

Abstract
Following myocardial infarction (MI), timely resolution of inflammation promotes wound healing and scar formation while limiting excessive tissue damage. Resolution promoting factors (RPFs) are agents that blunt leukocyte trafficking and inflammation, promote necrotic and apoptotic cell clearance, and stimulate scar formation. Previously identified RPFs include mediators derived from lipids (resolvins, lipoxins, protectins, and maresins), proteins (glucocorticoids, annexin A1, galectin 1, and melanocortins), or gases (CO, H2S, and NO). Matrix metalloproteinase-12 (MMP-12; macrophage elastase) has shown promising RPF qualities in a variety of disease states. We review here the evidence that MMP-12 may serve as a novel RPF with potential therapeutic efficacy in the setting of MI.
Keywords: inflammation, extracellular matrix, inflammation resolution, macrophage, neutrophil, fibroblast
Graphical abstract
1. Introduction
Myocardial infarction (MI) occurs due to blockage of the coronary artery that supplies the left ventricle (LV) with blood, leading to irreversible ischemic damage of the downstream myocardium. Within 30 minutes of ischemia, cardiomyocytes undergo necrosis that release damage-associated molecular factors that initiate an inflammatory cascade.(1) While inflammation is required to initiate a wound healing response, excessive inflammation can exacerbate tissue damage both in the infarct and remote areas of the LV. The infarcted portion of the LV undergoes a wound healing response and is eventually replaced with scar tissue. The wound healing cascade consists of three sequential but overlapping phases: inflammatory, proliferative, and maturation phases.(2, 3) For appropriate scar formation to occur, the necrotic myocytes in the ischemic area must be broken down and new extracellular matrix (ECM) synthesized in a balance of replacement scar formation that provides structure to the infarct while preventing the development of an overly stiff myocardium.(2, 3)
A promising therapeutic strategy for treating MI is to stimulate the production of endogenous inflammation resolution promoting factors (RPFs) to enhance wound healing, generate a stable scar, and limit progression to heart failure.(4-8) RPFs regulate leukocyte trafficking, blunt inflammatory mediator production, and promote necrotic cell clearance and tissue repair. The current RPF catalogue includes lipid mediators (resolvins, lipoxins, protectins, and maresins), proteins and bioactive peptides (glucocorticoids, annexin A1, galectin 1, and melanocortins), and gases (NO, H2S, and CO).(9-11)
Of the RPFs identified to date, lipid mediators are the most studied. In particular, immune cells rapidly produce resolvins after injury as a means to dampen inflammation.(9-16) Lipid mediator RPFs are enzymatically produced from polyunsaturated fatty acids and are rapidly produced by immune cells.(9-15, 17) Of the polyunsaturated fatty acid-derived resolvins, resolvin D1 binds to the formyl peptide receptor 2, resolvin E1 binds to the chemoattractant receptor 23, and both resolvins bind the leukotriene B4 receptor BLT1.(9) In post-MI mice, resolvin D1 treatment stimulates an earlier neutrophil exit from the LV infarct and spleen, reduces pro-fibrotic collagen I α and tenascin C, and improves fractional shortening.(15) Resolvin D1 also reduces macrophage numbers and stimulates a shift from pro- to antiinflammatory phenotype by downregulating the CC chemokine receptor 5 and CXC motif chemokine ligand Cxcl5 while upregulating mannose receptor c-type 1, arginase-1, and chitinase-like protein 3 (Ym1).(15) Annexin A1 is a small protein expressed by macrophages and neutrophils that serves as an RPF after MI by binding to the formyl peptide receptor-2 and inhibiting neutrophil pro-inflammatory actions.(18) These results provide a template for studies of other RPFs and serve as literature controls for comparisons (Table 1).
Table 1.
Criteria for classification as a resolution promoting factor (RPF) and comparison of matrix metalloproteinase (MMP)-12 to known RPFs
| MMP-12 | Resolvins | Annexin A1 | |
|---|---|---|---|
| Increases after MI | Yes(8) | Yes(91) | Yes(92) |
| Regulates leukocyte trafficking | Yes | Yes | Yes |
| ↑ monocyte/ neutrophil chemotaxis after injury(42, 44) | ↓ neutrophil transendothelial migration(93)
↓ neutrophil chemotaxis from spleen(15) |
↓ CD68+ macrophages after MI(92)
↓ neutrophil transendothelial migration(94) |
|
| Reduces proinflammation | Yes | Yes | Yes |
| ↓ IL1rl, IL6ra, IL11, and Cxcr5 after MI(8)
↓ IFN-α, CCL2, complement proteins, NETS, OPN, CD 14 (46, 48, 49, 51, 52) |
↓ cytokine and chemokines(95)
↓ NF-κB, STAT3, MAPK activity(95) |
↓ pro-inflammatory and pro-thrombotic cytokines(94) | |
| Increases antiinflammation | Yes | Yes | Yes |
| ↑ IL-13 activity(55, 56)
↑ TGF-β1 activity(25) ↑ IL-8 secretion(96) ↑ Th2 cell activity(58) ↑ globular adiponectin(60) |
↑ SIRT1 expression(95)
↑ M2 markers—Mrc1, Arg1, Ym1(15) |
↑ IL-10 expression(92)
↑ TGF-β secretion(97) Activation of lipoxin A4 receptor(98) |
|
| Promotes cell clearance | Yes | Yes | Yes |
| ↑ neutrophil clearance from arthritic joints(48)
↑ neutrophil apoptosis after MI(8) ↑ macrophage phagocytosis(65) |
↑ neutrophil efflux after MI(99) ↑ neutrophil apoptosis(100) |
↑ macrophage phagocytosis of apoptotic neutrophils(101) | |
| Promotes scar formation and angiogenesis | Yes | No | Yes |
| ↑ degradation of myocyte debris(73)
↑ ECM turnover and remodeling(8, 25) ↓ tissue degradation by other MMPs(8, 55) ↑ TGF-β1 activity in myofibroblasts(80) ↑ fibroblast proliferation(82) |
↓ fibrosis/collagen accumulation after MI(15) | ↑ fibroblast activation and collagen deposition(102) |
Arg 1- arginase 1; CCL- CC chemokine ligand; CD- cluster of differentiation; CXCR- CXC chemokine receptor; ECM- extracellular matrix; IFN- interferon; IL-interleukin; MAPK- mitogen activated protein kinase; MRC1- mannose receptor C type 1; NET- neutrophil extracellular trap; NF-κB- nuclear factor kB; OPN- osteopontin; Stat- signal transducer and activator of transcription; TGF- transforming growth factor; Ym1- chitinase-like protein 3
Wound healing following MI is a dynamic process that depends on a temporal succession of events, in which matrix metalloproteinases (MMPs) play critical roles.(19, 20) MMPs are a family of protease enzymes with a catalytic zinc ion. Collectively, MMPs degrade a number of ECM and intracellular proteins.(1, 3, 21, 22) Endogenous MMP inhibitors include alpha 2 macroglobulin in the circulation and tissue inhibitor of metalloproteinases (TIMPs) locally that provide feedback to temper MMP proteolytic activity.(3) MMPs and TIMPs are critically involved in ECM remodeling after MI, including having direct and indirect roles in inflammation modification. (3)
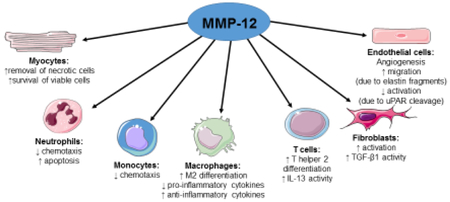
To date, no RPF has translated to the clinic, indicating a need to better understand the mechanisms involved and the need to identify additional RPF candidates. MMP-12 (macrophage metalloelastase) cleaves a number of MI-relevant ECM substrates including elastin, fibronectin, heparan sulfate, laminin, type IV collagen, and vitronectin.(3, 23) We have reported that MMP-12 inhibition after MI, using the selective phosphinic peptide inhibitor RXP 470.1, impaired CD44 and hyaluronan interaction to suppress neutrophil apoptosis and prolong inflammation, which worsened LV physiology.(8) This led to the concept that MMP-12 may serve a beneficial role in MI remodeling and may be working as a previously unidentified RPF. To demonstrate that MMP-12 is an RPF, there are a number of fulfillment criteria listed in Table 1 that should be met. Here, we summarize the current knowledgebase on MMP-12 in the post-MI myocardium, with the majority of information deriving from our own inhibition study. Where information is not currently available in the myocardium, we borrow from other fields to provide insight into possible MMP-12 roles in the post-MI LV (Figure 1).
Figure 1. Actions by MMP-12 as a potential resolution promoting factor following MI.

MMP-12 expression is regulated by several pathophysiological, pharmacological, and endogenous factors. MMP-12 has diverse actions on cardiomyocytes, neutrophils, monocytes and macrophages, lymphocytes, fibroblasts, and endothelial cells that may promote inflammation resolution and scar formation. Cell images were obtained from Servier Medical Art© (http://www.servier.com/).
2.1. MMP-12 is elevated after MI and with aging.
MMP-12 increases at MI day 1 and remains elevated through day 7.(8) Of interest, the LV MI neutrophil was identified as a novel MMP-12 source, as circulating blood neutrophils showed no expression and MI day 1 neutrophils isolated from the infarct zone showed robust expression. MMP-12 expression is induced by a number of MI relevant factors, including TGF-β1, IL-4, and hypoxia inducible factor (HIF)-1α, and is decreased by interferon gamma (IFN-γ).(24-27) In humans, MMP-12 is significantly elevated in the serum of patients with carotid atherosclerosis and ST-segment elevation MI, indicating MMP-12 persists through the pathological continuum initiating with atherosclerosis and culminating in MI. (28, 29)
Aging is an important modulator of MMP-12 at baseline and after MI. Aging stimulates LV hypertrophy, inflammation, and fibrosis, resulting in impaired diastolic function.(30-32) Older patients have a higher mortality rate after MI, in part due to baseline differences.(33) In the myocardium, MMP-12 increases in mice with age, and correlates positively with LV mass.(30, 34) MMP-12 increases in the insoluble fraction and decreases in soluble fraction, which may indicate MMP-12 is more associated with insoluble ECM substrates and less associated with soluble substrates such as tumor necrosis factor alpha (TNFα).(35) In response to pressure overload, MMP-12 increases to a higher extent in older mice compared to the younger cohort.(36) The higher MMP-12 in the older pressure-overloaded mice corresponds with less LV remodeling (less hypertrophy and dilation) and reduced mortality rates, indicating MMP-12 serves a protective role in aging hearts with pathology.(36) These effects may be due to MMP-12 cleavage of urokinase-type-plasminogen activator receptor on endothelial cells to inhibit pathological angiogenesis in response to LV hypertrophy.(36) MMP-12, therefore, associates with cardioprotective roles after MI and aging.
2.2. MMP-12 regulates leukocyte trafficking.
MI initiates a robust inflammatory response characterized by release of pro-inflammatory cytokines and chemokines and the influx of leukocytes.(2, 37-39) Monocyte infiltration begins first, followed by the influx of neutrophils at a much higher amplitude, such that by 24 h after MI neutrophils are the predominant leukocyte in the infarct region.(40) MMPs regulate the inflammatory response to MI by proteolytic processing chemotactic factors that stimulate leukocyte recruitment.(23, 41) MMP-12 roles in post-MI leukocyte infiltration have not been defined. In the smoke-induced emphysema mouse model, MMP-12 induces rapid monocyte and neutrophil influx into the lung through proteolytic processing of elastin, generating fragments that incite a strong chemoattractant response.(42) In a mouse model of lipopolysaccharide-induced lung injury, MMP-12 null mice had delayed neutrophil resolution.(43) One mechanism of action was through MMP-12 cleavage of the ELR+ motif at Glu-Leu amino acids to inactivate CXCL1, 2, 3, 5, and 8. MMP-12 also inactivates CCL2, 7, 8 and 13 to discontinue neutrophil recruitment. In the injured cornea, MMP-12 promotes epithelial proliferation and migration and early neutrophil infiltration to promote healing.(44, 45) In summary, MMP-12 is a critical regulator of leukocyte trafficking, and promotes both the initial influx and discontinuation of neutrophils into the site of injury. The net effect as assessed by MMP-12 null mice is to limit the chronic persistence of neutrophils.
2.3. MMP-12 turns off pro-inflammatory signaling.
MMP-12 inhibition after MI prolongs upregulation of pro-inflammatory molecules IL1rl, IL6ra, IL11, and Cxcr5, and worsens cardiac physiology (Table 2).(8) In addition to serving as a proteolytic mediator of inflammation signaling, MMP-12 is a transcription factor.(46) MMP-12 is endocytosed by cardiomyocytes and exerts transcriptional activity at the NFKBIA promoter, supporting interferon-alpha (IFN-α) production and anti-viral activity.(46) As a protease, MMP-12 later cleaves IFN-α to inhibit its activity and promote resolution of inflammation.(47) MMP-12 also possesses intracellular anti-bacterial properties in macrophages through its carboxy-terminal domain.(44) MMP-12 inactivates the complement cascade, one of the earliest stimuli to attract inflammatory cells during injury, and inactivates CCL2, a potent monocyte chemoattractant.(48, 49) Through cleavage of actin and fibrin, MMP-12 promotes clearance of neutrophil extracellular traps (NETs) that serve as neutrophil reservoirs following ischemic injury.(48, 50) In a mouse model of autoimmune encephalomyelitis, MMP-12 improves outcomes through proteolytic cleavage of osteopontin to reduce inflammation.(51) The CD14 receptor recognizes damage-associated molecular patterns released during MI, and MMP-12 sheds membrane bound CD14 to downregulate pro-inflammatory monocyte activity.(52, 53) In summary, MMP-12 turns off pro-inflammation by both direct cleavage of substrates and by serving as a transcription factor.
Table 2.
MMP-12 inhibitor treatment worsens MI day 7 LV geometry and physiology.Adapted from (8).
| MI day 7 Saline (vs day 0 no MI) |
MI day 7 MMP-12 inhibitor (vs day 0 no MI and Ml day 7 MI Saline) |
|
|---|---|---|
| Infarct wall thickness (mm) | ↓ | ↓↓ |
| End diastolic volume (μL_) | ↑ | ↑↑ |
| End systolic volume (μL) | ↑ | ↑↑ |
| Ejection fraction (%) | ↓ | ↓↓ |
| Remodeling index (end diastolic volume/ LV mass) | ↑ | ↑↑ |
2.4. MMP-12 stimulates anti-inflammatory signaling.
IL-13 is an anti-inflammatory cytokine expressed by T helper 2 (Th2) cells that promotes healing following MI by enhancing macrophage differentiation to a reparative phenotype.(54) While IL-13 has positive roles in MI remodeling, IL-13 is detrimental in lung injury. In a mouse model of bleomycin-induced lung injury, MMP-12 mediates many of the pro-fibrotic effects of IL- 13 and supports Th2 cell-derived IL-13 to promote pulmonary fibrosis.(55, 56) IL-13 positively feeds back to further induce MMP-12 expression.(57) MMP-12 is induced by transforming growth factor (TGF)β1, an anti-inflammatory and pro-fibrotic cytokine, and in turn MMP-12 stimulates TGFβ1-induced lung fibrosis.(25) In a mouse model of multiple sclerosis, MMP-12 confers protection by enhancing anti-inflammatory Th2 cell activation, and Th2 cell activation links to decreased MI risk.(58, 59) MMP-12 processes adiponectin from full-length to globular forms, which promotes nitric oxide production by endothelial nitric oxide synthase expression in endothelial cells, and promotes IL-10 expression while inhibiting pro-inflammatory cytokine expression in macrophages.(60, 61) In summary, MMP-12 stimulates anti-inflammatory signals, which in turn stimulate further MMP-12 production.
2.5. MMP-12 promotes cell clearance.
MMP-12 may also promote resolution of inflammation by mediating clearance of inflammatory cells. Neutrophil clearance is critical for inflammation resolution, as the persistent presence of neutrophils impairs resolution and healing.(4, 53, 62) Mice lacking MMP-12 show a continuation of neutrophils in arthritic joints.(48) A similar phenotype is seen in the post-MI LV where MMP-12 inhibition prevented neutrophil apoptosis and in vitro where MMP-12 directly stimulates neutrophil apoptosis by caspase 3 activation.(8) In tumor cells, MMP-12 promotes apoptosis by activating TNF-related apoptosis-inducing ligand.(63) MMP-12 also cleaves hyaluronic acid to promote its binding to CD44 in neutrophils, leading to their apoptosis.(8) At MI day 7, MMP-12 inhibition reduced CD44 and increased hyaluronic acid levels, halting neutrophil apoptosis due to inadequate CD44-hyaluronic acid binding.(8, 64) CD18 (β2 integrin), a cell adhesion molecule that suppresses neutrophil apoptosis, is elevated when MMP-12 is blocked.(8)
MMP-12 inhibition also reduces the phagocytic marker CD36 at MI day 7, decreasing the ability of macrophages to engulf apoptotic neutrophils.(8) Macrophage phagocytosis is a necessary component for optimal neutrophil removal and activation towards a pro-reparative state.(65, 66) In macrophages, the absence of MMP-12 prevents migration and ECM degradation.(3, 67) In the injured liver, MMP-12 is more highly expressed in Ly6C-low M2 macrophages, which play anti-inflammatory and pro-reparative roles following MI, compared to Ly6C-high M1 macrophages.(40, 68)
These results highlight the significant influence of MMP-12 on reducing apoptotic and necrotic cells within inflammed tissue.
2.6. MMP-12 promotes scar formation and angiogenesis.
Following MI, optimal scar formation requires an adequate inflammatory response, timely resolution of inflammation, and collagen synthesis and organization that does not infiltrate the remote, non-infarcted area.(39) Inadequate ECM synthesis paired with increased degradation can lead to LV rupture, while excessive ECM synthesis paired with insufficient breakdown can lead to elevated LV wall stiffness and progression to heart failure.(69) MMPs stimulates ECM degradation directly and ECM synthesis indirectly, and MMP-12 presents a unique protease mediating both processes.(2, 3)
MMP-12 has cardioprotective and wound healing properties after MI (Figure 2), as MMP-12 inhibition worsened MI outcomes including increased LV wall thinning, dilation, and hypertrophy.(8) In a mouse model of acetaminophen-induced liver injury, fibrinogen-induced MMP-12 protects against hepatocyte necrosis; whether MMP-12 is protective against myocyte necrosis following MI remains to be determined.(70) MMP-12 generates elastin-derived peptides with therapeutic relevance to the myocardium, as these peptides protect from ischemia by enhancing nitric oxide production.(71, 72)
Figure 2. Left ventricular remodeling following MI and potential therapeutic effects of MMP-12.

Following MI, inflammatory cells in the left ventricle clear dead myocytes and release matrix metalloproteinases (MMPs) that degrade the extracellular matrix (ECM), leading to wall thinning and LV dysfunction. MMP-12 may be beneficial after MI by preventing excessive inflammation while promoting more efficient removal of dead cells, decreasing other MMPs, and preserving ECM integrity.
In addition to potentially protecting viable myocardium from ischemic injury, there are other mechanisms by which MMP-12 may promote scar formation. MMP-12 degrades the sarcomeric protein titin to participate in the degradation of necrotic myocytes and contribute to their removal and replacement with scar tissue.(73) MMP-12 may limit excessive tissue degradation by other MMPs, as MMP-12 inhibition after MI results in compensatory upregulation of MMP-8, −10 and −14.(8) Mice deficient in MMP-12 have increased MMP-2, −9 and −13 following lung injury.(8, 55) MMP-12 cleaves fibronectin, whose fragments promote macrophage secretion of fibroblast growth factor-1, insulin-like growth factor-1, and leukemia inhibitory factor to protect cardiomyocytes from ischemic injury.(74, 75) MMP-12 promotes the motility of mesenchymal stem cells, which serve as a cardiac fibroblast source after MI.(76, 77)
MMP-12 influences cardiac fibroblasts, the major cell type responsible for ECM synthesis after MI.(78, 79) MMP-12 is expressed by myofibroblasts in the cornea to assist in the removal of damaged ECM components and support TGFβ1 activity.(80) Angiogenesis is a critical component of MI healing to re-vascularize the infarcted myocardium.(81) MMP-12 generated elastin fragments have pro-mitogenic effects on dermal fibroblasts and are chemotactic for endothelial cells, implicating a role for MMP-12 in scar formation and angiogenesis.(82) MMP-12 also promotes angiogenesis by degrading the basement membrane component collagen IV.(83) At the same time, fibroblast-derived MMP-12 inhibits angiogenesis by cleavage of urokinase-type-plasminogen activator receptor on endothelial cells.(84) This may be a later signal to turn off angiogenesis, when wound healing completes. Overall, MMP-12 promotes scar formation by regulating expression of other MMPs, promoting timely resolution of inflammation, and direct stimulation of fibroblasts and endothelial cells.
3.1. Therapeutic relevance.
There is strong logic for testing MMP-12 for RPF roles after MI. At the same time, both MMP-12 deletion and inhibition limit atherosclerosis progression,(23) which brings into question the clinical relevance of using MMP-12 to treat patients with MI. Acute (days) administration of MMP-12 in the immediate post-MI setting would target the initial inflammatory phase and stimulate efficient conversion of immune cells to repair phenotypes and limit the effects potentially seen with chronic (months) administration. MMP-12 is highly induced by statin use, which has anti-inflammatory properties. Statins are standard of care for patients with MI, and MI patients who discontinue statin use have a higher one-year mortality rate.(85, 86) Thus, the effects of commonly used pharmacological agents on MMP-12 should be taken into consideration when evaluating therapeutic efficacy.
A second strategy to promote MMP-12 beneficial effects while limiting detrimental effects would be to target a particular MMP-12 substrate, rather than MMP-12 itself. While the atherosclerosis environment is macrophage and smooth muscle cell predominant, the MI environment is dominated by neutrophils, macrophages, and fibroblasts. The substrate environment, therefore, is different for the two pathologies. A better understanding is needed to evaluate MMP-12 and MMP-12 substrate treatment timing and effect on different cell types. Following this process will allow us to develop a mechanistic foundation on which to test translational applications. With the recent development of a number of guidelines for cardiovascular research, experimental designs that incorporate rigor and reproducibility will enlighten us on the potential therapeutic relevance of MMP-12.(87-90)
3.2. Conclusions.
MMP-12 displays several features of an RPF in the post-MI setting, including blunting excessive infiltration and promoting clearance of inflammatory cells, preventing excessive cytokine and chemokine activity, promoting anti-inflammatory activities of macrophages and T cells, and stimulating ECM remodeling and fibroblast function to promote scar formation. Our studies and others build a foundation upon which to test MMP-12 as an RPF for the treatment of MI.
Acknowledgments.
We acknowledge funding from the American Heart Association 18POST34000039, National Institutes of Health under Award Numbers GM104357, GM114833, GM115428, HL051971, HL075360, HL105324, and HL129823, and from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development under Award Number 5I01BX000505. The content is solely the responsibility of the authors and does not necessarily represent the official views of the American Heart Association, the National Institutes of Health, or the Veterans Administration.
Footnotes
Disclosures. None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nielsen SH, Mouton AJ, DeLeon-Pennell KY, Genovese F, Karsdal M, Lindsey ML. Understanding cardiac extracellular matrix remodeling to develop biomarkers of myocardial infarction outcomes. Matrix Biol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frangogiannis NG. The mechanistic basis of infarct healing. Antioxid Redox Signal. 2006;8(11–12):1907–1939. [DOI] [PubMed] [Google Scholar]
- 3.DeLeon-Pennell KY, Meschiari CA, Jung M, Lindsey ML. Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. Prog Mol Biol Transl Sci. 2017;147:75–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rienks M, Carai P, Bitsch N, Schellings M, Vanhaverbeke M, Verjans J, Cuijpers I, Heymans S, Papageorgiou A. Sema3A promotes the resolution of cardiac inflammation after myocardial infarction. Basic research in cardiology. 2017;112(4):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kain V, Prabhu SD, Halade GV. Inflammation revisited: inflammation versus resolution of inflammation following myocardial infarction. Basic research in cardiology. 2014;109(6):444. [DOI] [PubMed] [Google Scholar]
- 6.Huang S, Frangogiannis NG. Anti-inflammatory therapies in myocardial infarction: failures, hopes and challenges. British journal of pharmacology. 2018;175(9):1377–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ingle KA, Kain V, Goel M, Prabhu SD, Young ME, Halade GV. Cardiomyocyte-specific Bmal1 deletion in mice triggers diastolic dysfunction, extracellular matrix response, and impaired resolution of inflammation. American journal of physiology Heart and circulatory physiology. 2015;309(11):H1827–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iyer RP, Patterson NL, Zouein FA, Ma Y, Dive V, de Castro Bras LE, Lindsey ML. Early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. International journal of cardiology. 2015;185:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serhan CN, Chiang N, Dalli J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Seminars in immunology. 2015;27(3):200–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serhan CN, Dalli J, Colas RA, Winkler JW, Chiang N. Protectins and maresins: New proresolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochimica et biophysica acta. 2015;1851(4):397–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haworth O, Buckley CD. Pathways involved in the resolution of inflammatory joint disease. Seminars in immunology. 2015;27(3):194–199. [DOI] [PubMed] [Google Scholar]
- 12.Colgan SP. Neutrophils and inflammatory resolution in the mucosa. Seminars in immunology. 2015;27(3):177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Croasdell A, Duffney PF, Kim N, Lacy SH, Sime PJ, Phipps RP. PPARgamma and the Innate Immune System Mediate the Resolution of Inflammation. PPAR research. 2015;2015:549691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duffield JS, Hong S, Vaidya VS, Lu Y, Fredman G, Serhan CN, Bonventre JV. Resolvin D series and protectin D1 mitigate acute kidney injury. Journal of immunology (Baltimore, Md : 1950). 2006;177(9):5902–5911. [DOI] [PubMed] [Google Scholar]
- 15.Kain V, Ingle KA, Colas RA, Dalli J, Prabhu SD, Serhan CN, Joshi M, Halade GV. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. Journal of molecular and cellular cardiology. 2015;84:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez EF, Kabarowski JH, Ingle KA, Kain V, Barnes S, Crossman DK, Lindsey ML, Halade GV. Obesity superimposed on aging magnifies inflammation and delays the resolving response after myocardial infarction. American journal of physiology Heart and circulatory physiology. 2015;308(4):H269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez EF, Kabarowski JH, Ingle KA, Kain V, Barnes S, Crossman DK, Lindsey ML, Halade GV. Obesity superimposed on aging magnifies inflammation and delays the resolving response after myocardial infarction. American journal of physiology: Heart and circulatory physiology. 2015;308(4):H269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin CX, Finlayson SB, Al-Sharea A, Tate M, De Blasio MJ, Deo M, Rosli S, Prakoso D, Thomas CJ, Kiriazis H, Gould E, Yang YH, Morand EF, Perretti M, Murphy AJ, Du XJ, Gao XM, Ritchie RH. Endogenous Annexin-A1 Regulates Haematopoietic Stem Cell Mobilisation and Inflammatory Response Post Myocardial Infarction in Mice In Vivo. Scientific reports. 2017;7(1):16615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindsey ML. Assigning matrix metalloproteinase roles in ischaemic cardiac remodelling. Nat Rev Cardiol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iyer RP, de Castro Bras LE, Jin YF, Lindsey ML. Translating Koch's postulates to identify matrix metalloproteinase roles in postmyocardial infarction remodeling: cardiac metalloproteinase actions (CarMA) postulates. Circulation research. 2014;114(5):860–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindsey ML, Iyer RP, Jung M, DeLeon-Pennell KY, Ma Y. Matrix metalloproteinases as input and output signals for post-myocardial infarction remodeling. Journal of molecular and cellular cardiology. 2016;91:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iyer RP, Jung M, Lindsey ML. Using the laws of thermodynamics to understand how matrix metalloproteinases coordinate the myocardial response to injury. Metalloproteinases Med. 2015;2:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luttun A, Lutgens E, Manderveld A, Maris K, Collen D, Carmeliet P, Moons L. Loss of Matrix Metalloproteinase-9 or Matrix Metalloproteinase-12 Protects Apolipoprotein E-Deficient Mice Against Atherosclerotic Media Destruction but Differentially Affects Plaque Growth. Circulation. 2004:01.CIR.0000121728.0000114930.DE. [DOI] [PubMed] [Google Scholar]
- 24.Oh H, Yang S, Park M, Chun JS. Matrix metalloproteinase (MMP)-12 regulates MMP-9 expression in interleukin-1beta-treated articular chondrocytes. J Cell Biochem. 2008; 105(6): 1443–1450. [DOI] [PubMed] [Google Scholar]
- 25.Kang HR, Cho SJ, Lee CG, Homer RJ, Elias JA. Transforming growth factor (TGF)- beta1 stimulates pulmonary fibrosis and inflammation via a Bax-dependent, bid-activated pathway that involves matrix metalloproteinase-12. J Biol Chem. 2007;282(10):7723–7732. [DOI] [PubMed] [Google Scholar]
- 26.Nelson MP, Christmann BS, Dunaway CW, Morris A, Steele C. Experimental Pneumocystis lung infection promotes M2a alveolar macrophage-derived MMP12 production. Am J Physiol Lung Cell Mol Physiol. 2012;303(5):L469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hiden U, Eyth CP, Majali-Martinez A, Desoye G, Tam-Amersdorfer C, Huppertz B, Ghaffari Tabrizi-Wizsy N. Expression of matrix metalloproteinase 12 is highly specific for non-proliferating invasive trophoblasts in the first trimester and temporally regulated by oxygen-dependent mechanisms including HIF-1A. Histochem Cell Biol. 2018;149(1):31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Wei G, Hu W, Li L, Ye Y, Wang H, Wan W, Li R, Li L, Ma L, Meng Z. Expression of matrix metalloproteinases-12 in ST-segment elevation myocardial infarction: A case-control study. Medicine (Baltimore). 2017;96(40):e8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu W, Wei R, Wang L, Lu J, Liu H, Zhang W. Correlations of MMP-1, MMP-3, and MMP- 12 with the degree of atherosclerosis, plaque stability and cardiovascular and cerebrovascular events. Exp Ther Med. 2018;15(2):1994–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meschiari CA, Ero OK, Pan H, Finkel T, Lindsey ML. The impact of aging on cardiac extracellular matrix. Geroscience. 2017;39(1):7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toba H, de Castro Bras LE, Baicu CF, Zile MR, Lindsey ML, Bradshaw AD. Increased ADAMTS1 mediates SPARC-dependent collagen deposition in the aging myocardium. Am J Physiol Endocrinol Metab. 2016;310(11):E1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiao YA, Ramirez TA, Zamilpa R, Okoronkwo SM, Dai Q, Zhang J, Jin YF, Lindsey ML. Matrix metalloproteinase-9 deletion attenuates myocardial fibrosis and diastolic dysfunction in ageing mice. Cardiovasc Res. 2012;96(3):444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mangion K, Berry C. Survival in the elderly after acute myocardial infarction: room for more improvement. Age Ageing. 2014;43(6):739–740. [DOI] [PubMed] [Google Scholar]
- 34.Lin J, Davis HB, Dai Q, Chou YM, Craig T, Hinojosa-Laborde C, Lindsey ML. Effects of early and late chronic pressure overload on extracellular matrix remodeling. Hypertens Res. 2008;31(6):1225–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindsey ML, Goshorn DK, Squires CE, Escobar GP, Hendrick JW, Mingoia JT, Sweterlitsch SE, Spinale FG. Age-dependent changes in myocardial matrix metalloproteinase/tissue inhibitor of metalloproteinase profiles and fibroblast function. Cardiovasc Res. 2005;66(2):410–419. [DOI] [PubMed] [Google Scholar]
- 36.Geng X, Hwang J, Ye J, Shih H, Coulter B, Naudin C, Jun K, Sievers R, Yeghiazarians Y, Lee RJ, Boyle AJ. Aging is protective against pressure overload cardiomyopathy via adaptive extracellular matrix remodeling. Am J Cardiovasc Dis. 2017;7(3):72–82. [PMC free article] [PubMed] [Google Scholar]
- 37.Frangogiannis NG. Cell biological mechanisms in regulation of the post-infarction inflammatory response. Current opinion in physiology. 2018;1:7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, Garrett MR, Ma Y, Harmancey R, Lindsey ML. Mapping macrophage polarization over the myocardial infarction time continuum. Basic research in cardiology. 2018;113(4):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mouton AJ, Rivera OJ, Lindsey ML. Myocardial infarction remodeling that progresses to heart failure: a signaling misunderstanding. American journal of physiology Heart and circulatory physiology. 2018;315(1):H71–H79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204(12):3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phatharajaree W, Phrommintikul A, Chattipakorn N. Matrix metalloproteinases and myocardial infarction. Can J Cardiol. 2007;23(9):727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, Mecham RP, Senior RM, Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. The Journal of clinical investigation. 2006;116(3):753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dean RA, Cox JH, Bellac CL, Doucet A, Starr AE, Overall CM. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, −7, −8, and −13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood. 2008;112(8):3455–3464. [DOI] [PubMed] [Google Scholar]
- 44.Wolf M, Maltseva I, Clay SM, Pan P, Gajjala A, Chan MF. Effects of MMP12 on cell motility and inflammation during corneal epithelial repair. Exp Eye Res. 2017;160:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lyu J, Joo CK. Wnt-7a up-regulates matrix metalloproteinase-12 expression and promotes cell proliferation in corneal epithelial cells during wound healing. J Biol Chem. 2005;280(22):21653–21660. [DOI] [PubMed] [Google Scholar]
- 46.Marchant DJ, Bellac CL, Moraes TJ, Wadsworth SJ, Dufour A, Butler GS, Bilawchuk LM, Hendry RG, Robertson AG, Cheung CT, Ng J, Ang L, Luo Z, Heilbron K, Norris MJ, Duan W, Bucyk T, Karpov A, Devel L, Georgiadis D, Hegele RG, Luo H, Granville DJ, Dive V, McManus BM, Overall CM. A new transcriptional role for matrix metalloproteinase-12 in antiviral immunity. Nat Med. 2014;20(5):493–502. [DOI] [PubMed] [Google Scholar]
- 47.Dandachi NG, Shapiro SD. A protean protease: MMP-12 fights viruses as a protease and a transcription factor. Nat Med. 2014;20(5):470–472. [DOI] [PubMed] [Google Scholar]
- 48.Bellac CL, Dufour A, Krisinger MJ, Loonchanta A, Starr AE, Auf dem Keller U, Lange PF, Goebeler V, Kappelhoff R, Butler GS, Burtnick LD, Conway EM, Roberts CR, Overall CM. Macrophage matrix metalloproteinase-12 dampens inflammation and neutrophil influx in arthritis. Cell Rep. 2014;9(2):618–632. [DOI] [PubMed] [Google Scholar]
- 49.Chan MF, Li J, Bertrand A, Casbon AJ, Lin JH, Maltseva I, Werb Z. Protective effects of matrix metalloproteinase-12 following corneal injury. J Cell Sci. 2013;126(Pt 17):3948–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ge L, Zhou X, Ji WJ, Lu RY, Zhang Y, Zhang YD, Ma YQ, Zhao JH, Li YM. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. American journal of physiology Heart and circulatory physiology. 2015;308(5):H500–509. [DOI] [PubMed] [Google Scholar]
- 51.Goncalves DaSilva A, Liaw L, Yong VW. Cleavage of osteopontin by matrix metalloproteinase-12 modulates experimental autoimmune encephalomyelitis disease in C57BL/6 mice. Am J Pathol. 2010;177(3):1448–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Senft AP, Korfhagen TR, Whitsett JA, Shapiro SD, LeVine AM. Surfactant protein-D regulates soluble CD14 through matrix metalloproteinase-12. Journal of immunology (Baltimore, Md : 1950). 2005;174(8):4953–4959. [DOI] [PubMed] [Google Scholar]
- 53.Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, Henry J, Cates CA, Deleon-Pennell KY, Lindsey ML. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res. 2016;110(1):51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hofmann U, Knorr S, Vogel B, Weirather J, Frey A, Ertl G, Frantz S. Interleukin-13 deficiency aggravates healing and remodeling in male mice after experimental myocardial infarction. Circ Heart Fail. 2014;7(5):822–830. [DOI] [PubMed] [Google Scholar]
- 55.Madala SK, Pesce JT, Ramalingam TR, Wilson MS, Minnicozzi S, Cheever AW, Thompson RW, Mentink-Kane MM, Wynn TA. Matrix metalloproteinase 12-deficiency augments extracellular matrix degrading metalloproteinases and attenuates IL-13- dependent fibrosis. Journal of immunology (Baltimore, Md : 1950). 2010;184(7):3955–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lanone S, Zheng T, Zhu Z, Liu W, Lee CG, Ma B, Chen Q, Homer RJ, Wang J, Rabach LA, Rabach ME, Shipley JM, Shapiro SD, Senior RM, Elias JA. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and −12 in IL-13-induced inflammation and remodeling. The Journal of clinical investigation. 2002;110(4):463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pouladi MA, Robbins CS, Swirski FK, Cundall M, McKenzie AN, Jordana M, Shapiro SD, Stampfli MR. Interleukin-13-dependent expression of matrix metalloproteinase-12 is required for the development of airway eosinophilia in mice. Am J Respir Cell Mol Biol. 2004;30(1):84–90. [DOI] [PubMed] [Google Scholar]
- 58.Weaver A, Goncalves da Silva A, Nuttall RK, Edwards DR, Shapiro SD, Rivest S, Yong VW. An elevated matrix metalloproteinase (MMP) in an animal model of multiple sclerosis is protective by affecting Th1/Th2 polarization. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005; 19(12):1668–1670. [DOI] [PubMed] [Google Scholar]
- 59.Engelbertsen D, Andersson L, Ljungcrantz I, Wigren M, Hedblad B, Nilsson J, Bjorkbacka H. T-helper 2 immunity is associated with reduced risk of myocardial infarction and stroke. Arterioscler Thromb Vasc Biol. 2013;33(3):637–644. [DOI] [PubMed] [Google Scholar]
- 60.Suzuki M, Mihara M. Adiponectin induces CCL20 expression synergistically with IL-6 and TNF-alpha in THP-1 macrophages. Cytokine. 2012;58(3):344–350. [DOI] [PubMed] [Google Scholar]
- 61.Hui X, Lam KS, Vanhoutte PM, Xu A. Adiponectin and cardiovascular health: an update. British journal of pharmacology. 2012;165(3):574–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma Y, Yabluchanskiy A, Lindsey ML. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenesis Tissue Repair. 2013;6(1): 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dandachi N, Kelly NJ, Wood JP, Burton CL, Radder JE, Leme AS, Gregory AD, Shapiro SD. Macrophage Elastase Induces TRAIL-mediated Tumor Cell Death through Its Carboxy-Terminal Domain. Am J Respir Crit Care Med. 2017;196(3):353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huebener P, Abou-Khamis T, Zymek P, Bujak M, Ying X, Chatila K, Haudek S, Thakker G, Frangogiannis NG. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. Journal of immunology (Baltimore, Md : 1950). 2008; 180(4):2625–2633. [DOI] [PubMed] [Google Scholar]
- 65.Vos CM, van Haastert ES, de Groot CJ, van der Valk P, de Vries HE. Matrix metalloproteinase-12 is expressed in phagocytotic macrophages in active multiple sclerosis lesions. J Neuroimmunol. 2003;138(1–2):106–114. [DOI] [PubMed] [Google Scholar]
- 66.Frodermann V, Nahrendorf M. Neutrophil-macrophage cross-talk in acute myocardial infarction. Eur Heart J. 2017;38(3):198–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shipley JM, Wesselschmidt RL, Kobayashi DK, Ley TJ, Shapiro SD. Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc Natl Acad Sci U S A. 1996;93(9):3942–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, Hartland SN, Snowdon VK, Cappon A, Gordon-Walker TT, Williams MJ, Dunbar DR, Manning JR, van Rooijen N, Fallowfield JA, Forbes SJ, Iredale JP. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. 2012;109(46):E3186–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yabluchanskiy A, Li Y, Chilton RJ, Lindsey ML. Matrix metalloproteinases: drug targets for myocardial infarction. Curr Drug Targets. 2013;14(3):276–286. [PMC free article] [PubMed] [Google Scholar]
- 70.Kopec AK, Joshi N, Cline-Fedewa H, Wojcicki AV, Ray JL, Sullivan BP, Froehlich JE, Johnson BF, Flick MJ, Luyendyk JP. Fibrin(ogen) drives repair after acetaminophen- induced liver injury via leukocyte alphaMbeta2 integrin-dependent upregulation of Mmp12. J Hepatol. 2017;66(4):787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Robinet A, Millart H, Oszust F, Hornebeck W, Bellon G. Binding of elastin peptides to S-Gal protects the heart against ischemia/reperfusion injury by triggering the RISK pathway. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21(9):1968–1978. [DOI] [PubMed] [Google Scholar]
- 72.Harris LK, Smith SD, Keogh RJ, Jones RL, Baker PN, Knofler M, Cartwright JE, Whitley GS, Aplin JD. Trophoblast- and vascular smooth muscle cell-derived MMP-12 mediates elastolysis during uterine spiral artery remodeling. Am J Pathol. 2010;177(4):2103–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vassiliadis E, Rasmussen LM, Byrjalsen I, Larsen DV, Chaturvedi R, Hosbond S, Saabye L, Diederichsen AC, Genovese F, Duffin KL, Zheng Q, Chen X, Leeming DJ, Christiansen C, Karsdal MA. Clinical evaluation of a matrix metalloproteinase-12 cleaved fragment of titin as a cardiovascular serological biomarker. J Transl Med. 2012;10:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Di Sabatino A, Jackson CL, Pickard KM, Buckley M, Rovedatti L, Leakey NA, Picariello L, Cazzola P, Monteleone G, Tonelli F, Corazza GR, MacDonald TT, Pender SL. Transforming growth factor beta signalling and matrix metalloproteinases in the mucosa overlying Crohn's disease strictures. Gut. 2009;58(6):777–789. [DOI] [PubMed] [Google Scholar]
- 75.Trial J, Rossen RD, Rubio J, Knowlton AA. Inflammation and ischemia: macrophages activated by fibronectin fragments enhance the survival of injured cardiac myocytes. Exp Biol Med (Maywood). 2004;229(6):538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee SJ, Jung YH, Oh SY, Yong MS, Ryu JM, Han HJ. Netrin-1 induces MMP-12-dependent E-cadherin degradation via the distinct activation of PKCalpha and FAK/Fyn in promoting mesenchymal stem cell motility. Stem Cells Dev. 2014;23(16):1870–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carlson S, Trial J, Soeller C, Entman ML. Cardiac mesenchymal stem cells contribute to scar formation after myocardial infarction. Cardiovasc Res. 2011;91(1):99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Squires CE, Escobar Gp, Payne JF, Leonardi RA, Goshorn DK, Sheats NJ, Mains IM, Mingoia JT, Flack EC, Lindsey ML. Altered fibroblast function following myocardial infarction. Journal of molecular and cellular cardiology. 2005;39(4):699–707. [DOI] [PubMed] [Google Scholar]
- 79.Ma Y, Iyer RP, Jung M, Czubryt MP, Lindsey ML. Cardiac Fibroblast Activation Post- Myocardial Infarction: Current Knowledge Gaps. Trends Pharmacol Sci. 2017;38(5):448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Iwanami H, Ishizaki M, Fukuda Y, Takahashi H. Expression of matrix metalloproteinases (MMP)-12 by myofibroblasts during alkali-burned corneal wound healing. Curr Eye Res. 2009;34(3):207–214. [DOI] [PubMed] [Google Scholar]
- 81.Lindsey ML, Iyer RP, Zamilpa R, Yabluchanskiy A, DeLeon-Pennell KY, Hall ME, Kaplan A, Zouein FA, Bratton D, Flynn ER, Cannon PL, Tian Y, Jin YF, Lange RA, Tokmina-Roszyk D, Fields GB, de Castro Bras LE. A Novel Collagen Matricryptin Reduces Left Ventricular Dilation Post-Myocardial Infarction by Promoting Scar Formation and Angiogenesis. J Am Coll Cardiol. 2015;66(12):1364–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heinz A, Jung MC, Duca L, Sippl W, Taddese S, Ihling C, Rusciani A, Jahreis G, Weiss AS, Neubert RH, Schmelzer CE. Degradation of tropoelastin by matrix metalloproteinases--cleavage site specificities and release of matrikines. FEBS J. 2010;277(8):1939–1956. [DOI] [PubMed] [Google Scholar]
- 83.Narimiya T, Wada S, Kanzaki H, Ishikawa M, Tsuge A, Yamaguchi Y, Nakamura Y. Orthodontic tensile strain induces angiogenesis via type IV collagen degradation by matrix metalloproteinase-12. J Periodontal Res. 2017;52(5):842–852. [DOI] [PubMed] [Google Scholar]
- 84.Serrati S, Cinelli M, Margheri F, Guiducci S, Del Rosso A, Pucci M, Fibbi G, Bazzichi L, Bombardieri S, Matucci-Cerinic M, Del Rosso M. Systemic sclerosis fibroblasts inhibit in vitro angiogenesis by MMP-12-dependent cleavage of the endothelial cell urokinase receptor. J Pathol. 2006;210(2):240–248. [DOI] [PubMed] [Google Scholar]
- 85.Arikan MC, Shapiro SD, Mariani TJ. Induction of macrophage elastase (MMP-12) gene expression by statins. J Cell Physiol. 2005;204(1):139–145. [DOI] [PubMed] [Google Scholar]
- 86.Daskalopoulou SS, Doonan RJ, Delaney JA, Pilote L. Different patterns of statin use in patients with acute myocardial infarction. Curr Vasc Pharmacol. 2014;12(6):885–892. [DOI] [PubMed] [Google Scholar]
- 87.Lindsey ML, Gray GA, Wood SK, Curran-Everett D. Statistical considerations in reporting cardiovascular research. American journal of physiology Heart and circulatory physiology. 2018;315(2):H303–H313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brooks HL, Lindsey ML. Guidelines for authors and reviewers on antibody use in physiology studies. American journal of physiology Heart and circulatory physiology. 2018;314(4):H724–H732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lindsey ML, Kassiri Z, Virag JAI, de Castro Bras LE, Scherrer-Crosbie M. Guidelines for measuring cardiac physiology in mice. American journal of physiology Heart and circulatory physiology. 2018;314(4):H733–H752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lindsey ML, Bolli R, Canty JM Jr., Du XJ, Frangogiannis NG, Frantz S, Gourdie RG, Holmes JW, Jones SP, Kloner RA, Lefer DJ, Liao R, Murphy E, Ping P, Przyklenk K, Recchia FA, Schwartz Longacre L, Ripplinger CM, Van Eyk JE, Heusch G. Guidelines for experimental models of myocardial ischemia and infarction. American journal of physiology Heart and circulatory physiology. 2018;314(4):H812–h838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Halade GV, Dorbane A, Ingle KA, Kain V, Schmitter JM, Rhourri-Frih B. Comprehensive targeted and non-targeted lipidomics analyses in failing and non-failing heart. Analytical and bioanalytical chemistry. 2018;410(7):1965–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Qin CX, Finlayson SB, Al-Sharea A, Tate M, De Blasio MJ, Deo M, Rosli S, Prakoso D, Thomas CJ, Kiriazis H, Gould E, Yang YH, Morand EF, Perretti M, Murphy AJ, Du XJ, Gao XM, Ritchie RH. Endogenous Annexin-A1 Regulates Haematopoietic Stem Cell Mobilisation and Inflammatory Response Post Myocardial Infarction in Mice In Vivo. Scientific reports. 2017;7(1):16615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Calder PC. Omega-3 fatty acids and inflammatory processes: from molecules to man. Biochem Soc Trans. 2017;45(5):1105–1115. [DOI] [PubMed] [Google Scholar]
- 94.Ansari J, Kaur G, Gavins FNE. Therapeutic Potential of Annexin A1 in Ischemia Reperfusion Injury. Int J Mol Sci. 2018;19(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhuo Y, Zhang S, Li C, Yang L, Gao H, Wang X. Resolvin D1 Promotes SIRT1 Expression to Counteract the Activation of STAT3 and NF-kappaB in Mice with Septic- Associated Lung Injury. Inflammation. 2018. [DOI] [PubMed] [Google Scholar]
- 96.Le Quement C, Guenon I, Gillon JY, Lagente V, Boichot E. MMP-12 induces IL-8/CXCL8 secretion through EGFR and ERK1/2 activation in epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1076–1084. [DOI] [PubMed] [Google Scholar]
- 97.Leoni G, Nusrat A. Annexin A1: shifting the balance towards resolution and repair. Biol Chem. 2016;397(10):971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gavins FN, Sawmynaden P, Chatterjee BE, Perretti M. A twist in anti-inflammation: annexin 1 acts via the lipoxin A4 receptor. Prostaglandins Leukot Essent Fatty Acids. 2005;73(3–4):211–219. [DOI] [PubMed] [Google Scholar]
- 99.Halade Gv, Kain V, Serhan CN. Immune responsive resolvin D1 programs myocardial infarction-induced cardiorenal syndrome in heart failure. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2018:fj201701173RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sansbury BE, Spite M. Resolution of Acute Inflammation and the Role of Resolvins in Immunity, Thrombosis, and Vascular Biology. Circulation research. 2016;119(1):113–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yona S, Buckingham JC, Perretti M, Flower RJ. Stimulus-specific defect in the phagocytic pathways of annexin 1 null macrophages. British journal of pharmacology. 2004;142(5):890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lai T, Li Y, Mai Z, Wen X, Lv Y, Xie Z, Lv Q, Chen M, Wu D, Wu B. Annexin A1 is elevated in patients with COPD and affects lung fibroblast function. Int J Chron Obstruct Pulmon Dis. 2018;13:473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]