

Abstract
Once dismissed as vestigial organelles, primary cilia have garnered the interest of scientists, given their importance in development/signaling, and for their implication in a new disease category known as ciliopathies. However, many, if not all, “cilia” proteins also have locations/functions outside of the primary cilium. These extraciliary functions can complicate the interpretation of a particular ciliopathy phenotype: it may be a result of defects at the cilium and/or at extraciliary locations, and it could be broadly related to a unifying cellular process for these proteins, such as polarity. Assembly of a cilium has many similarities to the development of other polarized structures. This evolutionarily preserved process for the assembly of polarized cell structures offers a perspective on how the cilium may have evolved. We hypothesize that cilia proteins are critical for cell polarity, and that core polarity proteins may have been specialized to form various cellular protrusions, including primary cilium.
Keywords: primary cilia, extraciliary, ciliopathy, polarity, cytoskeleton, trafficking, development
1. Introduction
Primary cilia are complex organelles, localized singularly on the surface of most cells, where they, in part, function as an immotile cellular compartment for signaling. Researchers have identified sensory roles for primary cilia[1] and have implicated this organelle in embryological development.[2] Proteins important for the assembly, maintenance, or function of the primary cilium are termed cilia or ciliogenesis proteins. Traditionally, cilia proteins consisted of proteins that localize to the cilium and/or some proteins at the centrosome. Typically, proteins that localize to the centrosome can be important for ciliogenesis, while proteins that localize to the cilium may be important for ciliogenesis, but could also represent components of ciliary signaling pathways. Defects in these cilia/ciliogenesis proteins are now linked to a group of human disorders named ciliopathies.[3] However, many ciliogenesis proteins also have extraciliary localizations, with some not even localizing to the cilium/centrosome at all, but still affecting ciliogenesis. Therefore, the use of “ciliopathy” is potentially confounding because it suggests that the loss of primary cilia is the primary driver of the pathology seen in ciliopathies.
While the centrosome is important for ciliogenesis (the mother centriole becomes the base of the cilium called the basal body), it is not the only function for this organelle. Centrosomes are better known for their role as the microtubule organizing center (MTOC) of the cell, which acts as the nucleation center from which microtubules emerge.[4] The MTOC organizes the mitotic/meiotic spindle apparatus for proper chromosomal segregation during cell division. The centrosome is also necessary for organization of microtubules in non-dividing cells, allowing for proper vesicle transport, movement of organelles throughout the cell, and establishment of cell shape and polarity.[4] However, it is not yet known whether all centrosomal and cilia proteins are polarity determinants. Structural centrosomal proteins, for example, may not participate in the polarity pathway directly, but the loss of such a crucial structure that is central in forming cellular asymmetry may impede polarity and even cell survival. Furthermore, defects in centrosomal proteins have been linked to diseases (i.e., microcephaly or cancer) that have not yet been linked to polarity, although such a possibility still exists.[4]
Because primary cilia are dynamic organelles, they are responsive to a wide range of stimuli (i.e., ligands/drugs, temperature, and changes in extracellular environment).[5–7] Hemi-Parkinsonian mice (with a unilateral depletion of dopamine in the striatum) have longer primary cilia in the striatum of the depleted hemisphere, demonstrating that merely changing the extracellular milieu of the brain can produce alterations in primary cilia morphology.[7] Furthermore, dynamic changes in primary cilia are observed depending on the surface to which the cell adheres. For instance, cells grown on hard glass coverslips have fewer cilia than cells plated on soft polyacrylamide-coated coverslips.[8] Such alterations in cilia dynamics in response to external factors can complicate the interpretation of cilia studies, especially where a mutation/defect may also affect the extracellular environment of a cell. A further difficulty in studying the function of primary cilia is that many primary cilia proteins localize at extraciliary sites (i.e., localizations not at the centrosomes, basal body or primary cilium) and/or have extraciliary functions (Box 1). Therefore, it is possible that there are important distinctions between “cilia” proteins, non-ciliary localizations/functions of “cilia” proteins, and the primary cilium itself as an organelle that require further elucidation.
Box 1. Primary cilia proteins can have extraciliary sites and functions.
Proteins that were once known for their ciliary localization and functions are becoming increasingly linked to functions outside of the cilium (reviewed in[9]). For example, Ift88 was the first primary cilia protein linked to a disease, polycystic kidney disease.[19] In addition to ciliary dysfunction, Ift88 mouse mutants have various defects in mitosis (a stage of the cell cycle in which primary cilia are not even present to exert any functional influences).[20] Arl13b often localizes to the primary cilium, but also outside the cilium, localizing to endocytic vesicles.[21] Conversely, proteins that were not previously known to be ciliary proteins (i.e., located in/at the primary cilium) are increasingly being observed to be linked to ciliogenesis. For instance, centrosome and spindle pole associated protein 1 (CSPP1) was initially observed at centrosomes and mitotic spindle poles, and only subsequently shown to also localize to primary cilia and be critical for ciliogenesis.[22,23] Further studies have now demonstrated that CSPP1 also localizes to kinetochores,[24] desmosomes,[25] centriolar satellites,[26] and the nucleus.[27] Such studies indicate that the primary cilium and ciliogenesis are just one of many localizations and functions for CSPP1. There are many other examples of cilia proteins with extraciliary functions at extraciliary sites (see review[9]). Primary cilia proteins have now been observed at the Golgi apparatus,[28–31] nucleus,[27] vesicles,[21,32] cell junctions,[25] and mitotic spindles.[20,24] More importantly, these proteins are known to have a range of extraciliary functions that include: cytoskeletal remodeling of the actin[33–36] and microtubule[37–39] cytoskeletons, centrosomal positioning,[40] mitotic spindle orientation,[41,42] phosphoinositide conversion,[43] and endocytic and exocytic vesicular trafficking.[21,32] Interestingly, this wide range of extraciliary functions can be unified under the theme of polarity.
To attempt to elucidate the function of the primary cilium as an organelle, and to distinguish it from “cilia” proteins that have both ciliary and extraciliary roles, we recently conducted a review of the cilia literature. This revealed that many cilia proteins have extraciliary functions that included such processes as cell cycle regulation, cytoskeletal regulation (both microtubules and actin), and trafficking (reviewed in[9]). Interestingly, many cilia proteins are found in cells that do not contain cilia, such as T-cells, where these proteins function to assemble an immune synapse (a structure used for cell communication, activation, and secretory events)(reviewed in[9]). Since cilia proteins localize in and are functional in non-ciliated cells, this suggests that “cilia” proteins may not be utilized solely for primary cilia function, but may instead have other general cellular roles. In fact, the many extraciliary localizations and functions of cilia proteins ascertained from the literature appeared to have in common a role in cellular polarity. Here, we propose that “cilia” proteins are not involved solely in centrosome repositioning and ciliogenesis (or ciliary function), but instead may have a more general role in establishing polarized cellular structures.
The importance of polarity in ciliogenesis is not novel (reviewed in[10]). Nodal cilia, a type of motile 9+0 cilia, are important for planar cell polarity (PCP), a process describing how multiple cells can establish polarity across a tissue plane. The rotary movement of nodal cilia establishes chemical gradients that guide organ growth during embryological development. For this reason, loss of nodal cilia, and with it, normal PCP protein function, is associated with situs inversus (a condition where organs grow in the wrong direction). As would be expected, PCP proteins have roles in ciliogenesis (and ciliopathies), providing a further link between polarity proteins and ciliogenesis proteins.[11–17] Given the relationship between tissue-level polarity and nodal cilia, it is not surprising that cellular-level polarity also plays a role in primary cilia ciliogenesis.[10]
The construction and assembly of a cilium occurs through various different processes, each with multiple steps (reviewed in[18]). Briefly, cilia formation generally requires: (1) repositioning of the mother centriole, (2) trafficking (often vesicular) of appropriate components to the centriole, (3) maturation of the centriole to become the basal body, and (4) the growth of a microtubule axoneme from the basal body. These steps are important for the unidirectional movement of the centrioles as well as the asymmetric division of PCP proteins within a cell. Thus, the cellular processes that allow for the establishment of cell polarity play an important underlying role in both ciliogenesis and PCP.
The relationship between polarity and ciliogenesis is recognized in the cilia field. However, instead of arguing that polarity is an underlying process important for ciliogenesis, we aim to present a hypothesis that cilia/ciliogenesis proteins are not merely dependent on polarity proteins, but many are likely polarity proteins themselves. We further hypothesize that polarity driven pathways critical for the construction of a primary cilium (a polarized structure itself) are likely also important for the assembly of other polarized cellular protrusions. Thus, cilia proteins may not be ciliogenesis-specific, but instead, are general polarity-establishing proteins that are important for polarity-genesis (in which ciliogenesis is a consequence of polarity)(Fig. 1).
Figure 1. The phenotype of a “ciliopathy” may be determined by both the ciliary and extraciliary functions of “cilia” proteins.
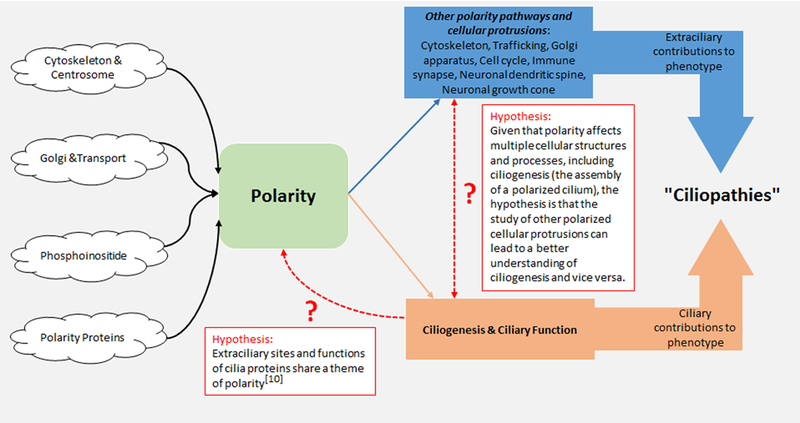
“Cilia” proteins have both ciliary and extraciliary localizations and functions, both of which may contribute to the ultimate “ciliopathy” phenotype. The primary cilium itself is important for signaling, but extraciliary functions are more varied from “cilia” protein to “cilia” protein. The diverse extraciliary functions of different “cilia” proteins may contribute to the variable phenotypes observed in “ciliopathies” that result from mutations in different genes. Furthermore, defects in certain extraciliary functions may result in an inability to form a cilium, and thus, indirectly impair cilia signaling. Such extraciliary sites and functions of cilia proteins share a theme of polarity. Moreover, given that polarity affects multiple cellular structures and processes, including ciliogenesis (the assembly of a polarized cilium), studying other polarized cellular protrusions may lead to a better understanding of primary cilia and ciliogenesis.
2. Could cilia/ciliogenesis proteins be polarity proteins themselves, instead of merely functioning downstream of polarity proteins: A polarization hypothesis for “cilia” proteins
Cell polarity is a coordinated and well-conserved process that requires proper cytoskeletal rearrangement, Golgi apparatus processing, vesicular trafficking, and phosphoinositide signaling.[44–46] Ciliogenesis is also dependent on these processes, and cilia proteins have been identified that function at each of these levels.[9,44–46] Centrosomal repositioning is important for cell migration (reviewed in[47]), but it is also important to help move the centriole to the plasma membrane where it can dock and become the basal body, an important initial step for at least one type of ciliogenesis (reviewed in[18]). Asymmetric vesicular transport is one way to divide PCP proteins to different hemispheres of a cell to establish cell polarity,[48–50] while directed vesicular trafficking helps target proteins to the cilium.[51,52] Thus, processes important for the establishment of polarity are also important for assembling cilia.
Interestingly, some traditional polarity proteins are localized inside the primary cilium, with loss of these polarity proteins negatively affecting ciliogenesis.[53] Conversely, knockdown of cilia proteins can result in loss of apical-basal polarity,[54] failure of centrosome docking,[40] loss of the ciliary phosphoinositide code (a lipid code on plasma membranes that is important for signaling and polarity),[43] and various microtubule and actin defects ([34,37]; reviewed in[9]). We propose that because the primary cilium is a polarized cell structure, then “cilia” proteins are likewise variably important in the function of other polarized cellular protrusions and possibly have localizations at these sites. Such a polarity theme is consistent with a hypothesis proposed by Jekely and Arendt in 2006 regarding the evolution of primary cilia.[55]
2.1. Can we learn from evolutionary origins?
Jekely and Arendt[55] proposed that given the sequence homology shared by intraflagellar transport (IFT) proteins, coat protein complexes (i.e., COPI), and clathrin coat proteins,[56] the IFT system evolved as a form of coated vesicular transport. They hypothesized that this primitive IFT system may have been instrumental in establishing an early-polarized patch of membrane on the cell, which was the initial step in cilium evolution. Later, this patch of membrane gradually protruded to allow better access to the extracellular space, and eventually compartmentalized for more efficient signaling (e.g., primary cilium) and beating/motion (e.g., motile cilia). Now, a decade later, new studies justify an update on their original hypothesis.
2.2. Ciliogenesis and neurite development, two polarized structures/events with a theme of polarity?
Cell polarization is an evolutionarily well-conserved process that is dependent on the operation and coordination of multiple proteins and pathways, and has critical functions in cellular physiology.[46] By viewing the primary cilium as a polarized structure, comparisons can be drawn between the primary cilium and the development of other polarized structures, such as the neuronal axon, in an attempt to understand how a primary cilium might have initially formed (Fig. 2). In doing so, many similarities between neuronal development and Jekely and Arendt’s hypothesis on cilium evolution become apparent.
Figure 2. Primary cilia formation hypothesis as compared to stages of neuron development.
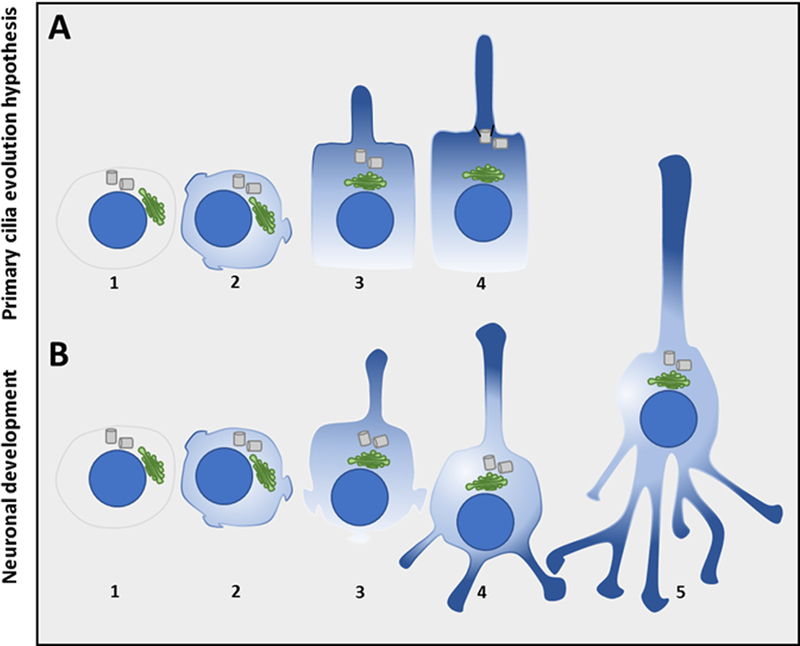
A1/B1, Cells are round, but some regions of the plasma membrane may exhibit some polarity. A2/B2, Some cellular protrusions/neurites begin to form at polarized membrane patches. A3/B3, The centrosome reorients to the base of a neurite. Importantly, the Golgi apparatus also reorients to this neurite, and provides preferential vesicular delivery here, stabilizing this polarized protrusion as the cilium or axon. A4, The centrosome docks to the base of the cilium, enclosing it as a distinct “closed” organelle/compartment. B4, The remaining neurites develop into dendrites, and the axon elongates (albeit an open compartment). B5, The neuron matures and forms synapses. The graduations of light blue to dark blue represent establishment of polarity. Blue circle: nucleus; green: Golgi apparatus; gray cylinders: centrioles.
While Jekely and Arendt[55] suggested that the initial establishment of a polarized patch of membrane was likely mediated by the IFT system for ciliary formation, we hypothesize a variation on their hypothesis based on neurite development.[57,58] When neurons are dissociated and grown in vitro, they undergo five distinct stages of development (reviewed in[58]). At Stage-1, neurons are round and have a surrounding lamellipodium. At Stage-2, multiple small neurites begin to emerge and grow from the cell body. The successful determination of one of the neurites to become the axon defines Stage-3. Then, the remaining neurites branch and become dendrites (Stage-4). Lastly, the axon matures and forms synapses (Stage-5). Stages 1 through 3 bare a remarkable resemblance to the first step of Jekely and Arendt’s cilium hypothesis,[55] and may provide further insights into the evolutionary origin of the cilium.
Before any morphological polarity is apparent in Stage-1 neurons, many aspects of the neuron already display preferential polarization.[59] Tubulin and actin localize more to one side of the round neuron. Trafficking of transferrin receptors, and the uptake and distribution of endocytic vesicles also preferentially localizes to one side of this early neuron. While Jekely and Arendt[55] proposed that the establishment of an initial polarized patch of membrane was mediated by IFT and the Golgi apparatus in ciliary formation, neurite polarization studies suggest a different order of events. F-actin condensation, microtubule polarization, and endocytotic and exocytotic polarized trafficking all precede the reorientation of the Golgi apparatus to the base of the first neurite. N-cadherin also condenses at the site where the first neurite will emerge.[60] This led to the hypothesis that the initial establishment of a polarized patch of neuronal membrane involves N-cadherin restricting F-actin to one pole of the neuron. In fact, N-cadherin and F-actin co-localize at the site of the first neurite. Interestingly, in Drosophila neurons, cadherin accumulation may be due to phosphoinositide recruitment and signaling.[61] Therefore, using neuron development as a guide, we hypothesize that primary cilia may have formed in a similar manner with the initial establishment of a polarized membrane according to the following sequence of events: (1) differential phosphoinositide clustering and signaling, (2) phosphoinositide mediated clustering of cadherins to a membrane patch, (3) subsequent polarized cytoskeletal rearrangement, and (4) reorienting of the Golgi apparatus and vesicular transport to the polarized pole (Fig. 2). An important test for such a hypothesis will be the continued and detailed examination of each of these steps in the formation of a primary cilium.
After the establishment of a polarized neuronal membrane, one neurite stabilizes, allowing it to protrude and become the axon. Experiments utilizing fluorescence recovery after photobleaching, which followed the reorientation of the Golgi apparatus to the base of the first neurite, demonstrated that tubulin and actin turnover in the undifferentiated neurites occurred at similar rates.[59] However, the neurite closest to the Golgi apparatus had the highest rate of transferrin receptor trafficking,[59] and was most likely to develop into the axon.[62] Thus, while the Golgi apparatus (and with it the IFT system) may not be critical in the initial establishment of a polarized membrane patch in neurons, it may be necessary for the stabilization of the polarized neurite to become an axon. We propose that during the evolution of primary cilia, the reorientation of the Golgi apparatus and IFT system towards the initial polarized membrane patch likely aided in the protrusion and stabilization of the initial cilium. Continued studies are needed that perform long-term live imaging studies of forming primary cilia, examining whether the Golgi and IFT system proteins are recruited to docked centrosomes and whether their loss can affect ciliogenesis. Interestingly, many Arf family proteins that are important for Golgi-mediated vesicular transport are known to be important for ciliogenesis and migration.[63,64] Lastly, studies determining whether loss of Golgi and IFT proteins can affect ciliary dynamics (i.e., the ability or speed in which the cilium can respond to stimuli) would aid in our understanding of ciliary stabilization and transduction.
The establishment of cell polarity and phosphoinositide coding along the plasma membrane allows for the asymmetric distribution of proteins into different regions of a cell, and establishes an actin cortex.[44] The actin cortex is important for basal body docking as loss of the actin cortex results in a failure of the basal body to dock to the plasma membrane, and subsequently, a failure in ciliogenesis.[65] Thus, the establishment of polarity and an actin cortex may have preceded and may be necessary for the docking of the mother centriole. Interestingly, the importance of proper actin regulation has been shown to be necessary for ciliogenesis (reviewed in[9]). None of this polarized rearrangement and trafficking would be possible, however, without the cell first defining polarized regions. In fact, polarity-establishing proteins are increasingly being found to be important for ciliogenesis (see below).[53,66,67]
The primary cilium is different from a growing axon, especially in that the mother centriole migrates and serves as the base of the cilium, and subsequently forms a closed compartment to the cell body. Conversely, the axon remains open and continuous with the cytoplasm. Further studies are required to determine why one type of cellular protrusion becomes a closed structure, while another remains open. For primary cilia, the docking of the mother centriole at the base of the cilium likely contributes to the closed nature of the cilium.
2.3. Polarity proteins are ciliogenesis proteins
The major groups of polarity proteins include Par complex proteins and Crumbs (reviewed in[67]). Crumbs3 (CRB3) and the polarity complex (Par3, Par6, and aPKC) all localize to the primary cilium axoneme and are essential for ciliogenesis.[53] Loss of two apical determining polarity proteins, Pard3 and PKC, produced shorter and fewer cilia, and decreased apical domain size.[54] This demonstrates that polarity proteins are important for ciliogenesis, but the reverse may also be likely true and should be examined further. Mutations in Talpid3, a centrosomal protein, cause the ciliopathy, Joubert syndrome (JBTS), and defects in ciliogenesis.[66,68,69] Whereas Talpid3 mutants still form centrosomes, these centrosomes fail to migrate to the apical surface of ependymal cells, disrupting ciliogenesis.[40] Thus, loss of Talpid3 results in a polarization defect and defective ciliogenesis, likely due to the failure of centrosome migration to the apical surface of the cell. Centrosome migration defects have been found in studies of other cilia proteins, including CEP164[8] and MYH10[35] with future examples likely.
2.4. Polarity and ciliogenesis require phosphoinositide signaling
The plasma membrane does not have a uniform composition. Instead, phosphoinositides and their regulators differentially localize to parts of the membrane.[44] Certain RhoGTPases regulate the phosphorylation status of phosphoinositides,[70] and this consequently determines what proteins are capable of binding to the phosphoinositide, allowing for a polarized distribution of proteins on the membrane.[44] Important enzymes for establishing this polarity include Src homology 2 domain-containing inositol 5-phosphatase 2 (SHIP2) and cell division control protein 42 (CDC42). SHIP2 is particularly important for determining basolateral polarity and has recently been associated with cilia regulation.[71] CDC42 is one of the main RhoGTPases important for polarity,[72] and similarly has been linked to ciliogenesis. Zebrafish with mutant cdc42 display ciliopathy phenotypes, and Cdc42 knockout mice have decreased numbers of cilia.[73] Receptor composition on the primary cilium is also regulated by phosphoinositides,[74] further suggesting that phosphoinositides play a key role in establishing polarity.
In the primary cilium, the transition zone membrane contains phosphatidylinositol (4,5)-bisphosphate (PIP2), allowing binding of the Tubby transport system.[75] The remainder of the cilium membrane comprises phosphatidylinositol 4-phosphate (PI4P). This differential expression of phosphoinositides within the cilium membrane allows for a “code” that promotes distinctive binding of proteins[76]. In primary cilia, inositol polyphosphate-5-phosphatase E (INPP5E) converts PIP2 into PI4P, establishing the polarity of the cilium membrane. Mutations in INPP5E are linked to JBTS, defects in cell polarity, and defects in the actin cortex.[43] Polycystic kidney disease mouse models also display a loss of intrinsic polarity that is hypothesized to contribute to cyst formation;[77] however, cyst formation remains an area of controversy.[78]
2.5. Polarity and ciliogenesis require cytoskeletal regulation
Cytoskeletal rearrangement is another important aspect of establishing cell polarity. For a cell to polarize, the cell has to break symmetry and maintain a new asymmetry; a function attributed in part to the cytoskeleton.[45] Recently, a number of cilia proteins have been found to directly interact with and regulate the microtubule and actin cytoskeletons in addition to functioning at cilia.[33–39] Furthermore, many traditional microtubule and actin regulating proteins have now been found to be important for ciliogenesis.[9] Knockdown of ciliogenesis proteins can also result in loss of apical basal polarity and failure of centrosome docking.[40,54] Lastly, it is increasingly being recognized that both microtubule and actin cytoskeletal regulation are critical to normal ciliogenesis.[37,38,79,80] Many ciliogenesis proteins in fact have extraciliary functions that involve microtubule and actin regulation (reviewed in[9]).
While more studies are needed, it is not hard to imagine why cytoskeletal regulation is important for ciliogenesis. As previously mentioned, an important step of ciliogenesis involves the reorientation and migration of the centriole, a process dependent on proper cytoskeletal function (reviewed in[81]), polarity determinants, and its effector proteins. Loss of any of these aforementioned proteins, regardless of their specific individual role in cytoskeletal regulation, ultimately lead to a common defect of loss of polarity and ciliogenesis. A proper microtubule cytoskeleton is also necessary for the IFT system, which operates on microtubules, to allow for directed transport of proteins and other cargo to different regions of the cell. Katanins, important for ciliogenesis, create a free pool of tubulin that can be used for microtubule growth.[82] Therefore, it is not surprising that loss of microtubule and actin regulation can negatively affect both polarization and ciliogenesis.
2.6. Polarity and ciliogenesis require vesicular trafficking
A main argument in Jekely and Arendt’s[55] hypothesis of cilium evolution depends on the fact that well-conserved[83] IFT proteins, found in the ciliary axoneme, share sequence homology with COP1 and clathrin coat proteins.[56] This strongly suggests that IFT has a role in vesicular transport. Indeed, multiple cilia proteins participate in vesicular transport. Of note, intraflagellar transport protein 20 (IFT20) localizes to the Golgi apparatus,[28] centrioles, and ciliary axonemes.[18] IFT20 also participates in vesicular trafficking at the primary cilium[28] and at the immune synapse (Box 2).[32] Another cilia protein that is important for endosomal recycling is Arl13b.[21] Therefore, endosomal trafficking is a potentially important extraciliary function for some cilia proteins.
Box 2. The curious case of the aciliate T-cell with “cilia” proteins that function at a non-cilium structure, the immune synapse.
Numerous studies have noted the many similarities between immune synapses and primary cilia (e.g., reviewed in[86,87]). Briefly, when T-cells encounter a target cell, these two cells form a structure called the immune synapse, allowing for cell activation and directed communication. Both genesis of the primary cilium and the construction of an immune synapse require centrosome polarization at the membrane,[88–90] docking of the mother centriole, and reliance on vesicular transport to the cilium or synapse (reviewed in[86,91]). These two distinct structures commonly utilize IFT20 transport,[28,32] Rab8,[51,92] and Rab29.[93] These similarities led Stinchombe and Griffiths[94] to ask whether “cilia” proteins may have more universal functions than previously thought since cilia proteins seem to be functional and present in T-cells (one of the few cell types that do not have primary cilia). They concluded that a central theme between immune synapses and primary cilia is the polarization of the centrosome. This complicates our nomenclature of “cilia” proteins as these proteins are increasingly being localized and functional in non-ciliated cells.
Jekely and Arendt found that cilia and the IFT system are not only well-conserved evolutionarily, but were likely present in the earliest eukaryotic ancestor. However, the simultaneous evolution of a complex vesicular trafficking system and a new organelle is unlikely. We hypothesize that the IFT system likely evolved first, and was later adopted to form a cilium. Cells that then had both an IFT system and a cilium were co-selected. However, if this were the case, then the IFT system must have been functional and important before a cilium evolved.
One possible function for a primitive IFT system would be motility. Continuing with our theme of trying to learn about cilium formation through examination of other polarized structures, we hypothesize that the growth cone may potentially be a good model to understand early IFT function. While growth cones are a specialized structure for just one cell type, neurons, they employ the basic components of cell migration, including lamellipodia, filopodia, and vesicular trafficking. In growth cones, membrane is endocytosed and redistributed to the side of a turning growth cone.[84] Thus, it is possible that the IFT system may have initially allowed for primitive cell turning, and was later used to form a protrusional motile cilium or flagellum (perhaps after cells became capable of compartmentalizing this protrusion/cilium structure) that allowed for more complex and directed motility, offering an evolutionarily selective advantage. Curiously, Arl13b is important for cell migration and localizes to the edge of lamellipodia and filopodia.[85] Future studies will be needed to determine whether the IFT system, which is also critical for both vesicular trafficking and ciliogenesis, is similarly localized to the leading edge and necessary for cell migration. Additional experiments will be necessary to see which cilia proteins are involved in cell migration and vesicular transport, which cilia proteins localize to lamellipodia and filopodia, and how knockdown of cilia proteins affects the leading edge of migrating cells.
2.8. Neuronal growth cones and dendritic spines share proteins with primary cilia
If cilia proteins are important for polarity, then they are likely important at many other polarized structures, such as neuronal growth cones and dendritic spines. While MYH10[34,35] and MyoVa[95] are general cytoskeletal proteins that have wide-ranging functions throughout the cell, both have critical roles in ciliogenesis, and have functions within growth cones[96,97] and dendritic spines.[98,99] The growth cone is structurally polarized with three domains: (1) a central domain composed of microtubules, (2) a transition zone made of actin and microtubules, and (3) a peripheral zone that is largely occupied by actin with rare invasion of dynamic microtubules.[100] Growth cones are also polarized in that actin and tubulin are largely oriented in the same direction (plus ends pointing out).[100] Growth cones also utilize RhoGTPase signaling, a mechanism used in polarity determination. Another polarity protein, CDC42, also functions at the growth cone by activating Par6/Par3 to regulate actin dynamics in the growth cone.[101] Additionally, expression of a dominant negative form of CDC42 can activate exocytosis;[102] thus, CDC42 may have multiple roles in the growth cone. Future studies are required for determining whether other growth cone proteins are critical for ciliogenesis and vice versa.
A case can be made that dendritic spines are also polarized structures. The polarity proteins, Par3 and Par6, appear to function in dendritic spines. Loss of Par3 results in the development of immature, filopodia-like dendritic spines.[103] While overexpression of Par6 or enzymatic activation of aPKC promotes spine development.[104] Septin7, which localizes within primary cilia axonemes,[105] can be found at the base of dendritic spines.[106,107] Septins function in part as diffusion barriers,[108] and may aid in creating a polarized membrane within dendritic spines. Indeed, Ewers and colleagues were able to show that Septin7 prevented the diffusion of GluA2 receptors into spines.[109]
Since multiple ciliogenesis and polarity proteins are found at growth cones and dendritic spines, we hypothesize that this is not a coincidence, but is indicative of the possibility that all these proteins function similarly and involve a polarity pathway. Many of these proteins have long been recognized as polarity proteins that function at other polarized structures (growth cones and dendritic spines), but are now also known to be important for ciliogenesis, supporting the idea that the cilium is just another polarized structure. In fact, our laboratory demonstrated that Ahi1, a protein implicated in JBTS, localizes to mouse growth cones.[110] It will be important to see how many other cilia proteins, particularly ciliopathy proteins, can be found in growth cones and dendritic spines where they could affect the function of these structures.
Given that polarity proteins and cilia proteins are both found in growth cones, dendritic spines, and at cilia, it becomes more difficult to distinguish which proteins are cilia proteins and which are polarity proteins. Proteins that localize to and function at the centrosome are likely important for both polarity and ciliogenesis as the centrosome is important in both processes. However, it is more difficult to understand how proteins (i.e., Arl13b) that localize to the cilium specifically and not to centrosomes, can affect both polarity and ciliogenesis. This likely will become easier to understand as extraciliary functions of “cilia” proteins are further elucidated, and are taken into consideration in accounting for the many steps involved in ciliogenesis. In our recent review[9], we documented that many cilia proteins have extraciliary functions and that they could possibly be unified by a theme of polarity. Arl13b, for example, may not be a centrosome protein or a cytoskeletal protein, but it has an important role in vesicular trafficking that may operate at a different step of the ciliogenesis pathway. Thus, even “cilia”-specific proteins may be important for polarity. This begs the question whether all cilia proteins are involved in establishing polarity? Given the co-occurrence of “cilia” and polarity proteins at many polarized structures, we hypothesize that ciliogenesis proteins are simply polarity proteins that may be found at other polarized structures. We would also predict that many more cilia proteins will be localized at growth cones and dendritic spines (and possibly vice versa).
2.9. Other implications for cilia proteins affecting polarity
If cilia proteins are polarity proteins, then defects in “cilia” proteins should yield a host of polarity defects as well. While this manuscript mainly deals with single cell polarity, it is important to recognize that single cell polarity contributes to these processes. Proper establishment of apical and basal polarity within a cell or tissue can stabilize and affect the PCP system[111], which is often linked back to nodal cilia[112] and ciliopathies.[77] Furthermore, misorientation of the mitotic spindle or impaired polarization can lead to improper asymmetric cell division.[113] These separate processes are all important, but further elaboration into these topics was beyond the scope of this paper.
3. What is a ciliopathy?
Ciliopathies are difficult to define. The term “ciliopathies” implies that the main defect observed in the disease is cilia-related, but such a claim is difficult to make given the large number of extraciliary sites and functions that have been identified for these “cilia” proteins (Box 3).[9] The claim that “ciliopathies” are diseases that result from defects in “cilia” proteins can be made, but the label of “cilia” proteins is insufficient. Such a discussion may ultimately be futile because it may not even be possible to have a disease that only affects primary cilia. Defects in primary cilia or ciliogenesis will more than likely be accompanied by a whole host of other extraciliary defects. We propose that it may be easier to see ciliopathies as defects in polarity.
Box 3. So what is a primary cilium for?
The answer is simple; the primary cilium is a signaling organelle. Some GPCRs involved in signaling localize to ciliary membranes, and the sonic hedgehog (Shh) pathway (critical for development) also signals through the primary cilium[114]. The primary cilium is responsive to exogenous and endogenous signals. Recently, roles for primary cilia have expanded to include ciliary involvement in endocytosis and exocytosis signaling.[115] Studies in humans and in such organisms as Chlamydomonas and C. elegans have identified bioactive ectosomes that appear to be in close proximity to primary cilia. Isolation and study of these ectosomes reveal that they contain many cilia proteins, including polycystin-1 (PC1), polycystin-2 (PC2), and fibrocystin.[116] In Chlamydomonas and C. elegans, these bioactive ectosomes seem to induce various mating behaviors,[117–119] but much more research will be needed to determine their role in mammalian cells.
The potential contribution of extraciliary defects to the symptoms seen in ciliopathies further complicates its definition. For example, individuals with JBTS have decussational (midline axon crossing) defects of axonal pathways in the hindbrain.[120] While some studies have argued that the primary cilium is important in migration,[121,122] we propose that the direct effects of defective extraciliary functions of cilia proteins in growth cones is a more likely cause for the decussational defects seen in JBTS. Additionally, another feature of JBTS is the common presence of cognitive defects. Although the primary cilium may have a yet to be identified role in cognition, it is more likely that defects in “cilia” proteins at dendritic spines, a structure already implicated in cognition and plasticity,[123,124] are the cause of cognitive defects in JBTS. Such possibilities suggest that cilia may not be the sole mediator of these defects, and that some phenotypes of a ciliopathy may be caused by dysfunction of “cilia” proteins at other cellular sites or even at other polarized structures.
5. Caveats
Examining cilia proteins as a whole reveals a connection between ciliogenesis proteins and the establishment of cell polarity, but why do defects in processes that all lead to the same goal of polarity not result in the same disease? Similarly, why do defects in the same organelle (the cilium), result in a whole host of different disorders? Currently, no good explanation exists to answer this difficult question; however, differences in primary cilia themselves and/or differences in the cells that express them may offer some insight. Tubulin tyrosine ligase-like (TTLL) enzymes are now implicated in ciliogenesis,[125] but these enzymes exhibit tissue specificity.[126] Thus, it may be possible to knockdown primary cilia in one tissue, and not others by targeting TTLL enzymes. Furthermore, primary cilia do not have the same composition of receptors from cell type to cell type. Adenylyl cyclase 3 was recently reported to be localized mainly in primary cilia of cells of a mesenchymal lineage.[127] It is not clear why different cells can have such diverse proteins at their primary cilium, though it is possible each primary cilium is specialized for its own cell type and extracellular space. Lastly, our laboratory has found that targeted deletion of Ahi1 in mice differentially affects primary cilia number and length in different types of neurons (unpublished data). Therefore, primary cilia proteins are likely to have diverse roles in different cell and tissue types. This variability may contribute to the wide range of syndromes and symptoms observed in ciliopathies.
Another important caveat is that of DNA damage repair (DDR) enzymes. Recently, some cilia proteins have been found to be important in DDR, including but not limited to: NEK1,[128] NEK8,[129] MRE11,[130] ZNF423,[130] CEP164,[130,131] and OFD1.[132] Lack of knowledge regarding the role of DDR enzymes and cilia makes it difficult to understand how these proteins may fit in our polarity hypothesis. Some of the examples above are easy to explain within our model. NEK family proteins have been studied in DDR, but have also been studied in cell cycle regulation and ciliogenesis, two processes that require proper microtubule regulation; thus, it is now thought that the NEK family proteins are general microtubule proteins.[133] Therefore, it is possible that the DDR function of NEK proteins is not critical for ciliogenesis, but the other functions of NEKs are important for the assembly of cilia. CEP164 and OFD1 are centrosomal proteins, so it is not inconceivable that centrosome proteins (like the many described above) could play a role in polarity and ciliogenesis. However, the role of CEP164 in DDR is now under question as another laboratory has failed to find any change in DDR in their CEP164 studies.[134]
The more difficult examples to explain in our polarity hypothesis are MRE11 and ZNF423. MRE11 is a meiotic recombination protein, while ZNF423 is a zinc finger protein. There are numerous examples of cilia proteins being involved in cell cycle regulation, and there are many examples of cilia proteins that have been found to localize inside the nucleus, but those proteins are known to have functions outside of the nucleus and cilia as well. Little is known, however, about whether MRE11 and ZNF423 have roles outside of their main functions, or whether they have any functions that may affect the polarity pathway. One possibility is that DDR is important for transcriptional control of polarity components, though studies will need to be done to directly address this. Since both DDR and primary ciliogenesis are in part reactive to cellular stress response, given their roles in the cell cycle, it is not surprising that direct interactions have been identified between centrosomal and DDR proteins.[135] In fact, increased cellular stress (replication stress) could result in centrosome dysfunction (and likely polarity defects too) in tumorigenesis and ciliary dysfunction in ciliopathies.[135] Lastly, it is possible that DDR does not have any relationship to polarity, and that these DDR enzymes do not have any yet to be described function that could be linked to polarity. If this holds true, it could mean that multiple upstream pathways (i.e., polarity and DDR) are important for the end result of ciliogenesis. Many more studies are needed to understand whether DDR proteins affect polarity and ciliogenesis.
6. Conclusions
“Cilia” proteins do not just affect cilia. The increasing number of extraciliary sites being identified, the understanding that extraciliary sites may contribute to ciliopathy phenotypes, and the realization that “cilia” proteins are commonly important for polarity, all suggest a need to rethink the use of some terms (Fig. 1). It is difficult to justify the continued use of “cilia” proteins knowing that ciliogenesis is only one function of these proteins. After all, these “cilia” proteins are present and functional in non-ciliated T-cells. “Cilia” proteins are also found at a number of polarized structures like growth cones and dendritic spines.[34,35,95–99,110] Thus, we argue that “cilia” proteins are general polarity proteins that likely function at most, if not all, polarized structures. Importantly, we are not the first to call into question the use of the terms, “cilia” proteins and “ciliopathies”.
One possibility may be in the recognition that “cilia” proteins may be more accurately called “polarity” proteins. This is not to claim that “cilia” proteins are equivalent to Par proteins that help segregate and define different polarized areas of the cell. Instead, this is a recognition of the fact that to establish cell polarity, a plethora of proteins and pathways need to be coordinated and functioning properly to reach the goal of cell polarization. As such, ciliopathies may be primarily due to defects in polarity, which subsequently disrupts ciliogenesis.
Acknowledgements
We apologize to all those colleagues whose studies could not be cited due to reference limitations. We thank members of the Ferland laboratory (J. Bourgeois, J. Munoz-Estrada, J. Nalwalk, and A. Srinivasan) for helpful discussions and commenting on drafts of this manuscript. This work was supported in part by a grant from NIH/NINDS: R01NS092062 (RJF).
Footnotes
The authors have declared no conflicts of interest.
References
- [1].Satir P, Cilia 2017, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV, Nature 2003, 426, 83. [DOI] [PubMed] [Google Scholar]
- [3].Hildebrandt F, Benzing T, Katsanis N, N Engl J Med 2011, 364, 1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bettencourt-Dias M, Hildebrandt F, Pellman D, Woods G, Godinho SA, Trends Genet 2011, 27, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Miyoshi K, Kasahara K, Miyazaki I, Asanuma M, Biochem Biophys Res Commun 2009, 388, 757. [DOI] [PubMed] [Google Scholar]
- [6].Prodromou NV, Thompson CL, Osborn DP, Cogger KF, Ashworth R, Knight MM, Beales PL, Chapple JP, J Cell Sci 2012, 125, 4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Miyoshi K, Kasahara K, Murakami S, Takeshima M, Kumamoto N, Sato A, Miyazaki I, Matsuzaki S, Sasaoka T, Katayama T, Asanuma M, PLoS One 2014, 9, e97918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pitaval A, Tseng Q, Bornens M, Thery M, J Cell Biol 2010, 191, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hua K, Ferland RJ, Cell Mol Life Sci 2018, 75, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gerdes JM, Davis EE, Katsanis N, Cell 2009, 137, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Adler PN, Wallingford JB, Trends Cell Biol 2017, 27, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cui C, Chatterjee B, Lozito TP, Zhang Z, Francis RJ, Yagi H, Swanhart LM, Sanker S, Francis D, Yu Q, San Agustin J. T., Puligilla C, Chatterjee T, Tansey T, Liu X, Kelley MW, Spiliotis ET, Kwiatkowski AV, Tuan R, Pazour GJ, Hukriede NA, Lo CW, PLoS Biol 2013, 11, e1001720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gray RS, Abitua PB, Wlodarczyk BJ, Szabo-Rogers HL, Blanchard O, Lee I, Weiss GS, Liu KJ, Marcotte EM, Wallingford JB, Finnell RH, Nat Cell Biol 2009, 11, 1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Park TJ, Haigo SL, Wallingford JB, Nat Genet 2006, 38, 303. [DOI] [PubMed] [Google Scholar]
- [15].Toriyama M, Lee C, Taylor SP, Duran I, Cohn DH, Bruel AL, Tabler JM, Drew K, Kelly MR, Kim S, Park TJ, Braun DA, Pierquin G, Biver A, Wagner K, Malfroot A, Panigrahi I, Franco B, Al-Lami HA, Yeung Y, Choi YJ, G. University of Washington Center for Mendelian, Duffourd Y, Faivre L, Riviere JB, Chen J, Liu KJ, Marcotte EM, Hildebrandt F, Thauvin-Robinet C, Krakow D, Jackson PK, Wallingford JB, Nat Genet 2016, 48, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zilber Y, Babayeva S, Seo JH, Liu JJ, Mootin S, Torban E, Mol Biol Cell 2013, 24, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ross AJ, May-Simera H, Eichers ER, Kai M, Hill J, Jagger DJ, Leitch CC, Chapple JP, Munro PM, Fisher S, Tan PL, Phillips HM, Leroux MR, Henderson DJ, Murdoch JN, Copp AJ, Eliot MM, Lupski JR, Kemp DT, Dollfus H, Tada M, Katsanis N, Forge A, Beales PL, Nat Genet 2005, 37, 1135. [DOI] [PubMed] [Google Scholar]
- [18].Bernabe-Rubio M, Andres G, Casares-Arias J, Fernandez-Barrera J, Rangel L, Reglero-Real N, Gershlick DC, Fernandez JJ, Millan J, Correas I, Miguez DG, Alonso MA, J Cell Biol 2016, 214, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG, J Cell Biol 2000, 151, 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Delaval B, Bright A, Lawson ND, Doxsey S, Nat Cell Biol 2011, 13, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Barral DC, Garg S, Casalou C, Watts GF, Sandoval JL, Ramalho JS, Hsu VW, Brenner MB, Proc Natl Acad Sci U S A 2012, 109, 21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Patzke S, Hauge H, Sioud M, Finne EF, Sivertsen EA, Delabie J, Stokke T, Aasheim HC, Oncogene 2005, 24, 1159. [DOI] [PubMed] [Google Scholar]
- [23].Patzke S, Redick S, Warsame A, Murga-Zamalloa CA, Khanna H, Doxsey S, Stokke T, Mol Biol Cell 2010, 21, 2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhu L, Wang Z, Wang W, Wang C, Hua S, Su Z, Brako L, Garcia-Barrio M, Ye M, Wei X, Zou H, Ding X, Liu L, Liu X, Yao X, J Biol Chem 2015, 290, 27053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sternemalm J, Geimer S, Frikstad KA, Schink KO, Stokke T, Patzke S, PLoS One 2015, 10, e0134789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shearer R, Fristad KM., McKenna J, McCloy R, Deng N, Burgess A, Stokke T, Patzke S, and Saunders DN, (preprint). [DOI] [PMC free article] [PubMed]
- [27].Sternemalm J, Russnes HG, Zhao X, Risberg B, Nord S, Caldas C, Borresen-Dale AL, Stokke T, Patzke S, Br J Cancer 2014, 111, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Follit JA, Tuft RA, Fogarty KE, Pazour GJ, Mol Biol Cell 2006, 17, 3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang L, Li W, Ni J, Wu J, Liu J, Zhang Z, Zhang Y, Li H, Shi Y, Teves ME, Song S, Strauss JF 3rd, Zhang Z, Cytoskeleton (Hoboken) 2015, 72, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Baron Gaillard C. L., Pallesi-Pocachard E, Massey-Harroche D, Richard F, Arsanto JP, Chauvin JP, Lecine P, Kramer H, Borg JP, Le Bivic A, Mol Biol Cell 2011, 22, 4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Stoetzel C, Bar S, De Craene JO, Scheidecker S, Etard C, Chicher J, Reck JR, Perrault I, Geoffroy V, Chennen K, Strahle U, Hammann P, Friant S, Dollfus H, Nat Commun 2016, 7, 13586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Finetti F, Paccani SR, Riparbelli MG, Giacomello E, Perinetti G, Pazour GJ, Rosenbaum JL, Baldari CT, Nat Cell Biol 2009, 11, 1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yin Y, Bangs F, Paton IR, Prescott A, James J, Davey MG, Whitley P, Genikhovich G, Technau U, Burt DW, Tickle C, Development 2009, 136, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rao Y, Hao R, Wang B, Yao TP, PLoS One 2014, 9, e114087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hong H, Kim J, Kim J, Biochem Biophys Res Commun 2015, 461, 180. [DOI] [PubMed] [Google Scholar]
- [36].Ravanelli AM, Klingensmith J, Dev Biol 2011, 350, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Das A, Dickinson DJ, Wood CC, Goldstein B, Slep KC, Mol Biol Cell 2015, 26, 4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dacheux D, Roger B, Bosc C, Landrein N, Roche E, Chansel L, Trian T, Andrieux A, Papaxanthos-Roche A, Marthan R, Robinson DR, Bonhivers M, J Cell Sci 2015, 128, 1294. [DOI] [PubMed] [Google Scholar]
- [39].Sharp DJ, Ross JL, J Cell Sci 2012, 125, 2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Stephen LA, Davis GM, McTeir KE, James J, McTeir L, Kierans M, Bain A, Davey MG, Dev Dyn 2013, 242, 923. [DOI] [PubMed] [Google Scholar]
- [41].Hehnly H, Doxsey S, Dev Cell 2014, 28, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kitagawa D, Kohlmaier G, Keller D, Strnad P, Balestra FR, Fluckiger I, Gonczy P, J Cell Sci 2011, 124, 3884. [DOI] [PubMed] [Google Scholar]
- [43].Xu W, Jin M, Hu R, Wang H, Zhang F, Yuan S, Cao Y, J Am Soc Nephrol 2017, 28, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Krahn MP, Wodarz A, Essays Biochem 2012, 53, 15. [DOI] [PubMed] [Google Scholar]
- [45].Li R, Gundersen GG, Nat Rev Mol Cell Biol 2008, 9, 860. [DOI] [PubMed] [Google Scholar]
- [46].Campanale JP, Sun TY, Montell DJ, J Cell Sci 2017, 130, 1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Elric J, Etienne-Manneville S, Exp Cell Res 2014, 328, 240. [DOI] [PubMed] [Google Scholar]
- [48].Eaton S, Martin-Belmonte F, Cold Spring Harb Perspect Biol 2014, 6, a016899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Guo Y, Zanetti G, Schekman R, Elife 2013, 2, e00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Merte J, Jensen D, Wright K, Sarsfield S, Wang Y, Schekman R, Ginty DD, Nat Cell Biol 2010, 12, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Knodler A, Feng S, Zhang J, Zhang X, Das A, Peranen J, Guo W, Proc Natl Acad Sci U S A 2010, 107, 6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Westlake CJ, Baye LM, Nachury MV, Wright KJ, Ervin KE, Phu L, Chalouni C, Beck JS, Kirkpatrick DS, Slusarski DC, Sheffield VC, Scheller RH, Jackson PK, Proc Natl Acad Sci U S A 2011, 108, 2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Fan S, Hurd TW, Liu CJ, Straight SW, Weimbs T, Hurd EA, Domino SE, Margolis B, Curr Biol 2004, 14, 1451. [DOI] [PubMed] [Google Scholar]
- [54].Krock BL, Perkins BD, PLoS One 2014, 9, e104661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jekely G, Arendt D, Bioessays 2006, 28, 191. [DOI] [PubMed] [Google Scholar]
- [56].Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, Zuker CS, Cell 2004, 117, 527. [DOI] [PubMed] [Google Scholar]
- [57].Flynn KC, Bioarchitecture 2013, 3, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Schelski M, Bradke F, Mol Cell Neurosci 2017, 84, 11. [DOI] [PubMed] [Google Scholar]
- [59].Gartner A, Fornasiero EF, Valtorta F, Dotti CG, J Cell Sci 2014, 127, 4409. [DOI] [PubMed] [Google Scholar]
- [60].Gartner A, Fornasiero EF, Munck S, Vennekens K, Seuntjens E, Huttner WB, Valtorta F, Dotti CG, EMBO J 2012, 31, 1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pollarolo G, Schulz JG, Munck S, Dotti CG, Nat Neurosci 2011, 14, 1525. [DOI] [PubMed] [Google Scholar]
- [62].Calderon de Anda F, Gartner A, Tsai LH, Dotti CG, J Cell Sci 2008, 121, 178. [DOI] [PubMed] [Google Scholar]
- [63].Li Y, Ling K, Hu J, J Cell Biochem 2012, 113, 2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Casalou C, Faustino A, Barral DC, Small GTPases 2016, 7, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Antoniades I, Stylianou P, Skourides PA, Dev Cell 2014, 28, 70. [DOI] [PubMed] [Google Scholar]
- [66].Stephen LA, Tawamie H, Davis GM, Tebbe L, Nurnberg P, Nurnberg G, Thiele H, Thoenes M, Boltshauser E, Uebe S, Rompel O, Reis A, Ekici AB, McTeir L, Fraser AM, Hall EA, Mill P, Daudet N, Cross C, Wolfrum U, Jamra RA, Davey MG, Bolz HJ, Elife 2015, 4, e08077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Assemat E, Bazellieres E, Pallesi-Pocachard E, Le Bivic A, Massey-Harroche D, Biochim Biophys Acta 2008, 1778, 614. [DOI] [PubMed] [Google Scholar]
- [68].Bachmann-Gagescu R, Phelps IG, Dempsey JC, Sharma VA, Ishak GE, Boyle EA, Wilson M, Marques Lourenco C., Arslan M, G. University of Washington Center for Mendelian, Shendure J, Doherty, Hum Mutat 2015, 36, 831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Roosing S, Hofree M, Kim S, Scott E, Copeland B, Romani M, Silhavy JL, Rosti RO, Schroth J, Mazza T, Miccinilli E, Zaki MS, Swoboda KJ, Milisa-Drautz J, Dobyns WB, Mikati MA, Incecik F, Azam M, Borgatti R, Romaniello R, Boustany RM, Clericuzio CL, D’Arrigo S, Stromme P, Boltshauser E, Stanzial F, Mirabelli-Badenier M, Moroni I, Bertini E, Emma F, Steinlin M, Hildebrandt F, Johnson CA, Freilinger M, Vaux KK, Gabriel SB, Aza-Blanc P, Heynen-Genel S, Ideker T, Dynlacht BD, Lee JE, Valente EM, Kim J, Gleeson JG, Elife 2015, 4, e06602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Mack NA, Georgiou M, Small GTPases 2014, 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hamze-Komaiha O, Sarr S, Arlot-Bonnemains Y, Samuel D, Gassama-Diagne A, Cell Rep 2016, 17, 2738. [DOI] [PubMed] [Google Scholar]
- [72].Woods B, Lew DJ, Small GTPases 2017, 28, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Choi SY, Chacon-Heszele MF, Huang L, McKenna S, Wilson FP, Zuo X, Lipschutz JH, J Am Soc Nephrol 2013, 24, 1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Badgandi HB, Hwang SH, Shimada IS, Loriot E, Mukhopadhyay S, J Cell Biol 2017, 216, 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Mukhopadhyay S, Wen X, Chih B, Nelson CD, Lane WS, Scales SJ, Jackson PK, Genes Dev 2010, 24, 2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Nakatsu F, Dev Cell 2015, 34, 379. [DOI] [PubMed] [Google Scholar]
- [77].Fischer E, Legue E, Doyen A, Nato F, Nicolas JF, Torres V, Yaniv M, Pontoglio M, Nat Genet 2006, 38, 21. [DOI] [PubMed] [Google Scholar]
- [78].Nishio S, Tian X, Gallagher AR, Yu Z, Patel V, Igarashi P, Somlo S, J Am Soc Nephrol 2010, 21, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Cao J, Shen Y, Zhu L, Xu Y, Zhou Y, Wu Z, Li Y, Yan X, Zhu X, Nat Cell Biol 2012, 14, 697. [DOI] [PubMed] [Google Scholar]
- [80].Yan X, Zhu X, Exp Cell Res 2013, 319, 147. [DOI] [PubMed] [Google Scholar]
- [81].Dawe HR, Farr H, Gull K, J Cell Sci 2007, 120, 7. [DOI] [PubMed] [Google Scholar]
- [82].Ghosh DK, Dasgupta D, Guha A, ISRN Mol Biol 2012, 2012, 596289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].van Dam TJ, Townsend MJ, Turk M, Schlessinger A, Sali A, Field MC, Huynen MA, Proc Natl Acad Sci U S A 2013, 110, 6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Tojima T, Itofusa R, Kamiguchi H, J Neurosci 2014, 34, 7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Casalou C, Seixas C, Portelinha A, Pintado P, Barros M, Ramalho JS, Lopes SS, Barral DC, J Cell Sci 2014, 127, 2709. [DOI] [PubMed] [Google Scholar]
- [86].Finetti F, Onnis A, Baldari CT, Traffic 2015, 16, 241. [DOI] [PubMed] [Google Scholar]
- [87].Onnis A, Finetti F, Baldari CT, Front Immunol 2016, 7, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Geiger B, Rosen D, Berke G, J Cell Biol 1982, 95, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kupfer A, Dennert G, Singer SJ, Proc Natl Acad Sci U S A 1983, 80, 7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kupfer A, Dennert G, J Immunol 1984, 133, 2762. [PubMed] [Google Scholar]
- [91].Finetti F, Baldari CT, Immunol Rev 2013, 251, 97. [DOI] [PubMed] [Google Scholar]
- [92].Finetti F, Patrussi L, Galgano D, Cassioli C, Perinetti G, Pazour GJ, Baldari CT, J Cell Sci 2015, 128, 2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Onnis A, Finetti F, Patrussi L, Gottardo M, Cassioli C, Spano S, Baldari CT, Cell Death Differ 2015, 22, 1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Stinchcombe JC, Griffiths GM, Philos Trans R Soc Lond B Biol Sci 2014, 369, 20130463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Assis LH, Silva-Junior RM, Dolce LG, Alborghetti MR, Honorato RV, Nascimento AF, Melo-Hanchuk TD, Trindade DM, Tonoli CC, Santos CT, Oliveira PS, Larson RE, Kobarg J, Espreafico EM, Giuseppe PO, Murakami MT, Sci Rep 2017, 7, 43692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Wada F, Nakata A, Tatsu Y, Ooashi N, Fukuda T, Nabetani T, Kamiguchi H, Cell Rep 2016, 15, 1329. [DOI] [PubMed] [Google Scholar]
- [97].Bridgman PC, Dave S, Asnes CF, Tullio AN, Adelstein RS, J Neurosci 2001, 21, 6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Yoshii A, Zhao JP, Pandian S, van Zundert B, Constantine-Paton M, J Neurosci 2013, 33, 8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Hodges JL, Newell-Litwa K, Asmussen H, Vicente-Manzanares M, Horwitz AR, PLoS One 2011, 6, e24149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Geraldo S, Gordon-Weeks PR, J Cell Sci 2009, 122, 3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Solecki DJ, Govek EE, Hatten ME, J Neurosci 2006, 26, 10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Alberts P, Rudge R, Irinopoulou T, Danglot L, Gauthier-Rouviere C, Galli T, Mol Biol Cell 2006, 17, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Zhang H, Macara IG, Nat Cell Biol 2006, 8, 227. [DOI] [PubMed] [Google Scholar]
- [104].Zhang H, Macara IG, Dev Cell 2008, 14, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Ghossoub R, Hu Q, Failler M, Rouyez MC, Spitzbarth B, Mostowy S, Wolfrum U, Saunier S, Cossart P, Jamesnelson W, Benmerah A, J Cell Sci 2013, 126, 2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Tada T, Simonetta A, Batterton M, Kinoshita M, Edbauer D, Sheng M, Curr Biol 2007, 17, 1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Xie Y, Vessey JP, Konecna A, Dahm R, Macchi P, Kiebler MA, Curr Biol 2007, 17, 1746. [DOI] [PubMed] [Google Scholar]
- [108].Mostowy S, Cossart P, Nat Rev Mol Cell Biol 2012, 13, 183. [DOI] [PubMed] [Google Scholar]
- [109].Ewers H, Tada T, Petersen JD, Racz B, Sheng M, Choquet D, PLoS One 2014, 9, e113916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Doering JE, Kane K, Hsiao YC, Yao C, Shi B, Slowik AD, Dhagat B, Scott DD, Ault JG, Page-McCaw PS, Ferland RJ, J Comp Neurol 2008, 511, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Wu J, Mlodzik M, Trends Cell Biol 2009, 19, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Antic D, Stubbs JL, Suyama K, Kintner C, Scott MP, Axelrod JD, PLoS One 2010, 5, e8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Homem CC, Knoblich JA, Development 2012, 139, 4297. [DOI] [PubMed] [Google Scholar]
- [114].Mukhopadhyay S, Rohatgi R, Semin Cell Dev Biol 2014, 33, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Wang J, Barr MM, Cell Mol Neurobiol 2016, 36, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Hogan MC, Manganelli L, Woollard JR, Masyuk AI, Masyuk TV, Tammachote R, Huang BQ, Leontovich AA, Beito TG, Madden BJ, Charlesworth MC, Torres VE, LaRusso NF, Harris PC, Ward CJ, J Am Soc Nephrol 2009, 20, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Barr MM, Garcia LR, in WormBook, (Eds: Jorgensen EM, Kaplan JM), The C. elegans Research Community, 2006, doi/10.1895/wormbook.1.78.1.
- [118].Sherlekar AL, Janssen A, Siehr MS, Koo PK, Caflisch L, Boggess M, Lints R, PLoS One 2013, 8, e60597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Wood CR, Huang K, Diener DR, Rosenbaum JL, Curr Biol 2013, 23, 906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, Shugart YY, Ruvolo M, Walsh CA, Nat Genet 2004, 36, 1008. [DOI] [PubMed] [Google Scholar]
- [121].Higginbotham H, Eom TY, Mariani LE, Bachleda A, Hirt J, Gukassyan V, Cusack CL, Lai C, Caspary T, Anton ES, Dev Cell 2012, 23, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Higginbotham H, Guo J, Yokota Y, Umberger NL, Su CY, Li J, Verma N, Hirt J, Ghukasyan V, Caspary T, Anton ES, Nat Neurosci 2013, 16, 1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J, Trends Neurosci 2010, 33, 121. [DOI] [PubMed] [Google Scholar]
- [124].Ma L, Li Y, Wang R, Clin Chim Acta 2015, 451, 121. [DOI] [PubMed] [Google Scholar]
- [125].Rocha C, Papon L, Cacheux W, Marques Sousa P., Lascano V, Tort O, Giordano T, Vacher S, Lemmers B, Mariani P, Meseure D, Medema JP, Bieche I, Hahne M, Janke C, EMBO J 2014, 33, 2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Pathak N, Austin CA, Drummond IA, J Biol Chem 2011, 286, 11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Antal MC, Benardais K, Samama B, Auger C, Schini-Kerth V, Ghandour S, Boehm N, PLoS One 2017, 12, e0170756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Thiel C, Kessler K, Giessl A, Dimmler A, Shalev SA, von der Haar S, Zenker M, Zahnleiter D, Stoss H, Beinder E, Abou Jamra R., Ekici AB, Schroder-Kress N, Aigner T, Kirchner T, Reis A, Brandstatter JH, Rauch A, Am J Hum Genet 2011, 88, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Choi HJ, Lin JR, Vannier JB, Slaats GG, Kile AC, Paulsen RD, Manning DK, Beier DR, Giles RH, Boulton SJ, Cimprich KA, Mol Cell 2013, 51, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, Wang H, Hurd TW, Zhou W, Cluckey A, Gee HY, Ramaswami G, Hong CJ, Hamilton BA, Cervenka I, Ganji RS, Bryja V, Arts HH, van Reeuwijk J, Oud MM, Letteboer SJ, Roepman R, Husson H, Ibraghimov-Beskrovnaya O, Yasunaga T, Walz G, Eley L, Sayer JA, Schermer B, Liebau MC, Benzing T, Le Corre S, Drummond I, Janssen S, Allen SJ, Natarajan S, O’Toole JF, Attanasio M, Saunier S, Antignac C, Koenekoop RK, Ren H, Lopez I, Nayir A, Stoetzel C, Dollfus H, Massoudi R, Gleeson JG, Andreoli SP, Doherty DG, Lindstrad A, Golzio C, Katsanis N, Pape L, Abboud EB, Al-Rajhi AA, Lewis RA, Omran H, Lee EY, Wang S, Sekiguchi JM, Saunders R, Johnson CA, Garner E, Vanselow K, Andersen JS, Shlomai J, Nurnberg G, Nurnberg P, Levy S, Smogorzewska A, Otto EA, Hildebrandt F, Cell 2012, 150, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Sivasubramaniam S, Sun X, Pan YR, Wang S, Lee EY, Genes Dev 2008, 22, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Abramowicz I, Carpenter G, Alfieri M, Colnaghi R, Outwin E, Parent P, Thauvin-Robinet C, Iaconis D, Franco B, O’Driscoll M, Hum Mol Genet 2017, 26, 19. [DOI] [PubMed] [Google Scholar]
- [133].Fry AM, O’Regan L, Sabir SR, Bayliss R, J Cell Sci 2012, 125, 4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Daly OM, Gaboriau D, Karakaya K, King S, Dantas TJ, Lalor P, Dockery P, Kramer A, Morrison CG, J Cell Sci 2016, 129, 1769. [DOI] [PubMed] [Google Scholar]
- [135].Johnson CA, Collis SJ, Cilia 2016, 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]