

Abstract
Multifunctional optogenetic systems are in high demand for use in basic and biomedical research. Near-infrared-light-inducible binding of bacterial phytochrome BphP1 to its natural PpsR2 partner is beneficial for simultaneous use with blue-light-activatable tools. However, applications of the BphP1–PpsR2 pair are limited by the large size, multidomain structure and oligomeric behavior of PpsR2. Here, we engineered a single-domain BphP1 binding partner, Q-PAS1, which is three-fold smaller and lacks oligomerization. We exploited a helix–PAS fold of Q-PAS1 to develop several near-infrared-light-controllable transcription regulation systems, enabling either 40-fold activation or inhibition. The light-induced BphP1–Q-PAS1 interaction allowed modification of the chromatin epigenetic state. Multiplexing the BphP1–Q-PAS1 pair with a blue-light-activatable LOV-domain-based system demonstrated their negligible spectral crosstalk. By integrating the Q-PAS1 and LOV domains in a single optogenetic tool, we achieved tridirectional protein targeting, independently controlled by near-infrared and blue light, thus demonstrating the superiority of Q-PAS1 for spectral multiplexing and engineering of multicomponent systems.
Expression in the brain of optogenetic tools based on microbial opsins has allowed precise spatiotemporal control of various processes in the nervous system1. In turn, natural diversity of nonopsin photoreceptors laid the basis for development of optogenetic tools that utilize light-triggered protein conformation changes or light-controlled protein–protein interactions (PPIs)2.
Light-triggered PPIs exploited in the nonopsin optogenetic tools include homodimerization, heterodimerization and oligomerization. Homodimerization of a small light–oxygen–voltage (LOV)-domain-containing protein, called Vivid, is used for light-controlled transcription3. A LOV2 domain of phototropin 1 from Avena sativa (AsLOV2) and a modified PDZ domain (ePDZ) are combined into an optogenetic system based on heterodimerization4. AsLOV2-based optogenetic tools enable light control of nuclear–cytoplasmic protein shuttling5–7. Cryptochrome 2 (CRY2) from Arabidopsis thaliana is another photoreceptor, which initially was applied to PPI heterodimerization approaches8. Later, its natural oligomerization ability was used in optogenetic clustering approaches9. Further tuning of the engineered light-activatable systems led to a design of the new generation of dimerizers for advanced control of the protein localization10, cell signaling11 and recombinase activity12. All these optogenetic systems sense 440–480 nm light. Therefore, for simultaneous use with the blue-light-controlled optogenetic tools, systems sensing light in a different spectral range are required.
A class of photoreceptors called phytochromes stands apart from other photosensing proteins because of their ability to absorb far-red or near-infrared (NIR) light. All phytochromes utilize heme-derived linear tetrapyrrole compounds as their light-sensing chromophores. A red-light-triggered interaction of a plant phytochrome B (PhyB) and the phytochrome-interacting factor 6 (PIF6) from Arabidopsis is successfully applied to transcriptional control13, cell signaling14 and protein localization15. Unlike plant phytochromes, which use phytochromobilin or phycocyanobilin tetrapyrroles as a chromophore, a subclass of bacterial phytochrome photoreceptors (BphPs) incorporate biliverdin IXα (BV) tetrapyrrole16,17. As BV has the largest electron-conjugated system, it absorbs the most NIR-shifted light among all chromophores found in phytochromes. This makes BphPs the favorable templates to develop fluorescent proteins for applications in mammals18,19. BphPs exist in two interconvertible states, traditionally called the red-absorbing (Pr; absorbs at 660–700 nm) and the far-red-absorbing (Pfr; absorbs at 740–780 nm). Upon NIR illumination, BphP-bound BV isomerizes via the fourth D-ring rotation around its 15–16 double bond. This Z–E isomerization results in the subsequent structural changes in a photosensory core module and an output domain of BphP.
Recently, the first PPI-based optogenetic system that uses BphP from Rhodopseudomonas palustris, called RpBphP1 (hereafter termed BphP1), was developed. The NIR light-triggered PPI of BphP1 with its natural binding partner RpPpsR2 (hereafter called PpsR2) allows precise control of gene transcription20. BphP1, serving as a light-sensing element of the BphP1–PpsR2 optogenetic system, belongs to noncanonical bathy BphPs, which in darkness adopt the Pfr state. Under NIR light of 740–780 nm, it undergoes the Pfr→Pr photoconvertion, resulting in the reversible binding of PpsR2 (ref. 21).
PpsR2 is a natural regulator of photosynthetic gene expression22. Although no structural data are available for PpsR2, a structure and an oligomeric state of its close homolog RsPpsR from Rhodobacter sphaeroides are characterized23,24. It was shown that RsPpsR contains three PAS domains, two of which (N-PAS and PAS2) are involved in tetramer formation in solution and octameric assembly upon binding to a DNA target. Taking into account its high sequence identity to RsPpsR, it seems likely that PpsR2 forms oligomers too21. Moreover, a Cys439 amino acid residue in the HTH domain of PpsR2 may form an intermolecular disulfide bond21. Altogether, these exchanges may lead to undesired PPIs in the BphP1–PpsR2 optogenetic system and, consequently, a reduction in its functionality and dynamic range.
Here, we focused on an improvement of the BphP1–PpsR2 pair to enable its new applications. First, we studied the BphP1–PpsR2 behavior in vitro and revealed a minimal part of PpsR2 required for the PPI. We deleted domains involved in the PpsR2 oligomerization, resulting in a single-domain 17-kDa protein, termed Q-PAS1, which efficiently interacted with BphP1 in vitro and in mammalian cells. We then designed a Q-PAS1-based chimeric transcription factor and applied the BphP1–Q-PAS1 pair to transcription activation via either its intracellular localization or oligomerization state. Next, we used the BphP1–Q-PAS1 pair to inhibit gene expression and to regulate its epigenetic state in a light-controlled manner. Lastly, we developed a multispectral system for tridirectional subcellular protein targeting, and demonstrated an absence of the spectral crosstalk between the BphP1–Q-PAS1 pair and two blue-light-activatable optogenetic systems.
RESULTS
Minimal fragment of PpsR2 interacting with BphP1
PpsR2 consists of four domains and an α-helical Q-linker (Fig. 1a), similarly to the recently crystallized RsPpsR protein23, which shares 30% identity with PpsR2. However, its BphP1-interacting fragment is still unknown. To determine a minimal PpsR2 part able to bind BphP1 in a light-dependent manner, we generated a series of PpsR2 deletion mutants and purified them from bacteria (Fig. 1a). mRuby2 tag was added for their visualization.
Figure 1. Characterization of BphP1 interaction with PpsR2 deletion mutants in vitro.
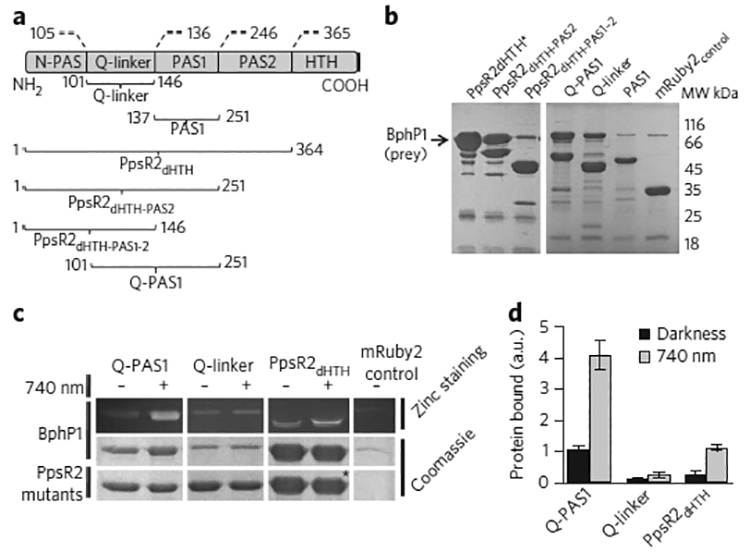
| (a) Domain structure of the PpsR family of proteins and PpsR2 mutants. Amino acid numbers are noted according to a R. palustris PpsR2 sequence (Genbank ANB32144.1). (b) BphP1 interaction with different PpsR2 deletion mutants. Black arrow indicates BphP1 position. (c) BphP1 interactions with PpsR2 mutants containing Q-linker in the presence or absence of 740 nm light. Top and middle panels, bound BphP1 (prey); bottom panel, Q-PAS1, Q-linker and PpsR2dHTH (bait). The bands marked with an asterisk contain the overlaid PpsR2dHTH-mRuby2 and BphP1 proteins, which have similar electrophoretic mobility. (d) Intensities of protein bands in pull-down analysis. Data were normalized to the band intensity of the sample containing Q-PAS1 in darkness. Error bars represent s.e.m.; n = 3 experiments. a.u., arbitrary units. Uncut gel images are provided in Supplementary Figure 4.
We first studied dimerization behavior of the mutants using a mild chemical crosslinking with carbodiimide (EDC) and N-hydroxysuccinimide (NHS). We found that all constructs containing a Q-linker formed dimers (Supplementary Results, Supplementary Fig. 1). Furthermore, we tested the ability of the deletion mutants to interact with BphP1 upon NIR 740 nm illumination in a pull-down assay using untagged BphP1 as a prey and the Strep-tagged PpsR2 mutants as the baits (Fig. 1b). Among them, only the deletion mutants containing both the α-helical Q-linker and the PAS1 domain efficiently bound BphP1; however, the weaker binding was also observed with the Q-linker alone.
Next, to study the light sensitivity of the interaction, we performed the pull-down experiments both in darkness and under NIR illumination. The purified Q-PAS1, Q-linker and PpsR2dHTH fragments interacted with BphP1 in both dark and light conditions; however, the interaction was substantially more efficient under light (Fig. 1c). Both the light-to-dark ratio and the amount of bound protein were maximal for the Q-PAS1 fragment, which showed a more than four-fold light-to-dark binding difference (Fig. 1d; Supplementary Fig. 2). Because both Q-PAS1 (Supplementary Fig. 1) and BphP1 (ref. 21) were able to dimerize, we analyzed their oligomerization states upon NIR illumination. Crosslinked Q-PAS1 and BphP1 alone migrated as bands of 100 kDa (for Q-PAS1–mRuby) and 162 kDa, respectively, which corresponded to their homodimers (Supplementary Figs. 3 and 4). However, when we mixed Q-PAS1 and BphP1 together, a heterodimer band of ~130 kDa, corresponding to a BphP1–Q-PAS1 complex with the 1:1 stoichiometry, was clearly detectable.
Thus, we mapped the minimal BphP1-interacting module as the Q-PAS1 fragment, which has a molecular mass of 17 kDa—three-fold smaller than that of wild-type PpsR2. Q-PAS1 effectively interacts with BphP1 upon 740 nm illumination and lacks the other domains, which may cause PpsR2 to form tetramers and octamers23.
Comparison of Q-PAS1- and PpsR2-based systems
We next checked whether the BphP1–Q-PAS1 pair could interact in a light-dependent manner in mammalian cells. For this we first tested whether Q-PAS1 could activate gene transcription using an approach reported for the original BphP1–PpsR2 system20. In this system, BphP1 was fused to a DNA-binding domain of tetracycline repressor (TetR) while PpsR2 was fused to a nuclear localization signal (NLS) and a VP16 transcriptional activation domain. Under NIR illumination, NLS–PpsR2–VP16 interacts with BphP1–TetR and transports it to the nucleus, where the protein complex initiates transcription of a reporter gene downstream of the tetO elements (Supplementary Fig. 5). We have replaced full-length PpsR2 with smaller Q-PAS1. The resulting BphP1–Q-PAS1 system showed ~10-fold increase in production of a secreted embryonic alkaline phosphatase (SEAP) reporter upon illumination, which was comparable to that of the original PpsR2-based system (Supplementary Fig. 6). We then compared two bicistronic plasmids encoding these two BphP1–PpsR2 and BphP1–Q-PAS1 transcription activation systems. With our BphP1–Q-PAS1 system, the SEAP level was several-fold higher and detected 8 h earlier than it was with the original BphP1–PpsR2 system (Supplementary Fig. 7). Perhaps the substantially smaller size of Q-PAS1 and its inability to form oligomers favor the formation of the BphP1–QPAS1 complexes and facilitate their transport to the nucleus.
Transcription inhibition by Q-PAS1 relocalization
Similarly to TetR-based transcription regulation, the GAL4–UAS system is widely used to control gene expression25,26. However, the GAL4–UAS system was not yet applied in optogenetic approaches using bacterial phytochromes. Furthermore, most transcription activation systems exploit light-driven nuclear import of transcription factor. Since their performance can be impaired by poor efficiency of nuclear transport27, we constructed a transcription inhibition system based on the light-triggered relocalization of a transcription factor from the nucleus by caging it at the plasma membrane.
For this, we fused Q-PAS1–VP16 with GAL4 truncated to 1–148 amino acid residues (GAL4(148)) that contain the DNA-binding and dimerization domains (Fig. 2a). BphP1 fused to mCherry–CAAX anchored to the plasma membrane via a -CAAX sequence (Fig. 2b). In the absence of interaction with BphP1–mCherry–CAAX, transfection with GAL4(148)–Q-PAS1–VP16 resulted in efficient EGFP reporter expression, detected by flow cytometry (Fig. 2c). In cells co-expressing GAL4(148)–Q-PAS1–VP16 and BphP1–mCherry–CAAX, NIR illumination induced binding of the Q-PAS1 construct to BphP1, thus caging it at the plasma membrane, which caused 4.6-fold reduction in the EGFP level (Fig. 2d, left bars). We did not observe the reduction in the reporter level in the absence of BphP1–mCherry–CAAX, which excluded artificial effects of NIR light on the cells (Supplementary Fig. 8). We also confirmed the transcription inhibition by live-cell epifluorescence microscopy. Similarly, the observed EGFP level in the cytoplasm was substantially lower in the illuminated cells (Fig. 2e). Because the VP16 sequence contained an NLS, which increased the transactivator concentration in the nucleus, we removed it to enhance the dark-to-light difference. As a result, the light-induced transcription inhibition of our system increased to eight-fold (Fig. 2d, right bars).
Figure 2. Transcription inhibition via light-induced relocalization approach.
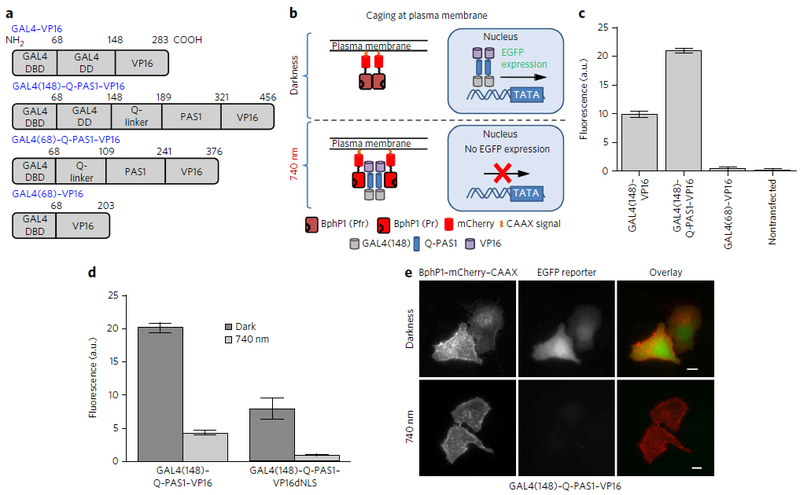
| (a) Domain structure of the GAL4–VP16 and GAL4–Q-PAS1–VP16 mutants. GAL4 DNA-binding domain, GAL4 dimerization domain, Q-linker and PAS1 domain of PpsR2, and VP16 transactivation domain are labeled as DBD, DD, Q-linker, PAS1 and VP16, respectively. Numbers refer to amino acid residue position in the corresponding construct. (b) Schematic representation of light-induced transcription inhibition via GAL4(148)–Q-PAS1–VP16 relocalization to the plasma membrane. (c) EGFP expression in HeLa cells cotransfected with different transcription activators and a pG5–EGFP (5× UAS) reporter plasmid. Constructs are marked according to a. Data were normalized to the EGFP expression level achieved with GAL4–VP16. Error bars represent s.e.m.; n = 3 experiments. (d) Comparison of EGFP expression levels launchedby the GAL4(148)–Q-PAS1–VP16 and GAL4(148)–Q-PAS1–VP16dNLS constructs. Data were normalized to the EGFP expression level detected with GAL4(148)–Q-PAS1–VP16dNLS in darkness. Error bars represent s.e.m.; n = 3 experiments. (e) Images of live HeLa cells cotransfected with GAL4(148)–Q-PAS1–VP16, BphP1–mCherry–CAAX and pG5–EGFP reporter plasmid in darkness and after 740 nm illumination. Scale bars, 10 μm. In d and e, the samples were incubated either in darkness or under 740 nm pulsed light (30 s on and 180 s off) of 0.2 mW cm−2 started 6 h after transfection. Analysis was performed 24 h after transfection.
Transcriptional inhibition by disruption of DNA binding
We next used the ability of Q-PAS1 to form heterodimers with BphP1 as a means to achieve better transcription inhibition. GAL4(148) binds DNA as a homodimer, and deletion of its dimerization domain (66–100 amino acids) abolishes this binding28. Indeed, the truncated GAL4(68) fused with VP16 failed to induce EGFP reporter expression, in contrast to GAL4(148)–VP16 (Fig. 2a,c). We hypothesized that Q-PAS1 itself could determine dimerization behavior of the GAL4-containing transcription factor (GAL4–Q-PAS1–VP16) and, consequently, control its ability to bind DNA in a light-dependent manner (Fig. 3a).
Figure 3. Light-induced dissociation of chimeric transcription factor.
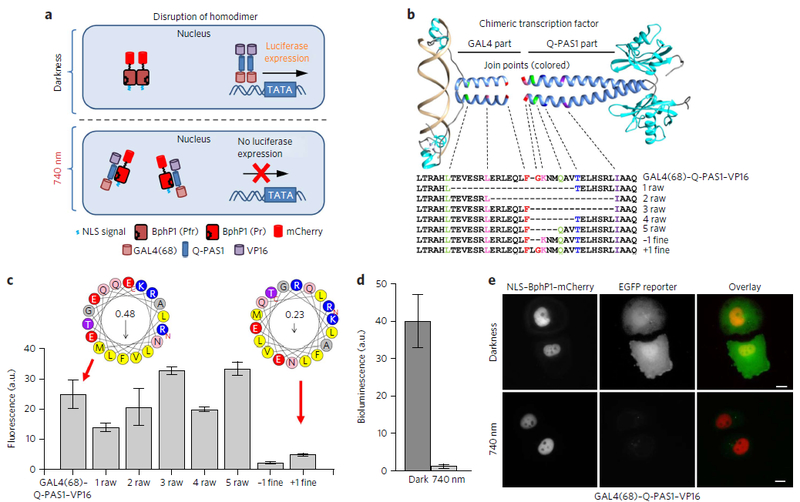
| (a) Schematic representation of light-induced transcription inhibition via disruption of the GAL4(68)–Q-PAS1–VP16 dimer. (b) Structure and sequence alignment of engineered chimeric transcription factors consisting of the Q-PAS1 and GAL4(68) parts fused together. The GAL4(DBD) structure in complex with DNA (PDB ID: 3COQ) and a modified RsPpsR structure (PDB ID: 4HH2)were used for visualization. Join points of the α-helical fusion regions for different constructs are shown with dashed lines and colors. (c) Comparison of the EGFP reporter levels for the different GAL4–Q-PAS1–VP16 fusion constructs co-expressed with NLS–BphP1–mCherry. For each construct, the EGFP induction level was normalized to the EGFP level in cells transfected with pG5–EGFP (5× UAS) reporter plasmid only. Helical wheels represent the α-helical joining regions of chimeric transcription factors with either preserved (like GAL4(68)–Q-PAS1–VP16) or disrupted (+1 fine) coiled-coil motif. The numbers in circles are the calculated hydrophobic moments. (d) Fluc expression in HeLa cells cotransfected with GAL4(68)–Q-PAS1–VP16, NLS–BphP1–mCherry and pFR–Luc reporter plasmid. Error bars represent s.e.m.; n = 3 experiments. (e) Images of live HeLa cells cotransfected with GAL4(68)–Q-PAS1–VP16, NLS–BphP1–mCherry and pG5–EGFP reporter plasmid in darkness and after 740 nm illumination. Scale bars, 10 μm. In d and e the samples were incubated either in darkness or under 740 nm pulsed light (30 s on and 180 s off) of 0.2 mW cm−2 started 6 h after the transfection. Analysis was performed 48 h after the transfection.
Both the Q-linker and the GAL4(50–72) fragments naturally form heptad coiled-coil motifs (amino acid residues in coiled-coil pattern are sequentially marked from “a” to “g”) stabilized by hydrophobic amino acids at the positions “a” and “d”. To test whether retaining the heptad repeat in a hybrid α-helical linker connecting the GAL4 and PAS1 domains is important for functionality of a chimeric transcription factor, we made several constructs with different positions of a fusion between the C-terminal GAL4(68) and the Q-linker helices (Fig. 3b). The ‘raw’ fusion constructs contained the deletions in the GAL4 and/or the Q-PAS1 parts that did not break the motif. By contrast, the ‘fine’ fusions differed from the GAL4(68)–Q-PAS1 construct by the insertion or deletion of 1 amino acid residue at the join point, resulting in the disruption of the coiled-coil motif (Fig. 3b). For all fusions, the probability of forming the coiled-coil motif was predicted using a MARCOIL algorithm29 (Supplementary Fig. 9). We analyzed the amphiphilicity of the helix joining regions using a HeliQuest software30 (Fig. 3c).
We transfected HeLa cells with different GAL4–Q-PAS1–VP16 fusions and NLS–BphP1–mCherry and checked reporter expression in darkness and under NIR illumination. We observed that even in darkness the ‘fine’ GAL4–Q-PAS1–VP16 fusions exhibited a low transcription activation, suggesting that their ability to bind DNA was impaired (Fig. 3c; Supplementary Table 1). By contrast, the ‘raw’ constructs activated transcription in darkness to a similar level as initial GAL4(68)–Q-PAS1–VP16, but their light-induced transcription inhibition was lower (Supplementary Fig. 10). This result suggested that the ‘raw’ deletions did not affect the DNA binding but likely impaired interaction of the ‘raw’ chimeric transcription factors with BphP1. The GAL4(68)–Q-PAS1–VP16 fusion construct showed the highest 40-fold light-induced inhibition of the firefly luciferase (Fluc) reporter bioluminescence (Fig. 3d). We also confirmed the transcription inhibition using live-cell epifluorescence microscopy. We observed a substantially higher cytoplasmic fluorescence signal of the EGFP reporter in cells kept in darkness than in those kept under 740 nm illumination (Fig. 3e).
Light control of chromatin epigenetic state
We next tested whether the BphP1–Q-PAS1 interaction could regulate chromatin epigenetic state. For this, we engineered a NIR-light-controlled site-targeted epigenetic modifier using a mSin interaction domain (SID). SID domains are found in Mad proteins which are able to recruit a mSin3–HDAC2 (histone deacetylase 2) complex to a gene target, resulting in histone deacetylation, heterochromatin formation and, consequently, downregulation of the gene transcription31. Their small size and robustness make the SID domains popular in synthetic biology32,33. We used GAL4(148)–Q-PAS1–VP16 as a transcription activator and BphP1–SID as an epigenetic modifier (Supplementary Fig. 11a). As was shown (Fig. 2c), GAL4(148)–Q-PAS1–VP16 efficiently activated gene transcription. However, under NIR illumination, BphP1–SID was recruited to the reporter gene, resulting in a 5.5-fold transcription inhibition (Supplementary Fig. 11b). To confirm that the transcriptional inhibition was due to histone deacetylation, we used an HDAC2 inhibitor, trichostatin A. In this case, we did not observe any difference between the light and dark samples (Supplementary Fig. 12). The BphP1–mCherry construct lacking the SID domain did not show any inhibition, which additionally proved the involvement of histone deacetylase (Supplementary Fig. 13). The inhibition level achieved by the SID recruitment was comparable to that previously reported34.
Transcription activation via recruitment to nucleus
Since performance of an optogenetic pair can depend on the fusion partners, we next tested the robustness of the BphP1–Q-PAS1 system by swapping Q-PAS1 with BphP1 in a transcription activation application. For this, we fused NLS–Q-PAS1 with a GAL4 transcription factor and BphP1 with a VP16 transactivator. We also designed a single BphP1–Q-PAS1-encoding plasmid using a self-cleaving T2A peptide that is ten-fold smaller than the internal ribosome entry site (IRES) sequence of 585 bp used in the TetR-based constructs (Supplementary Figs. 6,7). Together with the small size of Q-PAS1 and deletion of the mCherry tag from BphP1, these improvements resulted in the coding sequence of 3.7 kbp for the final enhanced BphP1–Q-PAS1 transcription activation construct, which is 2.5 kbp smaller than the original BphP1–PpsR2 system (Fig. 4a).
Figure 4. Light-controlled activation of reporter expression.
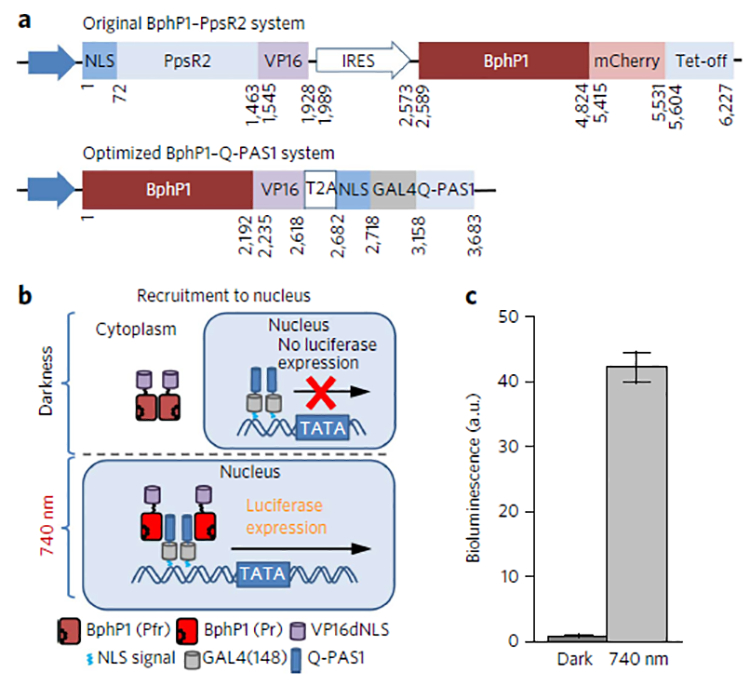
| (a) Constructs encoding the original BphP1–PpsR2 and the enhanced 1.7-fold smaller BphP1–Q-PAS1 transcription activation systems. IRES, ribosome entry site. (b) Schematic representation of light-controlled transcription activation of luciferase reporter via recruitment to nucleus of BphP1–VP16 transactivator. (c) Luciferase reporter expression in HeLa cells cotransfected with an optimized BphP1–Q-PAS1 system and a pFR–Luc reporter according to b. The samples were incubated either in darknessor under 740 nm pulsed light (30 s on and 180 s off) of 0.2 mW cm−2 started 6 h after the transfection. Analysis was performed 48 h after the transfection. Data were normalized to the bioluminescence signal of the cells kept in darkness. Error bars represent s.e.m.; n = 3 experiments.
For transcription activation, we exploited a NIR-light-induced recruitment of BphP1–VP16 fusion to the nucleus-localized NLS–GAL4(148)–Q-PAS1 construct (Fig. 4b). We tested the enhanced BphP1–Q-PAS1 system using a bioluminescence assay. Expression of the Fluc reporter in HeLa cells was 42-fold higher under NIR illumination than in darkness (Fig. 4c), showing the independence of the BphP1–Q-PAS1 system from the fusion partners.
Multiplexing with blue-light optogenetic tools
We next compared the BphP1−Q-PAS1 optogenetic pair with a LightON transcription regulation system activatable by blue light3 and analyzed their spectral crosstalk. The LightON system is based on the light-inducible homodimerization of the Vivid-type LOV domain. In HeLa cells LightON activation with 460 nm light resulted in a 39-fold increase of the Fluc signal compared to the dark-treated cells, whereas under 740 nm illumination, the contrast between the dark and light states’ signal was only 2.9 (Fig. 5a). In cells transfected with the BphP1–Q-PAS1 system, we detected a 42-fold activation under 740 nm illumination and a 2.5-fold activation under blue light as compared to darkness. We concluded that because of the very low cross-activation of both systems with 460 nm and 740 nm light we could combine LOV-based optogenetic elements with Q-PAS1 in a single system.
Figure 5. Spectral multiplexing of the BphP1–Q-PAS1 system with blue-light-activatable tools.
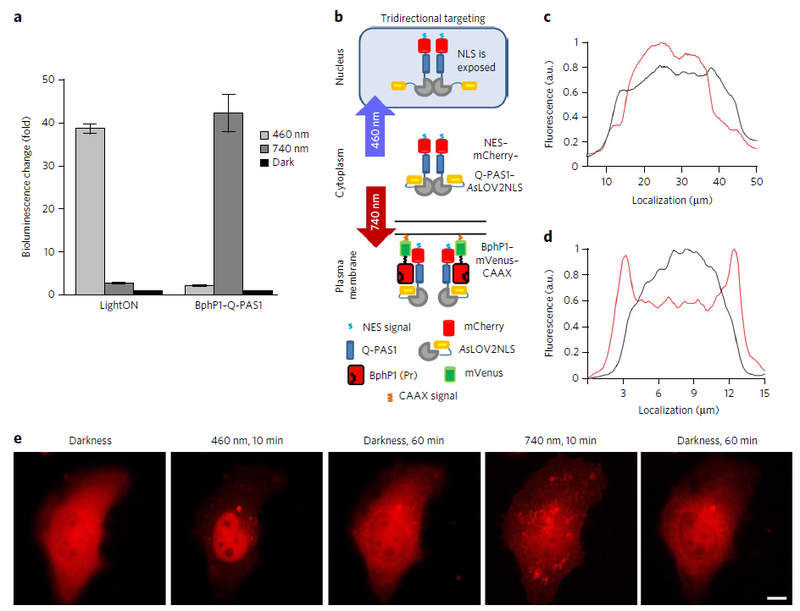
| (a) Light-to-dark ratio of luciferase reporter bioluminescence detected in HeLa cells cotransfected with the LightON and BphP1–Q-PAS1 transcription activation systems, both GAL4–UAS based. Samples were illuminated either by continuous 460 nm light of 1 mW cm−2 or by 740 nm pulsing light of 1 mW cm−2. In the latter case, 30 s light pulses were alternated with 180 s of darkness. Illumination started 6 h after the transfection and continued for 48 h. Data were normalized to the signal of dark sample. Error bars represent s.e.m.; n = 3 experiments. (b) Schematic representation of light-controllable tridirectional subcellular targeting using the merged BphP1–Q-PAS1 and AsLOV2cNLS systems, resulting in the NES–mCherry–Q-PAS1–AsLOV2cNLS construct, termed iRIS. (c) Intensity profile of mCherry fluorescence of iRIS of the cell before (black line) and after (red line) 10 min of 460 nm illumination. (d) Intensity profile of mCherry fluorescence of iRIS of the cell before (black line) and after (red line) 10 min of 740 nm illumination. (e) Sequential targeting of iRIS from a cytoplasm to the nucleus and to the plasma membrane in a single cell after the indicated illumination (10 min of 460 nm of 1 mW cm−2 or 740 nm of 1 mW cm−2 light) and dark relaxation periods. Scale bar, 10 μm.
For this application, we used an AsLOV2cNLS system6, which is based on structural changes in a single LOV2 domain. In LOV2, illumination with blue light leads to unwinding of the caged Jα-helix and exposure of the fused C-terminal peptide epitope, thus making it available for PPI. We combined Q-PAS1 with AsLOV2cNLS to enable its tridirectional relocalization between the cytoplasm, nucleus and plasma membrane (Fig. 5b). A nuclear exclusion signal (NES) and mCherry were added to the N terminus. The resulting NES–mCherry–Q-PAS1–AsLOV2cNLS construct, called iRIS (near-infrared-blue-light-inducible shuttle), was cloned with the BphP1–mVenus–CAAX fusion in a bicistronic vector via IRES2.
In darkness, HeLa cells transfected with the bicistronic plasmid showed even distribution of iRIS through cytoplasm. Blue light uncaged the NLS of the AsLOV2cNLS part, resulting in translocation of iRIS to the nucleus (Fig. 5c; Supplementary Fig. 14a). NIR light targeted iRIS to the plasma membrane, decorating the cell surface and filopodia (Fig. 5d; Supplementary Fig. 14b).
Finally, we performed tridirectional subcellular targeting in a single cell. The same cells were sequentially illuminated with blue light, kept in darkness, illuminated with NIR light and again kept in darkness. iRIS showed the respective sequential changes in the cell localization pattern, from cytoplasmic to nuclear and plasma membrane (Fig. 5e; Supplementary Video 1). Intensity profile measured using an epifluorescence microscope indicated 30–40% changes in its cytoplasmic fluorescence (Supplementary Fig. 15). In darkness, iRIS dissociated from the membrane-tethered BphP1–mVenus–CAAX and returned to the cytoplasm (Supplementary Fig. 16). The observed half-time of the dissociation was ~4.4 min in darkness, which shortened to ~2.3 min upon 620 nm illumination (Supplementary Fig. 17). These data demonstrated the effective multiplexing of the blue and NIR optogenetic tools with minimal spectral crosstalk as well as the possibility of combining the blue and NIR-light-sensing elements into a single dual-color optogenetic system.
DISCUSSION
Starting from the full-length PpsR2 protein of 50 kDa, we designed the single-domain Q-PAS1 fragment of 17 kDa, which efficiently interacted with BphP1 under NIR light. The Q-PAS1 protein was three-fold smaller and lacked the undesired oligomerization behavior inherent to the PpsR2 protein family23. This was achieved by deleting of the N-PAS and PAS2 domains, which were both reported to be responsible for tetramer and octamer formation. The latter makes Q-PAS1 additionally advantageous over PpsR2 in optogenetic applications where an oligomeric state of the targeted protein is important.
Moreover, the Q-PAS1 protein has the N-terminal α-helical Q-linker, which is a convenient structural element for attaching various functional fusions. Our analysis of the PpsR2 deletion mutants revealed the Q-linker as the essential element for efficient PPI of PpsR2 with BphP1. Importantly, this shows the functional conformity of R. sphaeroides AppA–PpsR24 and R. palustris BphP1–PpsR2 signaling schemes. The Q-linker and PAS1 domain likely form a homodimerization interface critical for PpsR2 functioning. We hypothesized that the same interface is also involved in PPIs with the HOS domain of BphP1 in the NIR-light-converted Pr state, resulting in the BphP1–Q-PAS1 heterodimerization (Supplementary Fig. 3).
Merging of the BphP1–Q-PAS1 pair with the common GAL4–UAS gene expression system resulted in several NIR optogenetic tools compatible with a variety of existing cellular and animal models that utilize the GAL4–UAS elements to control transcription events3,35–38. Moreover, in contrast to the reported TetR–tetO-based BphP1–PpsR2 gene transcription activation system, the BphP1–QPAS1-GAL4 systems we designed allowed both gene transcription activation and gene transcription inhibition (which is summarized in Supplementary Table 2). By tuning the junction between α-helices of the Q-linker and the GAL4 DNA binding domain, we engineered a chimeric transcription factor that interacts efficiently with BphP1 and enables a 40-fold dark-to-light contrast of gene transcription regulation (Fig. 3d). In this engineering approach, we successfully reconstructed a fully functional so-called signal helix, which is able to transfer the signal (for example, structural rearrangement due to photon absorption or PPI) from a sensing part of the protein or PPI complex to the effector part23,39. These data also demonstrated the possibility to use Q-PAS1 as a transferable dimerizing element controlled by NIR-light-sensing BphP1. The large size, multidomain structure and oligomerization of the PpsR family of proteins23 make the use of full-length PpsR2 (Supplementary Fig. 18) suboptimal in similar applications.
Moreover, the three-fold smaller Q-PAS1 and several sequence optimizations made it possible to design a fairly small vector carrying the fully functional BphP1–Q-PAS1 transcriptional control system of just 3.7 kbp (Fig. 4a). Being 1.7-fold smaller in size than the reported BphP1–PpsR2 transcription activation construct20, the BphP1–Q-PAS1 system can be used with gene delivery methods, which are sensitive to an insert size.
Nonopsin photoreceptors laid a basis for the development of versatile optogenetic tools for protein subcellular targeting. However, currently all these tools are only bidirectional, either recruiting the protein of interest from the cytoplasm to a specific compartment4–7,10,15 or translocating it between two different compartments40 (one in darkness and another in lit conditions). This limitation results from the monochromatic nature of the available protein-targeting tools as they sense light in the same spectral region. By integrating the blue-light-sensitive AsLOV2 and NIR-light-sensitive BphP1–Q-PAS1 elements into a single iRIS tool, we have achieved, for the first time, the tridirectional dual-light-controllable protein translocation between the cytoplasm, nucleus and plasma membrane (Fig. 5b–e, Supplementary Figs. 14–15). Furthermore, we observed the negligible light cross-activation between the LOV-based LightON and BphP1–Q-PAS1 transcription activation systems (Fig. 5a), confirming the spectral compatibility of the LOV- and BphP1-based systems.
Among future applications, the iRIS tool could be used for more stringent regulation of protein localization. For example, a transcription factor would be caged at the plasma membrane using iRIS. This caging could act synergistically with NES to additionally reduce the background activity of the transcription factors in the nucleus. In other applications, where a signaling protein activity depends on its localization, the nuclear targeting by iRIS could decrease its concentration at the plasma membrane. Furthermore, it is well known that some proteins are active in two distinct compartments41. The functionality of iRIS would be used to light-control activity of these proteins too. For this, various targeting signals could be used instead of the -CAAX sequence, present in the current iRIS variant, to localize a protein of interest to mitochondria, endoplasmic reticulum, peroxisome or allow its secretion.
In conclusion, the natural conservancy of the helix–PAS fold provides a wide field of possible future Q-PAS1 applications. Its structure allows the design of signal helices, transferring structural changes from various sensing domains to output modules of BphP1-controlled optogenetic systems. The spectral compatibility of the NIR chromophore of BphP1 with blue-light-sensing domains and the small size of Q-PAS1 make them a pair of choice for multiplexing with other optogenetic tools and for engineering of multicomponent optogenetic assemblies. These multifunctional optogenetic systems will enable an advanced control of cell metabolism and gene regulation in synthetic biology and biomedical research42,43.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
ONLINE METHODS
Design of bacterial and mammalian plasmids.
The PpsR2 and BphP1 genes were kindly provided by M. Papiz and E. Giraud, respectively. The pG5–EGFP and pFR–Luc reporter plasmids were from Systasy and Agilent Technologies, respectively. The GAL4 and VP16 sequences were amplified from a pGV-ER plasmid (Systasy). The pKA-100, pAL149-RpBphP1 (encoding BphP1), pKA-138.2 (encoding PpsR2–mRuby2), pKA-207 (bicistronic vector encoding BphP1–TetR and NLS–PpsR2–VP16) and pTRE-Tight-SEAP (expressing secreted embryonic alkaline phosphatase) plasmids were constructed earlier20. Truncated forms of a PpsR2 protein (Q-linker, PAS1, Q-PAS1, PpsR2dHTH–PAS2, PpsR2dHTH–PAS1–2 and PpsR2dHTH) were obtained using site- directed mutagenesis by inverse PCR of the PpsR2 gene. A GAL4(148)–Q-PAS1–VP16 fusion was constructed by cloning of a Q-PAS1 fragment between a GAL4(148) and a VP16 transactivation domain.
The GAL4(68)–Q-PAS1–VP16, 1–5 raw, and −1 and +1 fine mutants were generated from the GAL4(148)–Q-PAS1–VP16 construct by inverse PCR. The N-terminal part of Q-PAS1 in Figure 3b is shown as a helix for simplification; no structural data on this region is available. The C-terminal part of Q-PAS1 is a homology-based model built using MODELLER44 with the 4HH2 structure as a template. Models were visualized using Chimera 1.10.2 software45. The GAL4(148)–Q-PAS1–VP16 construct was used as a template to design a GAL4(148)–Q-PAS1–VP16dNLS fusion using a QuikChange kit (Stratagene).
To express BphP1 and truncated forms of PpsR2 in bacteria, a pBAD/His-B (Life Technologies/Invitrogen) and a pET22b (Novagen) were used. In the pET22b vector, an N-terminal pelB signal was replaced with a Strep-tag-II. A BphP1–mCherry–SID fusion was obtained by insertion of a SID fragment (amplified from Addgene #43882) into the pAL149–BphP1 plasmid. The pQP–-NLS–GAL4(148)–Q-PAS1 plasmid was obtained from the pQP–GAL4(148) construct by deletion of VP16 and insertion of NLS. A bicistronic vector pQP–T2A was obtained by insertion of BphP–mCherry–VP16dNLS upstream of the coding sequence in the construct pQP–NLS–GAL4(148)–Q-PAS1 via T2A peptide. After mCherry tag deletion the coding sequence was transferred to a backbone vector pSUB-CMV (kindly provided by the AAV Gene Transfer and Cell Therapy Core Facility). AsLOV2 sequence was amplified from the plasmid pKM546 (kindly provided by W. Weber and modified using mutagenesis, resulting in AsLOV2cNLS construct. The pQP-iRIS vector was obtained by insertion of AsLOV2cNLS instead of VP16 and NES–mCherry instead of GAL4(148) in the pQP–GAL4(148) plasmid. BphP1–mVenus–CAAX was inserted in MCS in the plasmid pIRES2–EGFP (Clontech) upstream of IRES2 and EGFP was swapped with NES–mCherry–Q-PAS1–AsLOV2cNLS. The plasmids designed in this study are summarized in Supplementary Table 3.
Protein expression and purification.
N-terminally 6× His-tagged BphP1 was expressed in E.coli LMG194 (Life Technologies/Invitrogen) together with hemeoxygenase from pWA23h plasmid for biliverdin IXα (BV) synthesis46. The bacterial cells were cultivated in RM medium with ampicillin, kanamycin and 0.02% rhamnose for 6–8 h followed by induction of the protein expression by 0.002% arabinose addition. The purification was performed using a Ni-NTA agarose (Qiagen). PpsR2 mutants fused with mRuby2 bearing a Streptag II at the N terminus and 6× His at the C terminus were expressed in E.coli BL21(DE3) (Agilent Technologies) grown in LB medium supplemented with ampicillin for 6 h, with 250 μM of IPTG induction. The proteins were purified sequentially with a Ni-NTA agarose (Macherey–Nagel) and Strep-Tactin sepharose (IBA Lifesciences).
In vitro pull-down assay.
For the in vitro pull-down assay, 1 μg of BphP1 protein and 2 μM of Strep-tag II fused bait proteins were incubated in 1 ml of PBS with 50 μl of Strep-Tactin slurry for various periods of time at room temperature. Pulled-down proteins were extensively washed with PBS, and the samples were resolved in SDS–PAGE gel. SDS–PAGE gel was prepared using 3% stacking gels and 12% resolving gel. For a zinc-induced fluorescence assay, 1 mM of ZnCl2 was added to the running buffer before electrophoresis. After electrophoresis, the gels with fluorescent BphP1-containing bands were photographed using a Benchtop-3 UV Transilluminator (Fisher Scientific), following by staining with Coomassie blue. BphP1 interactions with the PpsR2 mutants containing Q-linker were examined via pull-down assay for 2 h either in darkness or under NIR illumination with 740/25 nm custom-assembled LED array (LED Engin). All data were normalized to the intensity of the ‘Q-PAS1 darkness’ band.
Chemical crosslinking.
Chemical crosslinking was carried out in 20 μl reaction mix containing carbodiimide (EDC) and N-hydroxysuccinimide (NHS). For this purpose, 10 μg of BphP1 and Q-PAS1 incubated in 50 mM MES–NaOH, 1mM EDC, 5 mM NHS pH 6.0 for 30 min at room temperature. As a control, the same amounts of proteins were mixed in MES buffer without crosslinking reagents. The reaction was quenched by addition of 1× sample buffer, and the samples were analyzed at SDS–PAGE gel using the zinc-induced fluorescence assay and Coomassie blue staining.
Mammalian cell culture.
HeLa cells were purchased from the ATCC (CCL-2) and were not additionally authenticated or tested for mycoplasma contamination. Cells were cultured in DMEM medium with 10% FBS and penicillin- streptomycin mixture (all from Gibco) at 37 °C. For experiments, cells were plated on 6-well plates (Greiner Bio-One). Transient transfections were performed using an Effectene transfection reagent (Qiagen). The culture medium was changed 6 h after the transfection with the new one containing 25 μM of BV.
For light-induced relocalization of GAL4(148)–Q-PAS1–VP16 to plasma membrane, HeLa cells were cotransfected with plasmids encoding GAL4(148)–Q-PAS1–VP16 (or GAL4(148)–Q-PAS1–VP16dNLS), BphP1–mCherry–CAAX, and pG5–EGFP (or pFR–Luc) in a 1:1:3 ratio. The flow cytometry and bioluminescence analyses were performed 24 h after the cotransfection. To perform dimer disruption experiments, cells were cotransfected with plasmids expressing GAL4(68)–Q-PAS1-VP16 (or 1–5 raw, +1 and −1 fine tuning mutants), NLS–BphP1–mCherry and pG5–EGFP/pFR–Luc in a 1:1:3 ratio. Flow cytometry analysis, bioluminescence measurements and cell imaging were performed 48 h after the cotransfection. For light-controlled transcription activation, cells were cotransfected with the constructs pQP–T2A and pFR–Luc in a 3:1 ratio and analyzed 48 h after transfection. For comparison with a LightON system, cells were cotransfected with pQP–NLS–GAL4(148)-Q-PAS1, pBphP1–mCherry–VP16dNLS, and pFR–Luc plasmids in a 3:3:1 ratio and analyzed 48 h later.
Cell light activation and imaging.
For relocalization assay, dimer disruption, control of epigenetic state and transcription activation experiments, HeLa cells were illuminated with 740/25 nm of 0.2 mW cm−2 using the alternating cycles of light and darkness (30 s light and 180 s darkness) in CO2 incubator at 37 °C. The culture medium was changed 6 h after the transfection with the new one containing 25 μM of BV.
Epifluorescence microscopy was performed using an Olympus IX83 equipped with a 200 W metal halide arc lamp (Lumen 220PRO, Prior) and an opiMOS sCMOS camera (QImaging). Unless otherwise indicated, cells were imaged using either a 40×, 0.95 NA air or a 60×, 1.35 NA oil objective lens (UPlanSApo, Olympus). During imaging HeLa cells were incubated in a cell imaging medium (Life Technologies-Invitrogen) and kept at 37 °C. The data were analyzed using a SlideBook v. 6.0.8, and an ImageJ v. 1.50b software.
In spectral multiplexing, blue and NIR illumination was performed using the 460/20 nm and 740/25 nm custom-assembled LED arrays (LED Engin), respectively. During time-lapse imaging, blue-light-activatable proteins were irradiated with 10 min of 460 nm light of 1 mW cm−2. NIR light-sensing proteins were activated using 10 min of 740 nm irradiation of 1 mW cm−2. Focusing of the microscope was performed in a mCherry channel to prevent unspecific activation of blue and NIR light-sensing components. For dark relaxation experiments, cells were kept in darkness for 1 h.
Flow cytometry analysis.
Flow cytometry analysis of a light-induced EGFP expression was performed using Accuri C6 Flow Cytometer (BD Biosciences) equipped with a 488 nm laser and a 533/30 nm emission filter and with a 488 nm laser and a 610/20 nm emission filter. To calculate an efficiency of the light-induced EGFP expression in relocalization and light-induced activation experiments, a mean fluorescent intensity of EGFP positive cells was multiplied by a number of the positive cells and divided to a number of all events in live cells gate.
In dimer disruption experiments, efficiency of a light-induced EGFP expression was calculated as a ratio of two values, the first being a product of the mean EGFP fluorescence intensity of the EGFP positive cells and their number, and the second being a product of the mean mCherry fluorescence intensity of the mCherry positive cells and their number. Gates for the counting of EGFP positive cells were set using non-transfected cells as a negative control. Minimally, 5 × 104 cells were analyzed in each sample. The data were analyzed using a CFlow v. 1.0.264.15 software.
Firefly luciferase (Fluc) assay.
To measure Fluc activity, transfected HeLa cells grown in 24-well plates (Greiner Bio-One) and lysed 24 h (for relocalization and control of epigenetic state) or 48 h (for dimer disruption and light-induced activation) after the transfection. Cells were washed with PBS, and then 100 μl of lysis buffer (20 mM Tris–HCl pH 8.0, 10% glycerol, 0.1% β-mercaptoethanol, 0.1% Triton X-100, 1 mM PMSF) was added to each well of 24-well plate and incubated on ice for 30 min. 10 μl of cell lysate was mixed with 20 μl of Firefly Luc Assay reagent (NanoLight Technology) in wells of a 96-well half-area white plate (Costar). Bioluminescence signal of sample was then measured using a Victor X3 multilabel plate reader (PerkinElmer). Treatment with trichostatin A was performed as described47.
Transcription activation by the LightON system in HeLa cells was performed as described in the original paper3. In brief, HeLa cells were cotransfected with pGAVPO and pFR-Luc plasmids in a 1:1 ratio. Transfection proceeded for 6 h in darkness. After changing the culture medium, the cells were continuously illuminated with a 460/20 nm LED array of 1 mW cm−2, or remained in darkness for 48 h. Lysed cell samples were analyzed for luciferase activity as above.
Secreted embryonic alkaline phosphatase assay.
SEAP assay was performed using a Great EscApe fluorescent SEAP Assay kit (Clontech). Aliquots of cell culture media (25 μl) from wells of a 24-well plate were collected at each time point and stored at −20 °C until measurement. For kinetics studies, a culture medium was changed with a fresh medium at each time point. A fluorescence intensity of the SEAP reaction product was measured in white nonbinding clear bottom 96-well plates (Greiner Bio-One) using the Victor X3 multilabel plate reader. In all experiments, the investigators were not blinded.
Supplementary Material
Acknowledgments
We thank M. Papiz (Liverpool University, UK), E. Giraud (Institute for Research and Development, France), W. Weber (University of Freiburg, Germany) and F. Zhang (Broad Institute of MIT and Harvard, USA) for plasmids, and A. Leopold (University of Helsinki), A. Kaberniuk and A. Shemetov (both from Albert Einstein College of Medicine) for useful suggestions. We thank Biomedicum Imaging Unit, AAV Gene Transfer and Cell Therapy and Biomedicum Flow Cytometry core facilities staff (University of Helsinki) for the technical assistance. This work was supported by grants GM105997, GM108579 and NS099573 from the US National Institutes of Health, ERC-2013-ADG-340233 from the EU FP7 program, and 263371 and 266992 from the Academy of Finland.
Footnotes
Data availability. The authors declare that the main data supporting the findings of this study are available within the article and its Supplementary Information. Extra data are available from the corresponding author upon reasonable request.
Competing financial interests The authors declare no competing financial interests.
Additional information
Any supplementary information, chemical compound information and source data are available in the online version of the paper. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html. Correspondence and requests for materials should be addressed to V.V.V.
References
- 1.Deisseroth K Optogenetics. Nat. Methods 8, 26–29(2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shcherbakova DM, Shemetov AA, Kaberniuk AA & Verkhusha VV Natural photoreceptors as a source of fluorescent proteins, biosensors, and optogenetic tools. Annu. Rev. Biochem 84, 519–550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Chen X & Yang Y Spatiotemporal control of gene expression by a light-switchable transgene system. Nat. Methods 9, 266–269(2012). [DOI] [PubMed] [Google Scholar]
- 4.Strickland D et al. TULIPs: tunable, light-controlled interacting protein tags for cell biology. Nat. Methods 9, 379–384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niopek D et al. Engineering light-inducible nuclear localization signals for precise spatiotemporal control of protein dynamics in living cells. Nat. Commun 5, 4404(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yumerefendi H et al. Control of protein activity and cell fate specification via light-mediated nuclear translocation. PLoS One 10, e0128443(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niopek D, Wehler P, Roensch J, Eils R & Di Ventura B Optogenetic control of nuclear protein export. Nat. Commun 7, 10624(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kennedy MJ et al. Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods 7, 973–975 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taslimi A et al. An optimized optogenetic clustering tool for probing protein interaction and function. Nat. Commun 5, 4925(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H et al. LOVTRAP: an optogenetic system for photoinduced protein dissociation. Nat. Methods 13, 755–758 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guntas G et al. Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc. Natl. Acad. Sci. USA 112, 112–117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taslimi A et al. Optimized second-generation CRY2-CIB dimerizers and photoactivatable Cre recombinase. Nat. Chem. Biol 12, 425–430 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhoo SH, Davis SJ, Walker J, Karniol B & Vierstra RD Bacteriophytochromes are photochromic histidine kinases using a biliverdin chromophore. Nature 414, 776–779 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Toettcher JE, Weiner OD & Lim WA Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module. Cell 155, 1422–1434 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buckley CE et al. Reversible optogenetic control of subcellular protein localization in a live vertebrate embryo. Dev. Cell 36, 117–126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ulijasz AT & Vierstra RD Phytochrome structure and photochemistry: recent advances toward a complete molecular picture. Curr. Opin. Plant Biol 14, 498–506 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Piatkevich KD, Subach FV & Verkhusha VV Engineering of bacterial phytochromes for near-infrared imaging, sensing, and light-control in mammals. Chem. Soc. Rev 42, 3441–3452 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shcherbakova DM, Baloban M & Verkhusha VV Near-infrared fluorescent proteins engineered from bacterial phytochromes. Curr. Opin. Chem. Biol 27, 52–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shcherbakova DM & Verkhusha VV Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat. Methods 10, 751–754 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaberniuk AA, Shemetov AA & Verkhusha VV A bacterial phytochrome-based optogenetic system controllable with near-infrared light. Nat. Methods 13, 591–597 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bellini D & Papiz MZ Structure of a bacteriophytochrome and light-stimulated protomer swapping with a gene repressor. Structure 20, 1436–1446 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Braatsch S, Johnson JA, Noll K & Beatty JT The O2-responsive repressor PpsR2 but not PpsR1 transduces a light signal sensed by the BphP1 phytochrome in Rhodopseudomonas palustris CGA009. FEMS Microbiol. Lett 272, 60–64 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Heintz U, Meinhart A & Winkler A Multi-PAS domain-mediated protein oligomerization of PpsR from Rhodobacter sphaeroides. Acta Crystallogr. D Biol. Crystallogr 70, 863–876 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winkler A et al. A ternary AppA-PpsR-DNA complex mediates light regulation of photosynthesis-related gene expression. Nat. Struct. Mol. Biol 20, 859–867 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mallo M Controlled gene activation and inactivation in the mouse. Front. Biosci 11, 313–327 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Bacchus W, Aubel D & Fussenegger M Biomedically relevant circuit-design strategies in mammalian synthetic biology. Mol. Syst. Biol 9, 691(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whiteside ST & Goodbourn S Signal transduction and nuclear targeting: regulation of transcription factor activity by subcellular localisation. J. Cell Sci 104, 949–955 (1993). [DOI] [PubMed] [Google Scholar]
- 28.Hong M et al. Structural basis for dimerization in DNA recognition by Gal4. Structure 16, 1019–1026 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delorenzi M & Speed T An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 18, 617–625 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Gautier R, Douguet D, Antonny B & Drin G HELIQUEST: a web server to screen sequences with specific alpha-helical properties. Bioinformatics 24, 2101–2102 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Laherty CD et al. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 89, 349–356 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Xie M, Haellman V & Fussenegger M Synthetic biology-application-oriented cell engineering. Curr. Opin. Biotechnol 40, 139–148 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Blancafort P, Segal DJ & Barbas CF III. Designing transcription factor architectures for drug discovery. Mol. Pharmacol 66, 1361–1371 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Ayer DE, Laherty CD, Lawrence QA, Armstrong AP & Eisenman RN Mad proteins contain a dominant transcription repression domain. Mol. Cell. Biol 16, 5772–5781 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burcin MM, Schiedner G, Kochanek S, Tsai SY & O’Malley BW Adenovirus-mediated regulable target gene expression in vivo. Proc. Natl. Acad. Sci. USA 96, 355–360 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ornitz DM, Moreadith RW & Leder P Binary system for regulating transgene expression in mice: targeting int-2 gene expression with yeast GAL4/UAS control elements. Proc. Natl. Acad. Sci. USA 88, 698–702 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong Z, Peng J & Guo S Stable gene silencing in zebrafish with spatiotemporally targetable RNA interference. Genetics 193, 1065–1071 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewandoski M Conditional control of gene expression in the mouse. Nat. Rev. Genet 2, 743–755 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Anantharaman V, Balaji S & Aravind L The signaling helix: a common functional theme in diverse signaling proteins. Biol. Direct 1, 25(2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yumerefendi H et al. Light-induced nuclear export reveals rapid dynamics of epigenetic modifications. Nat. Chem. Biol 12, 399–401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arnoys EJ & Wang JL Dual localization: proteins in extracellular and intracellular compartments. Acta Histochem 109, 89–110 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Ausländer S & Fussenegger M Engineering gene circuits for mammalian cell-based applications. Cold Spring Harb. Perspect. Biol 8, a023895(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Müller K et al. Multi-chromatic control of mammalian gene expression and signaling. Nucleic Acids Res 41, e124(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Webb B & Sali A Protein structure modeling with MODELLER. Methods Mol. Biol 1137, 1–15 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Piatkevich KD, Subach FV & Verkhusha VV Far-red light photoactivatable near-infrared fluorescent proteins engineered from a bacterial phytochrome. Nat. Commun 4, 2153(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alland L et al. Identification of mammalian Sds3 as an integral component of the Sin3/histone deacetylase corepressor complex. Mol. Cell. Biol 22, 2743–2750 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.