra A: Fusion of a recombinant antibody fragment with a homo-amino-acid polymer: effects on biophysical properties and prolonged plasma half-life. Protein Engineering Design and Selection 2007, 20:273–284. [DOI] [PubMed] [Google Scholar]
- 122.Schlapschy M, Binder U, Börger C, Theobald I, Wachinger K, Kisling S, Haller D, Skerra A: PASylation: a biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Engineering Design and Selection 2013, 26:489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Floss DM, Schallau K, Rose-John S, Conrad U, Scheller J: Elastin-like polypeptides revolutionize recombinant protein expression and their biomedical application. Trends in Biotechnology 2010, 28:37–45. [DOI] [PubMed] [Google Scholar]
- 124.Cleland J: A placebo controlled single ascending dose phase 1 for safety, tolerability, pharmacokinetics, and pharmacodynamics after subcutaneous administration of VRS-859 in patients with type 2 diabetes mellitus. In American Diabetes Association 71st Scientific Sessions Edited by. Clinical Therapeutics/New Technology - Pharmacologic Treatment of Diabetes or its Complications; 2011. [Google Scholar]
- 125.Moore WV, Nguyen HJ, Kletter GB, Miller BS, Rogers D, Ng D, Moore JA, Humphriss E, Cleland JL, Bright GM: A randomized safety and efficacy study of somavaratan (VRS-317), a long-acting rhGH, in pediatric growth hormone deficiency. The Journal of Clinical Endocrinology and Metabolism 2016, 101:1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gregoriadis G, Jain S, Papaioannou I, Laing P: Improving the therapeutic efficacy of peptides and proteins: a role for polysialic acids. International Journal of Pharmaceutics 2005, 300:125–130. [DOI] [PubMed] [Google Scholar]
- 127.Fares FA, Suganuma N, Nishimori K, LaPolt PS, Hsueh AJ, Boime I: Design of a long-acting follitropin agonist by fusing the C-terminal sequence of the chorionic gonadotropin beta subunit to the follitropin beta subunit. Proceedings of the National Academy of Sciences of the United States of America 1992, 89:4304–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Patel J, Kothari R, Tunga R, Ritter N, Tunga B: Stability considerations for biopharmaceuticals:overview of protein and peptide degradation pathways. BioProcess International 2011, 9:20–31. [Google Scholar]
- 129.Arakawa T, Prestrelski S, Kenney W, Carpenter J: Factors affecting short-term and long-term stabilities of proteins. Adv Drug Deliv Rev 1993, 10:1–28. [DOI] [PubMed] [Google Scholar]
- 130.Cicerone M, Giri J, Shaked Ze, Roberts C: Protein stability — an underappreciated but critical need for drug delivery systems. Advanced Drug Delivery Reviews 2015, 93:1. [DOI] [PubMed] [Google Scholar]
- 131.Shusta EV, Kieke MC, Parke E, Kranz DM, Wittrup KD: Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency1. Journal of Molecular Biology 1999, 292:949–956. [DOI] [PubMed] [Google Scholar]
- 132.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL: Design of therapeutic proteins with enhanced stability. Proceedings of the National Academy of Sciences 2009, 106:11937–11942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liu Y, Caffry I, Wu J, Geng SB, Jain T, Sun T, Reid F, Cao Y, Estep P, Yu Y, et al. : High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. MAbs 2014, 6:483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Balcão VM, Vila MMDC: Structural and functional stabilization of protein entities: state-of-the-art. Advanced Drug Delivery Reviews 2015, 93:25–41. [DOI] [PubMed] [Google Scholar]
- 135.Lowe D, Dudgeon K, Rouet R, Schofield P, Jermutus L, Christ D: Aggregation, stability, and formulation of human antibody therapeutics. In Advances in Protein Chemistry and Structural Biology Edited by Donev R: Academic Press; 2011:41–61. vol 84.] [DOI] [PubMed] [Google Scholar]
- 136.Hötzel I, Theil F- P, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al. : A strategy for risk mitigation of antibodies with fast clearance. mAbs 2012, 4:753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kelley B: Industrialization of mAb production technology: The bioprocessing industry at a crossroads. mAbs 2014, 1:443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Singh SK: Impact of product-related factors on immunogenicity of biotherapeutics. Journal of Pharmaceutical Sciences 2011, 100:354–387. [DOI] [PubMed] [Google Scholar]
- 139.Yin L, Chen X, Vicini P, Rup B, Hickling TP: Therapeutic outcomes, assessments, risk factors and mitigation efforts of immunogenicity of therapeutic protein products. Cellular Immunology 2015, 295:118–126. [DOI] [PubMed] [Google Scholar]
- 140.Hermeling S, Crommelin DJA, Schellekens H, Jiskoot W: Structure-immunogenicity relationships of therapeutic proteins. Pharmaceutical Research 2004, 21:897–903. [DOI] [PubMed] [Google Scholar]
- 141.Deehan M, Garcês S, Kramer D, Baker MP, Rat D, Roettger Y, Kromminga A: Managing unwanted immunogenicity of biologicals. Autoimmunity Reviews 2015, 14:569–574. [DOI] [PubMed] [Google Scholar]
- 142.Roberts CJ: Therapeutic protein aggregation: mechanisms, design, and control. Trends in Biotechnology 2014, 32:372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ratanji KD, Derrick JP, Dearman RJ, Kimber I: Immunogenicity of therapeutic proteins: influence of aggregation. Journal of Immunotoxicology 2014, 11:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]