

Abstract
Hypertriglyceridemia is an independent risk factor for cardiovascular disease. Dietary interventions based on protein restriction (PR) reduce circulating triglycerides (TGs), but underlying mechanisms and clinical relevance remain unclear. Here, we show that 1 week of a protein-free diet without enforced calorie restriction significantly lowered circulating TGs in both lean and diet-induced obese mice. Mechanistically, the TG-lowering effect of PR was due, in part, to changes in very low–density lipoprotein (VLDL) metabolism both in liver and peripheral tissues. In the periphery, PR stimulated VLDL-TG consumption by increasing VLDL-bound APOA5 expression and promoting VLDL-TG hydrolysis and clearance from circulation. The PR-mediated increase in Apoa5 expression was controlled by the transcription factor CREBH, which coordinately regulated hepatic expression of fatty acid oxidation–related genes, including Fgf21 and Ppara. The CREBH-APOA5 axis activation upon PR was intact in mice lacking the GCN2-dependent amino acid–sensing arm of the integrated stress response. However, constitutive hepatic activation of the amino acid–responsive kinase mTORC1 compromised CREBH activation, leading to blunted APOA5 expression and PR-recalcitrant hypertriglyceridemia. PR also contributed to hypotriglyceridemia by reducing the rate of VLDL-TG secretion, independently of activation of the CREBH-APOA5 axis. Finally, a randomized controlled clinical trial revealed that 4–6 weeks of reduced protein intake (7%–9% of calories) decreased VLDL particle number, increased VLDL-bound APOA5 expression, and lowered plasma TGs, consistent with mechanistic conservation of PR-mediated hypotriglyceridemia in humans with translational potential as a nutraceutical intervention for dyslipidemia.
Keywords: Metabolism
Keywords: Lipoproteins

Dietary protein restriction modulates triglycerides levels via activation of a hepatic CREBH-APOA5 axis and by reducing liver VLDL-TG secretion.
Introduction
Hypertriglyceridemia is an independent risk factor for cardiovascular disease (1). When associated with other metabolic comorbidities, it often occurs concomitantly with high low-density lipoprotein (LDL) and low high-density lipoprotein (HDL) levels, constituting the denoted atherogenic lipid triad dyslipidemia (2–5). Despite this, existing therapeutic options are limited to modulation of triglyceride (TG) synthesis or reduction of very low–density lipoprotein (VLDL) secretion, thus highlighting the need for novel strategies targeting alternate mechanisms with hypotriglyceridemic potential, such as increasing peripheral VLDL-TG clearance (6).
Dietary restriction (DR), defined as reduced food intake without malnutrition, is widely acknowledged for its pleiotropic benefits, including longevity extension, reduced cancer development, resistance to oxidative stress, and improved metabolic fitness (7). In the latter context, DR has demonstrated efficacy in lowering circulating TGs and other cardio-metabolic risk factors in nonhuman primates (8) and humans (9). While impracticalities associated with long-term DR (defined here as greater than 1 week) limit direct clinical translation, we and others have demonstrated benefits from shorter periods of DR lasting a week or less (defined here as short-term DR), which includes increased surgical stress resistance and improved metabolic fitness in mice (10–14). However, the precise mechanisms through which DR exerts its beneficial effects on lipid metabolism are not known.
From a nutritional standpoint, many of the pleiotropic benefits of short-term DR can be triggered by restriction of dietary protein intake (protein restriction; PR) or essential amino acids (EAA), independent of overall calorie intake. These benefits include resistance to hepatic ischemia reperfusion injury (IRI), increased energy expenditure, and significant reduction of TGs, even during short-term PR interventions (13, 15–20). Amino acid–sensing pathways are implicated in controlling such systemic benefits in response to protein or EAA restriction. Amino acid insufficiency is detected via the general control nonderepressible 2 (GCN2) kinase, which activates the integrated stress response (ISR), as demonstrated by increased phosphorylation of the eukaryotic initiation factor 2α (eIF2α) and translation of activating transcription factor 4 (ATF4). A different eIF2α kinase, the protein kinase R–like (PKR-like) ER kinase (PERK), is activated upon dietary methionine restriction (21), suggesting redundancy with GCN2 in triggering the ISR upon PR. Amino acid sufficiency, on the other hand, is sensed through the mechanistic target of rapamycin complex 1 (mTORC1).
While GCN2 is required for benefits of individual EAA restriction against IRI (22), it is dispensable in the context of total PR, which instead requires downregulation of mTORC1 (23). In the context of lipid metabolism, both amino acid–sensing pathways are implicated in regulating the interplay between lipid and amino acid metabolism. GCN2 activation by single EAA deprivation suppresses lipid metabolism by repressing the induction of sterol regulatory element binding protein 1c–dependent SREBP1c-dependent (SREBP1c-dependent) lipogenic gene expression (24). Conversely, mTORC1 activation in the context of amino acid sufficiency promotes lipid synthesis via indirect regulation of SREBP1c (25, 26). Nonetheless, the role of these amino acid–responsive pathways in regulating the TG-lowering effects of PR have not been investigated.
Nutritional status is also a key determinant of VLDL-TG metabolism. TGs are transported primarily in chylomicrons in the fed state. In the fasted state, circulating TGs are transported predominantly in VLDL particles, which are assembled in the liver microsome compartment prior to secretion into circulation. Apolipoproteins expressed in liver are embedded onto VLDL particles to serve important roles in peripheral TG clearance. For instance, while some apolipoproteins serve as particle receptor ligands (e.g., APOB), while others act as coregulators of lipoprotein lipase (LPL) activity in periphery (e.g., APOC3, APOA5). Among these, APOA5 plays a pivotal role in facilitating TG hydrolysis from lipoprotein particles by stimulating LPL activity in the periphery (27, 28). Thus, mutations and polymorphisms affecting APOA5 function are common causes of hypertriglyceridemia (29–31).
Apoa5 expression is regulated by multiple transcription factors that also respond to host nutritional status. These include repression by the liver X-receptor (LXR)/SREBP-1c (32) and induction by peroxisome proliferator-activated receptor α (PPARα) (33), thyroid hormone receptor (34), and cAMP-responsive element–binding protein hepatocyte-specific (CREBH) (35, 36). CREBH is an ER-tethered transcription factor regulated by intramembrane proteolysis (37) through poorly characterized mechanisms modulated by feeding/fasting cycles (38). While originally described as an ER stress effector protein, the finding that CREBH-KO mice are hypertriglyceridemic with concomitant changes in apolipoprotein expression has suggested a major role for this transcription factor in controlling TG homeostasis (35, 39, 40). CREBH also regulates FGF21, a hormone that responds to nutritional cues, including protein deprivation (20) and sugar intake (41). FGF21 reduces TG levels (42) by increasing VLDL-TG disposal in white and brown adipose tissue (43). However, neither the effect of PR in regulating CREBH activation, nor the involvement of a potential CREBH-APOA5 axis in controlling TG levels during protein deprivation, have been investigated. Here, we sought to identify the mechanisms by which dietary protein intake exerts its hypotriglyceridemic effects and describe activation of hepatic and peripheral events controlling VLDL-TG metabolism, regulated through CREBH-APOA5–dependent and –independent mechanisms.
Results
Short-term dietary PR reduces circulating TGs independently of total caloric intake via effects on VLDL-TG levels.
To gain insight into the mechanism by which short-term DR reduces serum TG levels, we first explored the role of total protein and calorie intake in the hypotriglyceridemic effect of PR. To this end, B6D2F1 mice were fed with a complete (18% calories from protein) or protein-free (PF) diet (protein replaced by isocaloric sucrose) provided either ad libitum (AL) or restricted by 50% of total calories (calorie restriction; CR) for 1 week. The PF diet fed AL led to a more drastic reduction of serum TGs than 50% CR (Figure 1A). This effect was not further accentuated upon enforced food restriction in the PF-CR group (Figure 1A). Reduction of circulating TGs was confirmed using an unbiased serum lipidomic analysis, which revealed reduction in most individual TG species as a major change triggered by PR (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.99470DS1).
Figure 1. Short-term dietary protein restriction reduces circulating TGs independently of total caloric intake via effects on VLDL-TG levels.

B6D2F1 mice were fed the indicated complete (C, 18% protein content) or protein-free (PF, protein replaced with isocaloric sucrose) diet for 1 week prior to analysis. (A) Serum triglyceride (TG) concentrations in mice fed the indicated diet on an ad libitum basis (AL) or restricted daily (CR) by 50% (n = 5/group; 2-way ANOVA with Sidak post-hoc test between the indicated groups. (B) Serum TG in mice on the indicated diet (n = 4–5/group; 2-tailed Student’s t test between diets within fed or fasted state). (C) TG levels in FPLC fractions from pooled plasma samples (n = 5/group; VLDL, very low–density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; HDL, high-density lipoprotein). (D) Serum APOB-100 concentration representative of circulating VLDL particle number (n = 5/group; 2-tailed Student’s t test). (E) TG content of purified VLDL particles expressed per unit APOB-100, indicative of VLDL particle lipidation (n = 3/group; 2-tailed Student’s t test). Data expressed as mean ± SD; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Serum TGs levels were significantly reduced in PR mice in the fed and fasted states (Figure 1B), suggesting important changes in VLDL particles triggered during protein deprivation. This was confirmed in fractionation experiments using fast performance liquid chromatography (FPLC), which demonstrated a reduction of VLDL-TG levels upon PR (Figure 1C). Also, APOB-100, which is present at 1 molecule per VLDL particle and is, thus, representative of VLDL particle number, was reduced in serum upon PR (Figure 1D). Analysis of TG content of purified VLDL particles normalized to APOB-100 confirmed a significant reduction in the lipidation state of PR-derived VLDL particles (Figure 1E). Thus, TG lowering during PR is associated with a reduction in VLDL particle number and lipidation state.
PR alters peripheral VLDL-TG consumption via changes in hepatic apolipoprotein expression.
Along with VLDL particle lipidation and hepatic secretion, LPL-mediated TG hydrolysis in the periphery is a major determinant of circulating TG levels. To investigate the potential contribution of peripheral VLDL-TG consumption, we injected recombinant TG-rich lipoprotein particles containing radiolabeled TG (44) into mice fed a complete or PF diet for 1 week. We observed faster clearance of radio-labeled TG from circulation in PR mice (Figure 2A).
Figure 2. Protein restriction alters VLDL-TG consumption via changes in hepatic apolipoprotein expression.

(A and B) Time-dependent clearance of radiolabeled TG from the following circulating lipoprotein particles: (A) [3H]-triolein-labeled recombinant lipoprotein particles injected into mice fed a complete (C) or protein-free (PF) diet (n = 7/group); or (B) [3H]-palmitate-labeled and purified VLDL particles from mice on C or PF diets, and injected into complete-fed mice (n = 4/group; multiple t tests between diet groups with Holm-Sidak post-hoc test). (C) Hepatic mRNA expression of apolipoprotein-encoding genes from mice on the indicated diet (n = 4–11/group; 2-tailed Student’s t test). (D) Immunoblot of hepatic APOA5 protein expression. Below, quantitation normalized to tubulin and expressed in arbitrary units (AU, n = 5/group; 2-tailed Student’s t test). (E) Representative confocal microscopic images taken with a 63× objective from livers of mice on the indicated diet showing the ER marker KDEL (green), APOA5 (red), nuclei (DNA stained with DAPI, blue) superimposed to differential interference contrast (DIC) image and a composite image. Scale bar: 20 μm. (F) Circulating VLDL-bound APOA5 levels in mice on the indicated diet, expressed as the ratio of serum APOA5 to APOB-100 as measured by ELISA (n = 5/group; Mann-Whitney U test). (G) Correlation analysis between serum TG and APOA5. Each dot represents an individual mouse; r, Pearson’s coefficient. (H) Fasted serum TG levels in whole body APOA5-KO or control FVB WT mice on the indicated diet (n = 4/group; 2-way ANOVA with Sidak post-hoc test between the indicated groups). Data expressed as mean ± SD; **P < 0.01, ***P < 0.001; ****P < 0.0001.
To test whether diet-induced changes in VLDL particles themselves contributed to increased VLDL-TG clearance, we purified endogenously radiolabeled TG-containing VLDL particles from mice on complete or PF diets (Supplemental Figure 2A), and we injected them into recipient mice fed a complete diet. Radiolabeled VLDL-TG derived from PR mice cleared significantly faster from circulation than radiolabeled VLDL-TG derived from mice fed a complete diet (Figure 2B and Supplemental Figure 2B), suggesting the existence of intrinsic differences in VLDL particle composition themselves, as a contributing factor to TG lowering by PR.
The apolipoprotein composition of nascent lipoprotein particles plays an important role in regulating TG clearance in the periphery. We thus analyzed hepatic apolipoprotein gene expression as a function of diet, finding significant decreases in Apoa2, Apoc3, and Apoe. Of interest, we also observed a significant increase in mRNA expression of the hypolipidemic apolipoprotein gene Apoa5 upon PR (Figure 2C), which was also confirmed at the protein level (Figure 2D).
PR increased APOA5 localization in the ER of hepatocytes (Figure 2E), where it is suitable for incorporation into nascent VLDL particles. Serum APOA5 levels were not significantly higher after 1 week of PR (Supplemental Figure 2C); however, due to the reduced number of VLDL particles (Figure 1D), the APOA5/APOB ratio indicative of APOA5 occupancy per VLDL particle was significantly increased (Figure 2F) and negatively correlated with serum TG levels (Figure 2G). To measure the relative contribution of changes in APOA5 expression to PR-mediated TG lowering, we used whole-body APOA5-KO mice on an FVB genetic background and tested the hypotriglyceridemic response to PR. Interestingly, APOA5-KO serum TG levels were reduced by ~50% but remained significantly elevated relative to WT controls on a PF diet (Figure 2H). Taken together, these data point to a significant, although partial, contribution of APOA5 to the TG-lowering effects of PR.
CREBH regulates Apoa5 expression upon PR.
We next sought to determine the mechanism by which PR controls Apoa5 expression. In humans, APOA5 is regulated by PPARα (45), as well as CREBH (36). Here, we confirmed the presence of a highly conserved TCCACGTGGT-CREBH regulatory element in the promotor region of Apoa5 in mice and other mammals (Supplemental Figure 3, A and B). Immunoblotting with an antibody recognizing the amino-terminal fragment present in both the full-length, inactive (70 kDa) and the cleaved, active (45 kDa) forms of CREBH revealed an increase in the cleaved form in total liver lysates of mice on PR (Figure 3A). Cell fractionation experiments and confocal microscopy analysis confirmed nuclear enrichment of the cleaved form (Figure 3, B and C) compared with cytoplasmic fractions (Supplemental Figure 3C). Also, analysis of hepatic expression of direct CREBH targets including Pepck, Apoa4, Cyp4a10, and Fgf21 (35, 46–48) revealed an increase in livers of mice on PR (Figure 3D). Overexpression of HA-tagged active human CREBH (46) in human HepG-2 hepatoma cells resulted in nuclear localization (Figure 3E) and increased Apoa5 mRNA expression (Figure 3F). Finally, ChIP–quantitative PCR (ChIP-qPCR) analysis revealed enrichment of CREBH on the Apoa5 promoter in mouse livers independent of diet (Figure 3G). Together, these results are consistent with CREBH activation and regulation of APOA5 expression upon PR. We used whole-body CREBH-KO to explore the requirement of this transcription factor during PR. As predicted, hepatic Apoa5 mRNA was reduced in CREBH-KO mice relative to controls (Figure 3H). PR-induced Fgf21 expression was also significantly reduced (Figure 3I). Expression of PPARα targets Acadvl and Ctp1a, as well as Ppara mRNA itself, were reduced in CREBH-KO livers at baseline and/or upon PR (Figure 3J). These results are consistent with coordinated regulation of fatty acid oxidation (FAO) gene expression programs controlled by CREBH and PPARα.
Figure 3. CREBH regulates Apoa5 expression upon protein restriction.

(A–D) B6D2F1 mice were fed complete (C) or protein free (PF) diet for 1 week prior to analysis. (A) Immunoblot of CREBH full-length (FL) and cleaved fragment (CF) in liver extracts; 2 left lanes, CREBH-KO antibody specificity controls. Right, quantitation of CREBH CF normalized to FL and expressed in arbitrary units (AU; n = 5/group; 2-tailed Student’s t test). (B) CREBH CF immunoblot in liver nuclear extracts. Right, quantitation normalized to lamin A/C (n = 5/group; 2-tailed Student’s t test). (C) Representative confocal microscopic images (20× objective) of livers stained for CREBH amino-terminus (fire-red look-up intensity table visualization), DAPI-stained nuclei (DNA, blue) super-imposed onto the corresponding differential interference contract (DIC) image, and composite. Scale bars: 50 µm. Right micrograph, 10× digital magnification of indicated area. (D) Hepatic mRNA expression of CREBH targets Apoa4, Cyp4a10, Pepck, and Fgf21 (n = 5–10/group; 2-tailed Student’s t test). (E) Representative confocal microscopic images (63× objective) of HepG-2 cells transfected with hemagglutinin-tagged (HA-tagged) human CREBH CF (hCREBH [CF]-HA) vs. empty vector (Mock) and labeled with anti-HA antibody or DAPI (DNA) as indicated. Scale bars: 10 µm. (F) mRNA expression of human Apoa5 from cells transfected as in E; data in triplicate from one representative experiment of 3; 2-tailed Student’s t test. (G) ChIP of CREBH from livers of mice on the indicated diet, followed by qPCR analysis of the region flanking the predicted CREBH binding motif in the Apoa5 promoter (n = 3/group; 2-way ANOVA with Sidak post-hoc test between indicated groups). (H–N) CREBH-KO or WT control littermates were fed the indicated diet for 1 week prior to analysis. (H–J) Hepatic mRNA levels of Apoa5 (H), Fgf21 (I), and the Pparα targets Acadvl, Ctp1a, and Pparα itself (J) (n = 4/group; 2-way ANOVA with Sidak post-hoc test between indicated groups). (K) Immunoblot of hepatic APOA5; below, quantitation normalized to tubulin (n = 3/group; 2-way ANOVA with Sidak post-hoc test between indicated groups). (L–M) VLDL-bound APOA5 levels expressed as the ratio of serum APOA5 to APOB-100 (L) and serum FGF21 levels (M) (n = 4/group; 2-way ANOVA with Sidak post-hoc test between indicated groups). (N) Fasted serum TG concentrations pooled from 3 independent experiments including male and female WT and CREBH-KO mice (2-way ANOVA with Sidak post-hoc test between indicated groups). Data expressed as mean ± SD; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
The blunted capacity to upregulate APOA5 in CREBH-KO mice was also evident at the protein level (Figure 3K) and in serum (Supplemental Figure 3D), correlating with a significant reduction of APOA5 expression on the surface of VLDL particles (Figure 3L) without any significant change in VLDL particle number between genotypes (Supplemental Figure 3E). Interestingly, circulating FGF21 levels upon PR were also significantly decreased in PR-CREBH–KO vs. WT mice (Figure 3M). Reduced APOA5 and FGF21 levels correlated with a ~50% reduction of plasma TG in CREBH-KO mice (Figure 3N). Nonetheless, circulating TG levels remained significantly elevated relative to WT controls on a PF diet (Figure 3N). Together, these results are consistent with a partial role for CREBH in PR-mediated hypotriglyceridemia via coordinated transcriptional control of factors involved in systemic FAO, including hepatic PPARα, circulating FGF21, and VLDL-bound APOA5.
Role of GCN2, PERK, and the ISR in CREBH activation upon PR.
We next explored the role of amino acid–sensing pathways in regulating activation of the CREBH-APOA5 hepatic axis. Consistent with GCN2 activation, we observed increased eIF2α phosphorylation and ATF4 protein expression following a week on PR (Figure 4A and Supplemental Figure 4A). However, CREBH cleavage, APOA5 and FGF21 induction, and serum TG lowering upon PR were all intact in global GCN2-KO mice (Figure 4, B–E).
Figure 4. Role of GCN2, PERK, and the integrated stress response in CREBH activation upon protein restriction.

(A–I) WT or GCN2-KO mice were fed the indicated control (C) or protein-free (PF) diet for 1 week prior to analysis in the 4-hour fasted state. (A) Immunoblot of phospho- and total eIF2α in liver extracts of WT mice. Right, quantitation of phospho- eIF2α normalized to total expressed in arbitrary units (AU) (n = 5/group; 2-tailed Student’s t test). (B) Immunoblot of hepatic CREBH. Right, quantitation of CREBH cleaved form (CF) normalized to full-length (FL) (n = 3–7; 2-way ANOVA with Sidak post-hoc test between the indicated groups). (C) Immunoblot of hepatic APOA5 expression. Right, quantitation normalized to tubulin (n = 4–14/group; 2-way ANOVA with Sidak post-hoc test between the indicated groups). Due to the number of samples, blots in B and C were run on separate gels with the same complete WT control on each for between-blot normalization. (D) Serum FGF21 levels (n = 5/group, 2-way ANOVA with Sidak post-hoc test between the indicated groups). (E) Serum TG (n = 5/group; 2-way ANOVA with Sidak post-hoc test between the indicated groups). (F) Immunoblot of phospho- and total PERK in liver extracts. Right, quantitation of phospho-PERK normalized to total (n = 3/group; 2-tailed Student’s t test). (G) Immunoblot of phospho- and total eIF2α in liver extracts (n = 4–5/group; 1-way ANOVA with Tukey post-hoc test between the indicated groups). (H and I) Hepatic mRNA expression of Asns (H) and Atf4 (I) (n = 4–5/group; 2-way ANOVA with Sidak post-hoc test between the indicated groups). Data expressed as mean ± SD; *P < 0.05; **P < 0.01; ***P < 0.00; ****P < 0.0001.
CREBH activation was originally described in the context of the ER stress–inducing agent thapsagargin (37), which activates the ISR through the eIF2α kinase, PERK (21). Interestingly, PERK was activated upon PR (Figure 4F), even in the absence of GCN2 (Figure 4G), as reflected by increased eIF2α phosphorylation and expression of ATF4 target genes including asparagine synthetase (Asns) and Atf4 itself (Figure 4, H and I). Additionally, PR failed to activate ER stress markers IRE-1α, TRAF2, or JNK-1 (Supplemental Figure 4B), suggesting that CREBH activation upon PR does not require ER stress per se, but rather could result from ISR activation downstream of GCN2 and/or PERK, or via PERK activation independently of the ISR.
Constitutive mTORC1 signaling inhibits CREBH activation.
Signal transduction via mTORC1, determined based on S6 phosphorylation, was reduced following a week of PR (Figure 5A). To test the potential role of this signal transduction pathway in CREBH activation, we employed mice bearing hepatocyte-specific deletion of the mTORC1 negative regulator TSC1 (liver-specific TSC1–KO; LiTSC1-KO), which display constitutive and PR-recalcitrant hepatic mTORC1 activation (23). Importantly, LiTSC1-KO mice subjected to PR failed to induce CREBH cleavage (Figure 5B), Apoa5 gene expression (Figure 5C), or hepatic APOA5 protein expression (Figure 5D) upon PR. Inhibition of mTORC1 activity with rapamycin rescued Apoa5 mRNA expression in LiTSC1-KO mice (Figure 5E). However, hepatocyte-specific deletion of the activating mTORC1 subunit Raptor (LiRaptor-KO) was insufficient to activate CREBH on a complete diet, although it did not prevent CREBH activation upon PR (Figure 5F). Together, these data are consistent with mTORC1 as a negative regulator of CREBH activation by another trigger, such as PR.
Figure 5. Constitutive mTORC1 signaling inhibits CREBH activation.

(A) Immunoblot of phospho- and total S6 in liver extracts. Below, quantitation of phospho-S6 normalized to total and expressed in arbitrary units (AU); n = 4–5/group; 2-tailed Student’s t test. (B–E and G–K) Liver-specific TSC1–KO (LiTSC1-KO; TSC1fl/fl|Albumin-Cre+/–) mice and littermate controls (TSC1fl/fl|Albumin-Cre–/–) fed a complete (C) or protein-free (PF) diet for 1 week prior to analysis. (B) Immunoblot of CREBH full-length (FL) and cleaved fragment (CF) in liver extracts. Below, quantitation of CF normalized to FL (n = 3/group; 2-way ANOVA with Sidak post-hoc test between the indicated groups). (C) Hepatic Apoa5 mRNA expression (n = 7–8/group; 2-way ANOVA with Sidak post-hoc test between indicated groups). (D) Immunoblot of hepatic APOA5. Below, quantitation normalized to tubulin (n = 3/group; 2-way ANOVA with Sidak post-hoc test between indicated groups; interaction P = 0.01/30% variation; genotype P = 0.02/22% variation; diet P = 0.02/24% variation). (E) Apoa5 mRNA expression from livers of LiTSC1-KO mice injected once daily with rapamycin (1 mg/kg) or vehicle (Veh) for indicated days of treatment (Tx); n = 3–4/group; 1-way ANOVA with Dunnett post-hoc test compared with vehicle control. (F) Immunoblot of hepatic CREBH in mice with hepatocyte-specific Raptor deletion (LiRaptor-KO; Raptorfl/fl|Albumin-Cre+/–) vs. littermate controls (Raptorfl/fl|Albumin-Cre–/–). Below, quantitation of CREBH CF normalized to FL (n = 3/group; 2-way ANOVA with Sidak post-hoc test between indicated groups). (G) Serum TG (n = 13–14/group; 2-way ANOVA with Sidak post-hoc test between indicated groups). (H) VLDL-bound APOA5 levels expressed as the ratio of serum APOA5 to APOB-100 (n = 5–7/group; 2-way ANOVA with Sidak post-hoc test between indicated groups). (I) Correlation analysis between serum TG and circulating APOA5. Each symbol represents an individual mouse; r, Pearson’s coefficient. (J) TG content of VLDL particles purified from serum of WT and LiTSC1-KO mice expressed per unit APOB-100 indicative of VLDL particle lipidation (n = 3/group; 2-way ANOVA with Sidak post-hoc test between indicated groups). (K) Serum FGF21 levels (n = 4/group; 2-way ANOVA with Sidak post-hoc test between indicated groups). Data expressed as mean ± SD; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Reduced APOA5 in LiTSC1-KO mice correlated with hypertriglyceridemia at baseline that was refractory to the TG lowering effect of PR (Figure 5G). Also, LiTSC1-KO mice had lower VLDL-surface bound APOA5 expression (Figure 5H and Supplemental Figure 5, A and B), which negatively correlated with serum TG levels (Figure 5I). TG content of purified VLDL particles derived from LiTSC1-KO serum remained high after a week in PR (Figure 5J), consistent with decreased peripheral VLDL-TG hydrolysis as a result of decreased APOA5 lipoprotein surface expression. Surprisingly, despite the loss of CREBH induction and PR-recalcitrant hypertriglyceridemia, the induction of circulating FGF21 upon PR was normal (Figure 5K).
Finally, we considered the potential of mTORC1 activation to contribute to hypertriglyceridemia via effects on SREBP1c-dependent lipogenesis. However, reduction in hepatic SREBP1c protein and target lipogenic gene expression observed in WT mice upon PR (Supplemental Figure 5, C and D) was intact in LiTSC1-KO mice, with Fasn mRNA expression significantly reduced at baseline and further reduced upon PR (Supplemental Figure 5E). Taken together, these data suggest that increased mTORC1 signaling can prevent the hypotriglyceridemic effects of PR, in part, through effects on CREBH activation and APOA5 expression — but independently of hepatic lipogenesis and despite elevated circulating FGF21.
Reduced hepatic VLDL particle secretion rate contributes to TG lowering upon PR independently of CREBH-APOA5 activation.
The partial yet significant TG-lowering effects of PR observed in hypertriglyceridemic APOA5-KO and CREBH-KO models (Figure 2H and Figure 3N) suggested a mechanism of TG lowering independent of the increase in fat oxidation/consumption coordinately controlled by CREBH and PPARα and their downstream effectors including APOA5 and FGF21. To identify such a CREBH-APOA5–independent mechanism, we explored additional points controlling TG homeostasis.
Consistent with reduced steady-state circulating VLDL particle number upon PR in WT mice (Figure 1D), VLDL secretion rate — measured after tyloxapol administration — was also reduced upon PR (Figure 6A). Importantly, the magnitude of the PR-mediated reduction in VLDL secretion rate upon tyloxapol challenge was similar between CREBH-KO and control mice (Figure 6B). In contrast, VLDL secretion rate was not significantly different upon PR in LiTSC1-KO mice (Figure 6C), reminiscent of their recalcitrance to TG lowering upon PR (Figure 5G).
Figure 6. Reduced hepatic VLDL particle secretion rate contributes to TG lowering upon protein restriction independently of CREBH-APOA5 activation.

(A–C) Hepatic VLDL secretion rate measured following tyloxapol injection in mice fed a complete (C) or protein-free (PF) diet. (A) B6D2F1 WT mice (n = 14/group); (B) CREBH-KO and WT littermate control mice (n = 4/group); and (C) LiTSC1-KO (TSC1fl/fl|Albumin-Cre+/–) and control (Cont, TSC1fl/fl|Albumin-Cre–/–) littermate mice (n = 5–9/group). 2-tailed Student’s t tests using peak AUC from baselined TG values between diets within genotypes. Data expressed as mean ± SD; *P < 0.05; **P < 0.01; ***P < 0.001.
To better understand the CREBH-independent effects of PR on VLDL secretion rate, we looked at de novo VLDL particle biogenesis in WT mice. Hepatic free fatty acid (FFA) uptake (Supplemental Figure 6A), hepatic mRNA expression of genes involved in FFA esterification (Supplemental Figure 6B), total hepatic TG levels after tyloxapol (Supplemental Figure 6C), and hepatic levels of APOB and MTTP proteins involved in VLDL particle production (Supplemental Figure 6D) were all similar between diet groups, with the exception of Gpat1 mRNA, which was significantly reduced upon PR. Interestingly, incorporation of radiolabeled palmitate from a portal vein infusion of a lipid bolus into de novo–synthesized VLDL particles was significantly elevated upon PR (Supplemental Figure 6E), while the lipidation of VLDL particles in tyloxapol-treated animals was not significantly different between diet groups (Supplemental Figure 6F). These data are consistent with an intact ability to lipidate VLDL particles and suggests TG and FFA availability for particle incorporation as a limiting factor in VLDL biogenesis and secretion upon PR.
PR improves TG homeostasis in dyslipidemic models.
We next explored the hypotriglyceridemic effects of PR in different clinically relevant models of metabolic disease. We started with a diet-induced model of obesity and insulin resistance consisting of B6D2F1 mice fed for 3 months on a high-fat diet (HFD; 60% calories from fat, 20% from protein, 20% from carbohydrates). One week on a PF-HFD (60% calories from fat, 40% from carbohydrates; Supplemental Table 1) resulted in similar global metabolic effects, as observed in lean mice, including increased CREBH cleavage (Figure 7A), upregulation of hepatic APOA5 protein expression (Figure 7B), and reduced serum TGs under fed and fasted conditions (Figure 7C) relative to control mice maintained on the HFD.
Figure 7. PR improves TG homeostasis in dyslipidemic models.

(A–H) B6D2F1 mice (A–C and H) or low-density lipoprotein receptor KO (LDLR-KO, D–H) mice were fed a high-fat diet (HFD) for 3 months prior to switching half to an isocaloric protein-free HFD (PF-HFD) for 1 week. Metabolic and molecular analyses were performed after a 4-hour fast. (A and D) Immunoblot of CREBH full-length (FL) and cleaved form (CF) in liver extracts with quantitation below of CF normalized to full-length (arbitrary units [AU]; n = 4–5/group; 2-tailed Student’s t test). (B and E) Immunoblot of APOA5 in liver extracts with quantitation below normalized to tubulin; n = 3–4/group; 2-tailed Student’s t test. (C and G) Serum TG in fed or 4-hour–fasted states as indicated (n = 4–5/group; 2-tailed Student’s t test between diet groups within fed or fasted state). (F) Circulating VLDL-bound APOA5 levels expressed as the ratio of serum APOA5 to APOB-100 (n = 4/group; 2-tailed Student’s t test). (H) Serum FGF21 levels (n = 4/group; 2-tailed Student’s t test within genotype between diets). Data expressed as mean ± SD; *P < 0.05, **P < 0.01, ***P < 0.001.
Because HFD feeding resulted in only modest TG elevation in B6D2F1 mice, we combined HFD with a genetic model lacking LDLR receptor (LDLR-KO). After 3 months on a HFD, 1 week of PF-HFD feeding led to a significant induction of CREBH cleavage (Figure 7D), increased hepatic and VLDL-bound APOA5 expression (Figures 7, E and F), and reduced serum TGs (Figure 7G). Consistent with CREBH and PPARα activation, FGF21 levels were also significantly elevated in both dyslipidemic models (Figure 7H). Together, these results demonstrate that PR can activate CREBH under conditions of metabolic stress triggered by chronic HFD feeding, even in the absence of lipoprotein receptor–mediated lipoprotein uptake.
Moderate PR increases APOA5 expression and reduces TG levels in mice and humans.
Finally, we sought to test whether the increase in APOA5 expression and decrease in circulating TG levels observed in lean and obese rodents on a PF diet can be translated to humans at achievable levels of PR. To this end, we first titrated protein content in the rodent diet from 18%–0%, replacing the remaining calories with isocaloric sucrose. This resulted in a dose-dependent increase in CREBH cleavage (Figure 8A), APOA5 expression (Figure 8B), PERK phosphorylation (Figure 8C), ATF4 expression (Figure 8D), and serum FGF21 (Figure 8E) and a decrease in serum TGs (Figure 8F) within 1 week.
Figure 8. Moderate protein restriction increases APOA5 expression and reduces TG levels in mice and humans.

(A–F) Titration of protein restriction (18%–0% protein replaced with isocaloric sucrose) in B6D2F1 mice for 1 week. Immunoblot analyses of (A) CREBH full-length (FL) and cleaved form (CF), (B) APOA5, (C) PERK phosphorylation, and (D) ATF4 protein expression in whole liver extracts. For quantitation at right of each blot, samples were binned in 3 groups based on the level of dietary protein (high, 18%–10%; medium, 8%–6%; low, 4%–0%; n = 4–6/group; 1-way ANOVA with Dunnett post-hoc test compared with the high protein group). (E and F) Serum FGF21 (E) and TG (F) levels; 1-way ANOVA with Dunnett post-hoc test compared with the 18% protein group. (G–J) Effect of a low-protein diet in humans. Patients were randomized to control or protein restricted (PR, 7%–9% of energy from protein) diets; blood samples were taken before or after 4–6 weeks on the indicated diet (n = 19 per group; paired t test comparing parameter before and after diet). (G) Plasma APOB-100 concentration representative of circulating VLDL particle number. (H) Plasma APOA5. (I) VLDL-bound APOA5 levels expressed as the ratio of plasma APOA5 to APOB-100. (J) Plasma TG levels. Data expressed as mean ± SD; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.001.
Having established that benefits of a PF diet could be recapitulated at moderate levels of PR, we next measured plasma APOB-100, APOA5, and TGs in humans before and after 4–6 weeks of feeding on a customized isocaloric 8%–9% protein diet vs. control subjects who continued to eat normally. These samples were obtained as part of a clinical trial designed to test the effects of PR in patients with prostate cancer for approximately 1 month prior to prostatectomy relative to those maintained on their normal diet (49). As in rodents, PR significantly reduced plasma APOB-100 levels, indicative of a decrease in circulating VLDL particle number (Figure 8G). PR also significantly increased plasma APOA5 (Figure 8H) and the APOA5/APOB-100 ratio (Figure 8I), concomitant with a significant decrease in plasma TG levels (Figure 8J). Together, these data suggest that mechanisms of TG lowering by PR are conserved between mice and humans.
Discussion
A model for CREBH-dependent and -independent mechanisms of PR-mediated hypotriglyceridemia is presented in Figure 9. CREBH activation upon PR is part of a coordinated transcriptional response involving PPARα and mediating an overall increase in FAO on the organismal level through regulation of key players, including APOA5 and FGF21. PR also reduced the rate of VLDL secretion, resulting in VLDL-TG lowering independently of CREBH or activation of the FAO program it participates in.
Figure 9. Model of dietary protein control of TG homeostasis via effects on hepatic VLDL-TG metabolism.

Green lines denote pathways activated during protein restriction; red lines depict downregulated pathways. Dashed lines represent events for which no precise mechanism of regulation is known. Bold characters represent mediators activated during PR. Black arrows connect described effectors with the ultimate TG-lowering benefit of PR. FAO, fatty acid oxidation; ISR, integrated stress response.
APOA5 and FGF21 as effectors of PR-mediated TG lowering.
Here, we found that PR altered hepatic apolipoprotein expression, increasing the levels of APOA5 in liver and VLDL particles, where it stimulated TG clearance via stimulation of peripheral LPL activity (28, 50). Hepatic APOA5 can also reduce TG availability for lipoprotein lipidation by promoting intrahepatic TG lipolysis, thus decreasing the number and rate of VLDL particle export (28, 50, 51). However, future experiments are required to dissect the potential contribution of intrahepatic vs. VLDL-bound APOA5 in PR-mediated TG lowering.
Like APOA5, FGF21 is elevated upon PR (20, 52) — as well as in response to sugar (53) — and has an established role in promoting TG clearance via activation of white and brown adipose tissue (43) and increasing energy expenditure via central and peripheral mechanisms — for example, through increased expression of uncoupling proteins leading to energy dissipation as heat (54, 55). Also, like Apoa5, hepatic Fgf21 is controlled on the transcriptional level by CREBH and PPARα (48). Because FGF21 was not fully upregulated upon PR in CREBH-KO mice, the partial TG lowering in this model could be ascribed to the absence of a normal FGF21 response. However, the observation that FGF21 was normally elevated upon PR in the LiTSC1-KO model but failed to reduce hypertriglyceridemia argues that FGF21 expression alone is insufficient to lower circulating TGs. Considering the dysregulation of APOA5 at baseline and upon PR in the LiTSC1-KO model, and the severe hypertriglyceridemia of the APOA5-KO model, an interdependence between APOA5, FGF21, and perhaps other PPARα targets in TG lowering by PR seems likely. Future studies are required to sort out the relative contribution of each to TG lowering and how these factors interact with one another, as well as the contribution of other macronutrients such as sucrose.
Reciprocal regulation of pro-oxidative programs by PPARα and CREBH upon PR.
Like CREBH, PPARα regulates fat oxidation via transcriptional control of FAO machinery, FGF21, and even APOA5 in humans (mouse Apoa5 lacks a PPARα binding site; ref. 56). Activation of PPARα with fibrates is a pharmacological approach to treating hypertriglyceridemia, often in combination with statins. PPARα also regulates CREBH on the transcriptional level (38, 57) and is activated by PR (57), although — like CREBH — the mechanism of activation remains unclear.
Here, we found that PR is a powerful inducer of CREBH cleavage and activation, and that one of the genes that it regulates directly or indirectly encodes PPARα. Thus, these 2 transcription factors involved in promoting FAO appear to reciprocally regulate each other. This finding also suggests a potentially novel mechanism by which PR can activate PPARα. However, because CREBH is not absolutely required for basal or PR-mediated expression of these genes, some of the TG lowering observed upon PR could be due to residual APOA5, FGF21, and PPARα expression. Future experiments are, thus, required to better understand the relative contribution of these transcription factors to the coordinated expression of genes controlling FAO upon PR on the organismal level.
Mechanisms of CREBH regulation by dietary PR.
While links between dietary amino acid intake and hepatic lipid metabolism have been inferred based on the ability of methionine/choline-deficient diets to increase hepatic steatosis (58), Guo and Cavener first demonstrated a molecular link between amino acid sensing and lipid metabolism (24). They observed that GCN2-KO mice fed a leucine-deficient diet develop liver steatosis due to the failure to suppress SREBP1c-dependent lipogenesis. However, the circulating TG-lowering effects of leucine deficiency (24) or methionine restriction (21), as well as other metabolic responses to PR (17), remain mostly intact, if slightly delayed, in global GCN2-KO mice, similar to our findings here. Interestingly, the ER-bound eIF2α kinase PERK was also activated in liver upon PR, as reported previously upon methionine restriction (21). While this can explain the activation of the ISR in the absence of GCN2, it does not necessarily imply that the ISR is required for TG lowering. Future studies are required to better understand the mechanism of PR-mediated PERK activation and its requirement, if any, in TG lowering upon PR.
Although mTORC1 positively regulates the prolipogenic transcription factor SREBP-1c, LTSC1-KO mice with constitutive hepatic mTORC1 activity do not have fatty livers, likely as a result of compromised insulin signaling in this model (23, 26). Nonetheless, we report here that these mice are hypertriglyceridemic on a complete diet and that this is maintained on PR, correlating with the failure to activate CREBH or APOA5.
The molecular details of CREBH cleavage and activation remain unknown. CREBH was originally identified as an ER stress–responsive transcription factor (37, 46, 59) and was subsequently linked to carbohydrate and TG metabolism (35, 39), as well as fasting (47). PR did not appear to activate ER stress, but it did trigger the ISR through at least 2 different kinases, GCN2 and PERK. While the GCN2 kinase was not required for CREBH activation upon PR, future experiments are required to test the requirement for PERK and whether the ISR or some other ER-related function of PERK is involved.
Decreased hepatic VLDL secretion rate as a CREBH-independent mechanism of TG lowering by PR.
PR also resulted in a reduced VLDL secretion rate independently of CREBH that contributed to the partial hypotriglyceridemic effect observed during short-term PR in CREBH-KO and APOA5-KO models. Interestingly, in the LiTSC1-KO model, which was both hypertriglyceridemic and recalcitrant to PR-mediated TG lowering, basal VLDL secretion rate on a complete diet was lower than in WT and unchanged by PR, while VLDL particle lipidation remained high independently of diet. These data suggest that the change in VLDL secretion rate upon PR, rather than the basal rate per se, may be the critical factor in CREBH-independent TG lowering by PR.
Future experiments are also required to better understand the mechanism underlying the change in secretion rate induced by PR, as neither changes in hepatic TG nor APOB levels could readily account for this. PR mice also produced normally lipidated VLDL particles (albeit at a slower rate) when LPL was inhibited and had an increased capacity to incorporate radiolabeled palmitate into VLDL particles in the context of a bolus injection of lipid into the portal vein. Based on the increase in hepatic FAO controlled by the PPARα program, one possibility is that hepatic TGs are somehow sequestered from VLDL biosynthesis for lipid oxidation.
Translational potential.
Although reduced calorie, fat, and sugar intake and other behavioral modifications such as exercise are the first line of treatment of elevated triglyceride levels (6), the preclinical and clinical data presented here suggest that reduced protein intake can lower TGs, even without CR. This has important implications for translation due to the inherent difficulties associated with enforced food restriction. Most importantly, PR at sustainable levels of 7%–9% of total calorie intake was sufficient to reduce VLDL particle number and increase APOA5 expression concomitant with a reduction in TG levels in humans. However, future studies are required to determine the timing of onset of benefits, as previous studies have observed either no change (13) or increase in TG (60) within 3–7 days of low-protein (8%–10% calories) intake in people, with the additional caveat that both studies also reported a significant increase in total energy intake during this period.
Methods
Humans
Plasma samples from patients before and 4–6 weeks after PR were obtained from consented research volunteers registered under clinicaltrials.gov identifier NCT01692587 to test the effects of PR on prostate cancer growth as reported previously (49). After baseline screening, participants were allocated with a 1:1 ratio into a control (n = 19) or the PR diet group (n = 19). The control subjects continued their usual diet, while the volunteers randomized to the PR diet were fed customized isocaloric PR experimental meals prepared by the Metabolic Kitchen of Washington University CARS (St. Louis, Missouri, USA). In the PR menus, dietary protein intake was decreased to the dietary reference intake (DRI) for total protein. For adult males, the DRI is 56 g per day. To provide the participants with isocaloric menus, 3 calorie levels were available: 2,300 calories; 2,600 calories; and 2,900 calories. The menus were planned to provide a similar percent of calories from protein (7%–9%) for the 3 calorie levels. The intakes of protein were 56–60 g per day. Individualized energy intakes were estimated from a 4-day food diary and 2 formulas for estimating energy expenditure: Harris Benedict equation and Cunningham equation (include body composition and physical activity factors). The PR participants received a calorie level that was estimated to maintain body weight. Compliance with the isocaloric lower protein diet for volunteers randomized to PR was assessed by means of the daily meal checklist.
Mice
Young adult male B6D2F1/J (stock no. 100006) and LDLR-KO (stock no. 005061) mice were purchased from the Jackson Laboratory. APOA5-KO mice on a FVB/NJ background were provided in house. Congenic FVB/NJ controls for the experiments were purchased from the Jackson Laboratory (stock no. 001800). CREBH-KO mice (Creb3l3tm1.1Sad on a mixed 129/CD1/FVB/B6 background) were cryorecovered from a previously described strain (61) at the Jackson Laboratory and bred in our facility. LiTSC1-KO (TSC1fl/fl|Albumin-Cre+/–) and littermate control (TSC1fl/fl|Albumin Cre–/–); liver-specific Raptor–KO (Raptorfl/fl|Albumin-Cre+/–) and littermate control (Raptorfl/fl|Albumin-Cre–/–); and GCN2 WT and -KO littermates (all on a C57BL/6J background) were bred in our facility as described previously (23). Male mice were used in all experiments, except those involving LiTSC1-KO and CREBH-KO mice, in which both male and female mice were used.
Diets
Semipurified diets were purchased from Research Diets. The complete diet was based on D12450BS consisting of 20% calories from proteins derived from enzyme-hydrolyzed casein with additional cystine supplementation, 70% from carbohydrates, and 10% from fats (Supplemental Table 1). In the PF diet, the protein content was replaced with an isocaloric amount of sucrose (MilliporeSigma). For protein titration experiments, a casein-based amino acid formulation made from crystalline amino acids (Ajinomoto) was added at 18, 10, 6, 4, 2, or 0 g per 82 g of PF D12450BS base with sucrose added to 100 g (Supplemental Table 2). HFD (D12492) consisted of 20% calories from protein, 20% from carbohydrates, and 60% calories from fat. PF-HFD was formulated with 40% carbohydrate and 60% fat calories (Supplemental Table 1). All diets were prepared in a final 1% agar (MilliporeSigma, catalog A1296) formulation. Mice were acclimated on an agar-based complete diet for 2 days, followed by AL or restricted (percentage calculated based on intake of complete diet) access to complete or experimental diets for 7 days. For endpoints in the fed state, samples were taken at approximately 9 a.m. For endpoints in the fasted state, food was withdrawn at 9 a.m., and samples were taken approximately 4 hours later.
Body mass and composition analyses
Body mass was determined by measuring body weight on a daily basis at the time of feeding at approximately 5 p.m. Body composition was determined in awake animals using an EchoMRI analyzer system (EchoMRI).
TG measurements
Plasma and serum TG in humans and mice, respectively, were determined using a TG determination kit from MilliporeSigma (catalog TRO100-1KT). Liver TG levels were determined from the mice injected with tyloxapol to estimate hepatic VLDL-TG secretion. For this, approximately 100 mg of frozen liver was homogenized in 600 μl of PBS. TG levels were determined as above, directly from the liver homogenate. TG concentration values were then normalized per mg of tissue.
Recombinant lipoprotein particle labeling and clearance
For lipoprotein clearance experiments, recombinant lipoproteins containing [3H]-triolein (~0.13 μCi/g body weight) were prepared as previously reported (44) and injected via the tail vein into 4-hour–fasted mice, and blood was sampled for radioactivity at the indicated time points.
In vivo labeling, isolation, and clearance of VLDL-TG
Albumin conjugation.
Albumin was conjugated to [3H]-palmitate as follows: Delipidated endotoxin-free BSA (5 g, Thermo Fisher Scientific) was added to 25 ml of 0.9% NaCl solution, prewarmed at 37°C, and adjusted to pH 7.4. Separately, a 50-mM cold palmitate solution was prepared by adding 51.2 mg of palmitate to 4 ml of ethanol. The palmitate was suspended by stirring and heating the solution at 60°C, while adjusting the pH to 8.5 with NaOH. Then, 250 μl of this solution was mixed in a 50 ml conical tube with 1 mCi of tritiated palmitate (Perkin Elmer, catalog NET043001MC) and evaporated under nitrogen. When dried, the BSA solution (20 ml) was added to the hot-cold palmitate mixture, vortexed vigorously, and incubated overnight at 37°C. After filtration through a 0.22-μm filter, the resulting BSA-palmitate complex was analyzed for radioactivity in a Tri-Carb 4910TR liquid scintillation counter (PerkinElmer).
In vivo labeling.
For in vivo labeling, mice on control or PF diet for 1 week were fasted for 4 hours and then injected with tyloxapol at a dose of 500 mg/kg i.v. via the retro-orbital plexus. After 15 minutes, 100 μCi of the [3H]-palmitate-BSA conjugate in 200 μl was injected i.v. via the retro-orbital plexus. Forty-five minutes later, mice were terminally exsanguinated for VLDL particle isolation from serum.
Isolation.
VLDL particles were isolated from serum by density ultracentrifugation. Serum (50 μl minimum, 1 ml maximum) was adjusted to 1 ml final volume with PBS and a density of 1.006 gr/ml with KBr and centrifuged in 1.0 ml clear polycarbonate thick-wall tubes (Beckman Coulter, catalog 355657) using an Optima-XE ultra-centrifuge (Beckman Coulter) with a fixed angle aluminum Type 25 rotor (Beckman Coulter, catalog 347261) at 80,000 g at 14°C for 22 hours. The top 200 μl containing VLDL particles was collected after centrifugation.
In vivo clearance.
VLDL-TG hydrolysis and removal from circulation was determined by i.v. injection of 100,000 decays per minute (dpm) of purified VLDL-TG particles in a volume of 200 μl via the retro-orbital plexus. Blood was drawn at 0.5 minutes to obtain the maximum concentration of administered counts in circulation and, subsequently, at different time points for calculation of VLDL-TG removal from circulation in a T4910TR β scintillation counter (PerkinElmer).
Estimation of hepatic VLDL secretion rate
VLDL secretion rate was estimated by blocking peripheral LPL with tyloxapol (MilliporeSigma; 500 mg/kg in Dulbecco’s PBS (DPBS) i.v. via the retro-orbital plexus) in 4-hour–fasted mice and by subsequently assessing the rate of TG accumulation in serum over time.
Liver uptake of [3H]-palmitate in vivo
After 4 hours of fasting, mice were injected i.v. via the retro-orbital plexus with 200 μl of a 1:1 solution of PBS and [3H]-palmitate-BSA conjugate prepared as described above. After 15 minutes, mice were euthanized, and 120 mg of liver tissue was incubated in 1 ml of Solvable (Perkin-Elmer, catalog 6NE9100) at 65°C until fully dissolved. The digestion (400 μl) was added to 4 ml of radiation scintillation cocktail (EcoLume, MP Biomedicals) and measured in a Tri-Carb 4910TR liquid scintillation counter (PerkinElmer).
Hepatic VLDL lipidation capacity in vivo
After 4 hours of fasting, mice were injected with tyloxapol i.v. via the retroorbital plexus at a final concentration of 500 mg/kg. After 45 minutes, anesthetized mice were injected through the portal vein with 50,000 dpm of [3H]-palmitate-BSA conjugate prepared as described above, and serum samples were collected 10 and 15 minutes later. VLDL particles were isolated by ultracentrifugation as above. The VLDL particle fraction (50 μl) was added to 4 ml of scintillation fluid cocktail (EcoLume, MP Biomedicals) and read in a Tri-Carb 4910TR liquid scintillation counter (PerkinElmer). VLDL-TGs were then determined from the particles, and normalized VLDL incorporation index was obtained by dividing the cpm over the concentration of TGs in 50 μl of VLDL particles.
Lipidomic analysis
Serum lipidomic profiling was conducted using liquid chromatography separations coupled with positive ion mode mass spectrometry–based analysis on a Thermo Exactive Mass Spectrometer as described previously (62, 63).
FPLC analysis
Lipoprotein studies were performed as described previously (44). Briefly, pooled plasma was separated by FPLC (GE Healthcare) and the lipid concentration was determined in each fraction with Cobas Integra Reagent for triglyceride detection adapted to microtiter plates (Roche Diagnostics).
Determination of APOA5 and APOB
APOA5 and APOB-100 were measured by ELISA from mouse serum diluted in PBS (1:10 for APOA5, LS-F9345; 1:2,000 for APOB-100, LS-F5909) and human plasma diluted 1:50 for APOA5 (LS-F5818) and 1:200 for APOB-100 (LS-F22515, Lifespan Biosciences) according to the manufacturer’s instructions. Relative APOA5 amount per VLDL particle was obtained by dividing serum APOA5 by serum APOB concentration and was expressed in arbitrary units.
VLDL particle lipidation measurement
VLDL fractions were isolated as above. VLDL-TG concentration was determined and normalized to an equal concentration across samples (nTG expressed in mg/ml), by taking a given volume (v, expressed in µl), and diluting it with PBS. This solution was further diluted 1:4 (dil) with the diluent buffer provided by the APOB ELISA kit (LifeSpan Biosceince, catalog LS-F5909). That dilution (100 μl) was then used to determine the total APOB protein concentration by adding it undiluted to the ELISA kit and then measuring it following the manufacturer’s instructions. TG lipidation state of APOB-100–containing particles was determined using the following formula:
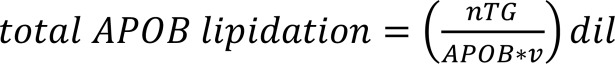 ,
,
where nTG refers to the normalized TG concentration (in mg/ml), APOB refers to the purified VLDL APOB concentration (expressed in μg/ml), and dil is the dilution factor (dil = 4).
Rapamycin treatment
Mice were injected with vehicle (0.25% PEG 400 in PBS) or rapamycin (1 mg/kg i.p.) daily for up to 4 days.
Immunoblotting
Snap-frozen tissues were homogenized in 1 ml of lysis buffer (10% glycerol, 1% NP-40, 1 mM MgCl2, 150 mM NaCl, 50 mM β-glycerophosphate, 20 mM Tris-HCl, pH 7.4) supplemented with a protease inhibitor and phosphatase inhibitor cocktails (Thermo Fisher Scientific) with an IKA T10 Ultra-Turrax tissue dispenser. Protein content was determined using the Pierce BCA protein assay kit (Thermo Fisher Scientific), and 40 μg of protein extract was resolved by SDS-PAGE and transferred to Immobilon-P polyvinylidene difluoride (PDVF) membranes (MilliporeSigma) for immunoblotting. Membranes were incubated overnight at 4°C with primary antibody diluted in antibody buffer (5% BSA, 0.1% sodium azide in TBST). After washing in TBST, membranes were incubated 90 minutes at room temperature with goat anti–mouse (catalog P0447) or goat anti–rabbit (catalog P0449) IgG conjugated with horseradish peroxidase (Dako) diluted 1:5,000 in 5% skim milk. After extensive washing with TBST, blots were developed using the Super Signal West Femto chemiluminescent substrate kit (Pierce, Thermo Fisher Scientific).
Antibodies
For a list of all primary antibodies, please refer to Supplemental Table 3.
Gene expression analysis
Frozen tissue samples were first homogenized in Qiazol (Qiagen) using a tissue grinder and then RNA was isolated using the chloroform:isopropanol precipitation method. cDNA was synthesized from 1 μg of total RNA by random hexamer priming using the Verso cDNA synthesis kit (Thermo Fisher Scientific). qPCR reactions were performed with 20 ng of cDNA as a template using Taq-Pro DNA polymerase (Danville Scientific) and SYBR green (Lonza) in a CFX384 touch Real-Time PCR detection system (Bio-Rad). The primers used in this study are provided in Supplemental Table 4. Gene expression was determined using the equation 2ΔΔCT, with Hprt for normalization. Data are expressed as fold change relative to the control group, which is typically WT mice fed a complete diet on an AL basis.
Cell transfection studies
Human HepG-2 hepatoma cells (ATCC HB-8065) cultured in optimized minimal essential media (Opti-MEM) supplemented with 10% FBS and 1× penicillin-streptomycin, were trypsinized and cultured at a density of 5 × 105 cells in 10-cm cell culture dishes for 2 days until they reached 90% confluence. For transfection, 25 μg of plasmid DNA containing an expression cassette coding for an hemagglutinin epitope linked to the amino-terminus active fragment of CREBH (amino acids 1-332, provided by Ann-Hwee Lee, from Cornell University, New York, New York, USA) vs. empty vector (46) was transfected in triplicate using lipofectamine (Thermo Fisher Scientific) according to the manufacturer’s instructions.
ChIP
ChIP analysis was performed using the ChIP-IT ChIP-Seq kit (Active Motif), following the manufacturer’s instructions, with the following specifications: a total of 300 mg of frozen liver tissues were homogenized in the kit’s buffer and sonicated in a 240 V, 50 Hz Misonix ultrasonic homogenizer at 25% of amplitude — pulsed 30 seconds on, 30 seconds off — for 10 minutes per tissue, using a 3.2 mm microtip probe. A total of 4 μg of Rb anti-CREBH antibody were used. After reversal of cross-linking and DNA purification, qPCR of the region flanking the TCCACGTGGT-CREBH binding site in the ApoA5 mouse promoter was performed using the following primers: forward: 5′-TTTACAGCAGTCATACATAGG-3′; reverse: 5′-ACCCTCCTTTCCTATATCC-3′.
Confocal microscopy
Frozen liver samples embedded in OCT and sectioned using a cryostat were fixed in 4% (v/v) paraformaldehyde (PFH) for 10 minutes, washed in DPBS, and permeabilized in 0.25% Triton X-100 in TBS. After washing in TBS, sections were blocked for 30 minutes with blocking buffer (10% [v/v] normal goat serum, 1% [w/v] BSA, glycine 0.3 M) and incubated overnight with primary antibody. After washing, sections were incubated with a secondary goat anti–rabbit Alexa 568 conjugated (also diluted in blocking buffer) for 2 hours at room temperature. After incubation, tissue sections were washed and mounted in Vectashield mounting medium containing DAPI (Vector Laboratories). Cells and tissues were visualized using a Zeiss LSM 880 laser scanning confocal microscope and analyzed using ImageJ (NIH) and Adobe Photoshop 11.0.2 (Adobe Systems).
Statistics
Statistical analyses were performed in GraphPad Prism 6.0 or Microsoft Excel using the indicated tests with P < 0.05 considered significant. Two-way ANOVA main effect statistics are contained in Supplemental Table 5.
Study approval
Studies involving human subjects were approved by the IRB of the Washington University in St. Louis (no. 201011804). All experiments involving mice were conducted with the approval of the Harvard Medical Area IACUC.
Author contributions
JHTV, LF, and JRM conceptualized and designed the research; JHTV, JSR, AB, MRM, AA, JH, CKO, BSK, LF, and JRM planned experiments; JHTV, JSR, AB, PKL, MRM, AA, VT, NV, BB, LEB, PM, KT, GSK, AL, RG, SSB, EH, ADB, RSF, GLA, and AT performed experiments; JHTV, JSR, AB, MRM, AA, JH, LF, and BSK interpreted the data; JHTV and JRM wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Gökhan Hotamisligil for insightful discussions and sharing of equipment and reagents; Suddha Biddinger for informative discussions; Frank Sacks for critical reading of the manuscript; and Ann-Hwee Lee and Kezhong Zhang for providing reagents. We acknowledge the assistance provided by the Dana-Farber/Harvard Cancer Center rodent histopathology core and the Microscopy Resources from the Quad (MicRoN) microscopy core facility at HMS. This work was supported by American Heart Association Grant-in-Aid 16GRNT27090006 to CKO; NIH (RO1DK090629, R01AG036712) and Charoen Pokphand Group to JRM; and Nutrition Obesity Research Center to BSK. The Fontana lab is supported by grants from the Bakewell Foundation, the Longer Life Foundation (an RGA/Washington University Partnership), and the National Center for Research Resources (UL1 RR024992). AB was supported by a Deutsche Forschungsgemeinschaft Research Fellowship (BA 4925/1-1) and the DZHK.
Version 1. 11/02/2018
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2018;3(20):e99470. https://doi.org/10.1172/jci.insight.99470.
Contributor Information
Justin S. Reynolds, Email: jsr4rr@gmail.com.
Alexander Bartelt, Email: abartelt@hsph.harvard.edu.
P. Kent Langston, Email: plangston@g.harvard.edu.
Alessandro Arduini, Email: aarduini@broadinstitute.org.
Valeria Tosti, Email: vtosti@dom.wustl.edu.
Nicola Veronese, Email: nicola.veronese@unipd.it.
Beatrice Bertozzi, Email: beatricebertozzi@hotmail.it.
Lear E. Brace, Email: learbrace@gmail.com.
Pedro Mejia, Email: mejia.jp@gmail.com.
Kaspar Trocha, Email: kmtrocha@gmail.com.
Alban Longchamp, Email: alban.longchamp@gmail.com.
Eylul Harputlugil, Email: eylulharputlugil@gmail.com.
Bruce S. Kristal, Email: bkristal@partners.org.
Luigi Fontana, Email: lfontana@dom.wustl.edu.
References
- 1.Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet. 2014;384(9943):626–635. doi: 10.1016/S0140-6736(14)61177-6. [DOI] [PubMed] [Google Scholar]
- 2.Christian JB, Arondekar B, Buysman EK, Jacobson TA, Snipes RG, Horwitz RI. Determining triglyceride reductions needed for clinical impact in severe hypertriglyceridemia. Am J Med. 2014;127(1):36–44.e1. doi: 10.1016/j.amjmed.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 3.Sarwar N, et al. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115(4):450–458. doi: 10.1161/CIRCULATIONAHA.106.637793. [DOI] [PubMed] [Google Scholar]
- 4.Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298(3):299–308. doi: 10.1001/jama.298.3.299. [DOI] [PubMed] [Google Scholar]
- 5.Hokanson JE, Austin MA. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk. 1996;3(2):213–219. doi: 10.1097/00043798-199604000-00014. [DOI] [PubMed] [Google Scholar]
- 6.Watts GF, Ooi EM, Chan DC. Demystifying the management of hypertriglyceridaemia. Nat Rev Cardiol. 2013;10(11):648–661. doi: 10.1038/nrcardio.2013.140. [DOI] [PubMed] [Google Scholar]
- 7.Speakman JR, Mitchell SE. Caloric restriction. Mol Aspects Med. 2011;32(3):159–221. doi: 10.1016/j.mam.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Colman RJ, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fontana L, Meyer TE, Klein S, Holloszy JO. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc Natl Acad Sci USA. 2004;101(17):6659–6663. doi: 10.1073/pnas.0308291101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mauro CR, et al. Preoperative dietary restriction reduces intimal hyperplasia and protects from ischemia-reperfusion injury. J Vasc Surg. 2016;63(2):500–9.e1. doi: 10.1016/j.jvs.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hine C, et al. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell. 2015;160(1-2):132–144. doi: 10.1016/j.cell.2014.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robertson LT, et al. Protein and Calorie Restriction Contribute Additively to Protection from Renal Ischemia Reperfusion Injury Partly via Leptin Reduction in Male Mice. J Nutr. 2015;145(8):1717–1727. doi: 10.3945/jn.114.199380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maida A, et al. A liver stress-endocrine nexus promotes metabolic integrity during dietary protein dilution. J Clin Invest. 2016;126(9):3263–3278. doi: 10.1172/JCI85946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell JR, et al. Short-term dietary restriction and fasting precondition against ischemia reperfusion injury in mice. Aging Cell. 2010;9(1):40–53. doi: 10.1111/j.1474-9726.2009.00532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malloy VL, Krajcik RA, Bailey SJ, Hristopoulos G, Plummer JD, Orentreich N. Methionine restriction decreases visceral fat mass and preserves insulin action in aging male Fischer 344 rats independent of energy restriction. Aging Cell. 2006;5(4):305–314. doi: 10.1111/j.1474-9726.2006.00220.x. [DOI] [PubMed] [Google Scholar]
- 16.Plaisance EP, et al. Dietary methionine restriction increases fat oxidation in obese adults with metabolic syndrome. J Clin Endocrinol Metab. 2011;96(5):E836–E840. doi: 10.1210/jc.2010-2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laeger T, et al. Metabolic Responses to Dietary Protein Restriction Require an Increase in FGF21 that Is Delayed by the Absence of GCN2. Cell Rep. 2016;16(3):707–716. doi: 10.1016/j.celrep.2016.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solon-Biet SM, et al. Dietary Protein to Carbohydrate Ratio and Caloric Restriction: Comparing Metabolic Outcomes in Mice. Cell Rep. 2015;11(10):1529–1534. doi: 10.1016/j.celrep.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hine C, et al. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell. 2015;160(1-2):132–144. doi: 10.1016/j.cell.2014.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laeger T, et al. FGF21 is an endocrine signal of protein restriction. J Clin Invest. 2014;124(9):3913–3922. doi: 10.1172/JCI74915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wanders D, et al. Role of GCN2-Independent Signaling Through a Noncanonical PERK/NRF2 Pathway in the Physiological Responses to Dietary Methionine Restriction. Diabetes. 2016;65(6):1499–1510. doi: 10.2337/db15-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng W, et al. Surgical stress resistance induced by single amino acid deprivation requires Gcn2 in mice. Sci Transl Med. 2012;4(118):118ra11. doi: 10.1126/scitranslmed.3002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harputlugil E, Hine C, Vargas D, Robertson L, Manning BD, Mitchell JR. The TSC complex is required for the benefits of dietary protein restriction on stress resistance in vivo. Cell Rep. 2014;8(4):1160–1170. doi: 10.1016/j.celrep.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo F, Cavener DR. The GCN2 eIF2alpha kinase regulates fatty-acid homeostasis in the liver during deprivation of an essential amino acid. Cell Metab. 2007;5(2):103–114. doi: 10.1016/j.cmet.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 25.Laplante M, Sabatini DM. mTORC1 activates SREBP-1c and uncouples lipogenesis from gluconeogenesis. Proc Natl Acad Sci USA. 2010;107(8):3281–3282. doi: 10.1073/pnas.1000323107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yecies JL, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011;14(1):21–32. doi: 10.1016/j.cmet.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Dijk KW, Rensen PC, Voshol PJ, Havekes LM. The role and mode of action of apolipoproteins CIII and AV: synergistic actors in triglyceride metabolism? Curr Opin Lipidol. 2004;15(3):239–246. doi: 10.1097/00041433-200406000-00002. [DOI] [PubMed] [Google Scholar]
- 28.Schaap FG, et al. ApoAV reduces plasma triglycerides by inhibiting very low density lipoprotein-triglyceride (VLDL-TG) production and stimulating lipoprotein lipase-mediated VLDL-TG hydrolysis. J Biol Chem. 2004;279(27):27941–27947. doi: 10.1074/jbc.M403240200. [DOI] [PubMed] [Google Scholar]
- 29.Kluger M, Heeren J, Merkel M. Apoprotein A-V: an important regulator of triglyceride metabolism. J Inherit Metab Dis. 2008;31(2):281–288. doi: 10.1007/s10545-008-0863-4. [DOI] [PubMed] [Google Scholar]
- 30.Nilsson SK, Heeren J, Olivecrona G, Merkel M. Apolipoprotein A-V; a potent triglyceride reducer. Atherosclerosis. 2011;219(1):15–21. doi: 10.1016/j.atherosclerosis.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 31.Pennacchio LA, et al. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294(5540):169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- 32.Jakel H, et al. The liver X receptor ligand T0901317 down-regulates APOA5 gene expression through activation of SREBP-1c. J Biol Chem. 2004;279(44):45462–45469. doi: 10.1074/jbc.M404744200. [DOI] [PubMed] [Google Scholar]
- 33.Prieur X, Coste H, Rodriguez JC. The human apolipoprotein AV gene is regulated by peroxisome proliferator-activated receptor-alpha and contains a novel farnesoid X-activated receptor response element. J Biol Chem. 2003;278(28):25468–25480. doi: 10.1074/jbc.M301302200. [DOI] [PubMed] [Google Scholar]
- 34.Prieur X, Huby T, Coste H, Schaap FG, Chapman MJ, Rodríguez JC. Thyroid hormone regulates the hypotriglyceridemic gene APOA5. J Biol Chem. 2005;280(30):27533–27543. doi: 10.1074/jbc.M503139200. [DOI] [PubMed] [Google Scholar]
- 35.Lee JH, et al. The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat Med. 2011;17(7):812–815. doi: 10.1038/nm.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song KH, Park AY, Kim JE, Ma JY. Identification and characterization of cyclic AMP response element-binding protein H response element in the human apolipoprotein A5 gene promoter. Biomed Res Int. 2013;2013:892491. doi: 10.1155/2013/892491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang K, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124(3):587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 38.Danno H, et al. The liver-enriched transcription factor CREBH is nutritionally regulated and activated by fatty acids and PPARalpha. Biochem Biophys Res Commun. 2010;391(2):1222–1227. doi: 10.1016/j.bbrc.2009.12.046. [DOI] [PubMed] [Google Scholar]
- 39.Zhang C, et al. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology. 2012;55(4):1070–1082. doi: 10.1002/hep.24783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cefalù AB, et al. Novel CREB3L3 Nonsense Mutation in a Family With Dominant Hypertriglyceridemia. Arterioscler Thromb Vasc Biol. 2015;35(12):2694–2699. doi: 10.1161/ATVBAHA.115.306170. [DOI] [PubMed] [Google Scholar]
- 41.von Holstein-Rathlou S, et al. FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet Taste Preference by the Liver. Cell Metab. 2016;23(2):335–343. doi: 10.1016/j.cmet.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kharitonenkov A, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115(6):1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schlein C, et al. FGF21 Lowers Plasma Triglycerides by Accelerating Lipoprotein Catabolism in White and Brown Adipose Tissues. Cell Metab. 2016;23(3):441–453. doi: 10.1016/j.cmet.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 44.Bartelt A, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011;17(2):200–205. doi: 10.1038/nm.2297. [DOI] [PubMed] [Google Scholar]
- 45.Vu-Dac N, et al. Apolipoprotein A5, a crucial determinant of plasma triglyceride levels, is highly responsive to peroxisome proliferator-activated receptor alpha activators. J Biol Chem. 2003;278(20):17982–17985. doi: 10.1074/jbc.M212191200. [DOI] [PubMed] [Google Scholar]
- 46.Xu X, Park JG, So JS, Hur KY, Lee AH. Transcriptional regulation of apolipoprotein A-IV by the transcription factor CREBH. J Lipid Res. 2014;55(5):850–859. doi: 10.1194/jlr.M045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee MW, et al. Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab. 2010;11(4):331–339. doi: 10.1016/j.cmet.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 48.Kim H, et al. Liver-enriched transcription factor CREBH interacts with peroxisome proliferator-activated receptor α to regulate metabolic hormone FGF21. Endocrinology. 2014;155(3):769–782. doi: 10.1210/en.2013-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fontana L, et al. Decreased Consumption of Branched-Chain Amino Acids Improves Metabolic Health. Cell Rep. 2016;16(2):520–530. doi: 10.1016/j.celrep.2016.05.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Merkel M, et al. Apolipoprotein AV accelerates plasma hydrolysis of triglyceride-rich lipoproteins by interaction with proteoglycan-bound lipoprotein lipase. J Biol Chem. 2005;280(22):21553–21560. doi: 10.1074/jbc.M411412200. [DOI] [PubMed] [Google Scholar]
- 51.Weinberg RB, et al. Structure and interfacial properties of human apolipoprotein A-V. J Biol Chem. 2003;278(36):34438–34444. doi: 10.1074/jbc.M303784200. [DOI] [PubMed] [Google Scholar]
- 52.Solon-Biet SM, et al. Defining the Nutritional and Metabolic Context of FGF21 Using the Geometric Framework. Cell Metab. 2016;24(4):555–565. doi: 10.1016/j.cmet.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 53.Potthoff MJ, et al. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci USA. 2009;106(26):10853–10858. doi: 10.1073/pnas.0904187106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu J, et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58(1):250–259. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Owen BM, et al. FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab. 2014;20(4):670–677. doi: 10.1016/j.cmet.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prieur X, et al. Differential regulation of the human versus the mouse apolipoprotein AV gene by PPARalpha. Implications for the study of pharmaceutical modifiers of hypertriglyceridemia in mice. Biochim Biophys Acta. 2009;1791(8):764–771. doi: 10.1016/j.bbalip.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 57.Contreras AV, Torres N, Tovar AR. PPAR-α as a key nutritional and environmental sensor for metabolic adaptation. Adv Nutr. 2013;4(4):439–452. doi: 10.3945/an.113.003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology. 2003;38(1):123–132. doi: 10.1053/jhep.2003.50307. [DOI] [PubMed] [Google Scholar]
- 59.Bailey D, Barreca C, O’Hare P. Trafficking of the bZIP transmembrane transcription factor CREB-H into alternate pathways of ERAD and stress-regulated intramembrane proteolysis. Traffic. 2007;8(12):1796–1814. doi: 10.1111/j.1600-0854.2007.00654.x. [DOI] [PubMed] [Google Scholar]
- 60.Gosby AK, et al. Raised FGF-21 and Triglycerides Accompany Increased Energy Intake Driven by Protein Leverage in Lean, Healthy Individuals: A Randomised Trial. PLoS One. 2016;11(8):e0161003. doi: 10.1371/journal.pone.0161003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luebke-Wheeler J, et al. Hepatocyte nuclear factor 4alpha is implicated in endoplasmic reticulum stress-induced acute phase response by regulating expression of cyclic adenosine monophosphate responsive element binding protein H. Hepatology. 2008;48(4):1242–1250. doi: 10.1002/hep.22439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bird SS, Marur VR, Sniatynski MJ, Greenberg HK, Kristal BS. Serum lipidomics profiling using LC-MS and high-energy collisional dissociation fragmentation: focus on triglyceride detection and characterization. Anal Chem. 2011;83(17):6648–6657. doi: 10.1021/ac201195d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bird SS, Marur VR, Sniatynski MJ, Greenberg HK, Kristal BS. Lipidomics profiling by high-resolution LC-MS and high-energy collisional dissociation fragmentation: focus on characterization of mitochondrial cardiolipins and monolysocardiolipins. Anal Chem. 2011;83(3):940–949. doi: 10.1021/ac102598u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.