

Abstract
Cardiac remodeling (CR) is a complex dynamic process common to many heart diseases. CR is characterized as a temporal progression of global adaptive and maladaptive perturbations. The complex nature of this process clouds a comprehensive understanding of CR, but greater insight into the processes and mechanisms has potential to identify new therapeutic targets. To provide a deeper understanding of this important cardiac process, we applied a new proteomic technique, PALM (Pulse Azidohomoalanine in Mammals), to quantitate the newly-synthesized protein (NSP) changes during the progression of isoproterenol (ISO)-induced CR in the mouse left ventricle. This analysis revealed a complex combination of adaptive and maladaptive alterations at acute and prolonged time points including the identification of proteins not previously associated with CR. We also combined the PALM dataset with our published protein turnover rate dataset to identify putative biochemical mechanisms underlying CR. The novel integration of analyzing NSPs together with their protein turnover rates demonstrated that alterations in specific biological pathways (e.g., inflammation and oxidative stress) are produced by differential regulation of protein synthesis and degradation.
Keywords: Cardiac remodeling, Isoproterenol, Azidohomoalanine, Proteomics, Metabolic labeling
1. Introduction
Heart failure (HF) and cardiac remodeling (CR) are both common stages of many heart diseases with increasing incidence and prevalence in the United States, posing major public health problems. CR is a multifactorial process and results not only from cardiac overload or injury but also from a complex interplay among genetic, inflammatory, biochemical, and neurohormonal alterations (Shimizu and Minamino 2016). Accordingly, the CR process involves prolonged over-stimulation of the sympathetic nervous system (SNS) followed by the release of catecholamines to increase both the heart rate and individual cellular contractility, thereby creating a compensatory adaptive response that transiently normalizes the biomechanical stress and optimizes cardiac contractility. Catecholamines are crucial stress hormones and regulators of heart function via stimulation of beta-adrenergic receptors (β-ARs). Chronic over- stimulation, however, augments the CR process, eventually leading to cardiac dysfunction and HF (Bristow, Ginsburg et al. 1982, Lohse, Engelhardt et al. 2003). CR in the early stages exhibits thickening of the ventricular and interventricular walls and is characterized by increasing size of the cardiomyocytes, changes in the organization of the sarcomeric structure, and increased protein synthesis (Drews, Tsukamoto et al. 2010). Similarly, persistent pharmacological β-AR stimulation with the agonist isoproterenol (ISO) can cause left ventricle hypertrophy and HF in laboratory animals (Rona, Chappel et al. 1959, Taylor and Tang 1984). Numerous studies have suggested that chronic ISO application stimulates structural and metabolic remodeling, which alters fatty acid utilization, glucose homeostasis, extracellular matrix turnover and mitochondrial function (Colucci 1997, van Bilsen, Smeets et al. 2004). To advance our knowledge in the underlying molecular mechanisms of CR and HF, it is important to identify how proteome alterations gradually manifest during these pathophysiological stages over time. Identification of proteins that drive CR may lead to novel therapeutic drug targets or clinical biomarkers for cardiac pathologies.
Quantitative proteomics has great potential to better understand the myriad of proteome alterations during the progression of ISO-induced CR. We recently developed the quantitative proteomic technique PALM (Pulse Azidohomoalanine Labeling in Mammals) to improve the temporal resolution of quantitative proteomics (McClatchy, Ma et al. 2015). PALM quantitates newly-synthesized proteins (NSPs). Since the NSP sub-proteome in theory is the first to respond to perturbations, quantitation of NSPs is more sensitive than quantitation of the whole proteome. PALM relies on azidohomoalanine (AHA), which is a non-canonical amino acid that is accepted by the endogenous methionine tRNA and inserted into proteins in vivo (Dieterich, Link et al. 2006). AHA is inserted into the mouse proteome through a customized AHA diet. The AHA diet is given to mice within a discrete time period to label NSPs in response to a perturbation. Proteins incorporating AHA can be covalently linked in vitro to a biotin-alkyne with click chemistry. Through standard biotin enrichment strategies, the static or “old” proteome is removed, and the remaining NSPs can be identified and quantitated using mass spectrometry (MS). In this study, we applied the PALM strategy to the ISO-induced CR mouse model, in which we quantify newly synthesized proteins through an acute phase, during which the CR process is at its highest rate, and a prolonged phase, during which the CR process has reached a plateau, to enhance our mechanistic understanding of cardiac proteome remodeling in this pathophysiological process. In the normal heart, homeostasis of continuous protein synthesis and degradation is essential to maintain cardiac function at the cellular and whole- organ level (Lam, Wang et al. 2014). Normal protein homeostasis through the protein turnover cycle is altered during CR and HF through factors such as hypertrophic signaling (Shimizu and Minamino 2016), calcium regulation (Goonasekera, Hammer et al. 2012), inflammatory reactions (Shimizu and Minamino 2016), and oxidative stress (Goirdano FJ et al, JCI 2005;115(3);500–508). For the first time, this study integrates two novel datasets using previously published protein turnover rates in the ISO-induced CR mouse model with the NSP quantitation in response to ISO treatment. This integrated analysis provides unique insight into the total balance of protein synthesis and degradation between two compared groups (i.e., sham group vs. ISO group) and highlights the mechanisms that underlie proteomic changes in CR and establishes a novel experimental and bioinformatic approach that is generally applicable to animal models of disease.
2. Methods:
2.1. Animals/Surgery/Tissue Collection
CR was induced by continuous ISO treatment for 14 days in male C57/BL6 wildtype mice (aged 8–12 weeks) using microosmotic pumps (Alzet, Model 1002). ISO was dissolved in phosphate- buffered saline 1X (PBS). The microosmotic pumps were implanted subcutaneously in the posterior neck area of the mouse under 2% isoflurane (vaporized in oxygen) anesthesia. One group of mice received continuous treatment of ISO with a dose of 15 mg/kg/day for a total period of 14 days. As a control, a second group of mice underwent sham treatment by vehicle (PBS).
AHA-Heavy pellets were fed to the ISO group and AHA-Light pellets to the control group. For efficient labeling as well as to study two different phases in the CR process, we collected samples on Day 4 and Day 14 in both the ISO group and the control group. To collect samples on Day 4, we commenced AHA labeling on Day 0. To collect samples on Day 14, we commenced AHA labeling on Day 10. To conduct our AHA labeling analyses after the 14-day period treatment, whole heart, skeletal muscle, liver, kidney, and brain were collected after sacrificing the mice. In contrast to sham treatment, the heart weight-body weight ratios (HW/BW) were increased in the ISO group on Day 4 and Day 14, confirming cardiac remodeling. Cardiac tissues were collected from left ventricle (LV) and right ventricle (RV) and rinsed in PBS. The septum was included as part of the left ventricle.
2.2. Assesment of mitochondrial function.
Mitochondria were isolated from mouse hearts followed by mitochondrial electron transport chain (ETC) activity assays as previously described(Deng, Zhang et al. 2011).
2.3. Tissue homogenization
LVs were transferred into Precellys CK14 tubes containing 1.4-mm ceramic beads, and 0.5-mm disruption beads (Research Products International Corp.) as well as 500-ul PBS were added. The homogenization was performed on Bertin Precellys bead-beating system: 6500 rpm, 3 times, 20 seconds internal. Then, the homogenates were sonicated for 30 secs after adding 0.5% SDS. A BCA protein assay was performed on each sample.
2.4. Click chemistry
First, 2.5-mg protein of LV from heavy AHA (H-AHA)-labeled ISO mice or sham mice and 2.5-mg protein of LV from light AHA (L-AHA)-labeled sham mice or ISO mice were mixed together as one biological replicate. For each biological replicate, the H-AHA/L-AHA mixture was divided into twenty aliquots (0.25 mg/aliquot). A click reaction was performed on each aliquot as previously published (Speers and Cravatt 2009). Briefly, for each click reaction, the following reagents were added in this order: 1) 30 ul of 1.7-mM TBTA, 2) 8 ul of 50-mM Copper Sulfate, 3) 8 ul of 5-mM Biotin-Alkyne (C21H35N3O6S, Click Chemistry Tools), and 4) 8 ul of 50-mM TCEP. PBS was then added to a final volume of 400 ul and incubated for 1 hour at room temperature. Methanol/Chloroform precipitation was performed, and the precipitated protein was combined so that there would only be one pellet per 5-mg starting material.
2.5. Digestion and biotin peptide enrichment
Biotin peptide enrichment was performed as previously described (Schiapparelli, McClatchy et al. 2014). Briefly, precipitated pellets were resuspended in 100-ul 8M urea and 100-ul 0.2% MS- compatible surfactant ProteaseMAX (Promega) in 50mM ammonium bicarbonate, then reduced, alkylated, and digested with trypsin as previously described (Schiapparelli, McClatchy et al. 2014). The digestion was then centrifuged at 13,000 × g for 10 min. The supernatant was transferred to a new tube, and the pellet was resuspended with PBS and centrifuged at 13,000 × g for 10 min. Supernatants were combined, and 150 ul of neutravidin agarose resin (Thermo Scientific) was added. The resin was incubated with the peptides for 2 hours at room temperature while rotating. The resin was then washed with 1-ml PBS, then PBS with 5% acetonitrile, PBS, and a final wash of distilled water. The peptides were eluted two times off the resin with 150-ul 80% acetonitrile, 0.2% formic acid, and 0.1% TFA on shaker for 5 min at room temperature, and another two times on a shaker at 70 °C. All elutions were transferred to a single new tube. Prior to MS analysis, the samples were dried with a speed-vac, and dried peptides were re-solubilized in buffer A (5% ACN, 95% water, 0.1% formic acid).
2.6. Mass spectrometry
Soluble peptides were pressure-loaded onto a 250-μm i.d. capillary with a kasil frit containing 2.5 cm of 10-μm Jupiter C18-A material (Phenomenex) followed by 2.5 cm of 5-μm Partisphere strong cation exchanger (Whatman). This column was washed with buffer A after loading. A 100-μm i.d. capillary with a 5-μm pulled tip packed with 15 cm of 4-μm Jupiter C18 material (Phenomenex) was attached to the loading column with a union, and the entire split-column (loading column-union-analytical column) was placed in line with an Agilent 1100 quaternary HPLC (Palo Alto). The sample was analyzed using MudPIT, which is a modified 11-step separation described previously (Washburn, Wolters et al. 2001). The buffer solutions used were buffer A, 80% acetonitrile/0.1% formic acid (buffer B), and 500-mM ammonium acetate/5% acetonitrile/0.1% formic acid (buffer C). Step 1 consisted of a 10 min gradient from 0–10% buffer B, a 50 min gradient from 10–50% buffer B, a 10 min gradient from 50–100% buffer B, and 20 min from 100% buffer A. Steps 2 consisted of 1 min of 100% buffer A, 4 min of 20% buffer C, a 5-min gradient from 0–10% buffer B, an 80-min gradient from 10–45% buffer B, a 10-min gradient from 45–100% buffer B, and 10 min of 100% buffer A. Steps 3–9 had the following profile: 1 min of 100% buffer A, 4 min of X% buffer C, a 5-min gradient from 0–15% buffer B, a 90-min gradient from 15–45% buffer B, and 10 min of 100% buffer A. The buffer C percentages (X) were 30, 40, 50, 60, 70, 80, and 100%, respectively, for the following 7-step analysis. In the final two steps, the gradient contained: 1 min of 100% buffer A, 4 min of 90% buffer C plus 10% B, a 5-min gradient from 0–10% buffer B, an 80-min gradient from 10–45% buffer B, a 10-min gradient from 45–100% buffer B, and 10 min of 100% buffer A. As peptides eluted from the microcapillary column, they were electrosprayed directly into an Elite mass spectrometer (ThermoFisher) with the application of a distal 2.4-kV spray voltage. A cycle of one full-scan FT mass spectrum (300–1600 m/z) at 240,000 resolution, followed by 20 data-dependent IT MS/MS spectra at a 35% normalized collision energy, was repeated continuously throughout each step of the multidimensional separation. Application of mass spectrometer scan functions and HPLC solvent gradients were controlled by the Xcalibur data system.
2.7. Data analysis
Both MS1 and MS2 (tandem mass spectra) were extracted from the XCalibur data system format (.RAW) into MS1 and MS2 formats using in-house software (RAW_Xtractor)(McDonald, Tabb et al. 2004). MS/MS spectra remaining after filtering were searched with Prolucid (Xu, Venable et al. 2006) against the UniProt_Mouse_03–25-2014 concatenated to a decoy database, in which the sequence for each entry in the original database was reversed (Peng, Elias et al. 2003). All searches were parallelized and performed on a Beowulf computer cluster consisting of 100 1.2-GHz Athlon CPUs (Sadygov, Eng et al. 2002). No enzyme specificity was considered for any search. The following modifications were searched for analysis: a static modification of 57.02146 on cysteine for all analyses, a differential modification of 452.2376 on methionine for AHA-biotin-alkyne, or 458.2452 for H-AHA-biotin-alkyne. Prolucid results were assembled and filtered using the DTASelect (version 2.0) program (Tabb, McDonald et al. 2002, Cociorva, D et al. 2007). DTASelect 2.0 uses linear discriminant analysis to dynamically set XCorr and DeltaCN thresholds for the entire dataset to achieve a user-specified false discovery rate (FDR). In DTASelect, the modified peptides were required to be fully tryptic, less than 5-ppm deviation from peptide match, and a FDR at the spectra level of 0.01. The FDRs are estimated by the program from the number and quality of spectral matches to the decoy database. For all datasets, the protein FDR was < 1% and the peptide FDR was < 0.5%. The MS data was quantified (i.e., generate heavy/light ratios) using the software pQuant(Liu, Song et al. 2014), which uses the DTASelect and MS1 files as the input. pQuant assigns a confidence score to each heavy/light ratio from zero to one. Zero, the highest confidence, means there is no interference signal, and one means the peptide signals are almost inundated by interference signals (i.e., very noisy). For this analysis, only peptide ratios with sigma less than 0.1 and intensity over 1e5 were allowed for quantification.
2.8. Pathway analysis
The significantly changed proteins, combined with unquantifiable large changes, were listed as input for functional pathway analysis performed by the software Ingenuity Pathway Analysis (IPA) (Calvano, Xiao et al. 2005). In addition, the significantly changed proteins combined with unquantifiable large changes of Day 4 and Day 14 were analyzed by STRING (Szklarczyk, Franceschini et al. 2015) using high confidence score settings and visualized by Cytoscape. Cell localization information was obtained from Uniprot (http://www.uniprot.org/).
2.9. Western blots
For total homogenates: Protein were resolved by NuPAGE 4–12% Bis-Tris gel (Life technologies) and transferred onto PVDF membranes using a semi-dry blotting apparatus (Life technologies). After blocking with 5% milk powder in PBS with 0.05% Tween 20, the membranes were incubated overnight at 4 °C with primary antibodies and then a HRP-conjugated secondary antibody.
For confirmation of protein levels in NSP datasets: 3mg proteins were taken as starting materials for each sample, followed with click reaction and Methanol/Chloroform precipitation. Then the precipitated protein was combined so that there would only be one pellet per 3-mg starting material. Precipitated pellets were resuspended with 100ul 8M urea and 10ul 20% SDS, boiled for 5 minutes and centrifuged at max speed for 10 minutes. The supernatant was transferred to a new 1.5ml eppendorf tube and incubated with 80ul neutravidin beads for two hours at room temperature while rotating. The resin was then washed with 1-ml PBS, then PBS with 5% acetonitrile, PBS, and a final wash of distilled water. The proteins were eluted off the resin by boiling in 50ul 4×sample buffer (Bio-Rad) and 2.5ul 20×reducing reagent (Bio-Rad). Protein were resolved by 4–12% gradient SDS-PAGE and transferred onto PVDF membranes using a semi-dry blotting apparatus (Life technologies). After blocking with 5% milk powder in PBS with 0.05% Tween 20, the membranes were incubated overnight at 4 °C with primary antibodies and then a HRP-conjugated secondary antibody. Protein bands were visualized by chemiluminescence. The following primary antibodies were used in this study: mouse monoclonal anti-GDF15 (sc-377195; Santa Cruz), monoclonal mouse anti-Galetin3 (sc-32790; Santa Cruz), mouse monoclonal anti-ApoA4 (sc-374543; Santa Cruz), mouse monoclonal anti- MYBPC3 (sc-137180; Santa Cruz) and rabbit polyclonal anti-NDUFVl (11238–1-AP; Proteintech). The following secondary antibodies were used in this study: HRP conjugated goat anti mouse and goat anti rabbit secondary antibodies.
2.10. Statistical analysis
One sample t-tests was employed to determine the significantly altered NSP at Day-4 ISO vs. Day-4 control and Day-14 ISO vs. Day-14 control. Significantly enriched pathways were determined by IPA. In Figure 4B, one-way ANOVA was performed, followed by Bonferroni’s post-hoc test, using the software Prism.
Figure 4:
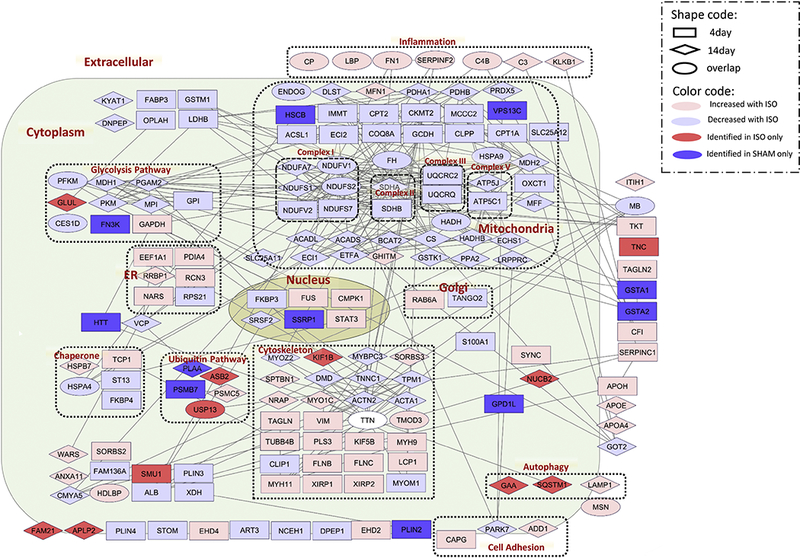
Functional CR network. The protein network and localization were analyzed by STRING and visualized by Cytoscape. The log ratio used in visualization was the average ratio of replicates. The white ellipse represents the protein Titin (TTN), which was found up-regulated at Day 4 of ISO but down-regulated at Day 14 of ISO.
3. Results
3.1. Experimental Design
Surgically implanted osmotic pumps delivered ISO (15 mg/kg/d) or PBS (i.e., control) to mice for 14 days (Figure 1A). The mice were fed a diet of either light AHA (L-AHA) or heavy AHA (H-AHA) for four days at two different time points. To determine the acute effects of ISO, mice were given the AHA diet during the first four days of ISO delivery (from Day 0 to Day 4), during which the cardiac remodeling process rate was at its maximum. To determine the prolonged effects of ISO, mice were given the AHA diet the last four days of the ISO treatment (from Day 10 to Day 14), during which the cardiac remodeling process was at a plateau. Mice were sacrificed at Day 4 or Day 14, and the LVs were quickly dissected, rinsed with PBS, and snap frozen. The LVs were homogenized. Before proteomic analysis, cardiac remodeling was confirmed in our ISO model. First, heart weight/body weight (HW/BW) ratio over 14 days of ISO was recorded. The HW/BW ratio in the ISO treated mice increased continuously whereas the HW/BW ratios in the control mice did not change over 14 days remaining steady at 4.5. Confirming cardiac ventricular remodeling, the HW/BW ratio in the ISO treated mice increased starting from Day 1 and was significantly higher versus the control group on days 5, 7, 10 and 14 (Figure 1B). We monitored the ejection fraction (EF) on the mouse hearts for every time point during ISO treatment. The EF was elevated starting from Day 1 compared to baseline on Day 0 due to beta-adrenergic over stimulation. We observed a downward trend of the EF on Day 14 suggesting that prolonged beta-adrenergic overstimulation (i.e., beyond 14 days) leads to deterioration of the heart function as previously reported(Drews, Tsukamoto et al. 2010, Lam, Wang et al. 2014) (Figure 1C). As the EF in our ISO model was still elevated, the mice were not in heart failure but rather displayed cardiac remodeling only. In addition, the activity of electron transport complex I(ETCI) from the OXPHOS system in mitochondria was analyzed. The ETCI activity decreased with ISO treatment indicating inhibition of mitochondrial function, specifically at the level of the OXPHOS(Lam, Wang et al. 2014)(Figure 1D). Finally, the homogenates were analyzed with immunoblots using two cardiac biomarkers, galectin-3 and growth differentiation factor - 15(GDF-15)(Savic-Radojevic, Pljesa-Ercegovac et al. 2017). Both of these proteins were increased in the ISO LV compared to the control LV suggesting cardiac dysfunction (Figure 1E). Overall, these experiments confirm the validity of the ISO treatment as a model for cardiac dysfunction.
Figure 1:
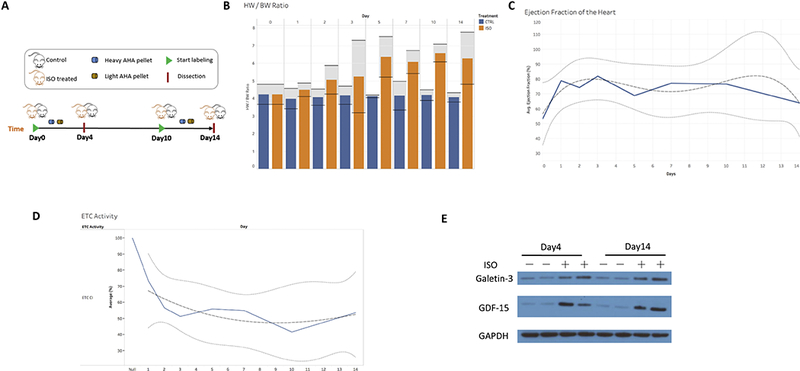
Confirmation of cardiac remodeling in ISO mouse model. A) Schematic workflow. Surgically implanted osmotic pumps delivered ISO (15 mg/kg/d) or saline (i.e., control) to mice for 14 days. The mice were fed a diet of either light AHA (L-AHA) or heavy AHA (H-AHA) for four days at different time points. Left ventricles (LVs) were dissected after labeling. B) HW/BW ratio and their 95% confidence intervals for each time point. The HW/BW ratio in the ISO treated mice increased continuously starting on Day 1, whereas the HW/BW ratios in the control mice did not change over 14 days remaining at 4.5. C) We monitored the cardiac function and displayed the ejection fraction (EF) for each day. Notably, the EF was elevated starting on Day 1 and remained elevated versus baseline on Day 0 due to the beta-adrenergic stimulation by ISO. As the EF was elevated during ISO treatment, the mice were not in heart failure but rather exhibited cardiac remodeling only. We observed a downward trend on Day 14 of ISO treatment indicating that prolonged beta-adrenergic stimulation (beyond 14 days) will eventually lead to cardiac heart failure. D) Mitochondrial complex I (ETC CI) activity was inhibited by ISO treatment over 14 days compared to baseline on day 0 demonstrating suppression of mitochondrial function at the level of ETC CI. The solid lines denote the regression. The dotted lines denote the 95% confidence interval. The dashed line denotes the trend. E) Mice model verification of cardiac remodeling. The levels of Galetin3 and GDF15 in total homogenates both control and ISO left ventricles were determined by western blotting.
Next, L-AHA and H-AHA tissues were mixed 1:1 (wt/wt). Day-4 ISO heart tissue was mixed with Day-4 control heart tissue, and Day-14 ISO heart tissue was mixed with Day-14 control heart tissue. Three biological replicates were analyzed for each L-AHA/H-AHA mixture (six mice per time point). Within each time point analysis, a label swap was performed between conditions to remove any possible effects of the heavy label on the proteome. Click chemistry was performed on these mixtures to covalently add a biotin-alkyne. After a tryptic digestion, the AHA peptides were isolated with neutravidin beads. These modified peptides were eluted from the beads and then identified by MS.
3.2. Identification of Newly-Synthesized Proteins at Day 4 and Day 14 of Beta-adrenergic-induced Cardiac Remodeling.
A similar number of NSPs were identified in the Day-4 and Day-14 analyses (Figure 2A and SuppTable 1). There were 921 and 740 NSPs identified in all three biological replicates for Day-4 and Day-14 analyses, respectively (Figure 2B and 2C), with 608 proteins identified at both time points in all biological replicates. Next, the ion chromatograms for the light and heavy AHA peptide pairs were extracted, and heavy/light ratios were calculated. There were 93 and 75 proteins that were statistically different between the ISO and control hearts in Day 4 and Day 14, respectively (Figure 2D and SuppTables 2 & 3). The significant proteins in Day-4 time points were evenly distributed over their fold change (ISO/control), while those in the Day-14 time points were not, with almost 70% of these proteins down-regulated in the ISO group. One caveat to MS quantitative analysis with heavy stable isotopes is that very large changes may prevent a heavy/light ratio from being generated (McClatchy, Liao et al. 2007). When there are very large changes between a light and heavy protein, only one is detected by the mass spectrometer due to its limited dynamic range. Thus, when only the abundance of one peptide (i.e., light or heavy) is measured, a light/heavy ratio cannot be calculated. To identify these unquantifiable large changes, the datasets were searched for proteins identified in all three biological replicates in one condition and not at all in the other (i.e., identified in three Day-4 ISO mice but never identified any of the Day-4 control mice) (SuppTable 4 & 5). In the Day-4 acute analysis, 3 proteins were identified in all three ISO mice but not in any control mice, suggesting that these proteins were up-regulated in CR. Conversely, 9 proteins were identified in all three Day-4 control mice but not in any of the Day-4 ISO mice, suggesting these proteins are down-regulated in CR. In the Day-14 analysis, 11 proteins were identified in all three ISO mice but not in any control mice, and 1 protein was identified in the control mice and not in the ISO mice. Finally, we confirmed the trend of three proteins, MYBPC3, NDUFV1, and APOA4, that were altered in our study (Figure 3A).
Figure 2:
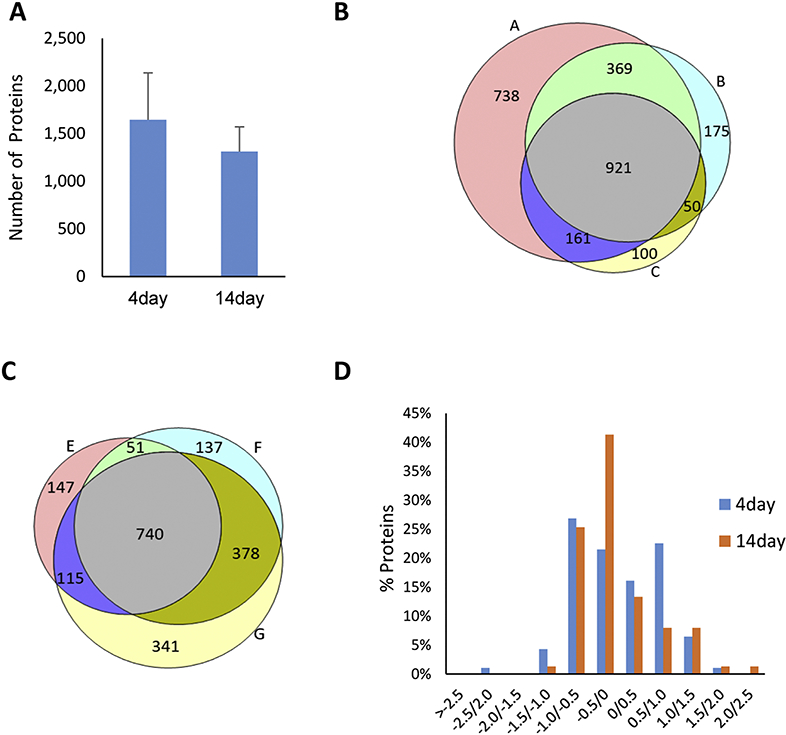
Protein identification of Day 4 and Day 14 time points. A) The number of NSPs identified at Day 4 and Day 14 are similar. Average protein numbers of three replicates for each time point were plotted. Error bar represents standard deviation. The overlap of protein identifications of three biological replicates are from (B) Day-4 (replicate A, B, and C) analysis and (C) Day-14 (replicate E, F, and G) analysis. D) Significant NSPs (p< 0.05 using a one sample t- test) were plotted in a histogram with their fold change (ISO/control) on the x-axis. There were 93 and 75 proteins that were significantly different between the ISO and control hearts in Day 4 and Day 14, respectively.
Figure 3:

NSP quantification A) Western blotting confirmation of MYBPC3, NDUFV1 and ApoA4 in NSP datasets. Taking GAPDH in input as internal standards, the three proteins showed consistent changes with the NSP datasets. N/A means the protein were not quantified at this time point. Bands were quantified using Image J software (National Institutes of Health, Bethesda, MD). B) Correlation of NSPs quantified at both acute and chronic analyses. The average of proteins ratios (ISO/control) calculated from 3 biological replicates of each time point were plotted to address correlation between the two datasets. C) NSPs quantified at both acute and chronic analyses. “*” represents an unquantifiable large change.
3.3. Functional Protein Network in Isoproterenol-induced Cardiac Remodeling
The proteins identified as unquantifiable large alterations were combined with the statistically significant proteins for further analysis. There were 266 proteins quantified confidently in both the Day-4 and Day-14 analyses (i.e., 6 MS analyses) with a correlation value of r=0.69, suggesting some similar individual protein perturbations between these time points (Figure 3B). Eighteen NSPs were statistically significant at both time points, indicating these changes occur during CR with the highest confidence (Figure 3C and SuppTable 6). These common significantly changed proteins all exhibited the same trend at both time points except for the protein titin (TTN). Next, a functional network for CR was generated using the STRING database to identify direct and indirect (i.e., functional) interactions within the dataset (Figure 4)(Szklarczyk, Franceschini et al. 2015). The proteins were annotated with multiple subcellular compartments, including the nucleus, mitochondria, endoplasmic reticulum, plasma membrane, and the extracellular matrix. The most striking trend from this network analysis was that 96% of the proteins that localized to the mitochondria were down-regulated in CR. In contrast, proteins involved in inflammation were all observed to be significantly up-regulated in the CR network. Next, pathway analysis was performed separately on the acute and prolonged effects of ISO (Figure 5A). The altered NSPs were significantly enriched in four pathways at both time points: mitochondrial dysfunction, oxidative phosphorylation, inflammatory response, and X receptor (LXR)/retinoid X receptors (RXR) activation. All of these pathways have previously been reported to be altered in cardiac remodeling (Zhou, Sucov et al. 1995, Epelman, Lavine et al. 2014, Brown, Perry et al. 2017). Ten additional pathways were significantly enriched, but these pathways were only significant at the Day-14 time point.
Figure 5:
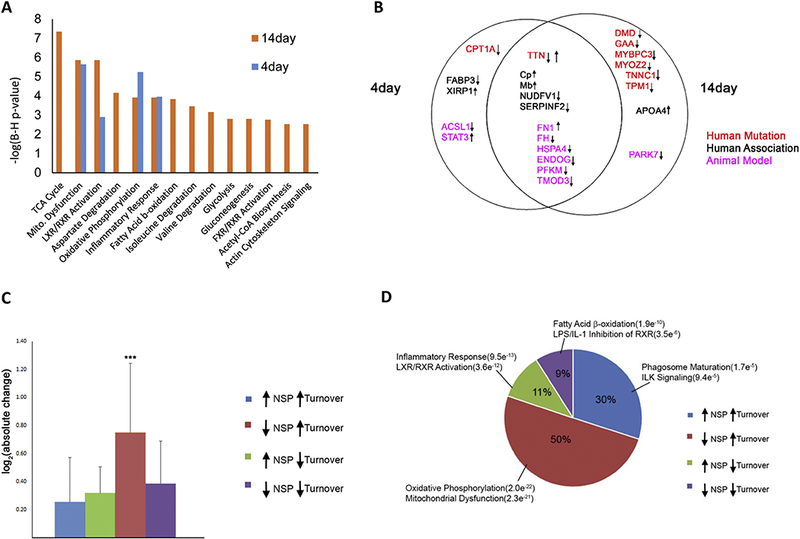
Comparison of acute and chronic ISO treatment. A) Pathway analysis on NSPs with significant changes and unquantifiable large changes at acute and chronic time points. B) Significantly altered NSPs previously associated with human cardiac diseases or animal models. Manual literature curation associated quantified NSPs in this study with previous reports of human genetic mutations causing cardiac disease (red), biomarker of or association with human cardiac disease (black), or association with animal models of cardiac disease (pink). Arrows indicate an up- or down-regulation of the NSP with ISO in this study. C) Percentage of trends identified from the comparison of NSP ratios and protein turnover rates in response to ISO. Each trend is annotated with the top two significantly enriched pathways. The number in parentheses represents the p-value for pathway enrichment. D) Comparison of the absolute NSP changes associated with each of the four NSP/turnover trends. One-way ANOVA analysis was performed, and a significant (p < 0.0001) difference was observed between the four trends. A Bonferroni’s Multiple Comparison post-hoc test was then performed, and a significant (***p<0.001) difference was observed in the “down NSP/up turnover” trend and each of the other trends.
3.4. Newly-Synthesized Proteins in CR Network-associated Cardiac Diseases.
Many biological pathways altered in our CR network have previously been associated with cardiac dysfunction. Next, we determined if specific proteins in our CR network have previously been associated with CR or other cardiac disorders. Twenty-five proteins were found to be directly linked to human cardiac disorders or animal models of cardiac disorders in previous publications (Figure 5B and SuppTable 7). Fifty percent of these proteins were significantly altered in both the acute (4 days) and prolonged (14 days) beta-adrenergic stimulation. Mutations in six genes, TTN, tropomyosin alpha-1 chain (TPM1); myozenin-2 (MYOZ2), troponin C (TNNC1); myosin-binding protein C (MYBPC3); and alpha-actinin-2 (ACTN2), reported to cause inherited human cardiomyopathy were all down-regulated in the CR network. The one exception was the contractile protein TTN, which was significantly up-regulated at Day 4 and down-regulated at Day 14. In addition, mutations in two other genes, lysosomal alpha- glucosidase (GAA) and dystrophin (DMD), correlated to Pompe Disease and Duchenne Muscular Dystrophy, respectively, have symptoms that also include cardiac dysfunction and heart failure (Lim, Li et al. 2014, van Westering, Betts et al. 2015). Other proteins in the CR network (i.e., fatty acid-binding protein (FABP3), fibronectin (FN1), protein deglycase DJ-1 (PARK7)) have been described as altered in or associated with human cardiomyopathy, but a direct genetic cause is not supported(McLean, Huang et al. 2008, Billia, Hauck et al. 2013, Mukherjee, Ong et al. 2015). Other proteins (i.e., long-chain-fatty-acid-CoA ligase 1 (ACSL1), ATP-dependent 6- phosphofructokinase (PFKM), fumarate hydratase (FH1)) have been associated with CR in animals(Garcia, Pujol et al. 2009, Ellis, Mentock et al. 2011, Ashrafian, Czibik et al. 2012).
3.5. Integration of Newly-Synthesized Proteins and Protein Turnover Rates.
To gain insight into how CR alters cardiac proteome homeostasis, the 14-day dataset was compared with our own published study, which quantified cardiac protein turnover rates using an identical 14-day ISO treatment (Lam, Wang et al. 2014). There were 270 proteins with quantified NSP (ISO/control) ratios and quantified protein turnover (ISO/control) rates (SuppTable 8). This comparison dataset was divided into four scenarios: 1) decreased NSP with increased turnover rate; 2) decreased NSP with decreased turnover rate; 3) increased NSP with decreased turnover rate; and 4) increased NSP with increased turnover rate. Changes in protein turnover rates can result from altered rates of degradation or synthesis. Since NSPs are linked directly to protein synthesis, this allowed us to postulate potential mechanisms for each of these observed trends (Table 1). Arguably, the simplest scenario for an increase in protein turnover is an increase in protein synthesis, and 30% of proteins in this comparison dataset exhibited this trend. On the other hand, a decrease in protein turnover could result from a decrease in protein synthesis, and this trend was observed in 9% of the proteins. The most frequently observed trend (50%) was a decrease in NSPs with ISO treatment coupled with an increase in turnover rate. We postulate that this correlates to an increase in protein degradation. The last trend, increased NSPs with decreased turnover rate, could represent a decrease in protein degradation but no change in synthesis. The absolute NSP changes were significantly the largest with the trend of a decrease in NSP with ISO treatment coupled with an increase in turnover rate (Figure 5C). Importantly, different biological pathways were enriched in each of the trends (Figure 5D). Overall, this analysis suggests that CR alters different signaling pathways employing different regulatory mechanisms.
Table 1:
Integration of protein turnover and PALM datasets for 14 days of ISO treatment. The table listed the four scenarios observed with this analysis and our proposed underlying mechanisms.
| Scenario | NSP | Turnover | Pro(2osed Mechanism |
|---|---|---|---|
| 1 | Increased | Increased | Increased translation |
| 2 | Decreased | Increased | Increased degradation |
| 3 | Increased | Decreased | Decreased degradation |
| 4 | Decreased | Decreased | Decreased translation |
4. Discussion
The cardiac proteome consists of thousands of proteins interacting in stable and transient complexes as well as numerous signaling pathways that maintain homeostasis and optimal cardiac function. CR is a process involved in many cardiac disorders and results not only from cardiac overload or injury but also from perturbations in a spectrum of biological processes, eventually leading to cell death (Shimizu and Minamino 2016). To study the temporal progression of alterations that underlie CR from this incredibly complex proteome, this study sought to reduce the complexity by quantifying the NSP sub-proteome using the methionine analog AHA. AHA has been routinely employed to identify and quantify NSPs by MS in cultured cells (Dieterich, Link et al. 2006, Zhang, Bowling et al. 2014). The PALM method was recently reported to incorporate AHA into mouse tissue proteomes during a discrete temporal window (McClatchy, Ma et al. 2015). The use of heavy stable isotopes, such as heavy amino acids and heavy water, have also been incorporated into proteomes to track changes in the synthesis of proteins(Schwanhausser, Gossen et al. 2009, De Riva, Deery et al. 2010). The advantage of employing AHA over these methods is that AHA labeled NSP can be separated from the “old” proteome through traditional biotin enrichment techniques and thereby increasing the signal of low abundant NSP. NSP that are labeled with heavy stable isotopes are unable to be separated from the whole proteome. With the limited dynamic range of the mass spectrometer, the abundant unlabeled “old” proteins will reduce the efficiency of NSP identification and quantification using these other methods. One advantage of heavy water over AHA is that heavy water can be used in humans while PALM is currently restricted to laboratory animals(Wang, Liem et al. 2014).
Taking advantage of the PALM strategy, the acute (i.e., Day 4) and prolonged (i.e., Day 14) proteomic changes were quantified in an ISO-induced mouse model of CR. Thousands of NSPs were identified and quantified from the LV, revealing temporal changes in CR. Accordingly, the proteins that were significantly altered between ISO and control hearts were assembled to form a temporal CR network. In correspondence with published studies on CR, a large variety of different protein functions were altered from multiple subcellular compartments. One important compartment in cardiac remodeling in the left ventricle is the extracellular matrix(ECM) which is produced by myofibroblasts(Hein and Schaper 2001). The ECM is mixture of structural, adhesive, fibrous, and cell surface proteins that are heavily glycosylated. When we compared our list of 2984 unique genes identified as NSP in our study, 65 could be annotated as ECM proteins or ECM related protein from a ECM database of 709 proteins(Liem, Murali et al. 2018). This suggests we did have low coverage of the ECM. This is consistent with numerous laboratories that have reported special enrichment techniques are required for comprehensive analysis of the insoluble ECM fraction (Lindsey, Jung et al. 2018). Five of the identified NSP-ECM proteins (APOA4, APOE, APOA4, FLNB, and MYH9) were significantly altered with ISO treatment. Interestingly, they were all up-regulated in the ISO left ventricles. Due to the low coverage of the insoluble ECM fraction, further investigation will be needed to determine if the NSP-ECM changes observed represent changes in ECM translation or changes in trafficking from the ECM fraction(Benesh, Miller et al. 2013). Nevertheless, many of the changes in or CR network has previously been associated with cardiac function. Mutated genes reported to cause inherited human cardiomyopathy were all down-regulated in the MCR network. This is consistent with the human mutations preventing expression or function of these proteins to promote cardiac dysfunction. In addition, FABP3(fatty acid-binding protein 3) is a heart-enriched cytosolic protein responsible for fatty acid transportation. It has been proposed as a useful acute biomarker for human cardiac dysfunction as it is detected in the blood hours after myocardial damage (McLean, Huang et al. 2008). FABP3 was significantly down-regulated in the Day-4 ISO hearts. Furthermore, newly synthesized FH1(fumarate hydratase) was reduced significantly in the MCR network. This protein regulates endogenous levels of fumarate, which is a metabolite of the citric acid cycle and reported to be cardioprotective. Knocking out the FH1 gene in mice increased fumarate levels, which was demonstrated to be cardioprotective against global cardiac ischemia (Ashrafian, Czibik et al. 2012). This correspondence with previously published cardiac disease research suggests great potential to increase the understanding of MCR from the proteins without previous links to cardiac dysfunction.
A unique aspect of our CR network is the temporal dynamics. CR involves a transition from adaptive or cardioprotective to maladaptive changes. One may hypothesize that the adaptive changes may be present in the acute phase while the maladaptive manifest in the prolonged phase. There is some evidence to support this hypothesis in our CR network. Six mutated genes known to cause inherited human cardiomyopathy were quantified as down- regulated by ISO in our CR network at Day-14 ISO treatment. This is consistent with the human mutations preventing expression or function of these proteins to cause cardiac dysfunction, implying more maladaptive changes occurred at the prolonged phase. In contrast, Xin actin- binding repeat-containing protein 1 (XIRP1) was observed within the acute phase of ISO treatment. XIRP1 has been reported to be decreased upon acute cardiac stress as a protective mechanism (Otten, van der Ven et al. 2010, Wang, Lin et al. 2014). TTN was unique in this dataset as it was significantly up-regulated at the acute timepoint and significantly down- regulated at the prolonged timepoint. This could represent a possible adaptive up-regulation of titin acutely induced by stress and possibly representing hypertrophy preceding the maladaptive decrease in titin function observed in inherited cardiac diseases. Overall, based on the protein synthesis profile, it appears there may be an early cardioprotective phase followed by a maladaptive phase in CR.
As we combined the NSP data with protein turnover rate measurements from in vivo ISO-treated mice compared to control mice, we identified a set of proteins falling into 4 distinct biological processes: Phagosome Maturation & Integrin-Linked Kinase Signaling, Oxidative Phosphorylation & Mitochondrial Function, Inflammatory Responses and Fatty Acid β-oxidation (Figure 5D). Proteins involving phagosome maturation and ILK signaling were significantly enriched, exhibiting increased NSP levels in parallel with protein turnover rate, indicating that these biological processes are up-regulated during ISO-induced CR. Phagosome maturation involves the activation of macrophages and neutrophils to degrade pathogens and cellular waste products in cardiomyocytes. Phagosomes degrade senescent cells and apoptotic cells to maintain cell homeostasis, and these groups of cells are affected during cardiac remodeling. In addition, ILK signaling is an essential process in the regulation of cardiac growth and hypertrophy. ILK is a multifunctional protein that physically links β-integrins with the contractile protein actin to regulate mechanoreceptors (Lu, Fedak et al. 2006, Nishimura, Kumsta et al. 2014). In our data, we observed an increase of NSP, and we observed that the rate of protein synthesis was greater than that of protein degradation (increased protein turnover, scenario 1 in Table 1) for proteins involved in phagosome maturation and ILK signaling, which falls in line with higher activities of these biological processes during ISO-induced CR compared to sham treatment. These findings imply that apoptosis as well as the activation of macrophages and neutrophils in cardiomyocytes are important factors within CR as previously reported and merit further study. (Narula, Haider et al. 1996, Olivetti, Abbi et al. 1997).
Our data also reveal that the majority (50%) of the protein dynamics detected are relevant to the biological processes of oxidative phosphorylation and mitochondrial function (Figure 5D); these proteins exhibited decreased levels of NSPs in parallel to a rate of protein synthesis greater than that of protein degradation (increased protein turnover rate, scenario 2 in Table 1). Perturbations in mitochondrial homeostasis and metabolism are essential mechanisms in the pathophysiology of cardiac remodeling and HF (Chaanine, Sreekumaran Nair et al. 2017). Previous observations suggest that mitochondrial biogenesis, signaling, and oxidative capacity are preserved or increased in the early stages of cardiac remodeling in order to match the energy demand imposed by a detrimental stressor such as ISO. Subsequently, the transition to CR and HF corresponds to a decrease in mitochondrial biogenesis and oxidative metabolism (Chaanine, Sreekumaran Nair et al. 2017). In our data, we observe a decreased level of NSPs involved in oxidative phosphorylation, but these proteins exhibited an increased rate of protein turnover. A rate of protein synthesis that is higher than the degradation rate while the level of NSPs is declining suggests an imbalance of mitochondrial homeostasis, with many dysfunctional proteins as well as impaired oxidative capacity in the face of high energy. These data fall in line with observations in HF patients and post-infarct mice with cardiac remodeling (Murphy, Ardehali et al. 2016, Chaanine, Sreekumaran Nair et al. 2017). Mitochondrial proteins may have a higher synthesis than degradation rate in CR (scenario 2 in Table 1); however, both the synthesis and degradation rates are most likely much lower compared to the sham group, clarifying the lower levels of NSPs. More specifically, most components of the respiratory chain, including electron transport chain (ETC) complex I (NDUFA7, NDUFA12, NDUFA13, NDUFS1, NDUFS2, NDUFS7, and NDUFV1) and complex V (ATP1A1, ATP1A2, ATP5A1, ATP5B, ATP5C1, ATP5F1, ATP5H, ATP5J, and ATP5O) exhibited this trend. Complex I - IV expression levels and activity appear to be decreased in HF patients (Murphy, Ardehali et al. 2016), falling in line with our data that shows a decrease in mitochondrial NSPs (scenario 2 in Table 1).
Since the initial observation by Levine et al., an extensive amount of information has been accumulating on the role of inflammation in the initiation and progression of HF due to ischemic injury, myocardititis, and long standing hypertension (Levine, Kalman et al. 1990). Many circulating pro-inflammatory cytokines such as TNF, IL-1, and IL-6 have been implicated in the process of CR and HF (Dick and Epelman 2016). The inflammatory response in CR is closely associated with the activation of the immune system. Notably, cardiomyocytes are a significant source of pro-inflammatory mediators that contribute to the progression of CR and HF. Our data confirm that proteins in the cardioprotective LXR activation pathway and inflammation pathways were enriched due to decreased degradation and a corresponding increase in NSPs (decreased turnover rate, scenario 3 in Table 1). Accordingly, inflammatory proteins may have a lower synthesis than degradation rate in CR; however, both the synthesis and degradation rates are most likely higher compared to the sham group, clarifying the higher levels of NSPs. Thus, these data suggest that inflammatory processes are increased in ISO-induced CR compared to sham treatment.
Interestingly, fatty acid oxidation has been reported to directly correlate with mitochondrial density and oxidative capacity, and a failing heart exhibits a metabolic shift towards glucose oxidation (Noordali, Loudon et al. 2017). Our data reveal that NSP levels in fatty acid β- oxidation are reduced while the protein degradation rate is higher than its synthesis, confirming that fatty acid oxidation is diminished during ISO-induced cardiac remodeling compared to sham treatment (scenario 4 in Table 1). This observation also corresponds to the reported reduction in the protein expression of this pathway in cardiac remodeling as the fuel source shifts from fatty acids to carbohydrates (Akhmedov, Rybin et al. 2015).
5. Conclusions
In conclusion, the balance between protein synthesis and degradation is the basis for homeostasis for any proteome, but the paucity of cellular proliferation in the heart increases the importance of this delicate balance in its response to stress. CR is a dynamic process that first attempts to restore homeostasis that was disrupted by physiological stressors but eventually produces deleterious changes to the proteome after prolonged stress, leading to cardiac dysfunction. Replicating known protein alterations in the cardiac literature provides confidence in the novel protein dynamics in our CR network, which will provide deeper insights into cardiac human physiology. The identification and timing of adaptive and maladaptive changes induced by CR is poorly understood. As our dataset provides a step to deconvolute the temporal dynamics of CR, future functional studies will be required to confidently assign adaptive/maladaptive labels to the proteins in our CR network. Our combination of NSP and turnover analysis suggests that the drugs that specifically manipulate translation and degradation (i.e., proteasomal or autophagic) processes could alter biological pathways differentially and possibly produce different clinical outcomes. Finally, we propose that the novel integration of NSP and turnover proteomic datasets could be applied to all animal models of disease to provide a greater understanding of the dysregulation of homeostasis in human diseases.
Supplementary Material
Supplementary Table 2: The NSPs that were significantly (p<0.05) altered in the Day-4 ISO analysis.
Supplementary Table 3: The NSPs that were significantly (p<0.05) altered in the Day-14 ISO analysis.
Supplementary Table 4: Large unquantifiable NSP changes in the Day-4 ISO analysis.
Supplementary Table 5: Large unquantifiable NSP changes in the Day-14 ISO analysis.
Supplementary Table 6: NSPs that were statistically significant with ISO at both the Day-4 and Day-14 analyses.
Supplementary Table 8: Quantified NSPs compared to their previously reported turnover rates.
Supplementary Table 7: Significantly altered NSPs in this study previously associated with cardiac dysfunction in the literature.
Supplementary Table 1: The number of identified newly synthesized proteins and peptides for each MS analysis.
Highlights.
We applied a new proteomic technique, PALM (Pulse Azidohomoalanine in Mammals), to quantitate the newly-synthesized protein (NSP) changes during the progression of isoproterenol (ISO)-induced cardiac remodeling in the mouse left ventricle.
The proteomic dataset revealed a complex combination of adaptive and maladaptive alterations at acute and prolonged time points including the identification of proteins not previously associated with cardiac remodeling.
The novel integration of the NSP dataset with previously published protein turnover rates in the ISO model demonstrated that alterations in specific biological pathways (e.g., inflammation and oxidative stress) are produced by differential regulation of protein synthesis and degradation.
Acknowledgements
The study was supported by NIH grants U54GM114833 (to P.P and J.R.Y), R35HL135772 (to P.P) and P41GM103533 (to J.R.Y.)
Footnotes
Competing Financial Interests
The authors declare no competing financial interests.
Disclosures
None
The raw MS data has been uploaded to ProteomeXchange (PXD010158).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhmedov AT, Rybin V and Marin-Garcia J (2015). “Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart.” Heart Fail Rev 20(2): 227–249. [DOI] [PubMed] [Google Scholar]
- Ashrafian H, Czibik G, Bellahcene M, Aksentijevic D, Smith AC, Mitchell SJ, Dodd MS, Kirwan J, Byrne JJ, Ludwig C, Isackson H, Yavari A, Stottrup NB, Contractor H, Cahill TJ, Sahgal N, Ball DR, Birkler RI, Hargreaves I, Tennant DA, Land J, Lygate CA, Johannsen M, Kharbanda RK, Neubauer S, Redwood C, de Cabo R, Ahmet I, Talan M, Gunther UL, Robinson AJ, Viant MR, Pollard PJ, Tyler DJ and Watkins H (2012). “Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway.” Cell Metab 15(3): 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesh EC, Miller PM, Pfaltzgraff ER, Grega-Larson NE, Hager HA, Sung BH, Qu X, Baldwin HS, Weaver AM and Bader DM (2013). “Bves and NDRG4 regulate directional epicardial cell migration through autocrine extracellular matrix deposition.” Mol Biol Cell 24(22): 3496–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billia F, Hauck L, Grothe D, Konecny F, Rao V, Kim RH and Mak TW (2013). “Parkinson- susceptibility gene DJ-1/PARK7 protects the murine heart from oxidative damage in vivo.” Proc Natl Acad Sci U S A 110(15): 6085–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC and Stinson EB (1982). “Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts.” N Engl J Med 307(4): 205–211. [DOI] [PubMed] [Google Scholar]
- Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, Cleland JG, Colucci WS, Butler J, Voors AA, Anker SD, Pitt B, Pieske B, Filippatos G, Greene SJ and Gheorghiade M (2017). “Expert consensus document: Mitochondrial function as a therapeutic target in heart failure.” Nat Rev Cardiol 14(4): 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF, Inflamm and P Host Response to Injury Large Scale Collab. Res (2005). “A network-based analysis of systemic inflammation in humans.” Nature 437(7061): 1032–1037. [DOI] [PubMed] [Google Scholar]
- Chaanine AH, Sreekumaran Nair K, Bergen RH 3rd, Klaus K, Guenzel AJ, Hajjar RJ and Redfield MM (2017). “Mitochondrial Integrity and Function in the Progression of Early Pressure Overload- Induced Left Ventricular Remodeling.” J Am Heart Assoc 6(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cociorva D, D LT and Yates JR (2007). “Validation of tandem mass spectrometry database search results using DTASelect.” Curr Protoc Bioinformatics Chapter 13: Unit 13 14. [DOI] [PubMed] [Google Scholar]
- Colucci WS (1997). “Molecular and cellular mechanisms of myocardial failure.” Am J Cardiol 80(11A): 15L–25L. [DOI] [PubMed] [Google Scholar]
- De Riva A, Deery MJ, McDonald S, Lund T and Busch R (2010). “Measurement of protein synthesis using heavy water labeling and peptide mass spectrometry: Discrimination between major histocompatibility complex allotypes.” Anal Biochem 403(1–2): 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng N, Zhang J, Zong C, Wang Y, Lu H, Yang P, Wang W, Young GW, Wang Y, Korge P, Lotz C, Doran P, Liem DA, Apweiler R, Weiss JN, Duan H and Ping P (2011). “Phosphoproteome analysis reveals regulatory sites in major pathways of cardiac mitochondria.” Mol Cell Proteomics 10(2): M110000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick SA and Epelman S (2016). “Chronic Heart Failure and Inflammation: What Do We Really Know?” Circ Res 119(1): 159–176. [DOI] [PubMed] [Google Scholar]
- Dieterich DC, Link AJ, Graumann J, Tirrell DA and Schuman EM (2006). “Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT).” Proc Natl Acad Sci U S A 103(25): 9482–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drews O, Tsukamoto O, Liem D, Streicher J, Wang Y and Ping P (2010). “Differential regulation of proteasome function in isoproterenol-induced cardiac hypertrophy.” Circ Res 107(9): 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JM, Mentock SM, Depetrillo MA, Koves TR, Sen S, Watkins SM, Muoio DM, Cline GW, Taegtmeyer H, Shulman GI, Willis MS and Coleman RA (2011). “Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs Fatty Acid oxidation and induces cardiac hypertrophy.” Mol Cell Biol 31(6): 1252–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ and Mann DL (2014). “Embryonic and adult- derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation.” Immunity 40(1): 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia M, Pujol A, Ruzo A, Riu E, Ruberte J, Arbos A, Serafin A, Albella B, Feliu JE and Bosch F (2009). “Phosphofructo-1-kinase deficiency leads to a severe cardiac and hematological disorder in addition to skeletal muscle glycogenosis.” PLoS Genet 5(8): e1000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goonasekera SA, Hammer K, Auger-Messier M, Bodi I, Chen X, Zhang H, Reiken S, Elrod JW, Correll RN, York AJ, Sargent MA, Hofmann F, Moosmang S, Marks AR, Houser SR, Bers DM and Molkentin JD (2012). “Decreased cardiac L-type Ca(2)(+) channel activity induces hypertrophy and heart failure in mice.” J Clin Invest 122(1): 280–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein S and Schaper J (2001). “The extracellular matrix in normal and diseased myocardium.” J Nucl Cardiol 8(2): 188–196. [DOI] [PubMed] [Google Scholar]
- Lam MP, Wang D, Lau E, Liem DA, Kim AK, Ng DC, Liang X, Bleakley BJ, Liu C, Tabaraki JD, Cadeiras M, Wang Y, Deng MC and Ping P (2014). “Protein kinetic signatures of the remodeling heart following isoproterenol stimulation.” J Clin Invest 124(4): 1734–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kalman J, Mayer L, Fillit HM and Packer M (1990). “Elevated circulating levels of tumor necrosis factor in severe chronic heart failure.” N Engl J Med 323(4): 236–241. [DOI] [PubMed] [Google Scholar]
- Liem DA, Murali S, Sigdel D, Shi Y, Wang X, Shen J, Choi H, Caufield JH, Wang W, Ping P and Han J (2018). “Phrase Mining of Textual Data to Analyze Extracellular Matrix Protein Patterns Across Cardiovascular Disease.” Am J Physiol Heart Circ Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JA, Li L and Raben N (2014). “Pompe disease: from pathophysiology to therapy and back again.” Front Aging Neurosci 6: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey ML, Jung M, Hall ME and DeLeon-Pennell KY (2018). “Proteomic analysis of the cardiac extracellular matrix: clinical research applications.” Expert Rev Proteomics 15(2): 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Song CQ, Yuan ZF, Fu Y, Chi H, Wang LH, Fan SB, Zhang K, Zeng WF, He SM, Dong MQ and Sun RX (2014). “pQuant improves quantitation by keeping out interfering signals and evaluating the accuracy of calculated ratios.” Anal Chem 86(11): 5286–5294. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Engelhardt S and Eschenhagen T (2003). “What is the role of beta-adrenergic signaling in heart failure?” Circ Res 93(10): 896–906. [DOI] [PubMed] [Google Scholar]
- Lu H, Fedak PW, Dai X, Du C, Zhou YQ, Henkelman M, Mongroo PS, Lau A, Yamabi H, Hinek A, Husain M, Hannigan G and Coles JG (2006). “Integrin-linked kinase expression is elevated in human cardiac hypertrophy and induces hypertrophy in transgenic mice.” Circulation 114(21): 2271–2279. [DOI] [PubMed] [Google Scholar]
- McClatchy DB, Liao L, Park SK, Venable JD and Yates JR (2007). “Quantification of the synaptosomal proteome of the rat cerebellum during post-natal development.” Genome Res 17(9): 1378–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchy DB, Ma Y, Liu C, Stein BD, Martinez-Bartolome S, Vasquez D, Hellberg K, Shaw RJ and Yates JR 3rd (2015). “Pulsed Azidohomoalanine Labeling in Mammals (PALM) Detects Changes in Liver-Specific LKB1 Knockout Mice.” J Proteome Res 14(11): 4815–4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald WH, Tabb DL, Sadygov RG, MacCoss MJ, Venable J, Graumann J, Johnson JR, Cociorva D and Yates JR 3rd (2004). “MS1, MS2, and SQT-three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications.” Rapid Commun Mass Spectrom 18(18): 2162–2168. [DOI] [PubMed] [Google Scholar]
- McLean AS, Huang SJ and Salter M (2008). “Bench-to-bedside review: the value of cardiac biomarkers in the intensive care patient.” Crit Care 12(3): 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee UA, Ong SB, Ong SG and Hausenloy DJ (2015). “Parkinson’s disease proteins: Novel mitochondrial targets for cardioprotection.” Pharmacol Ther 156: 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW 2nd, Kitsis RN, Otsu K, Ping P, Rizzuto R, Sack MN, Wallace D, Youle RJ, C. o. C. C. American Heart Association Council on Basic Cardiovascular Sciences, G. Council on Functional and B. Translational (2016). “Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association.” Circ Res 118(12): 1960–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW and Khaw BA (1996). “Apoptosis in myocytes in end-stage heart failure.” N Engl J Med 335(16): 1182–1189. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Kumsta C, Kaushik G, Diop SB, Ding Y, Bisharat-Kernizan J, Catan H, Cammarato A, Ross RS, Engler AJ, Bodmer R, Hansen M and Ocorr K (2014). “A dual role for integrin-linked kinase and beta1-integrin in modulating cardiac aging.” Aging Cell 13(3): 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordali H, Loudon BL, Frenneaux MP and Madhani M (2017). “Cardiac metabolism - A promising therapeutic target for heart failure.” Pharmacol Ther. [DOI] [PubMed] [Google Scholar]
- Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC and Anversa P (1997). “Apoptosis in the failing human heart.” N Engl J Med 336(16): 1131–1141. [DOI] [PubMed] [Google Scholar]
- Otten J, van der Ven PF, Vakeel P, Eulitz S, Kirfel G, Brandau O, Boesl M, Schrickel JW, Linhart M, Hayess K, Naya FJ, Milting H, Meyer R and Furst DO (2010). “Complete loss of murine Xin results in a mild cardiac phenotype with altered distribution of intercalated discs.” Cardiovasc Res 85(4): 739–750. [DOI] [PubMed] [Google Scholar]
- Peng J, Elias JE, Thoreen CC, Licklider LJ and Gygi SP (2003). “Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome.” J Proteome Res 2(1): 43–50. [DOI] [PubMed] [Google Scholar]
- Rona G, Chappel CI, Balazs T and Gaudry R (1959). “An infarct-like myocardial lesion and other toxic manifestations produced by isoproterenol in the rat.” AMA Arch Pathol 67(4): 443–455. [PubMed] [Google Scholar]
- Sadygov RG, Eng J, Durr E, Saraf A, McDonald H, MacCoss MJ and Yates JR 3rd (2002). “Code developments to improve the efficiency of automated MS/MS spectra interpretation.” J Proteome Res 1(3): 211–215. [DOI] [PubMed] [Google Scholar]
- Savic-Radojevic A, Pljesa-Ercegovac M, Matic M, Simic D, Radovanovic S and Simic T (2017). “Novel Biomarkers of Heart Failure.” Adv Clin Chem 79: 93–152. [DOI] [PubMed] [Google Scholar]
- Schiapparelli LM, McClatchy DB, Liu HH, Sharma P, Yates JR 3rd and Cline HT (2014). “Direct detection of biotinylated proteins by mass spectrometry.” J Proteome Res 13(9): 3966–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhausser B, Gossen M, Dittmar G and Selbach M (2009). “Global analysis of cellular protein translation by pulsed SILAC.” Proteomics 9(1): 205–209. [DOI] [PubMed] [Google Scholar]
- Shimizu I and Minamino T (2016). “Physiological and pathological cardiac hypertrophy.” J Mol Cell Cardiol 97: 245–262. [DOI] [PubMed] [Google Scholar]
- Speers AE and Cravatt BF (2009). “Activity-Based Protein Profiling (ABPP) and Click Chemistry (CC)- ABPP by MudPIT Mass Spectrometry.” Curr Protoc Chem Biol 1: 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ and von Mering C (2015). “STRING v10: proteinprotein interaction networks, integrated over the tree of life.” Nucleic Acids Res 43(Database issue): D447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb DL, McDonald WH and Yates JR 3rd (2002). “DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics.” J Proteome Res 1(1): 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PB and Tang Q (1984). “Development of isoproterenol-induced cardiac hypertrophy.” Can J Physiol Pharmacol 62(4): 384–389. [DOI] [PubMed] [Google Scholar]
- van Bilsen M, Smeets PJ, Gilde AJ and van der Vusse GJ (2004). “Metabolic remodelling of the failing heart: the cardiac burn-out syndrome?” Cardiovasc Res 61(2): 218–226. [DOI] [PubMed] [Google Scholar]
- van Westering TL, Betts CA and Wood MJ (2015). “Current understanding of molecular pathology and treatment of cardiomyopathy in duchenne muscular dystrophy.” Molecules 20(5): 8823–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Liem DA, Lau E, Ng DC, Bleakley BJ, Cadeiras M, Deng MC, Lam MP and Ping P (2014). “Characterization of human plasma proteome dynamics using deuterium oxide.” Proteomics Clin Appl 8(7–8): 610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Lin JL, Erives AJ, Lin CI and Lin JJ (2014). “New insights into the roles of Xin repeat- containing proteins in cardiac development, function, and disease.” Int Rev Cell Mol Biol 310: 89–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MP, Wolters D and Yates JR 3rd (2001). “Large-scale analysis of the yeast proteome by multidimensional protein identification technology.” Nat Biotechnol 19(3): 242–247. [DOI] [PubMed] [Google Scholar]
- Xu T, Venable JD, Park SK, Cociorva D, Lu B, Liao L, Wohlschlegel J, Hewel J and Yates JR (2006). “ProLuCID, a fast and sensitive tandem mass spectra-based protein identification program.” Molecular & Cellular Proteomics 5(10): S174–S174. [Google Scholar]
- Zhang G, Bowling H, Hom N, Kirshenbaum K, Klann E, Chao MV and Neubert TA (2014). “In-depth quantitative proteomic analysis of de novo protein synthesis induced by brain-derived neurotrophic factor.” J Proteome Res 13(12): 5707–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou MD, Sucov HM, Evans RM and Chien KR (1995). “Retinoid-dependent pathways suppress myocardial cell hypertrophy.” Proc Natl Acad Sci U S A 92(16): 7391–7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 2: The NSPs that were significantly (p<0.05) altered in the Day-4 ISO analysis.
Supplementary Table 3: The NSPs that were significantly (p<0.05) altered in the Day-14 ISO analysis.
Supplementary Table 4: Large unquantifiable NSP changes in the Day-4 ISO analysis.
Supplementary Table 5: Large unquantifiable NSP changes in the Day-14 ISO analysis.
Supplementary Table 6: NSPs that were statistically significant with ISO at both the Day-4 and Day-14 analyses.
Supplementary Table 8: Quantified NSPs compared to their previously reported turnover rates.
Supplementary Table 7: Significantly altered NSPs in this study previously associated with cardiac dysfunction in the literature.
Supplementary Table 1: The number of identified newly synthesized proteins and peptides for each MS analysis.