

Abstract
Objective
Gastric adenocarcinoma (GAC) is the third most common cause of cancer mortality worldwide. Accurate and affordable non-invasive detection methods have potential value for screening and surveillance. Herein, we identify novel methylated DNA markers (MDMs) for GAC, validate their discrimination for GAC in tissues from geographically separate cohorts, explore marker acquisition through the oncogenic cascade, and describe distributions of candidate MDMs in plasma from GAC cases and normal controls.
Design
Following discovery by unbiased whole methylome sequencing, candidate MDMs were validated by blinded methylation-specific PCR in archival case-control tissues from U.S. and South Korean patients. Top MDMs were then assayed by an analytically sensitive method (quantitative real-time allele-specific target and signal amplification) in a blinded pilot study on archival plasma from GAC cases and normal controls.
Results
Whole methylome discovery yielded novel and highly discriminant candidate MDMs. In tissue, a panel of candidate MDMs detected GAC in 92–100% of U.S. and S. Korean cohorts at 100% specificity. Levels of most MDMs increased progressively from normal mucosa through metaplasia, adenoma, and GAC with variation in points of greatest marker acquisition. In plasma, a 3 marker panel (ELMO1, ZNF569, C13orf18) detected 86% (95% CI 71–95%) of GACs at 95% specificity.
Conclusions
Novel MDMs appear to accurately discriminate GAC from normal controls in both tissue and plasma. The point of aberrant methylation during oncogenesis varies by MDM, which may have relevance to marker selection in clinical applications. Further exploration of these MDMs for GAC screening and surveillance is warranted.
Keywords: Stomach cancer, biomarkers, screening, carcinogenesis, DNA methylation
INTRODUCTION
While its incidence varies widely by country, gastric adenocarcinoma (GAC) represents the third most common cause of cancer-related death globally [1]. Currently, population-based screening is performed only in high-prevalence regions. Gastroscopy has become the test-of-choice given its sensitivity for early stage disease [2–4]. However, its limitations include patient non-compliance, invasiveness, and uncertain cost-effectiveness,[5] which pose challenges in population-based screening. An accurate, noninvasive detection method for GAC screening as a complement to endoscopy has potential to improve patient participation and overall effectiveness.
There may also be an important role for an accurate noninvasive detection tool in the postoperative surveillance of GAC. Current National Comprehensive Cancer Network guidelines recommend performing interval history and physical examinations; laboratory, imaging, and endoscopic evaluations are recommended only if clinically indicated.[6] To date, there are no compelling data that show a survival advantage with such reactive surveillance strategies[7] Furthermore, unlike surveillance algorithms with other types of cancer, routine tumor-specific blood tests are lacking with GAC.
Cancer-specific methylated DNA markers represent a rational class of candidate markers for GAC detection. Aberrant gene methylation contributes importantly to tumor development through silencing of tumor suppressor genes or stimulation of oncogene expression[8],[9]. Based on observations with GAC and other cancer types[10,11], aberrant methylation may occur early in oncogenesis with selected methylated DNA markers (MDMs). MDMs have been described which appear to discriminate GAC from normal tissue[9] or to predict GAC progression[12] or GAC risk.[13],[14] However, such studies have typically evaluated MDMs historically associated with GAC rather than selecting novel MDMs from a comprehensive discovery process[9].
Given its wide global variation in incidence, it is unclear if the molecular biology of GAC is similar across geographic regions. In South Korea, for example, GAC is the most common malignancy in men and fourth most common in women[15] with incidence rates more than six times higher than those in the U.S.[16] In the U.S, GAC incidence is much higher in immigrants from South Korea, Japan, and other high prevalence countries than in the general population [17]. We are unaware of prior studies that have evaluated the performance of GAC-specific MDMs across ethnically or geographically dissimilar populations.
In the evolution to GAC, acquisition of genetic and epigenetic abnormalities occurs during the phenotypic transformation from normal mucosa, to metaplasia, to adenoma/dysplasia, and finally to adenocarcinoma[18]. An understanding of when, during carcinogenesis, individual MDMs are acquired is relevant to the rational selection of marker panels tailored to specific early detection applications.
Testing of molecular markers in blood has not yet emerged as a viable or proven approach to GAC screening or surveillance. Early studies suggest that plasma assay of microRNA[19] and circulating cell-free mutant DNA[20] may have value in GAC detection. Few data are available on the use of MDMs for such[21]. Our group has recently identified discriminant MDMs for liver[22] and lung[23] cancer via rigorous whole methylome discovery efforts, and preliminary data suggest that top candidate markers applied to plasma are capable of highly accurate detection. It is unclear if such a regimented approach would yield similar results for GAC detection.
In this investigation, we sought to identify candidate markers for the accurate detection of GAC. Our specific aims were to 1) identify novel MDMs by whole-methylome sequencing that discriminate GAC from normal stomach at the tissue level, 2) validate the performance of selected candidate MDMs in tissues from geographically separate cohorts, 3) assess patterns of marker acquisition across the oncogenic cascade, and 4) apply top MDM candidates to archival plasma samples from GAC and healthy controls to explore the feasibility of this noninvasive detection approach.
MATERIALS AND METHODS
Study Design
This investigation had multiple sequential components (Figure 1). It began with a discovery step based on unbiased whole methylome sequencing using reduced representation bisulfite sequencing (RRBS). Regions demonstrating significant differential methylation were identified and technically validated as candidate MDMs by quantitative methylation-specific PCR (qMSP). Top candidate MDMs were chosen and subsequently applied to independent gastric tissues obtained from both U.S. and South Korean patients. Finally, we conducted a pilot investigation on the feasibility of a further refined set of MDMs in a case-control plasma pilot using quantitative allele-specific real-time target and signal amplification (QuARTS), a highly sensitive and specific analytical platform. All assays for tissue validation and plasma were performed in blinded fashion.
Figure 1. Sequential Study Design.
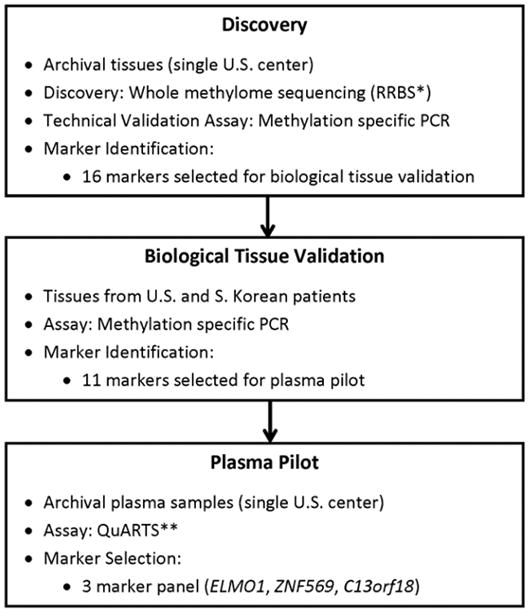
Abbreviations: RRBS* (reduced representation bisulfite sequencing), QuARTS** (quantitative allele-specific real-time target and signal amplification).
This retrospective study was conducted in accordance with the International Ethical Guidelines for Biomedical Research Involving Human Subjects. The protocol was approved by Institutional Review Boards at Mayo Clinic and Seoul National University College of Medicine (IRB No. 1409-101-610). Informed written consent had been obtained from patients to allow archiving of their biospecimens for future studies; these archives provided samples for the present study.
Biospecimen Sources
All GACs studied were classified according to AJCC criteria[24].
Discovery and Technical Validation
Frozen archival samples were obtained from histologically confirmed GAC tissues and control tissues (normal gastric mucosa from patients without GAC and normal colon mucosa taken at time of screening colonoscopy from patients without colorectal neoplasia). We also included white blood cell (buffy coat) DNA from patients without a history of cancer at any site. It is critical to eliminate methylated sequences in tumor DNA that overlap with those in buffy coat, as white blood cells are the major source of normal cell free DNA (cfDNA) in circulation. So, by design, our goal was to identify candidate tumor markers that avoid such potential confounding by white blood cells and, thus, have a better chance to achieve high specificity with plasma testing. This rationale and approach have been used by other groups seeking to find tumor specific markers for cancer detection who have used buffy coat as a control source at the discovery level[25]. Eliminating overlap with white blood cells also reduces the likelihood of gastric inflammation causing artefactual results. GAC, colonic mucosa, and buffy coat samples were obtained from the Biospecimens Archive Linking Investigators and Clinicians to GIH Cell Signaling Research Core at Mayo Clinic. Stomach tissue controls were derived from cancer free de-identified patient biopsies. An independent pathologist reviewed all tissues to confirm diagnosis and to mark slides for subsequent macro-dissection.
Biological Tissue Validation in U.S. and South Korea Samples
Case and control samples were obtained from independent gastric tissue sets from the U.S. and South Korea as a check on over-fitting from the discovery set and to evaluate the potential for geographic differences in MDM profiles with GAC.
For the U.S. cohort, archival paraffin-imbedded tissues from Mayo Clinic were used. Cases included samples from patients with pathologically-confirmed GAC and normal gastric mucosa from matched controls without GAC. Additionally, samples of histologically confirmed gastric metaplasia and gastric adenomas were studied. Dates of acquisition for archival U.S. samples ranged from January 1994–December 2013.
For the S. Korean cohort, frozen case tissues were obtained from patients with pathologically-confirmed GAC immediately following gastrectomy. Corresponding control specimens were obtained from the same surgical specimen after identification of tumor-free resection margin by intraoperative frozen-section. An effort was made to sample uninvolved control tissue of grossly normal-appearing gastric mucosa located as far as possible from tumor margins. Dates of acquisition for S. Korean archival tissues ranged from March 1999–December 2011.
For both cohorts, case tissues were sampled prior to neoadjuvant therapy. Patients with a history of gastrointestinal cancer, inflammatory bowel disease, or heritable cancer syndromes were excluded. As with Discovery, independent pathologists for both U.S and S. Korea cohorts reviewed tissues to confirm diagnosis and to mark slides to guide subsequent macro-dissection.
Plasma Pilot Study
Samples used in the plasma pilot comprised archival frozen plasma collected at Mayo Clinic from patients different than those involved in the tissue studies. Plasma samples from GAC cases without prior chemo or radiation therapy were provided by the Biospecimens Archive Linking Investigators and Clinicians to GIH Cell Signalling Research Clinical Core; samples from healthy controls without history of cancer at any site were enrolled from a separate registry. For each specimen, a total plasma volume of 2 mL was used for DNA extraction and bisulfite conversion. The clinical diagnosis and recorded tumor characteristics were based on the clinical records and pathology reports.
Assay Techniques
Discovery and Technical Validation
DNA was extracted from macro-dissected frozen tissues using the QIAamp DNA Mini kit (Qiagen, Valencia CA). RRBS libraries were prepared as previously described [26]. Sequencing was performed on the Illumina HiSeq 2000 by the Mayo Clinic Medical Genome Facility. SAAP-RRBS (streamlined analysis and annotation pipeline for reduced representation bisulfite sequencing) was used for sequence read assessment and clean-up, reference genome alignment, methylation status extraction, and CpG reporting and annotation[27]. Filtering criteria for marker selection are delineated in Statistical Methods below.
Verification of marker performance on an independent platform was assessed using qMSP on aliquots of the identical samples used for sequencing. Primers specific for post-bisulfite methylated sequences were designed and synthesized (IDT, Coralville, IA). Prior to use, qMSP assays were quality tested on bisulfite converted and unconverted methylation (+/−) controls to insure specific amplification. Optimal annealing temperatures for each assay were determined empirically. Assay standards were dilutions of a bisulfite converted fully methylated genomic DNA control. Patient DNA was bisulfite-converted using the Zymo EZ-96 DNA Methylation kit (Zymo Research, Irvine, CA) and amplified using the Roche 480 LightCycler (Mannheim Germany). Results were expressed as fractional methylation against a β-actin reference and analyzed by logistic regression.
Biological Tissue Validation in U.S. and South Korea Samples
Paraffin-embedded tissues were extracted using the QiaAmp FFPE Tissue Mini kit (Qiagen, Valencia CA). Pre-extracted DNA from South Korean samples was sent for analysis. MDMs meeting selection criteria after discovery (see Statistical Methods below) were assessed by targeted qMSP on the U.S. and S. Korean cohorts.
Plasma Pilot Study
Circulating free DNA (cfDNA) was purified from 2mL of plasma using an in-house method. Briefly, plasma samples were mixed with proteinase K and chaotropic solution which allowed the denaturation and subsequent binding of DNA to silica-coated magnetic particles. After washing, DNA was eluted and then bisulfite converted using an automated Hamilton Microlab STARlet system as described previously[28].
A multiplex PCR reaction was performed on bisulfite converted DNA. Candidate MDMs were assayed by the Quantitative Allele-Specific Real-time Target and Signal amplification (QuARTS®) method, as described[29]. QuARTS incorporates two enzymes (Flap endonuclease-1 and Taq polymerase) and requires perfect base pairing in the probe and primer regions to minimizing false signals due to un-methylated or partially methylated genes (Supplemental Figure 1). Because of the high analytical sensitivity and specificity of this platform (10 methylated fragments in a background of 1x105 unmethylated fragments), we felt the QuARTs platform was well-suited for assaying plasma samples where the majority of cfDNA is non-tumor derived[30]. QuARTS primers and probes were designed manually for each MDM, and assay performance was verified on positive and negative methylation controls. All amplifications were carried out on the 480 LightCycler (Roche).
Statistical Methods
Discovery and Technical Validation
Candidate MDMs were identified among differentially methylated regions (DMRs) according to following criteria: [case/control] methylation fold change (FC) >20, [case – control] absolute methylation difference (AMD) >0.10, area under the receiver-operator curve (AUC) >0.80, p-value <0.01, and control sample methylation <1.0%. These criteria identified hypermethylated candidates only. Statistical significance of methylated regions between GAC and controls was determined by fitting a logistic regression model to the methylation percentage per region. To account for varying read depths across individual subjects, an over-dispersed logistic regression model was used where the dispersion parameter was estimated by the Pearson Chi-square statistic of the residuals from the fitted model. Regions were ranked according to significance level. A second level of criteria was then applied to narrow the candidates further. DMR length had to be at least 50 bp and include a minimum of 5 CpGs and a maximum density of 25 CpGs/100bp. In addition, every unique CpG within a DMR had to demonstrate coordinated hypermethylation. These critera are essential for the construction of robust and functional amplification-based assays used in downstream validation and pilot studies. For the technical validation phase, qMSP results were normalized to input DNA using a CpG independent marker (β-actin) and analyzed by logistic regression. MDMs were ranked by AUC, FC, and AMD and compared to discovery metrics. We elected to carry forward MDM candidates that ranked highest based on discrimination metrics in both discovery and technical validation.
Biological Tissue Validation in U.S. and South Korean Samples
Continuous variables are summarized as medians with 25th (Q1) and 75th (Q2) percentiles whereas categorical variables are summarized as percentages of group total. The predictive accuracy of individual MDMs was estimated as the area under the ROC curve (AUC) with corresponding 95% confidence intervals. Association of MDM levels with patient characteristics was performed using Spearman’s correlation (ordinal characteristics) or the Wilcoxon Rank Sums test (categorical characteristics). GAC sensitivities for each MDM at selected specificities were calculated on U.S. and S. Korean tissues using within-group marker level cutoffs. The panel of MDMs was considered positive for GAC if the selected specificity level (e.g. 90%) of the most sensitive individual MDM was exceeded or if the level for any other MDM exceeded their corresponding 100% specificity cutoff. This simple approach ensured an overall panel specificity not higher than that for the most sensitive individual marker.
Plasma Pilot Study
Sensitivity of candidate markers was estimated with corresponding 95% confidence intervals at pre-determined specificity cutoffs of 95, and 100%. For each individual marker, the predictive accuracy was estimated as area under the ROC curve (AUC). To determine the optimal model for GAC discrimination using marker panels, regression partition trees (rPart) were used to identify the best predictive combinations of MDM levels[31]. The association of MDM levels with stage was estimated using Spearman’s correlation coefficient.
Power Assessment
For discovery, the minimum group sample size of 14 was determined to be sufficient to detect a fold-change of 3 or higher between GAC cases and controls (gastric, buffy coat, and normal colon) in the mean % methylation with 80% power and an overall false discovery rate of 5%. For this calculation, the percentage of truly differential CpG regions was varied from 5 to 10% as well as the variance inflation factor of the logistic model (1 to 3).
For the biological tissue validation and plasma pilot phases, the minimum group size of 35 was determined to be sufficient to detect an AUC of 0.85 or higher with 80% power and a one-sided significance level of 5% relative to a null hypothesis AUC of 0.70.
RESULTS
Discovery & Technical Validation in Tissue
In discovery, case tissues comprised 14 GACs (Table 1) and 42 control tissues (normal stomach mucosa from 6 patients, normal colon mucosa from 18 patients, and normal circulating white blood cells (buffy coat) from 16 healthy patients). Median age was 65 years (range 45–86) for GAC cases, 64 (51–80) for normal colon controls, and 54 (48–65) for normal buffy coats; women accounted for 57%, 61%, and 50% of samples, respectively. As normal stomach samples were derived from de-identified patient samples, age and sex data were not available.
Table 1.
Patient and tumor characteristics of gastric adenocarcinoma (GAC) in Discovery/Technical Tissue, Biological Tissue Validation and Plasma Pilot.
| Discovery/Technical Validation | Biological Tissue Validation | |||||||
|---|---|---|---|---|---|---|---|---|
| Cases | Buffy Coat | Normal Colona | U.S. | S. Koreab | Cases | Controls | ||
| (N=14) | (N=16) | (N=19) | Cases (N=35) | Normal Controls (N=35) | Cases (N=50) | (N=36) | (N=38) | |
| Patient Characteristics | ||||||||
| Sex of patient | ||||||||
| Male | 6 (43%) | 8 (50%) | 10 (53%) | 24 (69%) | 22 (63%) | 33 (66%) | 22 (61%) | 22 (58%) |
| Female | 8 (57%) | 8 (50%) | 9 (47%) | 11 (31%) | 13 (37%) | 17 (34%) | 14 (39%) | 16 (42%) |
| Age at Diagnosis (years) | ||||||||
| Median | 65 | 54 | 64 | 78 | 66 | 62 | 70 | 69 |
| Range | (45–86) | (48–65) | (51–80) | (35–88) | (44–88) | (38–86) | (23–88) | (42–86) |
| Tumor Characteristics | ||||||||
| Tumor Site | -- | -- | -- | -- | ||||
| GE junction | 0 (0%) | 0 (0%) | 2 (4%) | 4 (11%) | ||||
| Fundus/Cardia/Body | 8 (57%) | 18 (51%) | 14 (28%) | 24 (67%) | ||||
| Antrum/Pylorus | 5 (36%) | 16 (46%) | 31 (62%) | 5 (14%) | ||||
| Diffuse | 1 (7%) | 1 (3%) | 3 (6%) | 3 (8%) | ||||
| TNM Stagec | -- | -- | -- | -- | ||||
| 1 | 1 (7%) | 12 (34%) | 6 (12%) | 8 (22%) | ||||
| 2 | 6 (43%) | 14 (40%) | 8 (16%) | 13 (36%) | ||||
| 3 | 6 (43%) | 5 (14%) | 22 (44%) | 4 (11%) | ||||
| 4 | 1 (7%) | 4 (11%) | 14 (28%) | 11 (31%) | ||||
| Tumor Sized | -- | -- | ||||||
| ≤4 | 5 (36%) | 16 (46%) | 5 (10%) | 11 (31%) | ||||
| 4.1–7.9 | 5 (36%) | 14 (40%) | 29 (58%) | 12 (33%) | ||||
| 8–11.9 | 1 (7%) | 3 (9%) | 10 (20%) | 4 (11%) | ||||
| ≥12 cm | 3 (21%) | 2 (6%) | 6 (12%) | 9 (25%) | ||||
| Histologic Typee | -- | -- | -- | -- | ||||
| Intestinal | 9 (64%) | 25 (71%) | 31 (62%) | 17 (47%) | ||||
| Diffuse | 5 (36%) | 8 (23%) | 15 (30%) | 19 (53%) | ||||
| Mixed | 0 (0%) | 2 (6%) | 3 (6%) | - | ||||
| Undefined | 0 (0%) | 0 (0%) | 1 (2%) | - | ||||
Normal colon tissues were used in the Disovery/Technical Validation set to rule out sequences present in colon, as this would interfere with potential future stool application. Normal stomach tissues were tested, but were de-identified so that age/sex data could not be summarized.
Control samples were derived from endoscopic biopsy of histologically normal gastric mucosa synchronous to gastric adenocarcinoma.
TNM Stage (AJCC 7th edition)’,
Largest diameter,
Based on Lauren Classification.
From >3 million CpGs, >5 thousand differentially methylated regions between GAC cases and controls were identified by RRBS. Applying the stringent filters described in Statistical Methods, 22 regions of differential DNA methylation with highest discrimination were selected as candidate MDMs (Supplemental Table 1). Following subsequent technical validation by qMSP on all specimens from discovery, 16 MDMs held as candidates (Table 2). In comparisons between GAC and normal gastric tissue, candidate MDMs individually yielded AUCs ranging from 0.82–0.99 and fold changes from 3.7 to 883; comparisons of GAC against normal buffy coats showed AUCs of 0.89 to 0.99 and fold changes of 5.9 to greater than 30,000. ZNF569 and c13orf18 had lower FC numbers in technical validation compared to discovery, but were included because they excelled in other categories. For example, ZNF569 had a AUC of 0.99 when compared to WBC samples, a trait which would fit well in a liquid biopsy setting. Methylation of c13orf18 has been shown in preliminary data to be upper GI cancer specific and could aid in localizing a tumor in a screening setting[32] ().
Table 2.
Discrimination for gastric adenocarcinoma (GAC) of the top 16 methylated DNA marker candidates selected following discovery and technical validation.
| GAC vs Normal Mucosa | GAC vs Buffy Coat | |||||
|---|---|---|---|---|---|---|
| AUC | Fold-Change | p-value | AUC | Fold-Change | p-value | |
| RHGEF4 | 0.95 | 153 | < 0.0001 | 0.94 | 80 | < 0.0001 |
| ELMO1 | 0.95 | 208 | < 0.0001 | 0.99 | 3340 | < 0.0001 |
| ABCB1 | 0.97 | 48 | < 0.0001 | 0.89 | 14.2 | < 0.0001 |
| BMP3 | 0.92 | 120 | < 0.0001 | 0.86 | 5.92 | < 0.0001 |
| SP9 | 0.91 | 131 | < 0.0001 | 0.94 | 21381 | < 0.0001 |
| CLEC11A | 0.95 | 223 | < 0.0001 | 0.94 | 83.4 | < 0.0001 |
| EMX1 | 0.95 | 883 | < 0.0001 | 0.94 | 13621 | < 0.0001 |
| ST8SIA1 | 0.93 | 67.9 | < 0.0001 | 0.94 | 185 | < 0.0001 |
| CD1D | 0.91 | 237 | < 0.0001 | 0.87 | 135 | < 0.0001 |
| SFMBT2 | 0.99 | 711 | < 0.0001 | 0.98 | 1128 | < 0.0001 |
| CYP26C1 | 0.94 | 597 | < 0.0001 | 0.94 | 11243 | < 0.0001 |
| PKIA | 0.94 | 183 | < 0.0001 | 0.91 | 40.0 | < 0.0001 |
| NDRG4 | 0.88 | 288 | < 0.0001 | 0.89 | >30000 | < 0.0001 |
| ZNF569 | 0.88 | 3.71 | 0.0095 | 0.99 | 148 | < 0.0001 |
| PPP2R5C | 0.89 | 147 | < 0.0001 | 0.97 | 648 | < 0.0001 |
| C13ORF18 | 0.82 | 7.45 | < 0.0001 | 0.89 | 41.4 | < 0.0001 |
To lend confidence to our choices, we explored the functionality and pathway associations of the genes annotated to the DMR sequences. Most mapped to defined CpG islands in regulatory and non-coding regions; 25% were known transcription factors and another 25% operate in signaling pathways. Two genes (BMP3 and CLEC11A) function as growth factors. More than half of the markers have reported cancer associations (Supplemental Table 2).
Biological Tissue Validation in U.S. and South Korean Cohorts
Patient and Lesion Characteristics
In the U.S. cohort, cases included samples from 35 patients with GAC (Table 1). Controls included 35 separate sex-matched patients without GAC and with histologically-normal gastric epithelia. Additionally, 11 samples of gastric metaplasia and 20 gastric adenomas were studied.
For the S. Korean cohort, case tissues were obtained from 50 patients with GAC (Table 1). Of the synchronous gastric tissue samples available, 23 were interpreted as histologically normal mucosa and 15 as metaplasia. Clinical and tumor characteristics for U.S. and S. Korean patients studied are summarized (Table 1). Except for older median age of GAC cases in the U.S. cohort, characteristics are generally similar between cohorts.
In the U.S. cohort, 22 (63%), 4 (36%), and 9 (45%) of normal control tissues, metaplasia samples, and adenomas were from men, respectively; median ages for these sample types were 66 (53–99), 68 (38–78), and 70 years (25–90), respectively. Among adenomas, histologic features of high-grade dysplasia were present in 8 (40%) samples. In the S. Korean cohort, 17 (74%) of the 23 synchronous histologically-normal gastric mucosa and 10 (67%) of the 15 synchronous samples with metaplasia were from men, respectively; median ages were 64 (42–81) and 64 (38–86), respectively.
Assessment of Marker Discrimination in Separate Geographic Cohorts
GAC detection was high overall, and the discrimination with many of the MDMs selected from the discovery set was corroborated in these independent validation sets. For the top 10 MDMs, AUCs in the U.S. cohort ranged from 0.95–0.98 and in the S. Korean cohort from 0.66–0.96 (Table 3). At 90% and 100% specificities, the panel respectively detected 100% (95% CI 90–100%) and 100% (90–100%) of GACs in the U.S. cohort and 94% (83–99%) and 92% (81–98%) in the S. Korean cohort.,. Sensitivities of individual MDMs at a specificity of 90% are also shown in Table 3.
Table 3. Biological validation in tissues from U.S. and S. Korea patient cohorts.
Metrics are shown for discrimination of gastric adenocarcinoma from histologically normal gastric mucosa by the top 16 methylated DNA markers, including areas under the receiver operator curves (AUCs) and sensitivites at 90% specificity cuttoffs. Specificity cutoffs were based on marker levels in histologically normal mucosa from healthy controls in U.S. cohort and from biopsies synchronous with gastric cancer in S. Korea cohort.
| U.S. Cohort | South Korea Cohort | |||
|---|---|---|---|---|
|
| ||||
| AUC (95% CI) | Sensitivity, % (95% CI) | AUC (95% CI) | Sensitivity, % (95% CI) | |
|
| ||||
| ARGEF4 | 0.97 (0.93–1) | 94 (81–99) | 0.82 (0.72–0.92) | 72 (58–84) |
|
| ||||
| ELMO1 | 0.97 (0.93–1) | 94 (81–99) | 0.92 (0.86–0.99) | 88 (76–95) |
|
| ||||
| ABCB1 | 0.98 (0.96–1) | 91 (77–98) | 0.83 (0.74–0.92) | 78 (64–88) |
|
| ||||
| BMP3 | 0.94 (0.88–1) | 89 (73–97) | 0.84 (0.75–0.94) | 80 (66–90) |
|
| ||||
| SP9 | 0.95 (0.91–1) | 91 (77–98) | 0.66 (0.53–0.79) | 58 (43–72) |
|
| ||||
| CLEC11A | 0.95 (0.92–1) | 94 (81–99) | 0.78 (0.66–0.89) | 68 (53–80) |
|
| ||||
| EMX1 | 0.96 (0.91–1) | 91 (77–98) | 0.77 (0.65–0.89) | 76 (62–87) |
|
| ||||
| ST8SIA1 | 0.95 (0.89–1) | 91 (77–98) | 0.85 (0.76–0.93) | 72 (58–84) |
|
| ||||
| CD1D | 0.94 (0.88–0.99) | 89 (73–97) | 0.91 (0.84–0.98) | 86 (73–94) |
|
| ||||
| SFMBT2 | 0.93 (0.87–0.99) | 86 (70–95) | 0.96 (0.92–1) | 90 (78–97) |
|
| ||||
| CYP26C1 | 0.95 (0.89–1) | 91 (77–98) | 0.84 (0.74–0.93) | 74 (60–85) |
|
| ||||
| PKIA | 0.94 (0.89–1) | 89 (73–97) | 0.89 (0.82–0.97) | 80 (66–90) |
|
| ||||
| NDRG4 | 0.88 (0.81–0.96) | 77 (60–90) | 0.84 (0.77–0.92) | 74 (60–85) |
|
| ||||
| ZNF569 | 0.94 (0.88–1) | 91 (77–98) | 0.81 (0.70–0.91) | 70 (55–82) |
|
| ||||
| PPP2R5C | 0.90 (0.83–0.97) | 80 (63–92) | 0.68 (0.56–0.80) | 62 (47–75) |
|
| ||||
| C13ORF18 | 0.83 (0.72–0.93) | 74 (57–88) | 0.85 (0.78–0.92) | 72 (58–84) |
| Marker Panel | 0.99 (0.96–1) | 100 (90–100) | 0.95 (0.89–1) | 94 (83–99) |
While several markers exhibited similarly high discrimination for GAC between cohorts, others appeared to be more discriminant in the U.S cohort (Table 3). Compared to markers like ELMO1 that were similarly discriminant between cohorts (Figure 2A), MDMs with greater apparent discrimination in the U.S. cohort (e.g. ARGHEF4) had relatively higher background levels in S. Korean control samples (Figure 2B), perhaps reflecting molecular field effects in gastric mucosae synchronous with GAC. In contrast to differences observed in control tissues, levels of these MDMs in GAC tissues were generally similar between demographic cohorts. The distributions of most other MDMs showed clear separation between cases and control tissues from both cohorts (Figure 2C).
Figure 2. Distributions of selected methylated DNA markers in gastric adenocarcinoma (GAC) and gastric control tissues from U.S. and S. Korean cohorts.



(A) Two markers showing similarly high discrimination for GAC in geographic cohorts. Control tissues comprised histologically normal biopsies from healthy patients (Healthy) in the U.S. cohort and from mucosa synchronous (Sync) to GAC in the S. Korean cohort. (B) Two markers with relatively high control levels in the S. Korean samples. (C) Distributions of remaining six top markers. (Marker levels in each cohort were β-actin standardized and scaled according to their respective standard deviation. This allows data plots on the same vertical scale across cohorts and preserves the relative differences between cases and controls.)
For ELMO1, one of the overall top-performing markers in each cohort, performance at the tissue level was unaffected by patient age or sex or by tumor characteristics including stage, size, histology, and site. A minority of the other marker candidates was variably affected by patient and tumor covariates to a minimal degree (see Supplemental Table 3).
Marker Acquisition across the Oncogenic Cascade
For all MDM candidates, a significant trend in assayed copy number was observed along the progression from normal mucosa to GAC (p-value range <0.001–0.01), although inflection points of greatest proportional increase in marker acquisition varied by MDM (Figure 3). Most MDMs, including ELMO1, demonstrated the largest proportional increase at the transition from normal mucosa to metaplasia. For other markers, including BMP3, the most significant proportional increase was observed at the transition from metaplasia to adenoma. SFMBT2 was the only marker with the most significant change occurring between adenoma and cancer.
Figure 3. Tissue levels for selected methylated DNA markers across the oncogenic progression from normal mucosa to GAC.
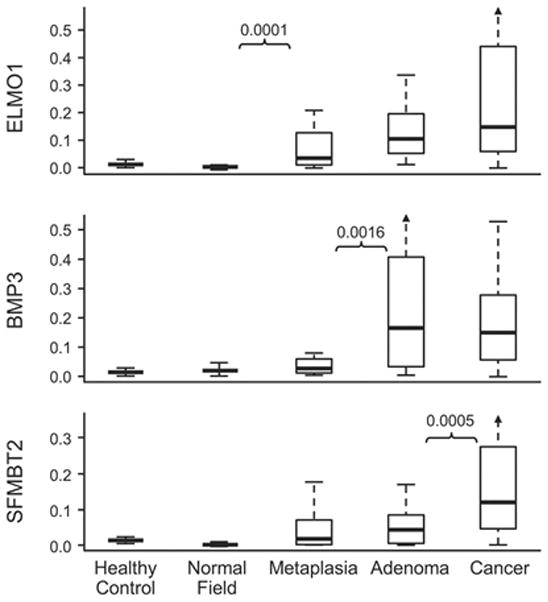
P-values are shown to indicate the step of greatest proportional increase for each marker. Marker levels are normalized to β-actin.
Plasma Pilot Study
Patient and Lesion Characteristics
Samples studied included archival plasmas from 74 patients (36 cases with pathologically-confirmed intact GAC and 38 controls who were age and sex-balanced healthy volunteers (Table 1). GACs reflected a range of stages, histologic types, and tumor locations. There were no significant differences in the age or sex distributions between cases and controls.
Marker Performance
We selected 12 MDMs (11 discriminate markers, 1 control marker) for plasma testing from the original 16 tissue MDMs (BMP3, NDRG4, EMX1, ABCB1, and SP9 were excluded). Receiver operating characteristic curves for top-performing individual MDMs and a 3-marker MDM panel are shown (Figure 4A). ELMO1 was the most discriminant with an AUC of 0.94 (95% CI 0.89–0.99). As with other MDMs, ELMO1 levels increased progressively from stages 1 to 4 with very low levels in controls (Figure 4B).
Figure 4. Detection of gastric adenocarcinoma (GAC) by assay of novel methylated DNA marker candidates in plasma.
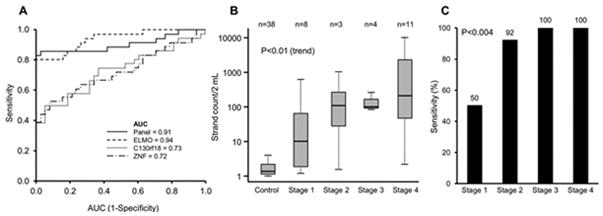
(A) Area under the reciever operating characteristic curves (AUCs) for three selected markers alone and in a combined panel. (B) Quantiation of methylated ELMO1 levels across GAC tumor stages. (C) Marker panel sensitivites at 100% specificity across tumor stages.
At 95% and 100% specificities, a 3-marker panel (ELMO1, ZNF569, and C13orf18) respectively detected 86% (95% CI 71–95%) and 83% (67–94%) of GAC cases GAC detection rates were influenced by tumor stage (Figure 4C). At 100% specificity, sensitivities at AJCC stages 1, 2, 3, and 4 were 50%, 92%, 100%, and 100%, respectively. Detection accuracy by the panel was not significantly affected by patient sex or age or by tumor size, site, or histology. At 95% specificity, the MDM panel detected 82% (14/17) of intestinal-type and 89% (17/19) of diffuse-type GAC (p=0.29).
DISCUSSION
An accurate blood test for GAC detection has potential value in screening or surveillance. The purpose of this study was to identify accurate candidate MDMs for GAC detection. We took a robust approach to find novel MDMs with high discrimination for GAC using whole methylome sequencing, show in validation studies on independent tissues that top MDMs are sensitive and specific for GAC across geographically diverse patient cohorts, observe that MDM levels are commonly elevated in GAC precursor lesions, and demonstrate proof-of-concept that high GAC detection rates can be achieved by a panel of MDMs assayed from plasma.
Applying stringent filtering criteria to the extensive data set created from the unbiased whole-methylome discovery, we identified novel MDMs that individually exhibited high discrimination for GAC relative to normal gastric mucosa or normal buffy coat (AUCs approaching 1.0 with desirably high tumor-to-background fold changes). These multiple filtering criteria coupled with marker validation through primer design and testing in biologically independent samples mitigate the risk of data over-fitting. Most of the top MDM candidates have not to our knowledge been reported with GAC (Supplemental Table 2). As aberrant hypermethlyation typically occurs at the promoter region and may influence gene function, it is of interest to note that four of the top MDM sequences were found on genes associated with transcriptional regulation, three with signal transduction, and two with cell growth. A few of the MDMs we identified with GAC (e.g. PPP255C, CYP26C1, and SFMBT2) have been reported with malignancies at other organ sites including lung[33] and colon[34]. Further studies are needed to evaluate the organ site specificity of MDM candidates.
To validate the accuracy of candidate MDMs, specific assays were designed to measure marker levels in gastric tissues from independent and geographically distinct patient cohorts in the U.S. and South Korea. Some (e.g. ELMO1 and SFMBT2) showed comparably high AUCs above 0.9, while others at first pass appeared to be more discriminant for GAC in U.S. than in S. Korean patients. However, while levels of candidate MDMs were similarly elevated in GAC tissues from both geographic cohorts, some marker levels were relatively much higher in control gastric mucosa obtained from S. Korean patients who harbored GAC than in control tissue obtained from U.S. patients with grossly normal stomachs. Our observation is consistent with recent findings by others showing elevated levels of some MDMs in gastric mucosa of those at high risk for GAC with highest levels seen in mucosa synchronous with GAC[35]. This phenomenon, also referred to as “field cancerization”, is well-recognized and involves aberrant methylation in normal-appearing mucosa adjacent to cancer[36]. These relatively higher levels of some MDMs in gastric mucosa synchronous to GAC may explain, in part, their lower AUCs seen in the S. Korean cohort. The study was powered to detect AUCs >0.85 within groups rather than to formally compare differences between groups, and it is also possible that some of the observed apparent differences were due to sample size.
Candidate MDMs were universally found in precursor lesions, and marker levels typically increased progressively along the oncogenic cascade from metaplasia through adenoma to GAC. Others have described MDMs in gastric metaplasia and adenomas[37]. We noted that the point along the oncogenic cascade of MDM acquisition or greatest proportional increase varied by individual marker. Similar findings with selected MDMs including accumulation of methylated ELMO1 during the histologic progression toward GAC have recently been reported by others[38]. Such variation in MDM acquisition across precursor lesions may be important in the selection of marker panels tailored to specific clinical applications, and may be especially germane to media containing exfoliated markers such as stool or gastric lavage.
In this initial study exploring the accuracy of novel MDMs for detection of GAC when assayed from plasma, a 3-marker panel achieved an overall sensitivity of 83% at a specificity cutoff of 100%. Sensitivities by the MDM panel were particularly high for the more advanced stages of GAC with 92% of stage 2 and 100% of both stage 3 and 4 lesions detected. The relatively lower sensitivity of 50% with stage 1 cancers is consistent with the generally less elevated MDM plasma levels that we observed in such cases. It remains to be determined if technical refinements can overcome this potential biological barrier and lead to improved detection of earliest stage GACs.
It is instructive to consider these early performance outcomes with plasma MDM testing against other blood tests for cancer screening or surveillance. Historically, some have advocated use of serum pepsinogen for GAC screening. Pepsinogen is a marker for gastric atrophy rather than GAC per se, and may identify those at increased risk[39]; pooled results across studies reveal sensitivities of 52–77% at specificities of 69–84%. Furthermore, given the biological nature of this marker, pepsinogen testing would have no role in a GAC surveillance application. The performance outcomes we observed on pilot plasma MDM testing would also, if corroborated in further studies, compare favorably to clinically available blood tests used to screen or surveil cancers in other organs, such as carcinoembryonic antigen[40] or methylated septin9[41] for colorectal cancer, alpha-fetoprotein (AFP) for hepatoma[42], or prostate specific antigen (PSA) for prostate cancer[43][44] detection. For example, reported sensitivity and specificity ranges at commonly used cutoffs are respectively 41–65% and 80–94% for detection of hepatoma with AFP[42] and 35–72% and 63–93% for detection of prostate cancer with PSA[43][44].
Several study limitations and interpretive precautions warrant mention. First, control tissues from normal stomachs were not evaluated in the S. Korean cohort and, as above, some of the elevated background MDM levels may reflect field cancerization in biopsies from normal-appearing mucosa synchronous to GAC rather than due to inherent differences in geographically separate populations. Second, a detailed history of tobacco exposure, Epstein-Barr virus status, or Helicobacter pylori status was not available on the majority of patients, so these covariates could not be evaluated with respect to MDM signatures. Epstein-Barr virus infection is an established risk factor for GAC[45] and has been associated with high rates of aberrant methylation on some genes[46]. Likewise, chronic H. pylori gastritis is a well-known risk factor for GAC[47] and may also be associated with aberrant methylation on some genes[14]. However, the filtering algorithm we followed to select candidate MDMs excluded markers present in normal buffy coat with the intent of eliminating those MDMs elevated in inflammatory cells. Furthermore, given their universally low background levels in control gastric mucosae from both geographic cohorts, some of the selected MDMs are unlikely affected by potential H. pylori gastritis. Third, while we describe several novel MDMs with comparably high discrimination for GAC in U.S. and S. Korean cohorts, findings cannot be extrapolated to populations in other regions without further study. Fourth, due to differences in the carcinogenesis pathways between intestinal and diffuse-type GAC[48], additional study is needed to determine how MDM detection of pre-malignant conditions varies between GAC subtypes. Fifth, levels of candidate MDMs were not assessed in metastatic lesions or in plasma from patients with distant recurrence, and both will need to be studied prior to a potential surveillance application. However, the observed sensitivity for stage IV disease was 100%, and the highest plasma concentrations of candidate MDMs were seen in the stage IV subset, which support pursuit of this application. And, finally, as the plasma study was performed in referred patients with primary GAC from a single institution, results will need to be corroborated in larger studies on screening, surveillance, or other intended-use populations.
Our study adds to the growing body of knowledge on molecular markers in GAC[48]. While standardized tumor-specific blood tests are available for screening or surveillance of other common malignancies, such tools are currently lacking for GAC. Based on the encouraging findings from this study, plasma assay of discriminant MDMs holds promise in filling this gap. Further studies using optimized marker panels and technical methods are indicated to extend and corroborate these early results.
Supplementary Material
Supplemental Table 1: Discrimination for gastric adenocarcinoma (GAC) by the top 22 methylated DNA marker candidates in Discovery (first three data columns) and Technical Validation (second three data columns)
Supplemental Table 2: Function of Genes Containing Differentially Methylated Regions Identified as Targets for Candidate Markers*
Supplemental Table 3: Covariate Effects on Tissue Levels of Methylated DNA Marker Candidates (Data shown represent p-values)
Supplemental Figure 1: Quantitative allele-specific real-time target and signal amplification (QuARTS) assay.
{kind=link}
Statement of Relevance.
Accurate noninvasive tools for detection of gastric adenocarcinoma (GAC) are lacking for both screening and surveillance. Based on findings from this study, plasma assay of novel methylated DNA markers (MDMs) holds promise in filling these clinical gaps. Most of the top MDM candidates identified have not to our knowledge been reported with GAC, but the biological functions of affected genes suggest potentially important roles in oncogenesis. Variation in MDM acquisition across precursor lesions may also be important in the selection of marker panels tailored to specific clinical uses. Our data on MDM discovery, tissue validation, and pilot plasma testing provide justification for larger clinical studies to further assess the value of MDM applications in screening and surveillance settings.
Acknowledgments
Authors thank Terri Johnson for her outstanding clerical assistance during study execution and with manuscript preparation and submission.
Funding and Grant Support: Tissue and blood specimens were provided by Clinical Core of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567). Exact Sciences advised on laboratory methodology and provided grant funding that covered most costs of logistics for sample procurement, sequencing, reagents for QuARTS assays, laboratory technician time, and statistician time but played no direct role in protocol development, study conduct, or data analysis. Funding was also provided by a generous grant from Eugene and Eva Lane.
Abbreviations
- GAC
Gastric adenocarcinoma
- MDM
Methylated DNA marker
- RRBS
Reduced representation bisulfite sequencing
- qMSP
Quantitative methylation-specific PCR
- QuARTS
Quantitative allele-specific real-time target and signal amplification
Footnotes
Conflicts of Interest: Mayo Clinic has licensed technology to Exact Sciences (Madison WI) related to this study. As co-inventors of such technology, Drs. Ahlquist & Kisiel, Ms. Yab, and Messrs. Taylor & Mahoney could share in potential royalties to Mayo Clinic in accordance with institutional policy. Dr. Ahlquist serves as scientific advisor to Exact Sciences, and Drs. Ahlquist & Kisiel are research collaborators with Exact Sciences. Drs. Lidgard & Allawi are employees at Exact Sciences. There are no conflicts of interest to declare for other authors.
References
- 1.Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19(8):1893–1907. doi: 10.1158/1055-9965.EPI-10-0437. [DOI] [PubMed] [Google Scholar]
- 2.Miyamoto A, Kuriyama S, Nishino Y, Tsubono Y, Nakaya N, Ohmori K, et al. Lower risk of death from gastric cancer among participants of gastric cancer screening in Japan: a population-based cohort study. Prev medicine. 2007;44(1):12–9. doi: 10.1016/j.ypmed.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 3.Choi KS, Jun JK, Suh M, Park B, Noh DK, Song SH, et al. Effect of endoscopy screening on stage at gastric cancer diagnosis: results of the National Cancer Screening Programme in Korea. Br J Cancer. 2015;112(3):608–12. doi: 10.1038/bjc.2014.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi KS, Kwak MS, Lee HY, Jun JK, Hahm MI, Park EC. Screening for gastric cancer in Korea: population-based preferences for endoscopy versus upper gastrointestinal series. Cancer Epidemiol Biomarkers Prev. 2009;18(5):1390–8. doi: 10.1158/1055-9965.EPI-08-0940. [DOI] [PubMed] [Google Scholar]
- 5.Dan YY, So JB, Yeoh KG. Endoscopic screening for gastric cancer. Clin Gastroenterol Hepatol. 2006;4(6):709–16. doi: 10.1016/j.cgh.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 6.National Comprehensive Cancer Network. [Accessed July 13, 2016];Gastric Cancer (Version 2.2016) https://www.nccn.org/professionals/physician_gls/pdf/gastric.pdf.
- 7.Bennett JJ, Gonen M, D’Angelica M, Jaques DP, Brennan MF, Coit DG. Is detection of asymptomatic recurrance after curative resection associated with improved survival in patients with gastric cancer? J Am Coll Surg. 2005;201(4):503–510. doi: 10.1016/j.jamcollsurg.2005.05.033. [DOI] [PubMed] [Google Scholar]
- 8.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3(4):253–66. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 9.Sapari NS, Loh M, Vaithilingam A, Soong R. Clinical potential of DNA methylation in gastric cancer: a meta-analysis. PloS One. 2012;7(4):e36275. doi: 10.1371/journal.pone.0036275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou XP, Zhang B, Zhang XQ, Chen M, Cao J, Liu WJ. Promoter hypermethylation of multiple genes in early gastric adenocarcinoma and precancerous lesions. Hum Pathol. 2009;40(11):1534–42. doi: 10.1016/j.humpath.2009.01.029. [DOI] [PubMed] [Google Scholar]
- 11.Okugawa Y, Grady WM, Goel A. Epigenetic Alterations in Colorectal Cancer: Emerging Biomarkers. Gastroenterology. 2015;149(5):1204–1225e12. doi: 10.1053/j.gastro.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nobili S, Bruno L, Landini I, Napoli C, Bechi P, Tonelli F, et al. Genomic and genetic alterations influence the progression of gastric cancer. World J Gastroenterol. 2011;17(3):290–9. doi: 10.3748/wjg.v17.i3.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin CM, Kim N, Park JH, Kang GH, Kim JS, Jung HC, et al. Prediction of the risk for gastric cancer using candidate methylation markers in the non-neoplastic gastric mucosae. J Pathol. 2012;226(4):654–65. doi: 10.1002/path.2990. [DOI] [PubMed] [Google Scholar]
- 14.Maekita T, Nakazawa K, Mihara M, Nakajima T, Yanaoka K, Iguchi M, et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res. 2006;12(3 Pt 1):989–95. doi: 10.1158/1078-0432.CCR-05-2096. [DOI] [PubMed] [Google Scholar]
- 15.Jung KW, Won YJ, Kong HJ, Oh CM, Cho H, Lee DH, et al. Cancer statistics in Korea: incidence, mortality, survival, and prevalence in 2012. Cancer Res Treat. 2015;47(2):127–41. doi: 10.4143/crt.2015.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karimi P, Islami F, Anandasabapathy S, Freedman ND, Kamangar F. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol Biomarkers Prev. 2014;23(5):700–13. doi: 10.1158/1055-9965.EPI-13-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim GH, Liang PS, Bang SJ, Hwang JH. Screening and surveillance for gastric cancer in the United States: Is it needed? Gastrointest Endosc. 2016;84(1):18–28. doi: 10.1016/j.gie.2016.02.028. [DOI] [PubMed] [Google Scholar]
- 18.Yuasa Y. Control of gut differentiation and intestinal-type gastric carcinogenesis. Nat Rev Cancer. 2003;3(8):592–600. doi: 10.1038/nrc1141. [DOI] [PubMed] [Google Scholar]
- 19.Konishi H, Ichikawa D, Komatsu S, Shiozaki A, Tsujiura M, Takeshita H, et al. Detection of gastric cancer-associated microRNAs on microRNA microarray comparing pre- and post-operative plasma. Br J Cancer. 2012;106(4):740–747. doi: 10.1038/bjc.2011.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsujiura M, Ichikawa D, Konishi H, Komatsu S, Shiozaki A, Otsuji E. Liquid biopsy of gastric cancer patients: Circulating tumor cells and cell-free nucleic acids. World J Gastroenterol. 2014;20(12):3265–3286. doi: 10.3748/wjg.v20.i12.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hibi K, Goto T, Shirahata A, Saito M, Kigawa G, Nemoto H, et al. Detection of TFPI2 methylation in the serum of gastric cancer patients. Anticancer Res. 2011;31(11):3835–3838. [PubMed] [Google Scholar]
- 22.Dukek BA, Kanipakam RV, Ghoz HM, Yab TC, Berger CK, Taylor WR, et al. DNA Methylation Markers for Detection of Hepatocellular Carcinoma: Discovery, Tissue Confirmation, and Exploratory Testing in Plasma. Hepatology. 2016;64(1):85A–86A. [Google Scholar]
- 23.Allawi HT, Giakoumopoulos M, Flietner E, Oliphant A, Volkmann C, Aizenstein B, et al. Abstract 712: Detection of lung cancer by assay of novel methylated DNA markers in plasma. Cancer Res. 2017;77(Supplement 13):712. [Google Scholar]
- 24.Edge S, Byrd DR, Compton CC, Fritz AG, Greene F, Trotti A, editors. AJCC Cancer Staging Manual. 7. Springer; New York: 2010. [Google Scholar]
- 25.Uehiro N, Sato F, Pu F, Tanaka S, Kawashima M, Kawaguchi K, Sugimoto M, Saji S, Toi M. Circulating cell-free DNA-based epigenetic assay can detect early breast cancer. Breast Cancer Res. 2016 Dec 19;18(1):129. doi: 10.1186/s13058-016-0788-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 2011;6(4):468–481. doi: 10.1038/nprot.2010.190. [DOI] [PubMed] [Google Scholar]
- 27.Sun Z, Baheti S, Middha S, Kanwar R, Zhang Y, Li K, et al. SAAP-RRBS: streamlined analysis and annotation pipeline for reduced representation bisulfite sequencing. Bioinformatics. 2012;28(16):2180–1. doi: 10.1093/bioinformatics/bts337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lidgard GP, Domanico MJ, Bruinsma JJ, Light J, Gagrat ZD, Oldham-Haltom RL, et al. Clinical performance of an automated stool DNA assay for detection of colorectal neoplasia. Clin Gastroenterol Hepatol. 2013;11(10):1313–1318. doi: 10.1016/j.cgh.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 29.Zou H, Allawi H, Cao X, Domanico M, Harrington J, Taylor WR, et al. Quantification of methylated markers with a multiplex methylation-specific technology. Clin Chem. 2012;58(2):375–83. doi: 10.1373/clinchem.2011.171264. [DOI] [PubMed] [Google Scholar]
- 30.Haber DA, Velculescu VE. Blood-based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014 Jun;4(6):650–61. doi: 10.1158/2159-8290.CD-13-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Recursive Partitioning and Regression Trees. Terry Therneau and Beth Atkinson and Brian Ripley. 2015 R package version 4.1–9. http://CRAN.R-project.org/package=rpart.
- 32.Kisiel JB, Yab TC, Ghoz HM, Foote PH, Devens ME, Mahoney DW, Smyrk TC, Boardman LA, Petersen GM, Buttar NS, Roberts LR, Lidgard GP, Ahlquist DA. Accurate site prediction of gastrointestinal cancer by novel methylated DNA markers: Discovery & Validation. AACR; Cancer Res; Proceedings of the American Association for Cancer Research; 2015 April 18–22; Philadelphia, PA. 2015. Abstract nr 4252. [Google Scholar]
- 33.Giakoumopoulos M, Sander T, Volkmann C, Oliphant A, Flietner E, Aizenstein B. A methylated DNA marker panel for the sensitive detection of lung cancer in tissue. J Clin Oncol. 2016;34(Suppl) abstract 8627. [Google Scholar]
- 34.Taylor WR, Kisiel JB, Yab TC, Mahoney DW, Smyrk TC, Boardman LA, et al. Discovery of Novel DNA Methylation Markers for the Detection of Colorectal Neoplasia: Selection by Methylome-Wide Analysis. Gastroenterology. 2014;146(5 Suppl):S-30. Abstract no. 109. [Google Scholar]
- 35.Yamashita S, Kishino T, Takahashi T, Shimazu T, Charvat H, Kakugawa Y, et al. Genetic and epigenetic alterations in normal tissues have differential impacts on cancer risk among tissues. Proc Natl Acad Sci USA. 2018 doi: 10.1073/pnas.1717340115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramachandran K, Singal R. DNA Methylation and Field Cancerization. Epigenomics. 2012;4(3):243–5. doi: 10.2217/epi.12.12. [DOI] [PubMed] [Google Scholar]
- 37.Kim TY, Lee HJ, Hwang KS, Lee M, Kim JW, Bang YJ, et al. Methylation of RUNX3 in various types of human cancers and premalignant stages of gastric carcinoma. Lab Invest. 2004;84:479–848. doi: 10.1038/labinvest.3700060. [DOI] [PubMed] [Google Scholar]
- 38.Pirini F, Noazin S, Jahuira-Arias MH, Rodriquez-Torres S, Friess L, Michailidi C, et al. Early detection of gastric cancer using global, genome-wide and IRF4, ELMO1, CLIP4 and MSC DNA methylation in endoscopic biopsies. Oncotarget. 2017 Mar 16; doi: 10.18632/oncotarget.16258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miki K. Gastric cancer screening using the serum pepsinogen test method. Gastric Cancer. 2006;9:245–253. doi: 10.1007/s10120-006-0397-0. [DOI] [PubMed] [Google Scholar]
- 40.Duffy MJ. Carcinoembryonic antigen as a maker for colorectal cancer: Is it clinically useful? Clin Chem. 2001;47(4):624–630. [PubMed] [Google Scholar]
- 41.Church TR, Wandell M, Lofton-Day C, Mongin SJ, Burger M, Payne SR, et al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut. 2014;63:317–325. doi: 10.1136/gutjnl-2012-304149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gupta S, Bent S, Kohlwes J. Test Characteristics of α-Fetoprotein for detecting hepatocellular carcinoma in patients with hepatitis C. Ann Intern Med. 2003;139:46–50. doi: 10.7326/0003-4819-139-1-200307010-00012. [DOI] [PubMed] [Google Scholar]
- 43.Adhyam M, Gupta AK. A review on the clinical utility of PSA in cancer prostate. Indian J Surg Oncol. 2012;3(2):120–129. doi: 10.1007/s13193-012-0142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoffman RM, Gilliland FD, Adams-Cameron M, Hunt WC, Key CR. Prostate-specific antigen testing accuracy in community practice. BMC Fam Pract. 2002;3:19. doi: 10.1186/1471-2296-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bae JM, Kim EH. Epstein-Barr Virus and Gastric Cancer Risk: A Meta-analysis with meta-regression of case-control studies. J Prev Med Public Health. 2016;49:97–107. doi: 10.3961/jpmph.15.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsusaka K, Funata S, Fukayama M, Kaneda A. DNA methylation in gastric cancer, related to Helicobacter pylori and Epstein-Barr virus. World J Gastroenterol. 2014;20(14):3916–3926. doi: 10.3748/wjg.v20.i14.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahn HJ, Lee DS. Helicobacter pylori in gastric carcinogenesis. World J Gastrointest Oncol. 2015;7(12):455–465. doi: 10.4251/wjgo.v7.i12.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu X, Meltzer SJ. Gastric cancer in the era of precision medicine. Cell Mol Gastroenterol Hepatol. 2017;3(3):348–358. doi: 10.1016/j.jcmgh.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Discrimination for gastric adenocarcinoma (GAC) by the top 22 methylated DNA marker candidates in Discovery (first three data columns) and Technical Validation (second three data columns)
Supplemental Table 2: Function of Genes Containing Differentially Methylated Regions Identified as Targets for Candidate Markers*
Supplemental Table 3: Covariate Effects on Tissue Levels of Methylated DNA Marker Candidates (Data shown represent p-values)
Supplemental Figure 1: Quantitative allele-specific real-time target and signal amplification (QuARTS) assay.