

Abstract
Decades of work have aimed to genetically reprogram T cells for therapeutic purposes1 using recombinant viral vectors, which do not target transgenes to specific genomic sites2,3. In addition, the need for viral vectors has slowed down research and clinical use as their manufacturing and testing is lengthy and expensive. Genome editing brought the promise of specific and efficient insertion of large transgenes into target cells through homology-directed repair (HDR)4,5. Here, we developed a CRISPR-Cas9 genome targeting system that does not require viral vectors, allowing rapid and efficient insertion of large DNA sequences (> 1kb) at specific sites in the genomes of primary human T cells, while preserving cell viability and function. This permits individual or multiplexed modification of endogenous genes. First, we apply this strategy to correct a pathogenic IL2RA mutation in cells from patients with monogenic autoimmune disease, demonstrating improved signaling function. Second, we replace the endogenous T cell receptor (TCR) locus with a new TCR redirecting T cells to a cancer antigen. The resulting TCR-engineered T cells specifically recognize tumour antigen and mount productive anti-tumour cell responses in vitro and in vivo. Taken together, these studies provide preclinical evidence that non-viral genome targeting can enable rapid and flexible experimental manipulation and therapeutic engineering of primary human immune cells.
The major barrier to effective non-viral T cell genome targeting of large DNA sequences has been the toxicity of the DNA6. While the introduction of short single-stranded oligodeoxynucleotide (ssODN) HDR templates does not cause significant T cell toxicity, it has been shown that larger linear double stranded (dsDNA) templates are toxic at high concentrations7,8. Contrary to expectations, we found that co-electroporation of human primary T cells with CRISPR-Cas9 ribonucleoprotein (Cas9 RNP9,10) complexes and long (>1kb) linear dsDNA templates reduced the toxicity associated with the dsDNA template (Extended Data Fig 1a-e). Cas9 RNPs were co-electroporated with a dsDNA HDR template designed to introduce an N-terminal GFP-fusion in the housekeeping gene RAB11A (Fig. 1a). Both cell viability and the efficiency of this approach were optimized by systematic exploration (Fig. 1b and Extended Data Fig. 1f-h) resulting in GFP expression in ~50% of both primary human CD4+ and CD8+ T cells. The method was reproducibly efficient with high cell viability (Fig. 1c, d, e). The system is also compatible with current manufacturing protocols for cell therapies. The method can be used with fresh or cryopreserved cells, bulk T cells or FACS-sorted sub-populations, and cells from whole blood or leukapheresis (Extended Data Fig. 2a-d).
Figure 1: Efficient non-viral genome targeting in primary human T cells.
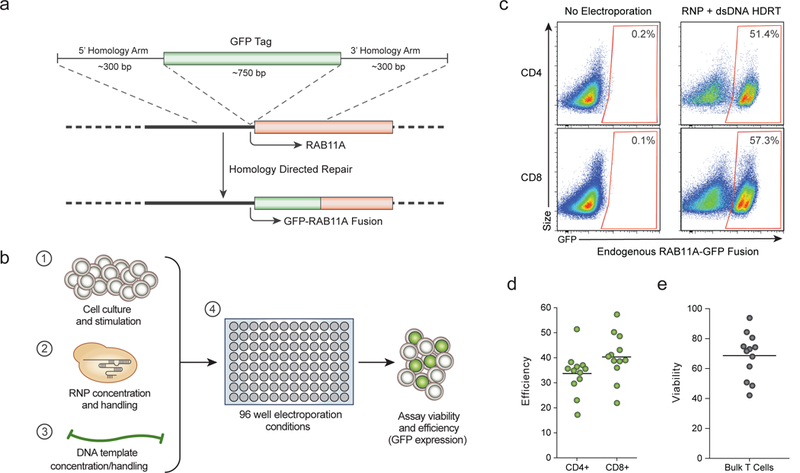
a, HDR mediated integration of a GFP fusion tag to the housekeeping gene Rab11A. b, Development and optimization of non-viral genome targeting for both cell viability and HDR efficiency. c, Insertion of a GFP fusion into the endogenous RAB11A gene using non-viral targeting in primary human CD4+ and CD8+ T cells. d, Average efficiency with the RAB11A-GFP HDR template was 33.7% and 40.3% in CD4+ and CD8+ cells respectively. e, Viability (number of live cells relative to non-electroporated control) after non-viral genome targeting averaged 68.6%. Efficiency and viability were measured 4 days following electroporation. Mean of n=12 independent healthy donors displayed (d-e). See also Extended Data Fig 1.
We next confirmed that the system could be applied broadly by targeting sequences in different locations throughout the genome. We efficiently engineered primary T cells by generating GFP fusions with different genes (Fig. 2a and Extended Data Fig. 2e-g). Live-cell imaging with confocal microscopy confirmed the specificity of gene targeting, revealing the distinct sub-cellular locations of each of the resulting GFP-fusion proteins11 (Fig. 2b). Appropriate chromatin binding of a transcription factor GFP-fusion protein was confirmed by performing genome-wide CUT & RUN12 analysis with an anti-GFP antibody (Fig. 2c and Extended Data Fig. 2h). Finally, we showed that gene targeting preserved the regulation of the modified endogenous gene. Consistent with correct cell-type specific expression, a CD4-GFP fusion was selectively expressed in the CD4+ population of T cells (Fig. 2d). Using HDR templates encoding multiple fluorescent proteins, we demonstrated that we could generate cells with bi-allelic gene targeting (Fig. 2e and Extended Data Fig. 3a-d) or multiplex modification of two (Fig. 2f and Extended Data Fig. 3e-h) or even three (Fig. 2g and Extended Data Fig. 3i) different genes13,14. These results show that multiple endogenous genes can be directly engineered without virus in T cells, and that gene and protein regulation are preserved.
Figure 2: Individual and multiplexed modification of endogenous T cell genes.
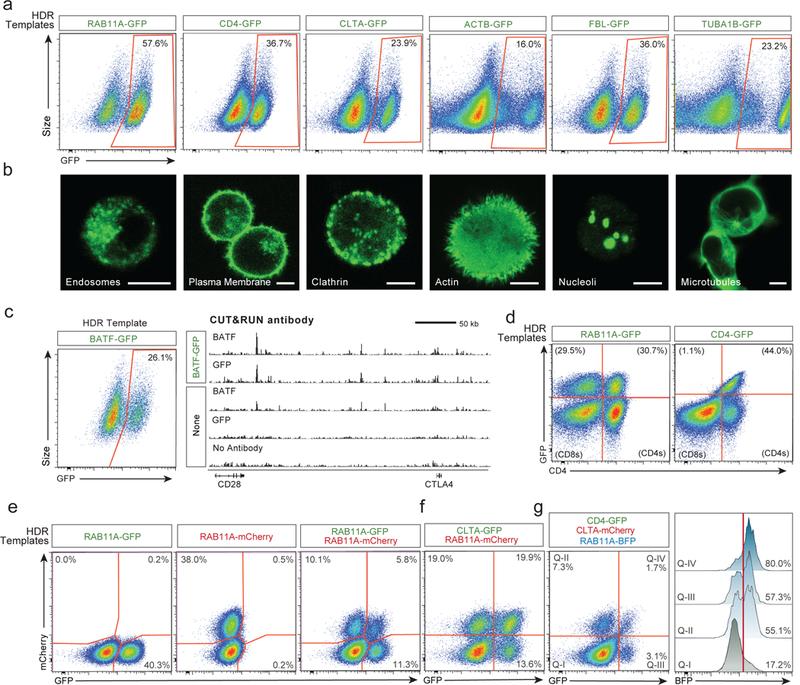
a, Non-viral genome targeting with GFP-fusion constructs into multiple endogenous genes. b, Confocal microscopy of live human T cells electroporated with the indicated HDR templates confirmed fusion-protein localization. Scale = 5 μm. c, GFP fused to the endogenous transcription factor BATF enabled genome-wide binding analysis (CUT&RUN) using anti-GFP or anti-BATF antibodies. d, RAB11A-fusions produced GFP positive CD4+ and CD8+ cells, whereas the CD4-fusions were selectively expressed in CD4+ cells. e, Bi-allelic non-viral genome targeting of two distinct fluorescent proteins into the same locus. f, Multiplexed non-viral genome targeting of HDR templates into two separate genomic loci. g, Simultaneous targeting of three distinct genomic loci. Cells positive for one (Q-II, Q-III) or two integrations (Q-IV), were highly enriched for a third HDR integration. One representative donor displayed from n=6 (a), n=4 (b, d-g), or n=2 (c) independent healthy donors. See also Extended Data Figs 2, 3.
For therapeutic use of genetically modified T cells, integrated sequences should be introduced specifically without unintended disruption of other critical genome sites15. We performed targeted locus amplification (TLA) sequencing16 and found no evidence of off-target integrations above the assay’s limit of detection (~1% of alleles) (Extended Data Fig. 4a-b). We further assessed potential off-target integrations at the single cell level by quantifying GFP+ cells generated using a Cas9 RNP that cuts outside the homology site. Similar to what has been described with viral HDR templates4,17, we found evidence to suggest that double-stranded templates could integrate independent of target homology18,19, albeit at low rates (Extended Data Fig. 4c-i). These rare events could be reduced almost completely by using single-stranded DNA templates20,21 (Extended Data Fig. 5a-d). As an additional safeguard that could be important for some applications, we demonstrated that efficient non-viral T cell genome targeting also could be achieved using either a single-stranded or double-stranded template with a Cas9 “nickase” engineered to reduce potential off-target double-stranded cuts22,23 (Extended Data Fig. 5e-h).
Having optimized this non-viral genome engineering approach in primary human T cells, we demonstrated its utility it in two different clinically relevant settings where targeted replacement of a gene would provide proof-of-principle that the method can be used to create therapeutically relevant gene modifications. Specifically, we tested the ability to rapidly and efficiently correct an inherited genetic alteration in T cells and we also tested the targeted insertion of the two chains of a TCR to redirect the specificity of T cells to recognize cancer cells.
We identified a family with monogenic primary immune deficiency with autoimmune disease caused by recessive loss-of-function mutations in the gene encoding the IL-2 alpha receptor (IL2RA)24 (Supplementary Table 4), which is essential for healthy regulatory T cells (Tregs)25 (Extended Data Fig. 6a-h). Whole exome sequencing revealed that the IL2RA-deficient children harboured compound heterozygous mutations in IL2RA (Fig. 3a and Extended Data Fig. 6i). One mutation, c.530A>G, creates a premature stop codon. With non-viral genome targeting, we were able to correct the mutation and observed IL2RA expression on the surface of corrected T cells from the patient (Fig. 3b). Long dsDNA templates led to efficient correction of the mutations. Because only two base pair changes were necessary (one to correct the mutation and one to silently remove the gRNA’s PAM sequence), a short single-stranded DNA (~120 bps) could also be used to make the correction. These single-stranded DNAs were able to correct the mutation at high frequencies, although here the efficiency of correction was lower than with the longer dsDNA template (Extended Data Fig. 7a, 8a). Correction was successful in T cells from all three siblings, but lower rates of IL2RA expression were seen in compound het 3, which could be due to altered cell-state associated with the patient’s disease or the fact she was the only sibling treated with immunosuppressive therapy (Supplementary Table 4 and Extended Data Fig. 8f). The second mutation identified, c.800delA, causes a frameshift in the reading frame of the final IL2RA exon. This frameshift mutation could be corrected both by HDR as well as by RNP cutting alone, presumably due to some of the small indels restoring the reading frame (Extended Data Fig. 8). Taken together, these data show that distinct mutations can be corrected in patient T cells using HDR template-dependent and non-HDR template-dependent mechanisms.
Figure 3: Monogenic autoimmune mutations corrected by non-viral genome targeting.
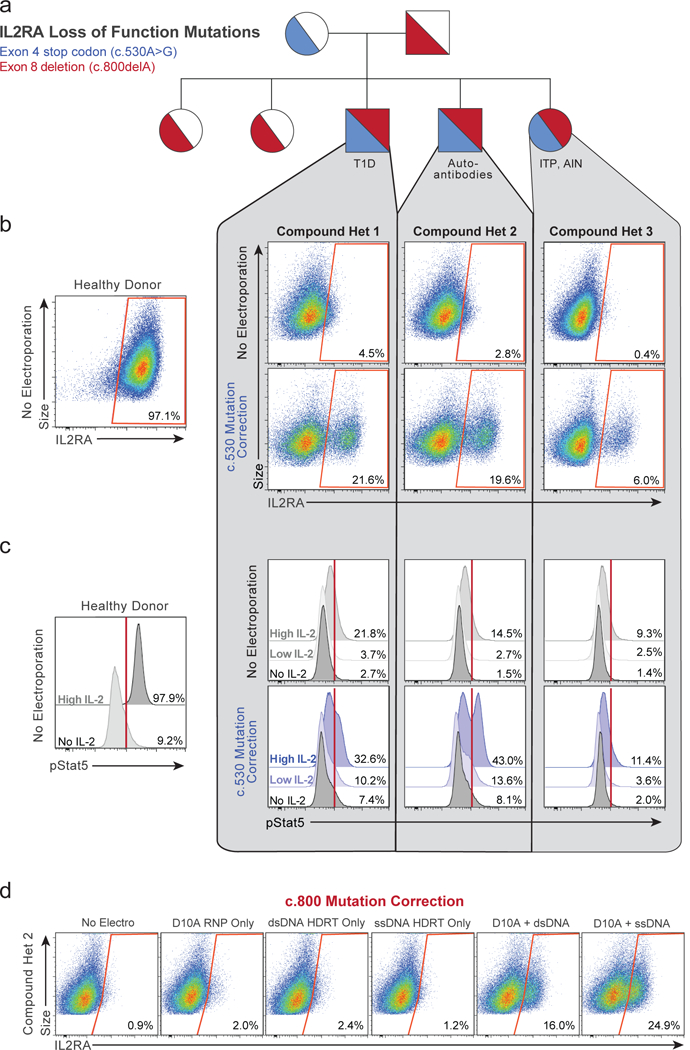
a, Pedigree of family with monogenic immune disease caused by compound heterozygous mutations in IL2RA (Supplementary Table 4). b, Correction of c.530A>G IL2RA mutation by non-viral genome targeting in three compound heterozygous siblings rescued IL2RA cell surface expression on CD3+ T cells 2 days following electroporation. c, 7 days after non-viral genome targeting, targeted unselected CD3+ T cells showed increased phosphorylation levels of Stat5 upon IL-2 stimulation compared to non-targeted controls. d, Non-viral genome targeting corrected the c.800delA mutation using D10A nickase and a long ssDNA HDR template. IL2RA surface expression measured after 9 days of ex-vivo expansion following electroporation (2 days following re-stimulation). n=3 compound heterozygous patients per correction. See also Extended Data Figs 6, 7, 8.
Mutation correction improved cell signalling function. Following correction of the c.530A>G IL2RA mutation, IL-2 treatment led to increased STAT5 phosphorylation, a hallmark of productive signalling (Fig. 3c and Extended Data Fig. 7c, 8c). In addition, following correction, we found that the modified T cells expressed both IL2RA and FOXP3, a critical transcriptional factor in Tregs (Extended Data Fig. 7d, 8d). We were also able to correct the IL2RA mutation in a sorted population of CD3+CD4+CD127loTIGIT+CD45RO+ Treg-like cells from a patient (Extended Data Fig. 7e-f), a strategy that could potentially be used in a gene-modified cell therapy for the children in this family. Cell-type specific and stimulus responsive expression of IL2RA is under tight control by multiple endogenous cis-regulatory elements that constitute a super-enhancer26,27. Therefore, effective therapeutic correction of the IL2RA defect is likely to depend on repairing the gene in its endogenous genomic locus; off-target effects should be avoided. We therefore demonstrated that the c.800delA mutation could also be repaired using Cas9 nickase combined with a single-stranded HDR template (Fig. 3d).
Non-viral genome targeting not only allows the correction of point mutations, but also enables integration of much larger DNA sequences. We were able to use a large DNA construct to rapidly reprogram the antigen specificity of human T cells, which is critical for many cellular immunotherapy applications. Recent work demonstrates that chimeric antigen receptors (CARs) have enhanced efficacy when they are genetically encoded in the endogenous TCR locus using CRISPR-Cas9 gene cutting and an adeno-associated virus vector as a repair template4. Targeting of specific TCR sequences to this locus is a more challenging problem because T cells must express paired TCR alpha (TCR-α) and beta chains (TCR-β) to make a functional receptor.
We developed a strategy to replace the endogenous TCR using non-viral genome targeting to integrate an approximately 1.5 kb DNA cassette into the first exon of the TCR-α constant region (TRAC) (Fig. 4a). This cassette encoded the full-length sequence of a TCR-β separated by a self-excising 2A peptide from the variable region of a new TCR-α, which encodes the full TCR-α sequence when appropriately integrated at the endogenous TRAC exon (Extended Data Fig. 9a-d). To test this strategy, we introduced a TCR-β and TCR-α pair (1G4) that recognizes the NY-ESO-1 tumour antigen28 into the TRAC locus of polyclonal T cells isolated from healthy human donors. Antibody staining for total TCR-α/β expression and NY-ESO-1-MHC dextramer staining for the NY-ESO-1 TCR expression revealed that non-viral genome targeting enabled reproducible replacement of the endogenous TCR in both CD8+ and CD4+ primary human T cells (Fig. 4b and Extended Data Fig. 9k). NY-ESO-1 TCR cells could also be generated with a similar targeting strategy at the TCR-β constant region (TRBC1/2) or with multiplexed simultaneous replacement of both endogenous TCR-α and TCR-β (Extended Data Fig. 9e-i). The majority of the T cells that did not express NY-ESO-1 TCR were TCR knockouts (Fig. 4b), presumably due to NHEJ events induced by the Cas9-mediated double-stranded breaks in TRAC exon 1. Up to ~70% of resulting TCR-positive cells recognized the NY-ESO-1 dextramer.
Figure 4: Replacement of the endogenous TCR by non-viral genome targeting.
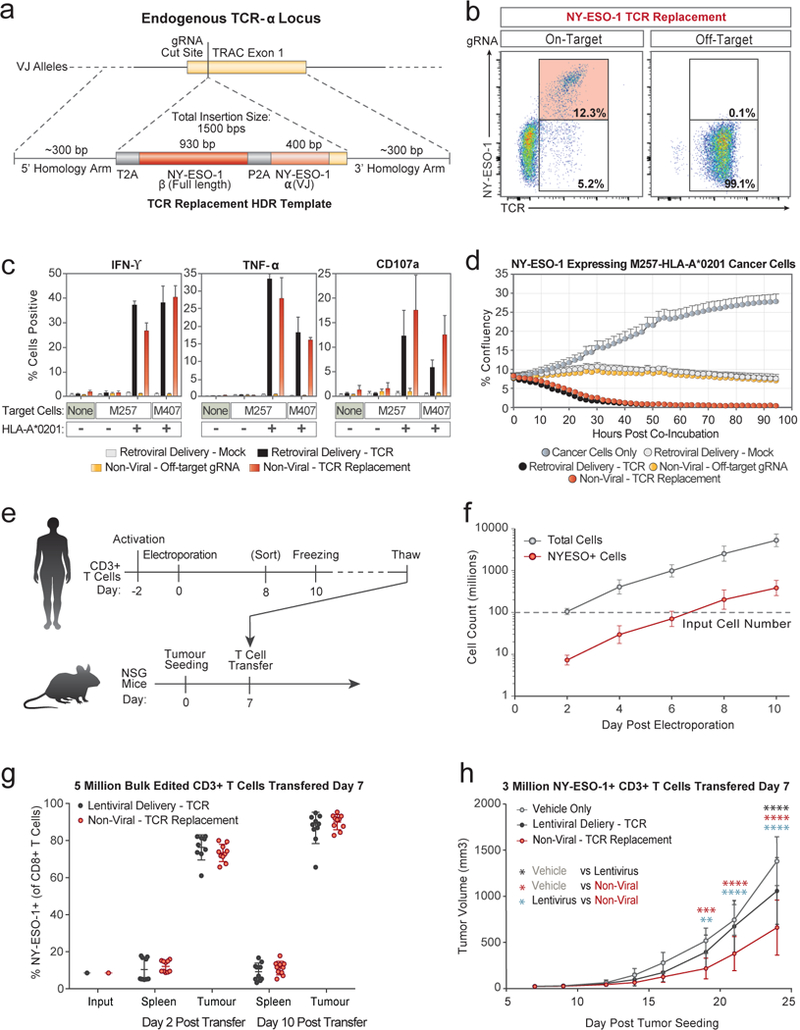
a, Schematic of HDR template used to replace the endogenous TCR. b, Non-viral genome targeting successfully replaced the endogenous TCR with the NY-ESO-1 antigen specific 1G4 TCR. c, Antigen-specific cytokine production and degranulation in CD8+ T cells with the replaced TCR. d, Antigen-specific target cell killing by CD8+ T cells with the replaced TCR. e, Melanoma tumour mouse xenograft model. f, Scalability of non-viral replacement of the endogenous TCR for adoptive cell therapy. g, Preferential in vivo localization of NY-ESO-1 TCR+ T cells to the tumour. h, Tumour growth following adoptive transfer of NY-ESO-1 TCR+ non-virally or lentivirally modified or vehicle alone (saline). One representative donor from n=6 (b) or n=2 (c, d) independent healthy donors with mean and standard deviation of technical triplicates (c, d). n=6 (f) or n=2 (g, h) independent healthy donors in 5 (g) or 7 mice (h) with mean and standard deviation (f-h). **P<0.01, ***P<0.001, ****P<0.0001 (Two Way ANOVA with Holm-Sidak’s multiple comparisons test). See also Extended Data Figs 9, 10.
Next, we assessed the tumour antigen-specific function of targeted human T cells. When the targeted T cells were co-cultured with two different NY-ESO-1+ melanoma cell lines, M257 and M407, the modified T cells robustly and specifically produced IFN-ɣ and TNF-α and induced T cell degranulation (measured by CD107a surface expression) (Fig. 4c). Cytokine production and degranulation only occurred when the NY-ESO-1 TCR T cells were exposed to cell lines expressing the appropriate HLA-A*0201 class I MHC allele required to present the cognate NY-ESO-1 peptide. Both the CD8+ and CD4+ T cell response was consistent across healthy donors, and was comparable to the response of T cells from the same healthy donor in which the NY-ESO-1 TCR was transduced by gamma retrovirus and heterologously expressed using a viral promoter (Fig. 4c and Extended Data Fig. 9j). NY-ESO-1 TCR knock-in T cells rapidly killed target M257-HLA-A*0201 cancer cells in vitro at rates similar to the positive control, retrovirally transduced T cells (Fig. 4d). Killing was selective for target cells expressing NY-ESO-1 antigen and the HLA-A*0201 allele, consistent across donors, and depended on the T cells being modified using both the correct gRNA and HDR template (Extended Data Fig. 9n-q).
Finally, we confirmed that non-viral genome targeting could be used to generate NY-ESO-1 TCR cells at scale and that these cells have in vivo anti-tumour function (Fig. 4e and Extended Data Fig. 10a). Given that knock-in efficiency was lower with non-viral targeting than with comparable sized AAV templates4, we first wanted to ensure that we could generate sufficient numbers of NY-ESO-1 positive cells for adoptive cell therapies. We electroporated 100 million T cells from six healthy donors, which after ten days of expansion yielded an average of 385 million NY-ESO-1 TCR T cells per donor (Fig. 4f and Extended Data Fig. 9i-m). NY-ESO-1 TCR knock-in T cells preferentially localized to, persisted at, and proliferated in the tumour rather than the spleen, similar to positive control lentivirally-transduced T cells (Fig. 4g and Extended Data Fig. 10b-f). Adoptive transfer of sorted NY-ESO-1 TCR T cells also reduced the tumour burden in treated animals (Fig. 4h).
Our therapeutic gene editing in human T cells is a process that takes only a short time from target selection to production of the genetically modified T cell product. In approximately one week, novel guide RNAs and DNA repair templates can be designed, synthesized, and the DNA integrated into primary human T cells that remain viable, expandable, and functional. The whole process and all required materials can be easily adapted to good manufacturing practices (GMP) for clinical use. Avoiding the use of viral vectors will accelerate research and clinical applications, reduce the cost of genome targeting, and potentially improve safety.
Looking forward, the technology could be used to “rewire” complex molecular circuits in human T cells. Multiplexed integration of large functional sequences at endogenous loci should allow combinations of coding and non-coding elements to be corrected, inserted, modified, and rearranged. Much work remains to be done to improve our understanding endogenous T cell circuitry if we are going to create synthetic circuits. Rapid and efficient non-viral tagging of endogenous genes in primary human cells will facilitate live-cell imaging and proteomic studies to decode T cell programs. Non-viral genome targeting provides an approach to re-write these programs in cells for the next generation of immunotherapies.
METHODS
Data reporting
No statistical methods were used to predetermine sample size. For all in vivo experiments experimental conditions were allocated randomly at the time of adoptive transfer, and experimental conditions were mixed among littermates. For in vivo tumour sizing experiments the investigator was blinded to experimental condition. No power analysis was used to determine sample sizes.
Antibodies
All antibodies used in the study for fluorescence activated cell sorting, flow cytometry, and cellular stimulations are listed in Supplementary Table 2.
Guide RNAs
All guide RNAs used in the study are listed in Supplementary Table 3.
Isolation of human primary T cells for gene targeting
Primary human T cells were isolated from healthy human donors either from fresh whole blood, residuals from leukoreduction chambers after Trima Apheresis (Blood Centers of the Pacific), or leukapheresis products (StemCell). Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood samples by Ficoll centrifugation using SepMate tubes (STEMCELL, per manufacturer’s instructions). T cells were isolated from PBMCs from all cell sources by magnetic negative selection using an EasySep Human T Cell Isolation Kit (STEMCELL, per manufacturer’s instructions). Unless otherwise noted, isolated T cells were stimulated as described below and used directly (fresh). When frozen cells were used, previously isolated T cells that had been frozen in Bambanker freezing medium (Bulldog Bio) per manufacturer’s instructions were thawed, cultured in media without stimulation for 1 day, and then stimulated and handled as described for freshly isolated samples. Fresh blood was taken from healthy human donors under a protocol approved by the UCSF Committee on Human Research (CHR #13-11950). Patient samples used for gene editing were obtained under a protocol approved by the Yale Human Investigation Committee (HIC). Additional leukapheresis products from healthy donors were collected either under UCLA Institutional Review Board (IRB) approval #10-001598 or purchased from AllCells, LLC. All patients and healthy donors provided informed consent.
Primary human T cell culture
Unless otherwise noted, bulk T cells were cultured in XVivo15 medium (STEMCELL) with 5% Fetal Bovine Serum, 50 mM 2-mercaptoethanol, and 10 mM N-Acetyl L-Cystine. Immediately following isolation, T cells were stimulated for 2 days with anti-human CD3/CD28 magnetic dynabeads (ThermoFisher) at a beads to cells concentration of 1:1, along with a cytokine cocktail of IL-2 at 200 U/mL (UCSF Pharmacy), IL-7 at 5 ng/mL (ThermoFisher), and IL-15 at 5 ng/mL (Life Tech). Following electroporation, T cells were cultured in media with IL-2 at 500 U/mL. Throughout the culture period T cells were maintained at an approximate density of 1 million cells per mL of media. Every 2-3 days post-electroporation additional media was added, along with additional fresh IL-2 to bring the final concentration to 500 U/mL, and cells were transferred to larger culture vessels as necessary to maintain a density of 1 million cells/mL.
RNP production
RNPs were produced by complexing a two-component gRNA to Cas9, as previously described10. Briefly, crRNAs and tracrRNAs were chemically synthesized (Dharmacon, IDT), and recombinant Cas9-NLS, D10A-NLS, or dCas9-NLS were recombinantly produced and purified (QB3 Macrolab). Lyophilized RNA was resuspended in 10 mM Tris-HCL (7.4 pH) with 150 mM KCl at a concentration of 160 μM, and stored in aliquots at −80C. crRNA and tracrRNA aliquots were thawed, mixed 1:1 by volume, and annealed by incubation at 37C for 30 min to form an 80 μNM gRNA solution. Recombinant Cas9 or the D10A Cas9 variant were stored at 40 μM in 20 mM HEPES-KOH pH 7.5, 150 mM KCl, 10% glycerol, 1 mM DTT, were then mixed 1:1 by volume with the 80 μM gRNA (2:1 gRNA to Cas9 molar ratio) at 37C for 15 min to form an RNP at 20 μM. RNPs were electroporated immediately after complexing.
Double stranded DNA HDRT production
Novel HDR sequences were constructed using Gibson Assemblies to insert the HDR template sequence, consisting of the homology arms (commonly synthesized as gBlocks from IDT) and the desired insert (such as GFP) into a cloning vector for sequence confirmation and future propagation. These plasmids were used as templates for high-output PCR amplification (Kapa Hotstart polymerase). PCR amplicons (the dsDNA HDRT) were SPRI purified (1.0X) and eluted into a final volume of 3 μL H2O per 100 μL of PCR reaction input. Concentrations of HDRTs were determined by nanodrop using a 1:20 dilution. The size of the amplified HDRT was confirmed by gel electrophoresis in a 1.0% agarose gel. All homology directed repair template sequences used in the study, both dsDNA and ssDNA, are listed in Supplementary Table 3.
Single stranded DNA HDRT production by exonuclease digestion
To produce long ssDNA as HDR templates, the DNA of interest was amplified via PCR using one regular, non-modified PCR primer and a second phosphorylated PCR primer. The DNA strand that will be amplified using the phosphorylated primer, will be the strand that will be degraded using this method. This makes it possible to prepare either a single-stranded sense or single-stranded antisense DNA using the respective phosphorylated PCR primer. To produce the ssDNA strand of interest, the phosphorylated strand of the PCR product was degraded by treatment with two enzymes, Strandase Mix A and Strandase Mix B, for 5 minutes (per 1kb) at 37C, respectively. Enzymes were deactivated by a 5 minute incubation at 80C. The resulting ssDNA HDR templates were SPRI purified (1.0X) and eluted in H2O. A more detailed protocol for the Guide-it™ Long ssDNA Production System (Takara Bio USA, Inc. #632644) can be found at the manufacturer’s website.
Single stranded DNA HDRT production by reverse synthesis
ssDNA HDR templates were synthesized by reverse transcription of an RNA intermediate followed by hydrolysis of the RNA strand in the resulting RNA:DNA hybrid product, as described21. Briefly, the desired HDR donor was first cloned downstream of a T7 promoter and the T7-HDR donor sequence amplified by PCR. RNA was synthesized by in vitro transcription using HiScribe T7 RNA polymerase (New England Biolabs) and reverse-transcribed using TGIRT-III (InGex). Following reverse transcription, NaOH and EDTA were added to 0.2 M and 0.1 M respectively and RNA hydrolysis carried out at 95C for 10 min. The reaction was quenched with HCl, the final ssDNA product purified using Ampure XP magnetic beads (Beckman Coulter) and eluted in sterile RNAse-free H2O. ssDNA quality was analysed by capillary electrophoresis (Bioanalyzer, Agilent).
Primary T cell electroporation
RNPs and HDR templates were electroporated 2 days following initial T cell stimulation. T cells were harvested from their culture vessels and magnetic anti-CD3/anti-CD28 dynabeads were removed by placing cells on an EasySep cell separation magnet for 2 minutes. Immediately prior to electroporation, de-beaded cells were centrifuged for 10 minutes at 90g, aspirated, and resuspended in the Lonza electroporation buffer P3 using 20 μL buffer per one million cells. For optimal editing, one million T cells were electroporated per well using a Lonza 4D 96-well electroporation system with pulse code EH115. Alternate cell concentrations from 200,000 up to 2 million cells per well resulted in lower transformation efficiencies. Alternate electroporation buffers were used as indicated, but had different optimal pulse settings (EO155 for OMEM buffer). Unless otherwise indicated, 2.5 μL of RNPs (50 pmols total) were electroporated, along with 2 μL of HDR Template at 2 μg/μL (4 μg HDR Template total).
The order of cell, RNP, and HDRT addition appeared to matter (Extended Data Fig. 1). For 96-well experiments, HDRTs were first aliquoted into wells of a 96-well polypropylene V-bottom plate. RNPs were then added to the HDRTs and allowed to incubate together at RT for at least 30 seconds. Finally, cells resuspended in electroporation buffer were added, briefly mixed by pipetting with the HDRT and RNP, and 24 μLs of total volume (cells + RNP + HDRT) was transferred into a 96 well electroporation cuvette plate. Immediately following electroporation, 80 μLs of pre-warmed media (without cytokines) was added to each well, and cells were allowed to rest for 15 minutes at 37C in a cell culture incubator while remaining in the electroporation cuvettes. After 15 minutes, cells were moved to final culture vessels.
Flow cytometry and cell sorting
Flow cytometric analysis was performed on an Attune NxT Acoustic Focusing Cytometer (ThermoFisher) or an LSRII flow cytometer (BD). Fluorescence activated cell sorting was performed on the FACSAria platform (BD). Surface staining for flow cytometry and cell sorting was performed by pelleting cells and resuspending in 25 μL of FACS Buffer (2% FBS in PBS) with antibodies at the indicated concentrations (Supplementary Table 2) for 20 minutes at 4C in the dark. Cells were washed once in FACS buffer before resuspension.
Confocal microscopy
Samples were prepared by drop casting 10 μl of a solution of suspended live T cells onto a 3×1” microscope slide onto which a 25 mm2 coverslip was placed. Imaging was performed on an upright configuration Nikon A1r laser scanning confocal microscope. Excitation was achieved through a 488 nm OBIS laser (Coherent). A long working distance (LWD) 60x Plan Apo 1.20 NA water immersion objective was used with additional digital zoom achieved through the NIS-Elements software. Images were acquired under “Galvano” mirror settings with 2x line averaging enabled and exported as TIFF to be analyzed in FIJI (ImageJ, NIH).
CUT&RUN
CUT&RUN was performed using epitope-tagged primary human T cells 11 days after electroporation and 4 days after re-stimulation with anti-CD3/anti-CD28 dynabeads (untagged cells were not electroporated). Approximately 20% and 10% of electroporated cells showed GFP-BATF expression as determined by flow cytometry in donor 1 and donor 2 samples, respectively. CUT&RUN was performed as described12, using anti-GFP (ab290), anti-BATF (sc-100974), and rabbit anti-mouse (ab46540) antibodies. Briefly, 6 million cells (30 million cells for anti-GFP CUT&RUN in GFP-BATF-containing cells) were collected and washed. Nuclei were isolated and incubated rotating with primary antibody (GFP or BATF) for 2 hours at 4C. BATF CUT&RUN samples were incubated an additional hour with rabbit anti-mouse antibody. Next, nuclei were incubated with proteinA-micrococcal nuclease (kindly provided by the Henikoff lab) for one hour at 4C. Nuclei were equilibrated to 0C and MNase digestion was allowed to proceed for 30 minutes. Solubilized chromatin CUT&RUN fragments were isolated and purified. Paired-end sequencing libraries were prepared and analysed on Illumina Nextseq machines and sequencing data was processed as described12. For peak calling and heatmap generation, reads mapping to centromeres were filtered out.
TLA sequencing and analysis
TLA sequencing was performed by Cergentis as previously described16. Similarly, data analysis of integration sites and transgene fusions was performed by Cergentis as previously described16. TLA sequencing was performed in two healthy donors, each edited at the RAB11A locus with either a dsDNA or ssDNA HDR template to integrate a GFP fusion (Fig. 1b). Sequencing reads showing evidence of primer dimers or primer bias (i.e. greater than 99% of observed reads came from single primer set) were removed.
In vitro Treg suppression assay
CD4+ T cells were enriched using the EasySep Human CD4+ T cell enrichment kit (STEMCELL Technologies). CD3+CD4+CD127loCD45RO+TIGIT+ enriched Treg-like cells from IL2RA-deficient subjects and HD as well as CD3+CD4+IL2RAhiCD127lo Tregs from IL2RA+/− individuals were sorted by flow cytometry. CD3+CD4+IL2RA-CD127+ responder T cells (Tresps) were labeled with CellTrace CFSE (Invitrogen) at 5 μM. Tregs and HD Tresps were co-cultured at a 1:1 ratio in the presence of beads loaded with anti-CD2, anti-CD3 and anti-CD28 (Treg Suppression Inspector; Miltenyi Biotec) at a 1 bead: 1 cell ratio. On days 3.5 to 4.5, co-cultures were analyzed by FACS for CFSE dilution. % inhibition is calculated using the following formula: 1 - (% proliferation with Tregs / % proliferation of stimulated Tresps without Tregs).
Sorting and TSDR analysis of corrected Tregs
Ex-vivo expanded Tregs and T effector cells from a healthy control and a patient with IL2RA compound heterozygous mutations (D6) were thawed and stained. Live cells were sorted based on expression of CD25 and CD62L markers directly into ZymoResearch M-digestion Buffer (2x) (cat# D5021-9) supplemented with proteinase K. The lysate was incubated at 65°C for greater than 2 hours and then frozen. Bisulfite conversion and pyrosequencing of the samples was performed by EpigenDx (assay ID ADS783-FS2) to interrogate the methylation status of 9 CpG sites intron 1 of the FOXP3 gene, spanning −2330 to −2263 from ATG.
Generation of retrovirally and lentivirally transduced control T cells
For retroviral infections, clinical grade MSGV-1-1G4 (NY-ESO-1 TCR transgene) retroviral vector (IUVPC, Indianapolis, IN) was used. For lentiviral production, HEK 293 cells were plated at 18 million cells in 15 cm dishes the night before transfection. Cells were transfected using the lipofectamine 3000 reagent following the manufacturer’s protocol (L3000001). Transfection media was changed the following day to fresh HEK 293 media (DMEM + 5% FBS + 1% pen/strep) with viral boost reagent per the manufacturer’s protocol at 500x (Alstem viral boost reagent #VB100). 48 hours after transfection the viral supernatant was collected, filtered, and the Alstem precipitation solution was added, mixed, and refrigerated at four degrees for four hours, concentrated by centrifugation, and the viral pellet was then resuspended at 100x in cold PBS following the manufacturer’s protocol (lentivirus precipitation solution #VC100).
T cells for viral infection were activated similarly to non-virally edited cells. Both retroviral and lentiviral transductions occurred 48 hours after TCR/cytokine stimulus, followed by expansion in IL-2 similarly to non-virally edited cells. For retroviral transduction, T cells were infected by spinoculation in retronectin (Clontech, Mountain View, CA) coated plates. Control mock-transduced T cells were also generated. For lentiviral transduction, viral concentrate was added to 1X final concentration.
Antigen specific TCR expression analysis
The expression of the NY-ESO-1 TCR was assessed in virally and non-virally modified cells with an NY-ESO-1 specific (SLLMWITQC) dextramer-PE (Immundex, Copenhagen, Denmark) according to the manufacturer’s protocol. Negative dextramer (Immudex, Copenhagen, Denmark) was used as a negative control.
T cell activation and cytokine production analysis
Melanoma cell lines were established from the biopsies of melanoma patients under the UCLA IRB approval #11-003254. Cell lines were periodically screened for mycoplasma contamination as well as authenticated using GenePrint® 10 System (Promega, Madison, WI), and were matched with the earliest passage cell lines. M257 (NY-ESO-1+ HLA-A*0201-), M257-A2 (NY-ESO-1+ HLA-A*0201+) and M407 (NY-ESO-1+ HLA-A*0201+) were cocultured 1:1 with the modified PBMCs in cytokine free media. The recommended amount per test of CD107a-APC-H7 (Supplementary Table 2) antibody was added to the coculture. After 1 hour, half the recommended amount of BD Golgi Plug and BD Golgi Stop (BD bioscience, San Jose, CA) was added to the coculture. After 6 hours, surface staining was performed followed by cell permeabilization using BD cytofix/cytoperm (BD bioscience, San Jose, CA) and intracellular staining according to manufacturer instructions (Supplementary Table 2). Negative dextramer and Fluorescence minus one (FMOs) staining were used as controls.
T Cell in vitro killing assay
M202-nRFP (NY-ESO-1-, HLA-A*0201+), M257-nRFP (NY-ESO-1+ HLA-A*0201-), M257-A2-nRFP (NY-ESO-1+ HLA-A*0201+), M407-nRFP (NY-ESO-1+ HLA-A*0201+), and A375-nRFP (NY-ESO-1+ HLA-A*0201+) melanoma cell lines stably transduced to express nuclear RFP (Zaretsky 2016 NEJM) were seeded approximately 16 hours before starting the coculture (~1500 cells seeded per well). Modified T cells were added at the indicated E:T ratios. All experiments were performed in cytokine free media. Cell proliferation and cell death was measured by nRFP real time imaging using an IncuCyte ZOOM (Essen, Ann Arbor, MI) for 5 days.
In vivo mouse solid tumour model
All mouse experiments were completed under a UCSF Institutional Animal Care and Use Committee protocol. We used 8 to 12 week old NOD/SCID/IL-2Rɣ-null (NSG) male mice (Jackson Laboratory) for all experiments. Mice were seeded with tumours by subcutaneous injection into a shaved right flank of 1×106 A375 human melanoma cells (ATCC CRL-1619). At seven days post tumour seeding, tumour size was assessed and mice with tumour volumes between 15-30 mm3 were randomly assigned to experimental and control treatment groups. Indicated numbers of T cells were resuspended in 100 μl of serum-free RPMI and injected retro-orbitally. For tumour sizing experiments, the length and width of the tumour was measured using electronic calipers and volume was calculated as v = 1/6 * π * length * width * (length + width) / 2. The investigator was blinded to experimental treatment group during sizing measurements. A bulk edited T cell population (5×106) or a sorted NY-ESO-1 TCR+ population (3×106) was transferred as indicated in figures and legends. For bulk edited T cell transfers, lentivirally edited cells generally had a higher percentage of NY-ESO-1 positive cells, so mock-infected cells were added to normalize the percentage of total T cells NY-ESO-1+ to equal that of the bulk population of non-virally edited T cells (~10% NY-ESO-1+). For sorted T cell transfers, NY-ESO-1+ T cells were FACS sorted eight days following electroporation, expanded for two additional days, and frozen (Bambanker freezing medium, Bulldog Bio). Non-virally or lentivirally modified human T cells were then thawed and rested in media overnight prior to adoptive transfer. For flow cytometric analysis of adoptively transferred T cells, single-cell suspensions from tumours and spleens were produced by mechanical dissociation of the tissue through a 70 μm filter. All animal experiments were performed in compliance with relevant ethical regulations per an approved IACUC protocol (UCSF), including a tumor size limit of 2.0 cm in any dimension.
Data and reagent availability
CUT&RUN data has been deposited in GEO as record GSE108600. TLA and amplicon sequencing data is available upon request. Source data for animal experiments (Fig. 4g, h and Extended Data Fig. 10) is provided. Plasmids containing the HDR template sequences used in the study are available through AddGene.
Extended Data
Extended Data Figure 1: Development of non-viral genome targeting in primary human T cells.
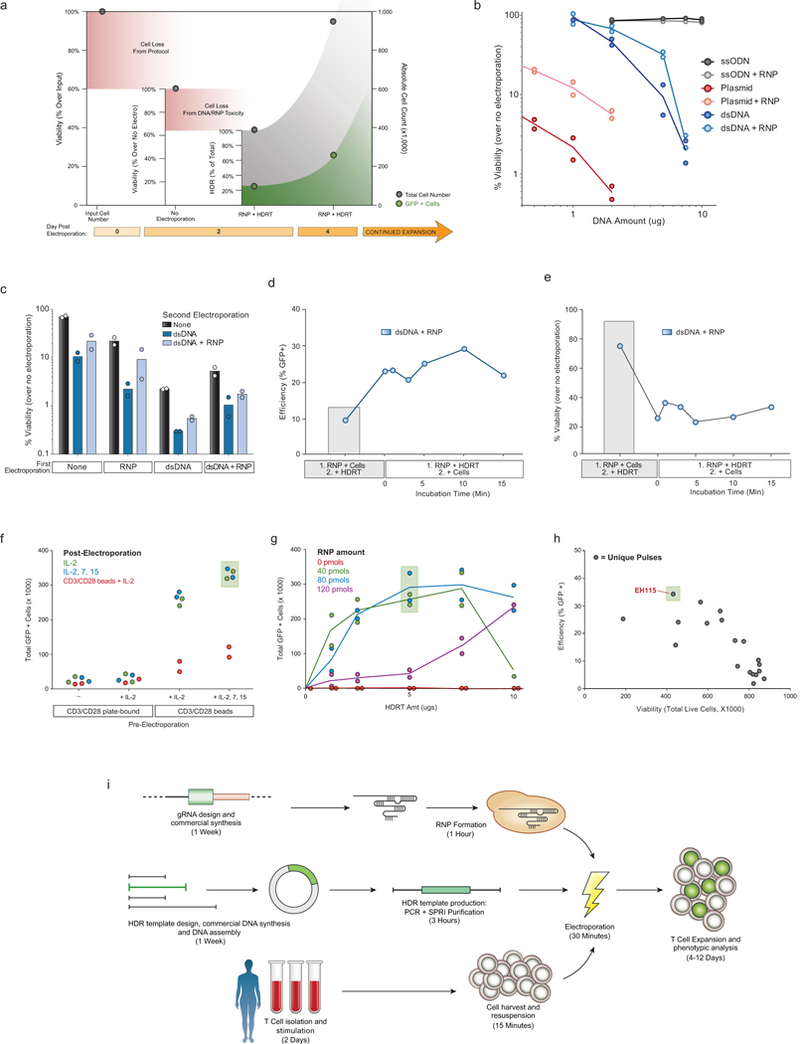
a, Except where explicitly noted otherwise, we use viability to refer to the number of live cells in an experimental condition (expressed as a %) relative to an equivalent population that went through all protocol steps except for the actual electroporation (No electroporation control). We use the term efficiency to refer to the percentage of live cells in a culture expressing the “knocked in” exogenous sequence (such as GFP). Finally, the total number of cells positive for the desired modification was calculated by multiplying the efficiency by the absolute cell count. Methodological changes that maximized efficiency were not always optimal for the total number of positive cells, and vice-versa. b, Double-stranded (ds)DNA, both circular (plasmid) and linear, when electroporated into primary human T cells, caused marked loss in viability with increasing amounts of template. Co-delivery of an RNP caused less reduction in viability post electroporation. Of note, in these experiments no loss in viability was seen with short single-stranded (ss)DNA oligo donor nucleotides (ssODNs). c, RNPs must be delivered concurrently with DNA to see increased viability. T cells from two donors were each electroporated twice with an eight-hour rest in between electroporations. While two electroporations so closely interspersed caused a high degree of cell death, delivery of the RNP and linear dsDNA template could be delivered separately. Initial RNP electroporation did not protect from the loss of viability if dsDNA was delivered alone in the second round of electroporation. d, We determined whether the order of adding reagents influenced targeting efficiency and viability. In wells where the RNP and the DNA HDR template were mixed together prior to adding the cells (1. RNP + HDRT; 2. + Cells), there was a marked increase in targeting efficiency. e, Note, with the high concentration of dsDNA used in these experiments, viability was higher if the RNP and cells were mixed first and the DNA template was added immediately prior to electroporation (1. RNP + Cells; 2. + HDRT). Taken together, these data likely suggest that pre-incubation of the RNP and HDR template, even for a short period, increased the amount of DNA HDR template delivered into the cell, which increased efficiency but decreased viability. However, viability after RNP and dsDNA HDR template pre-incubation was still higher than was observed with dsDNA HDR template electroporation by itself (b). 5 μg of dsDNA HDR temple was used in (c-e). f, Primary human T cells were cultured for 2 days using varying combinations of anti-CD3/CD28 T cell receptor (TCR) stimulation and cytokines prior to electroporation of RAB11A targeting RNP and HDR template, followed by varying culture conditions post-electroporation. g, Among the RNP and HDR template concentrations tested here, optimal GFP insertion into RAB11A was achieved at intermediate concentrations of the RNP and dsDNA HDRT. h, Arrayed testing of electroporation pulse conditions showed that, in general, conditions yielding higher HDR efficiency decreased viability. EH115 was selected to optimize efficiency, while still maintaining sufficient viability. i, Diagrammatic timeline of non-viral genome targeting. Approximately one week is required to design, order from commercial suppliers, and assemble any novel combination of genomic editing reagents (gRNA and the HDR template). Two days prior to electroporation, primary human T cells isolated from blood or other sources (Extended Data Fig. 2) are stimulated. dsDNA HDR templates can be made easily by PCR followed by a SPRI purification to achieve a highly concentrated and pure product suitable for electroporation. On the day of electroporation, the gRNA (complexed with Cas9 to form an RNP), the HDR template, and harvested stimulated T cells are mixed and electroporated, a process taking approximately 1.5 hours. After electroporation, engineered T cells can be readily expanded for an additional 1-2 weeks. Viability was measured 2 days following electroporation and GFP expression was measured at day 4. Graphs display mean (b, c, g, h) and/or individual donor values (b-h) in n=2 independent healthy donors (b-h). For d, e, and h one representative donor is shown.
Extended Data Figure 2: Non-viral genome targeting is consistent across T cell types and reproducible across target loci.
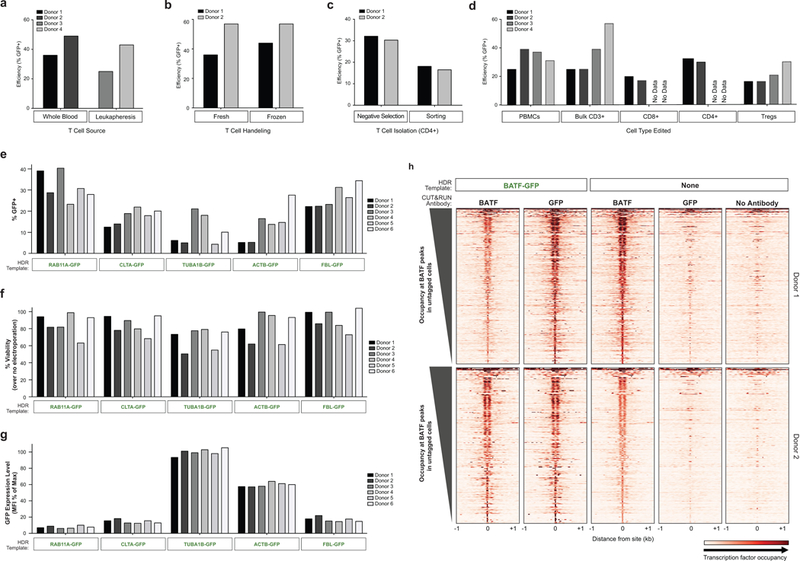
a, Efficient genome targeting was accomplished with a variety of T cell processing and handling conditions that are used with current manufacturing protocols for cell therapies. Non-viral genome targeting of a RAB11A-GFP fusion protein using a linear dsDNA HDR template was performed in bulk CD3+ T cells isolated from either whole blood draws or by leukapheresis. b, Targeting was similar either using bulk CD3+ T cells fresh after isolation or after cryopreservation (stored in liquid nitrogen and thawed prior to initial activation). c, CD4+ T cells isolated by fluorescent activated cell sorting (FACS) showed detectable GFP+ cells indicative of efficient editing, albeit at lower rates than targeting in CD4+ cells isolated by negative selection (potentially due to the added cellular stress of sorting). d, Using the same optimized non-viral genome targeting protocol (Methods), a variety of T cell types could be successfully edited, including peripheral blood mononuclear cells, without any selection (T cell culture conditions cause preferential growth of T cells from PBMCs). Sorted T cell subsets CD8+, CD4+, and CD4+IL2RA+CD127lo regulatory T cells (Tregs) could be successfully targeted with GFP integration. PBMCs were cultured for two days identically to primary T cells (Methods). Bulk CD3+ T cells were isolated by negative enrichment. The electroporations in panel d used only 2 μg of dsDNA HDR template, a concentration that was later found to be less efficient than the final 4 μg (contributing to the lower efficiencies seen compared to Fig. 1d). RAB11A-GFP template was used with on-target gRNA was used in a-d. e, Four days after electroporation of different GFP templates along with a corresponding RNP into primary CD3+ T cells from six healthy donors, GFP expression was observed across both templates and donors. f, High viability post-electroporation was similarly seen across target loci. g, The fusion tagged proteins produced by integrating GFP into specific genes localized to the subcellular location of their target protein (Fig. 2b), and were also expressed under the endogenous gene regulation, allowing protein expression levels to be observed in living primary human T cells. Note how GFP tags of the highly expressed cytoskeletal proteins TUBA1B (beta tubulin) and ACTB (beta actin) show consistently higher levels of expression compared to the other loci targeted across six donors. GFP mean fluorescent intensity (MFI) was calculated for the GFP+ cells in each condition/donor, and normalized as a percentage of the maximum GFP MFI observed in the experiment. h, Gene fusions not only permitted the imaging and analysis of expression of endogenous proteins in live cells, but also could be used for biochemical targeting of specific proteins. For example, ChIP-Seq, and more recently CUT & RUN, have been widely used to map transcription factor binding sites; however, these assays are often limited by the availability of effective and specific antibodies. As a proof-of-principle we used anti-GFP antibodies to perform CUT & RUN in primary T cells where the endogenous gene encoding BATF, a critical transcription factor, had been targeted to generate a GFP-fusion. Binding sites identified with anti-GFP CUT & RUN closely matched the sites identified with an anti-BATF antibody. Anti-BATF, anti-GFP, and no antibody heatmaps of CUT&RUN data obtained from primary human T cell populations electroporated with GFP-BATF fusion HDR template (untagged cells were not electroporated). Aligned CUT&RUN binding profiles for each sample were centered on BATF CUT&RUN peaks in untagged cells and ordered by BATF peak intensity in untagged cells. Experiment (h) was performed in two independent healthy donors.
Extended Data Figure 3: Bi-allelic and multiplexed non-viral genome targeting.
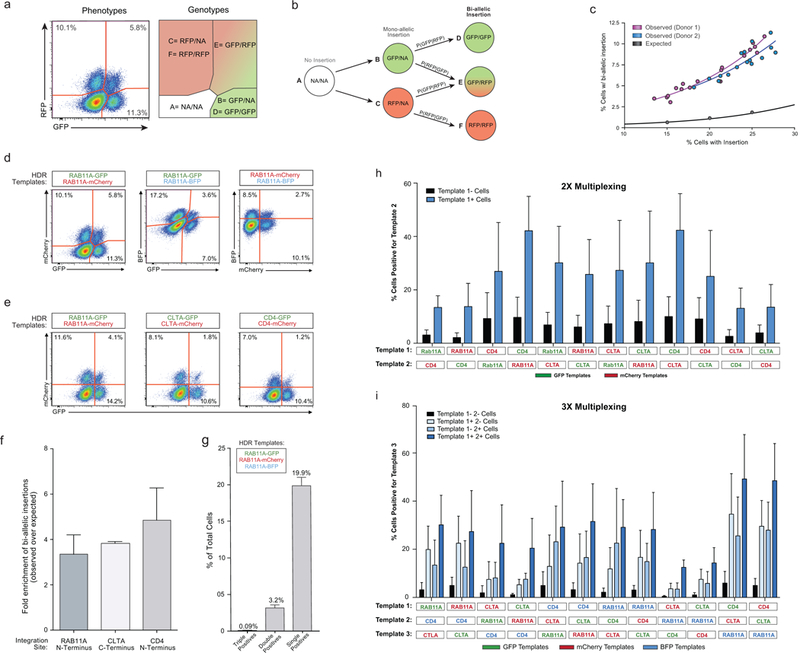
a, We wanted to confirm that we could generate cells with genome insertions in both alleles and quantify the frequency of bi-allelic modifications. Targeting the two alleles of the same gene with two distinct fluorophores would provide a way to quantify and enrich cells with bi-allelic gene modifications. The possible cellular phenotypes and genotypes when two fluorescent proteins are inserted into the same locus are displayed. Importantly, the number of cells that express both fluorescent proteins underestimates the percentage of cells with bi-allelic integrations because some cells will have inserted either GFP or mCherry on both alleles. We constructed a model to account for bi-allelic integrations of the same fluorescent protein (Supplementary Note 1). b, Diagram of bi-allelic integration model. The total percentage of cells with bi-allelic HDR integrations must be the sum of genotypes D, E, and F. While the proportion of cells with genotype E (dual fluor positives) is immediately apparent from the phenotypes, genotypes D and F are not. Our model allow for the de-convolution of the multiple genotypes in the single fluor positive phenotypes, and thus an estimation of the true percentage of cells bi-allelic for HDR. c, The observed level of bi-allelic integrations was higher in cells that acquired at least one integration than would be expected by chance. Individual points represent replicates where the combination of the genes encoding the fluorescent proteins was varied (either GFP + mCherry, GFP + BFP, or mCherry + BFP) as was the amount of the HDR template (3 to 6 μg). d, Bi-allelic HDR analysis was applied across a variety of fluorophore permutations inserted into the RAB11A locus. e, Dual fluorescence bi-allelic integrations were seen across target loci. f, The data also suggest that cells with one mono-allelic integration were more likely to have also undergone a second targeted bi-allelic integration, and this effect was observed across three genomic loci. While the total percentage of cells with an insertion varied with the efficiency of each target site, the fold enrichment in the observed percentage of homozygous cells over that predicted by random chance was largely consistent across loci. g, Co-delivery of three fluorescent-tags targeting the RAB11A locus resulted in only a few cells that expressed all three fluorophores, consistent with a low rate of off-target integrations. As a maximum of two targeted insertions are possible (at the locus’ two alleles; assuming a diploid genome), no cells positive for all three loci should be observed (triple positives). Indeed, while large numbers of single fluorophore integrations were observed (single positives), as well as cells positive for the various permutations of two fluorophores (double positives), there was a ~30 fold reduction in the number of triple positive cells compared to double positives. All flow cytometric analysis of fluorescent protein expression shown here was performed 4 days following electroporation. h, Multiplex editing of combinatorial sets of genomic sites would support expanded research and therapeutic applications. We tested whether multiple HDR templates could be co-delivered along with multiple RNPs to generate primary cells in which more than one locus was modified. Primary human T cells with two modifications were enriched by gating on the cells that had at least one modification, and this effect was consistent across multiple combinations of genomic loci. HDR template permutations from a set of six dsDNA HDR templates (targeting RAB11A, CD4, and CLTA; each site with GFP or RFP) were electroporated into CD3+ T cells isolated from healthy human donors. Four days after electroporation of the two indicated HDR templates along with their two respective on-target RNPs, the percentage of cells positive for each template was analysed by gating on cells either positive or negative for the other template. Not only was two-template multiplexing possible across a variety of template combinations, but gating on cells positive for one template (Template 1+ Cells, Blue) yielded an enriched population of cells more likely to be positive for the second template compared to cells negative for the first (Template 1- Cells, Black). 2 μg of each template, along with 30 pmols of each associated RNP, were electroporated for dual multiplexing experiments. i, We were also able to achieve triple gene targeting and could significantly enrich for cells that had a third modification by gating on the cells with two targeted insertions, an effect again consistent across target genomic loci. 1.5 μg of each template (4.5 μg total) were electroporated together with 20 pmols of each corresponding RNP (60 pmols total). Graphs display mean and standard deviation in n=4 (f-i) independent healthy donors. Other experiment (c-e) were performed in two independent healthy donors.
Extended Data Figure 4: Examination of off-target integrations with non-viral genome targeting.
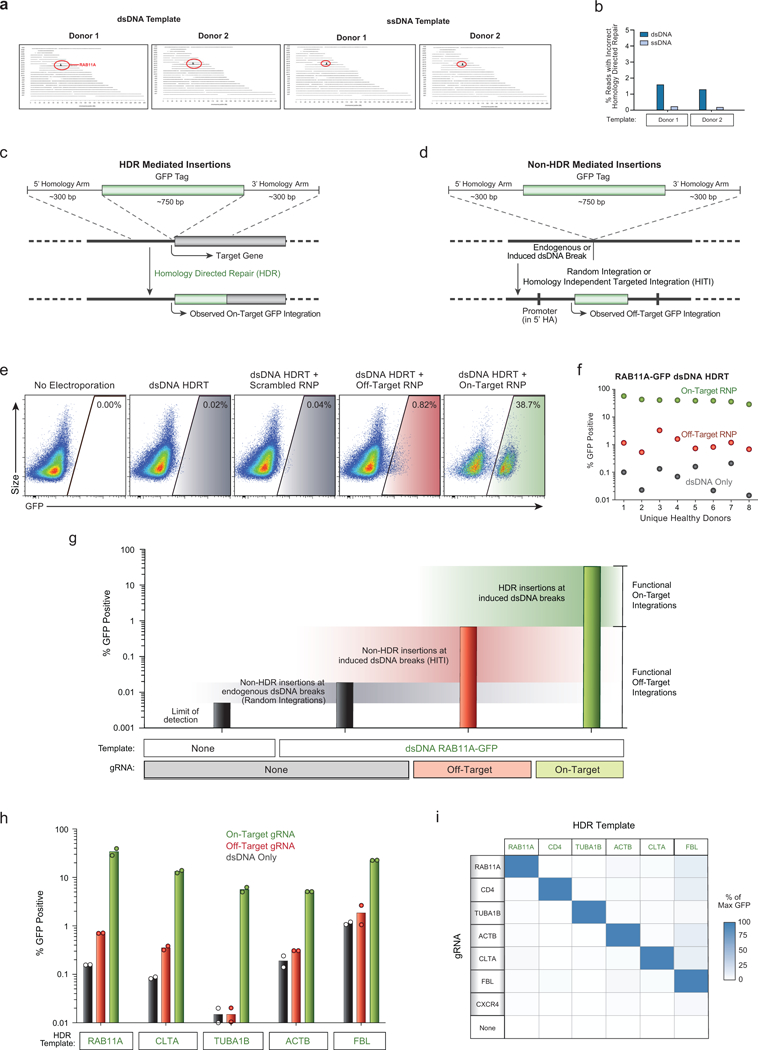
a, Results of targeted locus amplification (TLA) sequencing. No off-target integration sites were identified (assays limit of detection ~1% of alleles) with either a dsDNA or ssDNA HDR template in two healthy donors. The on-target RAB11A locus on chromosome 15 is indicated in red. b, The frequency of one observed incorrect HDR mediated integration at the target locus was reduced using a long ssDNA HDR template in two human blood donors (Supplementary Note 2). c, Diagram of HDR mediated insertions at the N-terminus of a target locus. The homology arms specify the exact sequence where the insert (a GFP tag in this case) will be inserted, allowing for scarless integration of exogenous sequences. Because a GFP fusion protein is created, GFP fluorescence will be seen as a result of the on-target integration, which is dependent on an RNP cutting adjacent to the integration site. d, dsDNA can be integrated via homology-independent repair mechanisms at off-target sites through either random integration at naturally occurring dsDNA breaks, or potentially at induced double stranded breaks, such as those at the off-target cut sites of the RNP. This effect can be harnessed to allow for targeted integration of a dsDNA sequence at a desired induced dsDNA break in quiescent cell types which lack the ability to do HDR, but crucially the entire sequence of the dsDNA template is integrated, including any homology arms. In the case that the homology arms contain a promoter sequence (such as for N-terminal fusion tags), these off target integrations can drive observable expression of the inserted sequence without the desired correct HDR insertion. e, We looked for unintended non-homologous integrations with the non-viral system using an N-terminal GFP-RAB11A fusion construct that contained the endogenous RAB11A promoter sequence within its 5’ homology arm. This construct could express GFP at off-target integration sites, which allowed us to assay for off-target events at the single cell level using flow cytometry. Inclusion of a gRNA designed to cut a genome region that is not the homologous region to the targeting sequence can be used to infer integration at an off-target cut site. f, While efficient GFP expression depended on pairing the HDR template with the correct gRNA targeting that site, rare GFP+ cells were observed when dsDNA HDR templates were delivered either alone (~0.1%) or with an “off target” Cas9 RNP (~1%). g, Quantification of different types of functional off-target integrations. The increase in the percentage of fluorescent cells over the limit of detection when the template alone is electroporated likely represents random integrations at naturally occurring dsDNA breaks (although cut-independent integration at the homology site is also possible in theory). Not every off-target integration will yield fluorescent protein expression, but the relative differences in functional off-target expression between different templates and editing conditions can be assayed. Inclusion of an RNP targeting CXCR4 (Off-Target) dramatically increased the observed off-target homology-independent integrations, likely through a homology independent insertion event. As expected, efficient GFP expression as expected was only seen with the correct gRNA sequence and HDR mediated repair. Bars represent observed GFP+ percentages from T cells from one representative donor electroporated with the indicated components. h, Comparisons of on-target GFP expression vs functional off-target integrations across five templates reveal HDR is highly specific, but that off-target integrations can be observed at low frequencies. i, A matrix of gRNAs and HDR templates were electroporated into CD4+ T cells from two healthy donors. The average GFP expression as a percentage of the maximum observed for a given template is displayed. Across six unique HDR templates and gRNAs, on-target HDR mediated integration was the by far most efficient. One HDR template, a C-terminal GFP fusion tag into the nuclear factor FBL, had consistently higher off-target expression across gRNAs, potentially due to a gene-trap effect as the 3’ homology arm for FBL contains a splice-site acceptor followed by the final exon of FBL leading into the GFP fusion. n=2 (a, b, h, i) or n=8 (e, f) independent healthy donors.
Extended Data Figure 5: Non-viral genome targeting using long ssDNA HDR templates and a Cas9 nickase.
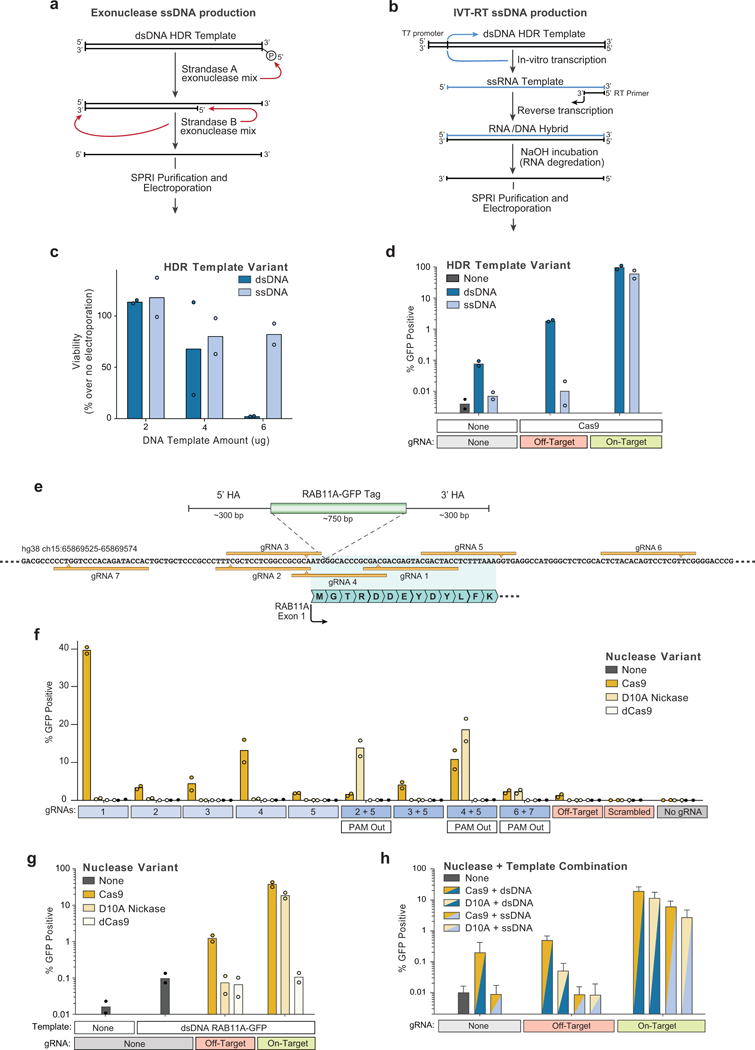
a, Long single-stranded (ss)DNA templates have potential to reduce homology-independent integrations while preserving on-target efficiency. One method to generate long ssDNA templates involves a two-step selective exonuclease digestion that specifically degrades one strand of a PCR product that has been labelled by 5’ phosphorylation, which can be easily added to a PCR primer prior to amplification. b, We also applied a second ssDNA production method based on sequential in vitro transcription (IVT) and reverse transcription (RT) reaction. A PCR product with a short T7 promoter appended serves as an IVT template to produce a ssRNA product. Following annealing of an RT primer and reverse transcription, an RNA/DNA hybrid can formed which can be transformed into a long ssDNA template by incubation in sodium hydroxide, which selectively degrades the RNA strand. c, At 4 days post-electroporation, varying concentrations of a long ssDNA HDR templates (~1.3 kb) did not show the decreased viability observed in CD3+ T cells electroporated with a linear dsDNA HDR template of the same length. d, Electroporation of a ssDNA HDR template reduced off-target integrations to the limit of detection (i.e. comparable to levels seen with no template electroporated) both with no nuclease added and at induced off-target dsDNA breaks (Off-target gRNA + Cas9). e, Diagram of the genomic locus containing the first exon of RAB11A. Use of spCas9 with an individual guide RNA (gRNA 1, “On-Target” in d) along with a dsDNA HDR template integrating a GFP in frame with RAB11A directly after the start codon results in efficient GFP expression (Fig. 1d). Use of a Cas9 nickase (D10A variant) with two gRNAs may reduce the incidence of off-target genome cutting. f, A series of individual gRNAs as well as dual gRNA combinations were tested for GFP insertion efficiency at the RAB11A N-terminal locus. As expected, no gRNAs showed appreciable levels of GFP insertion when using a nuclease dead Cas9 (dCas9). Multiple individual gRNAs that cut adjacent to the insertion site showed GFP integration when used with Cas9, but none were as efficient as gRNA 1. The D10A nickase showed little to no GFP integration with individual guides, but multiple two-guide combinations showed efficient GFP integration. Only in gRNA combinations where the two PAM sequences were directed away from each other (PAM Out) was GFP integration seen. g, GFP integration efficiencies as presented in (f) but graphed on a logarithmic scale reveal lower levels of functional off-target integrations when using the D10A nickase compared to spCas9 (with an individual “off-target” gRNA, targeting CXCR4), likely due to the requirement for the D10A nickase to have two gRNAs bound in close proximity to induce a dsDNA break. h, Long ssDNA templates (~1.3kb) could be successfully combined with Cas9 nickases (D10A) for targeted integration, similar to linear dsDNA templates. Here, long ssDNA HDR templates with D10A nickase showed lower efficiencies of GFP integration at the RAB11A site, but this appeared to be site specific; the combination of long ssDNA and D10A appeared to have higher efficiencies compared to dsDNA and Cas9 at a different site (Fig. 3d). n=2 (c, d, f, g) or n=3 (h) independent healthy donors with mean (c, d, f-h) and standard deviation (h).
Extended Data Figure 6: Reduced Treg frequencies and function in subjects with two loss of function IL2RA mutations.
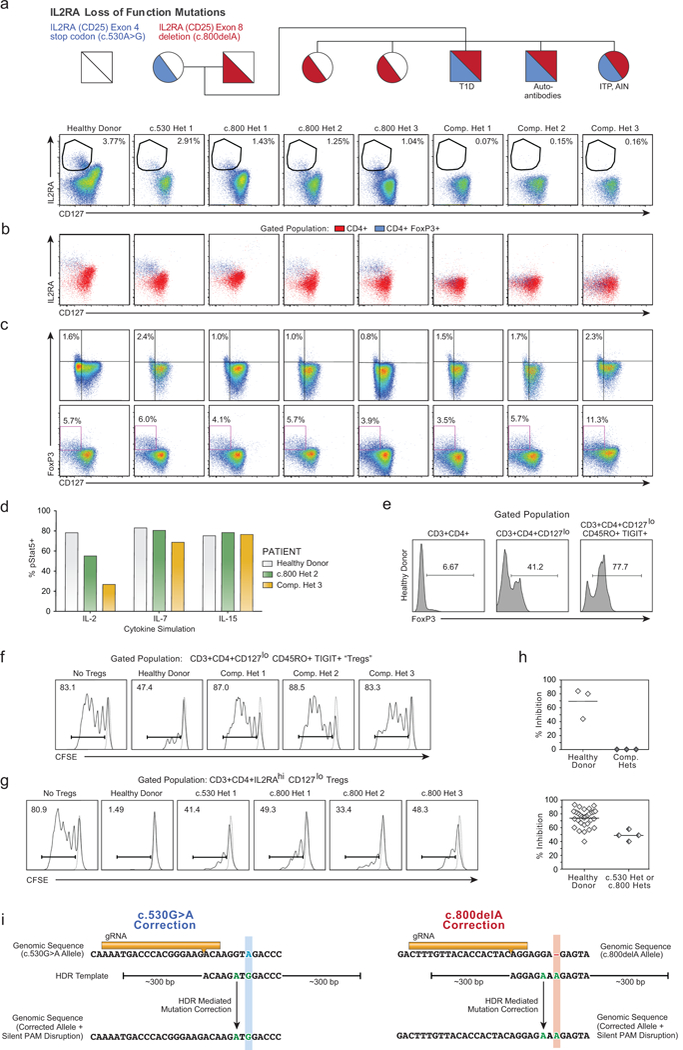
a, CD4+ T cells from a healthy donor and all family members, including IL2RA heterozygotes (c.530 het 1, c.800 hets 1-3) as well as compound heterozygous children (Comp. Hets 1-3), with loss-of-function IL2RA mutations were analysed by flow cytometry to assess the presence of IL2RAhiCD127lo Tregs. b, In healthy donors and individuals with only one IL2RA mutation, CD4+FoxP3+ T cells are predominantly IL2RAhiCD127lo. In the compound heterozygotes, a CD127loCD4+FoxP3+ population is present, but does not express high levels of IL2RA. c, Clinical phenotyping performed at two separate sites showed that compound heterozygotes have CD127loFoxP3+ cells. d, Deficiency in IL2RA surface expression in compound heterozygote 3 led to aberrant downstream signalling as measured by phosphorylated (p)Stat5 expression after stimulation with IL-2, but not IL-7 or IL-15. e, Due to the inability to sort IL2RAhi Tregs from the IL2RA-deficient compound heterozygotes, FoxP3+ cells were enriched from CD4+ using an alternate gating strategy that employed the surface markers CD127loCD45RO+TIGIT+. Intracellular FoxP3 staining of T cells from the indicated gated population is shown. f, While these CD3+CD4+CD127loCD45RO+TIGIT+ potential “Tregs” were highly enriched for FoxP3 and showed some suppressive capacity when cultured with CFSE-labelled stimulated responder T cells (Tresps) from healthy donors, CD3+CD4+CD127loCD45RO+TIGIT+ from the compound heterozygotes showed no suppressive ability. Stimulated Tresp population (Solid curves), non-stimulated Tresp (Dashed curve). g, Correction of either IL2RA mutation in the compound heterozygotes individually would still leave the other mutation, leaving the cells as single heterozygotes. To confirm that such a potential correction would result in some level of functional suppression, we assessed the suppressive ability of CD4+IL2RAhiCD127lo Tregs from the c.530 and c.800 single heterozygote family members as in (f). h, Dot plot summaries of Treg suppressive ability in cells from healthy donors [n=3 with single (top) or twelve (bottom) technical replicates], IL2RA-deficient compound heterozygotes (f, n=3 total patients) and IL2RA+/− c.530 or c.800 heterozygotes (g, n=4 total patients). While CD3+CD4+CD127loCD45RO+TIGIT+ “Tregs” from compound heterozygotes showed no suppressive ability, conventional CD4+IL2RAhiCD127lo Tregs from the single heterozygote family members showed some suppressive capacity, consistent with their lack of a pronounced clinical phenotype compared to the compound hets. Thus, correcting functional IL2RA expression on the surface of FOXP3+ T cells from these patients may represent a viable approach for developing an ex vivo gene therapy. Mean value is displayed. i, Initial genetic testing of the proband (Supplementary Note 3) using an in-house targeted next-generation sequencing multi-gene panel of over 40 genes known to be involved in monogenic forms of diabetes was negative. Subsequent exome sequencing in the trio of proband and parents revealed two causative mutations in the IL2RA gene. The mother possessed a single heterozygous mutation (c.530G>A) in exon 4 of IL2RA, resulting in a premature stop codon. The father possessed a single heterozygous mutation (c.800delA) in exon 8 of IL2RA, resulting in a frameshift mutation leading to a 95 amino acid long run-on. Sanger sequencing confirmed that the proband was a compound heterozygote with both mutations. A gRNA was designed to cut adjacent to the site of each mutation, 8 bps away for c.530 mutation (Blue), and 7 bps away for c.800 (Red). For each mutation, an HDR template was designed including the corrected sequence (Green) as well as a silent mutation in a degenerate base to disrupt the PAM sequence (“NGG”) for each guide RNA. Displayed genomic regions (not to scale) for c.530 mutation site (hg38 ch10:6021526-6021557) and c800 mutation site (hg38 ch10:6012886-6012917).
Extended Data Figure 7: HDR mediated correction of IL2RA c.530A>G loss of function mutation.
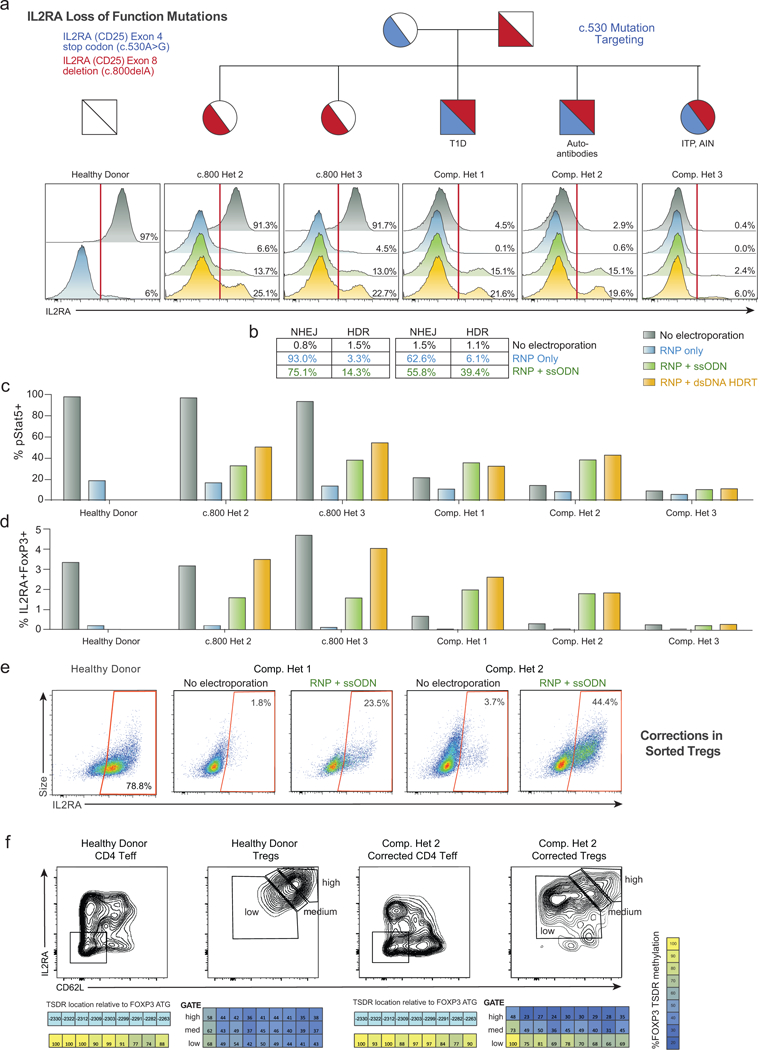
a,Unlike the gRNA targeting the c.800delA mutation at the C-terminus of IL2RA (Extended Data Fig. 8), the gRNA targeting the c.530A>G mutation (causing a stop codon in an interior exon) results in substantial (~90%) loss of IL2RA cell surface expression in a healthy donor and single heterozygotes (c.800 Het 2 and 3) 2 days following electroporation of the RNP alone (Blue) into CD3+ T cells. While starting from a very small IL2RA+ percentage, this reduction was observed in all three compound heterozygotes, potentially because a small amount of protein can be surface expressed from the c.800delA allele. This reduced IL2RA expression could be partially rescued by inclusion of an ssODN HDR template (Green) and even more substantially rescued using a large dsDNA HDR template (Yellow). Both template types contained the corrected sequence, a silent mutation to remove the gRNA’s PAM sequence, and either 60 bp (ssODNs) or ~300 bp (large dsDNA) homology arms (Extended Data Fig. 6i). b, Amplicon sequencing of the c.530 site in select patients shows the correlation between IL2RA cell surface expression and genomic correction. Small numbers of reads in the “No Electroporation” and “RNP only” conditions were called as HDR, potentially due to small amounts of cross-well contamination. c, Increased phosphorylated (p)Stat5 in response to IL-2 stimulation (200 U/mL) 7 days following electroporation in CD3+ T cells from compound heterozygote patients undergoing HDR mediated mutation correction compared to no electroporation or RNP only controls. pStat5+ cells correlated with increased IL2RA surface expression. d, Similarly, increased proportions of IL2RA+FoxP3+ cells are seen 9 days following electroporation in the HDR correction conditions in compound heterozygote patients. Lower percentages of correction were seen when targeting the c.530 mutation for HDR correction in compound heterozygote 3, potentially due altered cell-state associated with the patient’s disease or the patient’s immunosuppressive drug regimen (Supplementary Table 4). e, Mutation correction was possible in sorted Treg-like cells from the affected patients. CD3+CD4+CD127loCD45RO+TIGIT+ “Tregs”, a population highly enriched for FoxP3+ cells (Extended Data Fig. 6e), identified without the traditional Treg IL2RA surface marker (absent due to the causative mutations), were FACS sorted and underwent correction of the c.530A>G mutation using a Cas9 nuclease and short ssDNA HDR template (ssODN). After 12 days in culture, during which time the cells expanded >100 fold, greater than 20% (compound het 1) and 40% (compound het 2) of targeted cells expressed IL2RA on their surface, demonstrating functional correction and expansion of a therapeutically relevant cell type. In these experiments, expansion was less robust for cells from compound het 3. f, After 12 days in culture, corrected “Treg” cells from compound het 2, and a female healthy control, were sorted based on IL2RA and CD62L expression. Methylation of the TSDR (Treg-specific demethylated region) of FOXP3 intron 1 was analyzed in the indicated sorted cell populations by bisulfite sequencing (Epigendx). Due to X-chromosome inactivation, incomplete demethylation is observed in the control Treg populations from the female healthy donor. The sorted IL2RA-high CD62L-high population of corrected “Tregs” showed increasing TSDR demethylation, whereas similarly edited and expanded CD4+ T effector cells (Teff) did not show substantial TSDR demethylation in the healthy donor or in corrected cells from compound heterozygote 2. All electroporations were performed according to optimized non-viral genome targeting protocol (Methods). For ssODN electroporations, 100 pmols in 1 μL water were electroporated.
Extended Data Figure 8: HDR and non-HDR mediated correction of IL2RA c.800delA frameshift loss-of-function mutation.
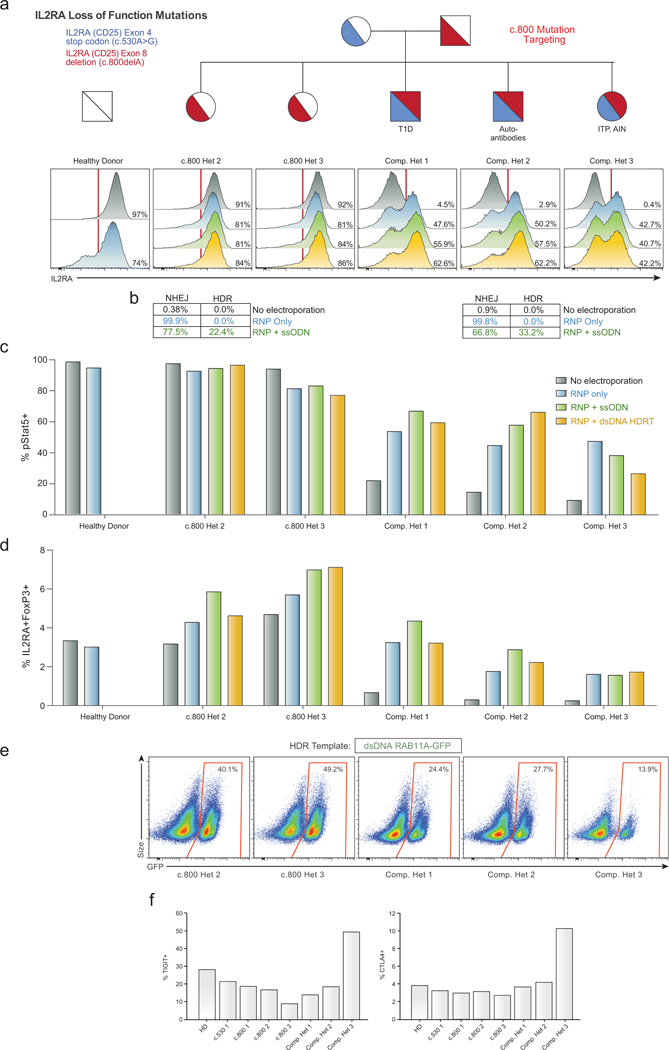
a,Histograms of IL2RA surface expression in CD3+ T cells in all children from a family carrying two loss-of-function IL2RA mutations, including three compound heterozygotes that express minimal amounts of IL2RA on the surface of the T cells (No electroporation, Grey). Two days following electroporation of an RNP containing a gRNA for the site of one of the two mutations, a one base pair deletion in the final exon of IL2RA (c.800delA) causing a run-on past the normal stop codon, CD3+ T cells from a healthy donor and single hets (c.800 Het 2 and 3) showed slight increases in IL2RA- cells (RNP only, Blue). This modest reduction is potentially due to the gRNA targeting the C-terminus of the protein where small indels may cause less pronounced loss of surface protein expression. Surprisingly, the RNP alone resulted in IL2RA surface expression in almost 50% of edited T cells in all three compound heterozygotes. In cells from two of the compound heterozygous children, increases in the percent of cells with IL2RA correction compared to RNP only could be achieved by inclusion of an ssODN HDR template sequence with the mutation correction (RNP+ssODN, Green), and further increased at this site when using a longer dsDNA HDR template to correct the mutation (RNP + dsDNA HDRT, Yellow) (Extended Data Fig. 6i). b, Amplicon sequencing was performed in select targeted patient cells. c, Stat5 phosphorylation (pStat5) in response to high dose IL-2 stimulation (200 U/mL) in targeted CD3+ T cells following 7 days of expansion post-electroporation. Increased numbers of pStat5+ cells correlated with increased IL2RA surface expression (a). d, Following 9 days of expansion post-electroporation, intracellular FoxP3 staining revealed an increased proportion of IL2RA+ FoxP3+ cells in CD3+ T cells compared to no electroporation controls. Electroporations were performed according to optimized non-viral genome targeting protocol (Methods). For ssODN electroporations, 100 pmols in 1 μL water were electroporated. e, Flow cytometric analysis of GFP expression 6 days following electroporation of a positive HDR control RAB11A-GFP dsDNA HDR template into CD3+ T cells from the indicated patients revealed lower GFP expression in the three compound heterozygotes compared to their two c.800 heterozygote siblings. Compared to a cohort of twelve healthy donors similarly edited (Fig. 1d), both c.800 heterozygotes as well as compound het 1 and 2 were within the general range observed across healthy donors, whereas compound het 3 had lower GFP expression than any healthy donor analysed. Of note, in compound het 3 HDR mediated correction at the c.530 mutation was substantially lower than the other two compound heterozygotes (Fig. 3b). IL2RA surface expression after electroporation of the c.800delA targeting RNP alone was similar though. Compared to HDR-mediated repair, NHEJ mediated frameshift correction at c.800delA may be less dependent on cell proliferation, consistent with compound het 3 being the only compound heterozygous patient on active immunosuppressants at the time of blood draw and T cell isolation (Supplementary Note 3). f, Altered cell-state associated with the patient’s disease could also contribute to diminished HDR rates. TIGIT and CTLA4 expression levels in non-edited, isolated CD4+ T cells from each indicated patient was measured by flow cytometry. Consistent with altered cell states and or/ cell populations, cells from compound het 3 had a distinct phenotype, with increased TIGIT and CTLA4 expression compared both to healthy donors, the single heterozygous family members, as well the other two compound heterozygous siblings.
Extended Data Figure 9: Endogenous TCR replacement strategy and functional characterization.
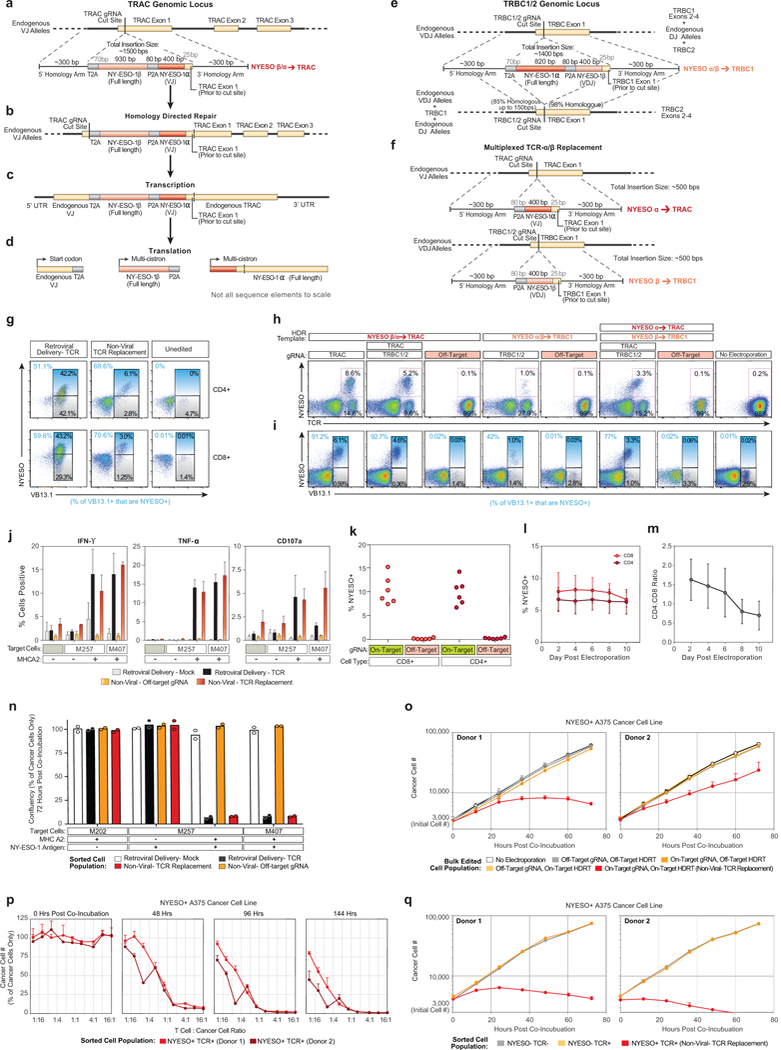
a-d, Schematic description of HDR template for endogenous TCR replacement by in-frame integration of a new TCR-β chain and a new variable region of a TCR-α chain at the TCR-α locus and the new TCR’s subsequent transcription and translation. e, HDR template for endogenous TCR replacement at the TCR-β locus. f, Multiplexed integration of a new TCR-α at the TCR-α locus and a new TCR-β at the TCR-β locus. Detailed description of TCR replacement strategy in Supplementary Note 4. g, TCR mispair analysis after retroviral delivery or non-viral TCR replacement of an NY-ESO-1 specific TCR in gated CD4+ or CD8+ T cells. With viral introduction of the new TCR, an infected cell will potentially express at least four different TCRs (new TCR-α + new TCR-β; new TCR-α + endogenous TCR-β; endogenous TCR-α and new TCR-β; endogenous TCR-α + endogenous TCR-β). Staining for the specific beta chain in the new introduced TCR (VB13.1) along with MHC-peptide multimer (NYESO) can provide a rough estimate of TCR mispairing by distinguishing between cells that predominantly expressed the introduced TCR (VB13.1+ NYESO+; new TCR-α + new TCR-β) vs those that expressed predominantly one of the potential mispaired TCRs (VB13.1+ NYESO-; endogenous TCR-α + new TCR-β). h, i, TCR replacement by targeting an entire new TCR into TRAC (a-d, also possible with a multiplexed knockout of TCR-β), an entire new TCR into TRBC1/2 (f), or multiplexed replacement with a new TCR-α into TRAC and a new TCR-β into TRBC1/2. j, Functional cytokine production was observed selectively following antigen exposure in gated CD4+ T cells, similarly to gated CD8+ T cells (Fig. 4c). k, Non-viral TCR replacement was consistently observed at four days post electroporation in both CD8+ and CD4+ T cells across a cohort of six healthy blood donors. l, In a second cohort of six additional healthy blood donors, 100 million T cells from each donor were electroporated with the NY-ESO-1 TCR replacement HDR template and on-target gRNA/Cas9 (Fig. 4f). The percentage of CD4+ and CD8+ T cells that were NY-ESO-1 TCR+ was consistent over ten days of expansion following electroporation. m, Over 10 days of expansion following non-viral genome targeting, CD8+ T cells showed a slight proliferative advantage over CD4+ T cells. n, The indicated melanoma cell lines were co-incubated with the indicated sorted T cell populations at a ratio of 1:5 T cells to cancer cells. At 72 hours post co-incubation the percent cancer cell confluency was recorded with by automated microscopy (where nuclear RFP marks the cancer cells). T Cells expressing the NY-ESO-1 antigen specific TCR, either by retroviral transduction (Black) or by non-viral knock-in endogenous TCR replacement (Red) both showed robust target cell killing only in the target cancer cell lines expressing both NY-ESO-1 and the HLA-A*0201 class I MHC allele. o, To ensure that target cell killing by non-viral TCR replacement T cells (Red) was not due to the either the gRNA or the HDR template used for TCR replacement alone, a matrix of on/off target gRNAs and on/off target HDR templates was assayed for target cell killing of the NY-ESO-1+ HLA-A*0201+ A375 cancer cell line (off-target gRNA and HDRT were specific for RAB11A-GFP fusion protein knock-in). Only cells with both the on-target gRNA as well as the on-target HDR template demonstrated target cell killing. p, Sorted NY-ESO-1+ TCR+ cells from a bulk T cell edited population (on-target gRNA, on-target HDR template) showed a strong dose-response effect for target cancer cell killing. Within 48 hours T cell to cancer cell ratios of 2:1 and greater showed almost complete killing of the target cancer cells. By 144 hours, T cell to cancer cell ratios of less than 1:16 showed evidence of robust target cell killing. q, Target cell killing by non-viral TCR replacement T cells was due specifically to the NY-ESO-1-recognizing TCR+ cell population observed by flow cytometry after non-viral TCR replacement (Fig. 4b). Starting with the bulk edited T cell population (all of which had been electroporated with the on-target gRNA and HDR template), we separately sorted three populations of cells: the NY-ESO-1+TCR+ cells (non-virally replaced TCR) (red), the NY-ESO-1-TCR- cells (TCR knockout) (grey), and the NY-ESO-1-TCR+ cells (those that retained their native TCR but did not have the NY-ESO specific knock-in TCR) (orange). Only the sorted NY-ESO-1+ TCR+ population demonstrated target cell killing (4:1 T cell to cancer cell ratio). One representative donor from n=2 (g, j) or n=3 (h, i) independent healthy donors with mean and standard deviation of technical triplicates (j). Mean and standard deviations of n=6 independent healthy donors (l, m) or of four technical replicates for n=2 independent healthy donors (o-q) are shown. Mean and individual values for n=2 independent healthy donors (n).
Extended Data Figure 10: In vivo functionality of T cells with non-viral TCR replacement.
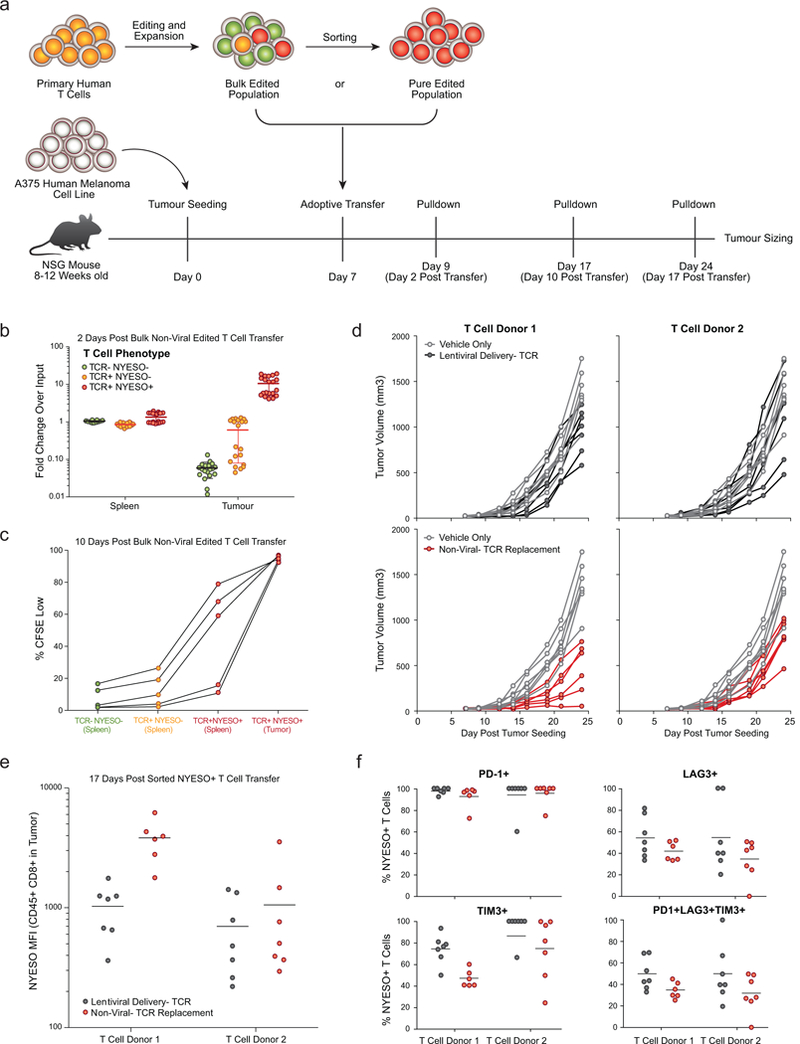
a,Diagram of in vivo human antigen specific tumour xenograft model. 8 to 12 week old NSG mice were seeded with 1×106 A375 cells (human melanoma cell line; NY-ESO-1 antigen+ and HLA-A*0201+) subcutaneously in a shaved flank. Primary human T cells edited to express an NY-ESO-1 antigen specific TCR were generated (either through lentiviral transduction or non-viral TCR replacement), expanded for 10 days following transduction or electroporation, and frozen. Either a bulk edited population was used (b,c) or a NY-ESO-1 TCR+ sorted population (d-f) was used. At seven days post tumour seeding, T cells were thawed and adoptively transferred via retro-orbital injection. b, Two days following transfer of 5×106 bulk non-virally targeted T cells (~10% TCR+ NYESO-1+ (Red), ~10% TCR+ NYESO-1- (Orange), and ~80% TCR- NYESO-1- (Green), see Fig 4b), NY-ESO-1+ non-virally edited T cells preferentially accumulated in the tumour vs. the spleen. n=5 mice for each of four human T cell donors. c, Ten days following transfer of 5×106 bulk non-virally targeted CFSE labeled T cells, NYESO-1 TCR+ cells showed greater proliferation than TCR- or TCR+NYESO-1- T cells, and showed greater proliferation (CFSE Low) in the tumour than in the spleen. At ten days post transfer TCR- and TCR+NYESO- T cells were difficult to find in the tumour (Fig 4g). d, Individual longitudinal tumour volume tracks for data summarized in Fig 4h. 3×106 sorted NY-ESO-1 TCR+ T cells generated either by lentiviral transduction (Black) or non-viral TCR replacement (Red) were transferred on day 7 post tumour seeding and compared to vehicle only injections until 24 days post tumour seeding. Note that the same data for vehicle control data are shown for each donor in comparison to lentiviral delivery (above) and non-viral TCR replacement (below). e,f, In these experiments, seventeen days following T cell transfer (d), non-virally TCR replaced cells appeared to show greater NY-ESO-1 TCR expression and lower expression of exhaustion markers. Transfer of both lentivirally transduced and non-viral TCR replaced cells showed significant reductions in tumour burden on day 24. In this experimental model, non-viral TCR replacement showed further reductions compared to the lentiviral transduction (Fig. 4h), potentially due to knockout of the endogenous TCR, endogenous regulation of the new TCR’s expression, some difference in the cell populations amenable to non-viral vs lentiviral editing, or confounding variables in cell handling between lentiviral transduction and non-viral genome targeting. n=4 (b), n=2 (d-f), or n=1 (c) independent healthy donors in 5 (b, c) or 7 mice (d-f) per donor with mean (b, e, f) and standard deviation (b).
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Marson Lab, Chris Jeans (QB3 MacroLab), Kyle Marchuk (UCSF BIDC), Jeffrey Bluestone and Qizhi Tang (UCSF Regulatory T Cell Therapy Program), Ryan Wagner (Parnassus CAT), the UCSF Flow Cytometry Core (NIH P30 DK063720 and 1S10OD021822-01), Lonza, Jacob Corn and Sarah Pyle for suggestions and assistance. This research was supported by NIH grants DP3DK111914-01 (A.M.), P50GM082250 (A.M.), R35 CA197633 (A.R.), K23 DK094866 (S.W.G.), T32GM007618 (T.L.R, J.H.), T32 DK007418 (T.L.R.), and P30 DK020595 (S.W.G.), the NIH NCI Intramural Program (A.L.F., S.H.H.), grants from the Keck Foundation (A.M.), National Multiple Sclerosis Society (A.M.; CA 1074-A-21), gifts from Jake Aronov, Galen Hoskin, the Jeffrey Modell Foundation (A.M), and awards from the Burroughs Wellcome Fund (A.M.) and the Ressler Family Fund (C.P.S., J.S., A.R.). A.M. is a Chan Zuckerberg Biohub investigator. A.R. is a Parker Institute for Cancer Immunotherapy member.
Footnotes
Competing Financial Interests
A.M. is a co-founder of Spotlight Therapeutics. A.M. serves as an advisor to Juno Therapeutics and PACT Pharma and the Marson laboratory has received sponsored research support (Juno Therapeutics, Epinomics, Sanofi) and a gift from Gilead. T.L.R., C.P.S., E.S., A.R., and A.M. are inventors on new patent applications related to this manuscript.
REFERENCES
- 1.Sadelain M, Rivière I & Riddell S Therapeutic T cell engineering. Nature 545, 423–431 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA & Restifo NP Adoptive cell transfer as personalized immunotherapy for human cancer. Science (80-. ). 348, 62–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verhoeyen E, Costa C & Cosset F-L Lentiviral vector gene transfer into human T cells. Methods Mol. Biol. 506, 97–114 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Eyquem J et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hale M et al. Homology-Directed Recombination for Enhanced Engineering of Chimeric Antigen Receptor T Cells. Mol. Ther. - Methods Clin. Dev. 4, 192–203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornu TI, Mussolino C & Cathomen T Refining strategies to translate genome editing to the clinic. Nat. Med. 23, 415–423 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y et al. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol. Ther. 13, 151–159 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hornung V & Latz E Intracellular DNA recognition. Nat. Rev. Immunol. 10, 123–130 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Kim S, Kim D, Cho SW, Kim J & Kim JS Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 24, 1012–1019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schumann K et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. 112, 10437–10442 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leonetti MD, Sekine S, Kamiyama D, Weissman JS & Huang B A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc. Natl. Acad. Sci. 113, E3501–E3508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skene PJ & Henikoff S An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bak RO et al. Multiplexed genetic engineering of human hematopoietic stem and progenitor cells using CRISPR/Cas9 and AAV6. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agudelo D et al. Marker-free coselection for CRISPR-driven genome editing in human cells. Nat. Methods 14, 615–620 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Lux CT & Scharenberg AM Therapeutic Gene Editing Safety and Specificity. Hematology/Oncology Clinics of North America 31, 787–795 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cain-Hom C et al. Efficient mapping of transgene integration sites and local structural changes in Cre transgenic mice using targeted locus amplification. Nucleic Acids Res. 45, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dever DP et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 539, 384–389 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murnane JP, Yezzi MJ & Young BR Recombination events during integration of transfected DNA into normal human cells. Nucleic Acids Res. 18, 2733–2738 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki K et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quadros RM et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol. 18, 92 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H et al. Design and specificity of long ssDNA donors for CRISPR-based knock-in. doi.org178905 (2017). doi:10.1101/178905 [Google Scholar]
- 22.Mali P, Esvelt KM & Church GM Cas9 as a versatile tool for engineering biology. Nat. Methods 10, 957–963 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ran FA et al. Double nicking by RNA-guided CRISPR cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharfe N, Dadi HK, Shahar M & Roifman CM Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proc. Natl. Acad. Sci. U. S. A. 94, 3168–3171 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakaguchi S, Sakaguchi N, Asano M, Itoh M & Toda M Immunologic Self-Tolerance Maintained by Activated T Cells Expressing 11-2 Receptor a-Chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases.. J. Immunol 155, 1152–1164 (1995). [PubMed] [Google Scholar]
- 26.Farh KK-H et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simeonov DR et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature (2017). doi:10.1038/nature23875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robbins PF et al. Single and Dual Amino Acid Substitutions in TCR CDRs Can Enhance Antigen-Specific T Cell Functions. J. Immunol. 180, 6116–6131 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.