

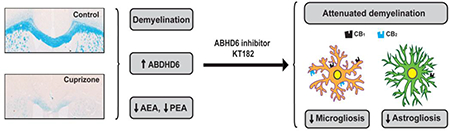
Abstract
Multiple sclerosis (MS) is a chronic demyelinating disease of unknown etiology in which tissue pathology suggests both immune-dependent attacks to oligodendroglia and primary oligodendrocyte demise. The endocannabinoid system has been crucially involved in the control of autoimmune demyelination and cannabinoid-based therapies exhibit therapeutic potential, but also limitations, in MS patients. In this context, growing evidence suggests that targeting the hydrolysis of the main endocannabinoid 2-arachidonoylglycerol (2-AG) may offer a more favorable benefit-to-risk balance in MS than existing cannabinoid medicines. Here we evaluated the modulation of endocannabinoid signaling and the therapeutic potential of targeting the 2-AG hydrolytic enzyme alpha/beta-hydrolase domain-containing 6 (ABHD6) in the cuprizone model of non-immune dependent demyelination. The concentrations of N-arachidonoylethanolamine (anandamide, AEA) and its congener N-palmitoylethanolamine (PEA) were reduced following 6 weeks of cuprizone feeding. Deregulation of AEA and PEA levels was not due to differences in the expression of the hydrolytic and biosynthetic enzymes fatty acid amide hydrolase and N-acylphosphatidylethanolamine-phospholipase D, respectively. Conversely, we measured elevated transcript levels of 2-AG hydrolytic enzymes monoacylglycerol lipase, ABHD6 and ABHD12 without changes in bulk 2-AG concentration. Upregulated CB1 and CB2 receptors expression, ascribed in part to microglia, was also detected in the brain of cuprizone-treated mice. Administration of an ABHD6 inhibitor partially attenuated myelin damage, astrogliosis and microglia/macrophage reactivity associated to cuprizone feeding. However, ABHD6 blockade was ineffective at engaging protective or differentiation promoting effects in oligodendrocyte cultures. These results show specific alterations of the endocannabinoid system and modest beneficial effects resulting from ABHD6 inactivation in a relevant model of primary demyelination.
Keywords: cuprizone, demyelination, ABHD6, cannabinoid receptors, oligodendrocyte, microglia
Graphical Abstract
1. Introduction
Multiple sclerosis (MS) is a chronic disease of the central nervous system (CNS) with pathological characteristics including inflammation, demyelination, gliosis and axonal degeneration [1]. Pathological assessment at the early stages of white matter demyelination indicates that actively demyelinating lesions are heterogeneous, and can be categorized in distinct patterns [2]. While experimental autoimmune encephalomyelitis (EAE) and Theiler’s murine encephalomyelitis virus infection (TMEV) effectively mimic the type I and II lesions showing mononuclear and T cell infiltration [3, 4], these models do not reproduce certain aspects of MS histopathology suggestive of immune-independent myelin damage which are present in approximately 25% of biopsied active lesions, classified as pattern III [2]. Cuprizone-induced primary demyelination is regarded as a valuable model to reproduce patterns III and IV of MS early demyelinating lesions, characterized by oligodendrocyte dystrophy and apoptotic death [5]. The copper chelator cuprizone inhibits mitochondrial function, resulting in oligodendrocyte damage and demyelination without major blood-brain barrier disruption or involvement of T cells [3, 6]. Oligodendrocytes undergo caspase 3 activation-mediated apoptosis after 1 week of cuprizone exposure, followed by severe brain demyelination between week 3 and 6 of continuous exposure to a cuprizone-containing diet [3, 6]. Demyelination is accompanied by a well-characterized sequence of events that participate to myelin damage and repair, including microglia activation, astrocyte proliferation and recruitment of oligodendrocyte progenitor cells (OPCs) [6, 7].
Endocannabinoids are bioactive lipids formed from plasma membrane precursors that bind and activate cannabinoid receptors CB1 and CB2. More than two decades of research have demonstrated that cannabinoid modulating drugs relieve symptomatology and attenuate neurodegeneration, demyelination and inflammatory responses in MS [8]. It is a consistent finding that administration of CB1/CB2 receptor agonists, such as the phytocannabinoid Δ9-tetrahydrocannabinol (Δ9-THC), exerts beneficial effects in experimental models of MS [9–11]. Beyond cannabis derivatives, a number of recent preclinical studies have put forward the potential of targeting endocannabinoid hydrolysis in MS. In particular, pharmacological blockade of monoacylglycerol lipase (MAGL), the main enzyme responsible for the hydrolysis of the endocannabinoid 2-arachidonoylglycerol (2-AG), fully mimics the protective and regenerative efficacy of exogenous cannabinoids in MS mouse models [12–14]. However, the use of Δ9-THC and MAGL inhibitors has been associated to the appearance of unwanted effects, including psychoactivity, desensitization of brain CB1 receptors and functional tolerance [15], which limits their utility in the clinical practice. By contrast, accumulating evidence suggests that selective inhibition of the minor 2-AG hydrolytic enzyme α/β-hydrolase domain-containing 6 (ABHD6) can provide therapeutic benefits in CNS inflammatory conditions, MS included, without producing cannabimimetic side effects [16–19].
The involvement of the endocannabinoid system in MS control is supported by a number of studies showing alterations in the levels of endocannabinoids and cannabinoid receptors in the EAE and TMEV mouse models [9, 20–22]. However, the activation state of the endocannabinoid system in animal models of primary demyelination has not been investigated. Here we studied possible changes in the expression of the main endocannabinoid signaling elements in the cuprizone model. We measured endocannabinoid levels and evaluated the expression of the main enzymes implicated in the synthesis and metabolism of these compounds. In addition, we analyzed the effects of ABHD6 blockade in this model of MS. Finally, we investigated the expression of the classical cannabinoid receptors CB1 and CB2 in damaged white matter following cuprizone feeding. Our results show specific alterations of the endocannabinoid system in a clinically relevant model of primary demyelination and subtle white matter protective, anti-inflammatory effects resulting from pharmacological ABHD6 blockade.
2. Materials and Methods
2.1. Reagents
KT182 [4-[3′-(Hydroxymethyl)[1,1′-biphenyl]-4-yl]-1H-1,2,3-triazol-1-yl](2-phenyl-1-piperidinyl)-methanone] was synthesized in B Cravatt´s laboratory at The Scripps Research Institute (La Jolla, CA, USA), as previously described [23]. For ABHD6 expression analysis we used a rabbit anti-ABHD6 antibody kindly provided by Dr J Mark Brown (Cleveland Clinic Lerner Research Institute, Cleveland, OH, USA) [24]. All other chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless stated otherwise.
2.2. Cuprizone model
Experimental demyelination was induced by feeding 8 week-old male C57BL/6 mice (Charles River, Barcelona, Spain) a diet containing 0.3% cuprizone (bis-cyclohexanone oxaldihydrazone) mixed into milled chow (Altromin, Lage, Germany) for 3 or 6 weeks. In experiments aimed at testing the effect of ABHD6 blockade in demyelination mice were daily treated with KT182 or vehicle for 21 days starting at the first day of cuprizone administration. The ABHD6 inhibitor was dissolved in 15% DMSO:4.25% polyethylene glycol 400:4.25% Tween-80:76.5% saline and administered intraperitoneally (i.p.) in a volume of 10 mL/kg. Animals were maintained under standard laboratory conditions at the Animal Facility of the University of the Basque Country (UPV/EHU) with food and water ad libitum. All animal procedures were carried out in compliance with the European Communities Council Directive of 22 September 2010 on the protection of animals used for scientific purposes (Directive 2010/63/EU), and approved by the local ethical committee of the UPV/EHU (Comité de Ética en Experimentación Animal, CEEA-UPV/EHU).
2.3. Measurement of endocannabinoid levels
Mice anesthetized with ketamine/xylacine (80/10 mg/kg, i.p.) were killed by decapitation and a single coronal slice containing the corpus callosum was trimmed between levels 1 and −1 bregma [25] using a David Kopf Instruments brain blocker. Tissue samples, stored at −80 ºC until the moment of analysis, were weighted and homogenized in an ice-cold glass dounce-homogenizer in a mixture 2:1:1 (v:v:v) of chloroform:methanol:Tris HCl 50 mM (pH = 7.5) in a volume of 4 ml for 80–120 mg of tissue sample. Then, 200 pmol of d8-arachidonoylethanolamine (d8-AEA), 200 pmol of d8–2-AG, and 200 pmol of d5-palmitoyletanolamide (d5-PEA) (Cayman Chemical, Ann Arbor, MI, USA) as internal standards were added. The organic and aqueous layers were separated by centrifugation (4500 x g, 2 min) and the organic layer transferred to a clean vial and dried under a stream of argon. The resulting fraction was reconstituted in 50 μL acetonitrile and analyzed by high-pressure liquid chromatography coupled to mass spectrometry (LC-MS). LC-MS analysis was performed using an Agilent 1200LC-MSD VL instrument. LC separation was achieved with a Zorbax Eclipse Plus C18 column (5 μm, 4.6 mm x 50 mm) together with a guard column (5 μm, 4.6 mm x 12.5 mm). The gradient elution mobile phases consisted of A (95:5 water:acetonitrile) and B (95:5 acetonitrile:water), with 0.1% formic acid as the solvent modifier. The gradient (flow rate of 0.5 mL/min) started at 0% B (for 5 min), increased linearly to 100% B over the course of 45 min, and decreased to 0% B for 10 minutes before equilibrating for 5 min with an isocratic gradient of 0% B. Mass spectrometry analysis was performed with an electrospray ionization source. The capillary voltage was set to 3.0 kV, and the fragmentor voltage was set at 70 V. The drying gas temperature was 350ºC, the drying gas flow rate was 10 L/min, and the nebulizer pressure was 20 psi. LC-MS measurements were made by selected ion monitoring in positive mode. Fractions were quantified by measuring the area under the peak and normalized using d8-AEA, d8–2-AG, or d5-PEA as internal standards. Absolute AEA, 2-AG, and PEA levels were estimated by comparison with the respective deuterated standard.
2.4. Flow cytometry analysis
Mice treated with cuprizone for 6 weeks and untreated controls were decapitated under isoflurane anesthesia (IsoVet®, B Braun, Barcelona, Spain), and forebrain cells purified according to previously described procedures [26]. Briefly, brain tissue was dissected and placed in enzymatic solution (116 mM NaCl, 5.4 mM KCl, 26 mM NaHCO3, 1 mM NaH2PO4, 1.5 mM CaCl2, 1 mM MgSO4, 0.5 mM EDTA, 25 mM glucose, 1 mM L-cysteine) with papain (20 U/mL) and DNAse I (150 U/μL, Invitrogen, Carlsbad, CA, USA) for digestion at 37ºC for 15 min. After homogenization, tissue clogs were removed by filtering the cell suspension through a 40 μm nylon strainer to a 50 mL Falcon tube and digestion quenched by addition of 5 mL of 20% heat inactivated fetal bovine serum in Hank´s Balanced Salt Solution (HBSS, Thermo Fisher Scientific, Waltham, MA, USA). For further enrichment of microglia, myelin was removed by using Percoll gradients (GE Healthcare Europe GmbH, Barcelona, Spain). For this purpose, cells were centrifuged at 200 x g for 5 min and resuspended in a 20% solution of isotonic Percoll in HBSS. Then, each sample was layered with HBSS and centrifuged for 20 min at 200 x g. Collected cells were washed in HBSS by centrifuging at 200 x g for 5 min and pellet was resuspended in 500 μL of sorting buffer (25 mM HEPES, 5 mM EDTA, 1% BSA, in HBSS). Cell suspensions were incubated with fluorochrome conjugated antibodies to CD11b (FITC; 1:100) and CD45 (PE; 1:100) (BioLegend, San Diego, CA, USA). Fc receptor Blocking Reagent was added to prevent nonspecific antibody binding to Fc receptors. Microglia were sorted as CD11b+ cells with low CD45 expression [27] using a BD FACS Jazz (2B/4YG) cell sorter and analyser (BD Bioscience, San José, CA, USA). Cells were collected in lysis buffer (Qiagen, Hilden, Germany) containing 1% β-mercaptoethanol and stored at −80ºC until processing. We confirmed that we had isolated a relatively pure population of microglia by nanofluidic qPCR analysis for the expression of several cell-type specific markers (Rbfox3, Aqp4, Gfap, Mbp, Olig2, Pdgfrα, Gpr34, P2ry12, Tmem119).
2.5. Nanofluidic qPCR
RNA from FACS-sorted microglia was extracted using the RNeasy plus Micro Kit (Qiagen). Following quality control of RNA samples (2100 Bioanalyzer, Agilent Technologies, Santa Clara, CA, USA), cDNA synthesis, pre-amplification and amplification steps were performed at the General Genomics Service of the UPV/EHU. For nanofluidic qPCR, the pre-amplified cDNA samples from purified microglia were measured with no reverse transcriptase and no template controls on 48.48 Dynamic Array chips and processed in the BioMark Real-Time PCR System (Fluidigm Corporation, San Francisco, CA, USA). We used commercial primers from IDT Integrated DNA Technologies (Leuven, Belgium) or Fluidigm Corporation targeting Cnr1 (Mm.PT.58.30057922), Cnr 2 (Mm.PT.58.41156189), Rbfox3 (Mm.PT.58.32889417), Aqp4 (Mm.PT.58.9080805), Gfap (Mm.PT.58.31297710), Olig2 (Mm.PT.58.42319010), Pdgfrα (Mm.PT.56a.5639577), Mbp (Mbp_31932_e3), Gpr34 (Gpr34_36763_i0), P2ry12 (P2ry6_37046_i1), Tmem119 (Mm.PT.58.6766267). Data preprocessing and analysis were completed using Fluidigm Melting Curve Analysis Software 1.1.0 and Real-time PCR Analysis Software 2.1.1 (Fluidigm Corporation) to determine valid PCR reactions. Data were corrected for differences in input RNA using the geometric mean of the reference genes Ppia (Ppia_10013_i2) and B2m (B2m_60287_i0), and relative expression values were calculated with the 2-ΔΔCt method.
2.6. Quantitative real-time PCR (RT-qPCR)
Anesthetized mice were transcardially perfused with cold RNase Free phosphate buffer saline (PBS, Ambion®, Thermo Fisher Scientific) for 30 s in order to remove blood cells from the brain. A single coronal slice containing the corpus callosum was trimmed between levels 1 and −1 bregma [25] and stored at −80ºC until analysis. Total RNA from brain samples and oligodendrocyte cultures was extracted using RNeasy Protect Mini Kit (Qiagen) and Absolutely RNA Miniprep Kit (Agilent), respectively. First strand cDNA synthesis was carried out with reverse transcriptase Superscript™ III (Invitrogen) using random primers. RT-qPCR was performed following MIQE guidelines (Minimal Information for Publication of Quantitative Real Time Experiments [28]. Primer pairs were designed with the PrimerExpress software (Applied Biosystems, Thermo Fisher Scientific) and were targeted to exon junctions to avoid amplification of contaminating genomic DNA. Primer sequences were as follows: mouse and rat Abhd6 forward 5´- ATC TAT TAT TGG TAC TGG CGG AGG-3´ and reverse 5´- TCA TGG TGT GCG TAG CGA AC-3´; mouse Abhd12 forward 5´- AGC TGT ACA AGG TGC TGA GTT CC −3´ and reverse 5´- CCC CAA CCT CTG TAG TCA AAG −3´; mouse Dagla forward 5´- GAC CTC AAG AAC TCG CAC GAG-3´and reverse 5´- GCG GAT AGC GAT TGC GTT-3´; mouse Faah forward 5´- GGC GAC TTT GTG GAT CCC T-3´and reverse 5´- CGA TAC ATC TCA ATC TCA TGC TGC-3´; mouse Magl forward 5´- GGA ACA AGT CGG AGG TTG ACC-3´ and reverse 5´- CAC TCT TGC GAC GGC ATT C −3´; mouse Mog forward 5´- CCA TCG GAC TTT TGA TCC TCA −3´and reverse 5´- GGT GCA GCC AGT TGT AGC AG −3´; Nape-pld forward 5´- GCA GCG GGT TTC GAC TTC T-3´and reverse 5´-TGG ATA CTG ATA GCT TGG CGC-3´; mouse Gapdh forward 5´-TGC AGT GCC AGC CTC GTC −3´and reverse 5´-GCC ACT GCA AAT GGC AGC-3´; mouse Hprt1 forward 5´- TAC TGT AAT GAT CAG TCA ACG GGG-3´ and reverse 5´-GTT GAG AGA TCA TCT CCA CCA ATA AC-3´; rat Gapdh forward 5´-GAA GGT CGG TGT CAA CGG ATT T-3´ and reverse 5´- CAA TGT CCA CTT TGT CAC AAG AGA A-3´. RT-qPCR was performed using SsoFastTM EvaGreen Supermix (Bio-Rad, Hercules, CA, USA). Amplifications were run in a CFX96™ Real Time System (Bio-Rad). Values were normalized to Gapdh and Hprt1 (brain tissue) or Gapdh (cultured cells) levels as endogenous references. The target mRNA quantity in each sample was determined from the relative standard curve and expressed in arbitrary units corresponding to the dilution factors of the standard RNA preparation (brain tissue) or using the 2-ΔΔCt algorithm (cultured cells).
2.7. Western blot analysis
Anesthetized mice were transcardially perfused with cold PBS containing 140 mM NaCl, .5 mM Na2HPO4 and 2.5 mM NaH2PO4 (pH = 7.4) for 30 s and the brains were rapidly removed. A single brain slice between levels 1 and −1 bregma [25] was obtained from each mouse and homogenized in 200 μL of RIPA with a protease inhibitor cocktail (Complete, Mini EDTA-free tablets, Roche, Mannheim, Germany). Tissue homogenates were centrifuged at 4°C for 10 min at 13,000 x g and protein content in the supernatant was quantified with the Qubit® Protein Assay Kit (Invitrogen). Tissue extracts (10 μg) were subjected to 10% SDS-PAGE and proteins were electroblotted to nitrocellulose membranes (Hybond ECL, GE Healthcare Europe GmbH). After blocking, membranes were probed with primary antibodies overnight at 4ºC followed by incubation with horseradish peroxidase-conjugated secondary antibodies (1:5000). Blots were developed using enhanced chemiluminiscence according to the manufacturer’s instructions (Super Signal West Dura, Pierce, Rockford, IL, USA). Protein bands were detected with a ChemiDoc™ XRS Imaging System (Bio-Rad). Signals were quantified using NIH Image-J Software (Bethesda, MD, USA) and values normalized to β-actin signal. Membranes were probed with rabbit anti-ABHD6 antibody provided by Dr J Mark Brown [24] (1:500), rabbit anti-ABHD12 (1:1000; Abcam, Cambridge, UK), rabbit anti-CB1 (1:500; ImmunoGenes, Budapest, Hungary); goat anti-FAAH (1:500; Santa Cruz Biotechnology, Dallas, TX, USA), rabbit anti-MAGL (1:1000; Santa Cruz Biotechnology), rabbit anti-β-actin (1:5000) or mouse anti-GAPDH (1:1000; Merck Millipore, Darmstadt, Germany). Western blot analysis of CB2 receptor expression was carried out using an antibody directed against residues 200–300 of rat CB2 receptor (ab45942, 1:500; Abcam, Cambridge, UK). This antibody recognizes a ~45KDa band according to the theoretical molecular weight of CB2 receptors, that is abolished by reabsorption with the immunizing peptide [29]. To further validate the anti-CB2 antibody used in western blot, we carried out immunocytochemistry experiments in HEK293 cells transfected with a bicistronic plasmid containing CB2 receptor and GFP cDNA, generated at Dr. Itsván Katona´s laboratory (Institute of Experimental Medicine, Budapest, Hungary). Showing that the antibody is sensitive to CB2 protein, incubation with the anti-CB2 resulted in reliable immunolabeling of CB2-GFP expressing cells that was absent from the non-expressing cells.
2.8. Histology, immunofluorescence and image analysis
Anesthetized mice were transcardially perfused with 0.1 M phosphate buffer (25 mM NaH2PO4•H2O; 75 mM Na2HPO4; pH = 7.4) followed by 4% paraformaldehyde (PFA) in the same buffer. After extraction, the brains were post-fixed in 4% PFA for 4 hours. Tissue samples for cryostat sectioning were cryoprotected overnight in 20% sucrose at 4ºC, embedded in Tissue-Tek OCT (Electron Microscopy Sciences, Hatfield, PA, USA) and stored at −80ºC until use. All histological analyses were carried out in serial coronal sections between levels 1 and −1 bregma [25]. Demyelination and glial reactions were evaluated in 10 μm-thick cryostat sections using luxol fast blue (LFB) and immunofluorescence standard protocols. Tissue sections were blocked for 60 min in Tris-HCl buffered saline (TBS) (100 mM Tris Base, 150 mM NaCl; pH = 7.4) supplemented with 5% normal goat serum (Vector Laboratories, Burlingame, CA, USA) and 0.2% Triton X-100. Sections were then incubated overnight at 4ºC with primary antibodies against myelin basic protein (MBP; 1:1000; Covance, Princeton, NJ, USA), GFAP for astrocytes (1:40; Merck Millipore), CD11b (1:100; Merck Millipore) for microglia/macrophages or NG2 (1:200; Merck Millipore) for OPCs. Optical images from tissue sections processed in parallel were acquired in the same session using a Zeiss Axioplan 2 microscope coupled to an Axiocam MRc5 digital camera (Zeiss, Oberkochen, Germany). Myelin damage was blind scored by two observers in both LFB and MBP stained sections using a 5-point scale ranging from 4 (normal myelin) to 0 (complete demyelination). Demyelination and glial reactivity were evaluated in two coronal sections separated 200 μm by examination of two digital 40X objective pictures from each slice including the middle line of the corpus callosum. Analysis of immunostained sections was carried out using microscope fluorescence intensity settings at which the control sections without primary antibody gave no signal. Threshold analysis of GFAP immunostained area was carried out in 16-bit grey scale transformed pictures and the values were referred to the total corpus callosum area in each optical section. CD11b- and NG2-positive cells were quantified by cell counting and data expressed as mean cell number per mm2 of corpus callosum area. Glial immunoreactivity was quantified using NIH Image J Software.
2.9. Immunostaining for ABHD6 and CB1 receptors
For ABHD6 expression analysis, 50 μm-thick vibratome sections containing the corpus callosum were pre-incubated in 1% H2O2 for 10 min at room temperature (RT) to inhibit endogenous peroxidase activity, and subsequently washed in TBS (pH = 7.4). Slices were blocked in 10% BSA, 0.1% sodium azide and 0.02% saponin in TBS for 30 minutes at RT. Tissue sections were subsequently incubated with rabbit anti-ABHD6 antibody [24] (1:250) for 48 h at 4°C. After primary antibody incubation, sections were treated with biotinylated anti-rabbit IgG (1:200; Vector Laboratories, Peterborough, UK) and then with avidin biotinylated-horseradish peroxidase complex (ABC; Elite, Vector Laboratories). Finally, the immunoperoxidase reaction was developed using 3, 3´diaminobenzidine (DAB) as a chromogen (Roche Diagnostics, Risch-Rotkreuz, Switzerland). Tissues were dehydrated and mounted in DPX. Threshold analysis of ABHD6 stained area was carried out in two digital 40X objective pictures including the middle line of the corpus callosum using NIH Image J Software.
For CB1 receptor localization studies, vibratome sections were rinsed in TBS (pH = 7.4) and incubated for 60 min at RT in a blocking-permeabilization solution containing 1% human serum (Vector Laboratories) and 0.3% Triton X-100. Tissue sections were subsequently incubated for 48 h at 4ºC with rabbit anti-CB1 receptor (ImmunoGenes, Budapest, Hungary) combined with rat anti-CD11b (1:100; Merck Millipore) or mouse anti-GFAP (1:40; Merck Millipore) in TBS supplemented with 0.1% Triton X-100. Following extensive washing, primary antibodies were detected by incubation with appropriate Alexa Fluor® 594 and 488 conjugated goat antibodies (1:400; Invitrogen, Thermo Fisher Scientific) for 2 h at RT in darkness. Hoechst 33258 (5 μg/mL) was used for chromatin staining. Sections were mounted in Vectashield (Vector Laboratories) mounting medium. Tissue sections from CB1 receptor knockout mice on a C56BL6 background bred at the Animal Facility of the UPV/EHU were processed in parallel, and used as internal control for the specificity of the immunostaining. Confocal microscopy of labelled tissue sections was performed using a Leica TCS Sp8 STED CW confocal laser scanning microscope. Threshold analysis of CB1 receptor immunostained area in the corpus callosum was carried out in two 40X objective confocal images acquired at 5 μm from tissue surface, using NIH Image J Software. The localization of CB1 receptor in astrocytes and microglia/macrophages was evaluated by examination of z-stacked confocal images acquired with a 40X objective at a 0.3 μm step-size through dual-labelled tissue sections. All histological analyses were carried out in at least 3 mice per group.
2.10. Oligodendrocyte cultures
Primary mixed glial cultures were prepared from newborn (P0-P2) Sprague-Dawley rats bred at the Animal Facility of the UPV/EHU, according to previously described procedures [30]. Briefly, forebrains were removed from the skulls and the cortices isolated and digested by incubation (15 min, 37°C) in HBSS 0.25% trypsin and 4% DNAse. Cells were dissociated by passage through needles (21 G and 23 G), centrifuged and resuspended in Iscove´s modified Dulbecco´s medium supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and antibiotic/antimycotic solution. Cells were seeded into poly-D-lysine-coated 75 cm2 flasks, and maintained in culture at 37°C and 5% CO2. After 10–15 days in culture, the flasks were shaken (400 rpm) for 2 h at 37°C to remove loosely adherent microglia. The remaining OPCs present on top of the confluent monolayer of astrocytes were dislodged by shaking overnight at 400 rpm. The cell suspension was then filtered through a 10 μM nylon mesh and preplated on bacterial grade Petri dishes for 2 h. The non-adherent OPCs that remained in suspension were recovered and plated again on bacterial grade Petri dishes for 1 h. The resulting enriched OPC suspension was centrifuged and resuspended in a chemically defined Dulbecco´s modified eagle´s medium that contained 100 μg/mL transferrin, 60 ng/mL progesterone, 40 ng/mL sodium selenite, 5 μg/mL insulin, 16 μg/mL putrescine and 100 μg/mL BSA. For excitotoxicity experiments, cells were plated in poly-D-lysine-coated 48-well culture dishes at a density of 8 × 103 cells/well. Alternatively, OPCs were plated onto poly-D-lysine-coated glass coverslips in 24-well culture dishes at density of 4 × 104 cells/well for western blot and 105 cells/well for qPCR analysis. Cells were cultured in the presence of the mitogenic factors PDGF-AA (5 ng/mL) and bFGF (5 ng/mL) for 2 days to expand their number and prevent their differentiation. The purity of the cultures was routinely assessed by examining cell morphologies under phase contrast microscopy and was confirmed by immunolabeling with cell-type specific antibodies against Olig-2 (1:1000; Merck Millipore); PDGFRα (1:200; Santa Cruz Biotechnology); GFAP (1:80; Merck Millipore) or Iba-1 (1:2000; Wako, Richmond, VA, USA). After 2 days in culture Olig-2+ cells represented 95 ± 0.2% of total cells, with the remaining cells being identified as GFAP+ astrocytes or Iba-1+ microglia (n = 7 cultures, 4 coverslips per culture; 15 microscopic fields per coverslip). In the same cultures, PDGFRα+ oligodendrocyte progenitors represented 92.5 ± 0.5% of total cells. For excitotoxicity experiments, OPCs were allowed to differentiate for 1–2 days in fresh medium lacking mitogenic factors and supplemented with the differentiating factors triiodothyronine (T3; 30 ng/mL) and thyroxine (T4; 40 ng/mL).
2.11. Excitotoxicity assays
Oligodendrocytes were pre-incubated for 30 min with drugs or vehicle [DMSO 0.1–0.2% (v/v) final concentration] and exposed to AMPA plus cyclothiazide (10 μM:100 μM; Abcam) for 15 min. The medium was subsequently replaced and cells were incubated overnight in the presence of MAGL or ABHD6 inhibitors. Oligodendrocyte viability was determined 24 h after the excitotoxic stimulus using MTT (CellTiter 96® Non-Radioactive Cell Proliferation Assay, Promega, Madison, WI, USA) in a Synergy-HT fluorimeter (Bio-Tek Instruments, Winooski, VT, USA) as described previously [31]. At least 3 independent experiments in triplicate were performed for each condition.
2.12. Differentiation assays
After the 2 day proliferation period, cell differentiation of OPCs was promoted by switching the cultures to fresh medium lacking mitogenic factors and supplemented with the differentiating factor T3 (30 ng/mL), in the presence or absence of experimental drugs for the times indicated. Drugs were replenished in the cultures every day. Differentiation was evaluated by western blot expression analysis of the oligodendrocyte differentiation marker MBP. Cells were harvested in 40 μl ice-cold electrophoresis sample buffer and were subjected to 10% SDS-PAGE as described above. Data are representative of at least 3 independent experiments.
2.13. Data analysis and statistics
Data are presented as means ± S.E.M. and n represents the number of animals or cultures tested. Statistical analyses were performed using GraphPad Prism 6 for Windows, version 5.0 (GraphPad Software Inc. San Diego, CA, USA). Results were analyzed by unpaired Student’s t-tests or univariate analysis of variance (ANOVA). In all instances, p values < 0.05 were considered statistically significant.
3. Results
3.1. Demyelination by cuprizone feeding disrupts endocannabinoid tone
Exposure to a cuprizone containing diet for 3 and 6 weeks induced effective demyelination of the forebrain as reflected by the progressive decline in Mog expression and by the loss of LFB myelin staining in the corpus callosum (Figures 1Band E). Consistent with previous reports [7], demyelination by cuprizone feeding was accompanied by a prominent glial reaction consisting in the recruitment of microglia, astrocytes and OPCs to the corpus callosum. Following 6 weeks of cuprizone administration, we measured significant increases in the presence of CD11b+ microglia/macrophages (1985 ± 210 vs 351 ± 87 cells/mm2 in control; p < 0.0001), GFAP+ astrocytes (immunostained area in arbitrary units was 0.35 ± 0.085 vs 0.023 ± 0.010 in control; p = 0.007) and NG2+ OPCs (2031 ± 126 vs 815.3 ± 47.02 cells/mm2 in control; p < 0.0001) (n = 5 control and 6 cuprizone-treated mice).
Figure 1.

Demyelination by cuprizone feeding disrupts endocannabinoid tone. (A) Effect of cuprizone feeding on brain endocannabinoid levels. Endogenous levels of AEA and PEA were significantly decreased following 6 weeks of cuprizone administration, whereas 2-AG levels were not significantly influenced (n = 8–9 mice). (B-C) RT-qPCR quantification of enzymes involved in endocannabinoid synthesis (Nape-pld, Dagla) (B) and metabolism (Mgll, Abhd6, Abhd12, Faah) (C) in brain tissue from mice fed a cuprizone containing diet for 6 weeks (n = 6–7 mice). Changes in Mog expression were analyzed in the same samples as internal control for the efficacy of cuprizone treatment. Expression levels were normalized to Gapdh and Hprt1 and presented relative to that measured in control mice. (D) Western blot analysis of endocannabinoid hydrolytic enzymes in brain samples from mice treated with cuprizone for 6 weeks (n = 4–5 mice). (E) Luxol fast blue (LFB) myelin staining and (F) immunoperoxidase labeling of ABHD6 in coronal brain sections from mice fed a cuprizone containing diet for 6 weeks and untreated controls. Analysis of immunostained area showed a substantial increase in ABHD6 expression in demyelinated corpus callosum (n = 3 mice). Scale bars: 500 μm (LFB) and 50 μm (ABHD6). CPZ, cuprizone. *p < 0.05 and ***p < 0.001 versus mice fed a control diet; Student´s t-tests.
To investigate whether the endocannabinoid system is modulated during primary demyelination, we first measured endocannabinoid concentrations in brain slices from mice fed a cuprizone-containing diet for 6 weeks. Brain levels of the main endocannabinoid 2-AG were not significantly influenced by cuprizone feeding (Figure 1A). By contrast, the concentrations of the endocannabinoid AEA and of the structurally related N-acylethanolamine PEA were significantly decreased in cuprizone treated mice as compared to untreated controls (Figure 1A). These results point to a selective deregulation of AEA and PEA production and/or metabolism in the brain of mice treated with cuprizone. We next sought for possible changes in the expression of the enzymes responsible for the synthesis and degradation of AEA and PEA in cuprizone-treated mice. The most widely studied pathway for the production of AEA and PEA is the one-step conversion of the corresponding Nacylphosphatidylethanolamines (NAPEs) by NAPE-phospholipase D (NAPE-PLD), while the major route for their metabolism involves enzymatic hydrolysis by the fatty acid amide hydrolase (FAAH) [32]. However, qRT-PCR analyses showed no significant alteration in the expression of Nape-pld or Faah at any of the time points tested (Figures 1B-C). Consistent with the absence of major changes in 2-AG levels, cuprizone feeding did not modulate the gene expression levels of the main enzyme involved in brain 2-AG production, namely diacylglycerol lipase-α (DGLα) (Figure 1B). Unexpectedly, the enzymes responsible for degradading 2-AG were upregulated at the transcriptional level during the time-course of cuprizone administration (Figure 1C). Following 6 weeks of toxin feeding, we measured a small but significant increase (21 ± 13%; p < 0.05) in Mgll expression levels and a more prominent augmentation of Abhd6 (86 ± 46%; p < 0.05) and Abhd12 (506 ± 70%; p < 0.001) levels. In order to determine whether these changes in gene expression levels are translated into protein increases, we performed western blotting in brain samples from the same cohorts of cuprizone-treated mice and untreated controls. We found a significant increase in ABHD12 (45 ± 17%; p < 0.05) expression without significant upregulation of MAGL levels (Figure 1D). Protein quantification also showed a non-significant upregulation of ABHD6 expression (36 ± 25%) by cuprizone feeding. To further analyze possible changes in ABHD6 levels following cuprizone intoxication we evaluated enzyme expression in damaged white matter by immunohistochemistry. Immunolabeling of ABHD6 in tissue sections evidenced a substantial increase in the expression levels of the enzyme in the demyelinated corpus callosum of mice treated with cuprizone for 6 weeks (Figure 1E-F). Collectively, these results suggest that primary demyelination is associated to deficits in endocannabinoid signaling involving decreased bulk AEA and PEA levels as well as an enhanced expression of 2-AG hydrolytic enzymes ABHD6 and ABHD12.
3.2. ABHD6 inactivation attenuates demyelination by cuprizone feeding
Blockade of ABHD6 has been recently postulated as a novel strategy to modulate the endocannabinoid system in MS based on results in the EAE model [17]. In this study, we sought to evaluate the effects of ABHD6 blockade in the cuprizone model of primary demyelination. With this aim, we used the recently characterized ABHD6 inhibitor KT182, which readily crosses the blood brain barrier and effectively targets the enzyme in brain tissue following intraperitoneal administration [23]. Daily prophylactic treatment with KT182 reduced demyelination of the corpus callosum after 3 weeks of cuprizone treatment, as determined by histological evaluation of LFB and MBP staining (Figures 2A-B). Amelioration of myelin pathology by KT182 was associated to a substantial suppression of inflammation evidenced by a reduced amount of both CD11b+-microglia/macrophages and GFAP+-astrocytes in the corpus callosum of mice treated with the ABHD6 inhibitor (Figure 2C). By contrast, administration of KT182 did not modulate the recruitment of NG2+ OPCs to the demyelinated corpus callosum (Figure 2C). Altogether, these data support the hypothesis that ABHD6 blockade engages myelin protective and anti-inflammatory effects during primary demyelination.
Figure 2.

ABHD6 inactivation attenuates demyelination by cuprizone feeding. (A) Analysis of luxol fast blue (LFB) staining and myelin basic protein (MBP) immunolabeling in corpus callosum slices from mice fed a cuprizone containing diet for 3 weeks and treated in parallel with KT182 (2 mg/kg) or vehicle, in comparison to control tissue sections. Administration of the ABHD6 inhibitor was associated to a significant attenuation of demyelination by cuprizone (n = 4–5 mice). Myelin damage was scored from 4 (normal myelin) to 0 (complete demyelination). Representative images showing LFB and MBP staining in the corpus callosum of control and cuprizone-treated mice that received KT182 or vehicle are shown in B. Scale bars: 500 μm (LFB) and 50 μm (MBP). (C) Quantification of glial reactivity in brain sections from cuprizone treated mice and untreated controls. Administration of the ABHD6 inhibitor was associated to a reduced presence of CD11b+ microglia/macrophages and GFAP+ astrocytes in demyelinated corpus callosum, without changes in the number of NG2+ OPCs (n = 4–5 mice). Representative images depict immunolabeling of CD11b, GFAP and NG2 in the corpus callosum cuprizone-treated mice that received KT182 or vehicle. Scale bars: 20 μm. CPZ, cuprizone. *p < 0.05, **p < 0.01 and ***p < 0.001; One-way ANOVA followed by Newman Keuls multiple comparison tests.
3.3. ABHD6 blockade does not modulate oligodendrocyte excitotoxicity and maturation
Protection from oligodendrocyte excitotoxicity is a relevant mechanism for the beneficial effects of MAGL inhibitors in MS animal models [12, 33]. In this regard, we have recently shown that 2-AG engages CB1 receptor mediated protective effects in cultured oligodendrocytes that are potentiated by pharmacological attenuation of MAGL activity [12]. Based on these results, we next sought to investigate whether inhibition of ABHD6 modulates excitotoxicity to oligodendrocytes in vitro. We first corroborated the expression of Abhd6 in cultured oligodendrocytes by qPCR analysis (Figure 3A). The ABHD6 inhibitor KT182 did not prevent oligodendrocyte death by activation of AMPA receptors at any of the concentrations tested (10 nM-1 μM) (Figure 3B). Confirming our previous observations [12], the MAGL inhibitor JZL184 effectively attenuated excitotoxicity in the same cultures and thus corroborated the efficacy of 2-AG production and signaling machinery in our experimental conditions (Figure 3C).
Figure 3.

Effects of ABHD6 blockade in cultured oligodendrocytes. (A) Abhd6, Abhd12 and Mgll expression levels in primary cultures of oligodendrocytes were determined by RT-qPCR (n = 3 cultures). Gene expression was normalized to Gapdh and presented relative to Mgll. (B, C) ABHD6 blockade did not prevent oligodendrocyte excitotoxicity. Graphs depict cell viability of oligodendrocytes exposed to AMPA plus cyclothiazide (10 μM:100 μM, 15 min) in the presence of the ABHD6 inhibitor KT182 or the MAGL inhibitor JZL184 (n = 4 cultures). (D-F) ABHD6 inactivation failed to promote OPC differentiation. (D) Western blot analysis of MBP expression in OPC cultures grown in the presence of T3 for 48–96 hours (n = 3 cultures). (E) Incubation of differentiating OPCs with the ABHD6 inhibitor KT182 did not increase the expression levels of MBP following 96 h of compound treatment at any of the concentrations tested (n = 3 cultures). (F) Exposure to the MAGL inhibitor JZL184 for 96 h enhanced MBP expression in the same cultures. *p < 0.05 versus vehicle-treated cells (V); One-way ANOVA followed by Newman Keuls multiple comparison tests (D) or Student´s t-tests (C, F).
Blockade of MAGL accelerates differentiation of OPCs by endogenously synthesized 2AG and promotes remyelination in vivo [13, 34]. In the cuprizone model, proliferating NG2+ OPCs differentiate even during continuous toxin feeding and newly generated mature oligodendrocytes reappear at low numbers already at week 5 [6]. Although the absence of significant alterations in the number of NG2+ cells in demyelinated corpus callosum from mice dosed KT182 (Figure 2C) suggests that accelerated differentiation is not a relevant mechanism of white matter protection by ABHD6 blockade, this possibility cannot be completely discarded based on this histological observation. To specifically address this issue, we evaluated the efficacy of KT182 to induce in vitro differentiation of rodent OPCs. Basal differentiation of OPCs was induced by mitogen withdrawal and addition of thyroid hormone T3 for 48–96 h and confirmed by western blot analysis of the mature oligodendrocyte marker MBP (Figure 3D). Incubation with increasing concentrations of the ABHD6 inhibitor failed to enhance the expression levels of MBP as determined by western blot analysis following 96 h of compound treatment (Figure 3E). In good agreement with previous reports [34], the MAGL inhibitor JZL184 induced substantial differentiation of OPCs at this time point (Figure 3F). Overall, these results show that blockade of ABHD6 activity in oligodendrocytes is incapable of engaging autocrine/paracrine cannabinoid receptordependent protective and maturation promoting mechanisms in these cells.
3.4. Demyelination by cuprizone upregulates CB1 and CB2 receptor expression
The protective efficacy of ABHD6 inhibitors in rodent models of CNS damage is normally attributed to the elevation of 2-AG levels engaging both CB1 and CB2 receptor-dependent [17, 19] and -independent [18, 35] mechanisms. As both cannabinoid receptors are crucially involved in regulating tissue damage and neuroinflammation in murine models of MS [10, 36–38], we next evaluated possible changes in the expression of cannabinoid CB1 and CB2 receptors induced by cuprizone feeding. qRT-PCR analyses showed significant increases in the expression levels of Cnr1 (47.2 ± 7.9%; p < 0.01) and Cnr2 (743 ± 53%; p < 0.001) (n = 7 mice) following 6 weeks of cuprizone feeding (Figure 4A). However, evaluation of protein expression levels by western blot did not reveal significant modulation of CB1 or CB2 receptors in brain samples from cuprizone-treated mice (Figure 4B).
Figure 4.

Modulation of CB1 and CB2 receptors by cuprizone feeding. (A) qRT-PCR analysis in brain samples showed upregulation of Cnr1 and Cnr2 transcripts following 6 weeks of cuprizone administration (n = 7 mice). Gene expression was normalized to Gapdh and Hprt1 and presented relative to levels in control mice. (B) Western blot analysis of CB1 and CB2 receptor expression in brain tissue from cuprizone-treated mice (n = 4–5 mice). (C) The expression of Cnr1 and Cnr2 was analyzed in microglia sorted from control and cuprizonetreated mice by nanofluidic qPCR (n = 3–5 mice). Gene expression was normalized to Ppia and B2m and presented relative to Cnr1 levels in microglia from control mice. (D) Confocal images and quantification of CB1 receptor immunostaining in the corpus callosum of cuprizone-treated mice and untreated controls (n = 6–8 mice). Brain sections from CB1 receptor knockout mice (n = 3) were used as internal controls for the specificity of the immunolabeling. (E) Single plane confocal images corresponding to immunostaining of CB1 receptors and CD11b or GFAP in the corpus callosum of mice fed a cuprizone containing diet for 6 weeks. The intensity of CB1 receptor staining in astrocytes (GFAP+, arrow) was always lower than in microglia/macrophages (CD11b+, arrowheads). CPZ, cuprizone 6 weeks. Scale bars: 20 μm. *p < 0.05, **p < 0.01 and ***p < 0.001 versus control wild-type mice; ###p < 0.001 versus control CB1 knockout mice; Student´s t-tests.
During neuroinflammation, activated microglia modulate CB1 and CB2 receptor expression in a context-specific manner [39, 40]. To further investigate the role of these proteins as regulators of microglial activity in the cuprizone model, we evaluated the expression of CB1 and CB2 receptors in microglial cells purified using flow cytometry. CB1 receptor transcript levels were significantly downregulated in microglia sorted from cuprizone-treated mice (p < 0.001), while CB2 receptor mRNA expression increased by 1.2 fold (p < 0.05) (Figure 4C). Although there is agreement that CB2 receptors are crucially involved in the control of microglia activation during neuroinflammation [39], the contribution of CB1 receptors to endocannabinoid mediated modulation of microglial cell function in vivo remains controversial. On the other hand, accumulating evidence suggests that astrocytes express CB1 receptors [41] amenable to regulate the function of these cells during inflammatory demyelination. Because both astrocytes and microglia are markedly upregulated by cuprizone feeding and both astroglial and microglial reactivity were attenuated by administration of the ABHD6 inhibitor KT182 (Figure 2C), we next analyzed the expression and localization of CB1 receptors with regard to either cell type in the demyelinated corpus callosum. Animals fed with control diet exhibited positive CB1 receptor immunolabeling in the corpus callosum as compared to CB1 receptor deficient mice used as internal controls for the specificity of the staining protocol (Figure 4D). Consistent with the results from gene expression studies in brain tissue, immunofluorescence analysis showed significant CB1 receptor upregulation following cuprizone intoxication (Figure 4D). Unexpectedly, high magnification confocal microscopy showed that CB1 receptors were predominantly expressed by the microglia/macrophage cell population in demyelinated corpus callosum, as the majority of CB1 receptor immunostaining appeared to lie on processes positive for the CD11b, and only scattered puncta had potential association with GFAP+ profiles (Figure 4E). As parenchymal microglia largely outnumber infiltrating macrophages during demyelination by cuprizone feeding [42], our histological observations suggest that CB1 receptor overexpression in damaged white matter is preferentially related to the microglia versus astrocytic pool of glial cells. Altogether, these observations support the possibility that both CB1 and CB2 receptors participate in the control of microglial activity and inflammation during demyelination by cuprizone feeding.
4. Discussion
The first goal of this study was to determine whether primary demyelination alters endocannabinoid levels and signaling machinery. Secondly, we sought to investigate the benefits of targeting the 2-AG hydrolytic enzyme ABHD6 during toxin-induced demyelination in vivo, and the potential mechanisms mediating such effects. Our work shows that the endocannabinoid system is relevant to the demyelination processes ensuing oligodendrocyte demise and that ABHD6 blockade exerts mild myelin protective effects associated to the control of glial reactivity.
In this study, we provide evidence that AEA and PEA, but not 2-AG, are reduced in the cuprizone model. The absence of major changes in 2-AG levels is in good agreement with findings in the spinal cord of EAE mice [43] and in cerebrospinal fluid from MS patients [22], though higher 2-AG levels have also been reported in the EAE model [9]. On the other hand, our data concerning reduced AEA levels in cuprizone-treated mice are in clear contrast with the increased concentration of this endocannabinoid reported in brain tissue, cerebrospinal fluid and plasma from MS patients [22, 44, 45]. It is worth mentioning, however, that lower levels of AEA and its congener PEA have also been reported in patients with MS [46]. Findings in animal models are equally controversial, as reduced [47], increased [9, 22] and unaltered [21] AEA concentrations have been shown in the EAE model. Discrepancies between studies addressing endocannabinoid levels in MS models and patients may be attributed to differences in the extent of tissue damage, targeted CNS region and time point of analysis. Importantly, elevation of AEA levels in MS patients has been associated to the presence of lymphocytes, as AEA is more abundant in active lesions than in quiescent ones and increased levels of the endocannabinoid have been measured in peripheral lymphocytes from MS patients [22, 44]. In this context, the absence of significant infiltration of peripheral immune cells in the brain of cuprizone treated mice will most likely contribute to the specific changes in endocannabinoid levels that we report in this animal model. Concerning PEA, increased levels of this endocannabinoid have been measured in chronic EAE and TMEV [9, 20], whereas both elevations [45] and reductions [46] have been reported in plasma and cerebrospinal fluid from MS patients. Regardless inconsistencies between studies, our results in the cuprizone model suggest that primary demyelination is associated to deficits in endocannabinoid signaling resulting from a selective deregulation of AEA and PEA levels, and support the potential of targeting endocannabinoid metabolism for myelin protective purposes.
AEA and PEA share common biosynthetic and metabolic pathways [48] and it seems plausible to propose that the observed parallelism in the evolution of both lipids may reflect changes in the activity of one or several enzymes commonly regulating their levels during demyelination. Elevated concentrations of AEA have been associated to increased NAPEPLD and reduced FAAH expression and/or activities in EAE mice and MS patients [9, 22]. On the other hand, FAAH expression is induced in hypertrophic astrocytes of MS lesions [49] pointing to an enhanced AEA hydrolytic activity in these cells. In the cuprizone model, however, we found no significant modulation of NAPE-PLD or FAAH expression levels despite the prominent astrocyte reaction associated to toxin-induced demyelination. Although our results do not exclude local changes in the activity of these enzymes that may contribute to the reduced AEA and PEA levels reported here, they also point to the possibility that cuprizone intoxication upregulates alternative endocannabinoid hydrolytic and oxygenating enzymes, such as N-acylethanolamine-hydrolyzing acid amidase (NAAA) and cyclooxygenase-2 (COX-2)[48]. In this regard, the existing literature is supportive of increased COX-2 activity mediating demyelination in the cuprizone model [50, 51]. Addressing the relative contribution of canonical and alternative pathways to endocannabinoid metabolism during primary demyelination is a pending issue in the MS field that will help elucidate the utility of targeting the endocannabinoid system with specific enzyme inhibitors in this medical condition.
When examining the consequences of ABHD6 inactivation in the cuprizone model we found a modest but significant attenuation of toxin-induced demyelination in mice dosed with KT182. This result is reminiscent of the beneficial effects associated to MAGL blockade in the cuprizone model [12], and support the utility of targeting 2-AG hydrolysis during primary demyelination. In terms of myelin protection, however, targeting ABHD6 (present report) seems to be less efficient than inactivating MAGL [12] against cuprizone intoxication. This observation is consistent with the minor role of ABHD6 in 2-AG metabolism (4%) as compared to MAGL [52]. At this point, it is also worth mentioning that blockade of ABHD6 blockade did not mimic the efficacy of MAGL inhibition at attenuating oligodendrocyte excitotoxicity or at promoting the in vitro differentiation of OPCs. From these observations, we can conclude that the weak enzymatic activity of ABHD6 in oligodendroglia is insufficient to promote 2-AG mediated biological effects through the activation of cannabinoid receptors present in these cells. In this scenario, manipulation of MAGL dependent 2-AG metabolism emerges as a unique opportunity to locally engage endocannabinoid signaling mechanisms and therapeutic benefits in myelinated white matter, as supported by recent evidence from in vivo studies in MS animal models [13, 14].
Our in vitro observations, together with the absence of changes in the number of NG2+ cells in mice treated with KT182, suggest that the myelin preserving effect of the ABHD6 inhibitor during cuprizone feeding does not result from direct, protective or remyelination promoting effects in oligodendrocyte lineage cells. Conversely, ABHD6 blockade was associated to a robust decrease in the inflammatory cellular infiltrate associated to cuprizone intoxication, as evidenced by the reduced recruitment of both microglia and astrocytes to the demyelinated corpus callosum. Whether attenuated reactivity of microglia and astrocytes results from cell-autonomous modulation or, indirectly, as a consequence of ABHD6 blockade and inhibition of 2-AG metabolism in other cell types such as neurons or oligodendroglial cells, remains to be addressed. Notably, ABHD6 inactivation exerts a more efficient control of demyelination associated astrogliosis in the cuprizone model as compared to MAGL blockade [12], despite the proposed relevance of astrocytic MAGL in the termination of 2-AG signaling [53]. These results show that each individual 2-AG hydrolytic enzyme allows for a differential control of endocannabinoid signaling at particular cell types during myelin damage and repair, and point to a relevant role of ABHD6 in the regulation of astrocytic functions during myelin pathology in vivo.
Supporting a role of CB1 and CB2 receptors in the cellular events governing primary demyelination, in this study we report a 8-fold increase in CB2 receptor gene expression and a more modest upregulation of CB1 receptor mRNA levels in brain tissue from cuprizonetreated mice. Concerning CB1 receptors, our observations contrast early reports showing decreased CB1 receptor expression in the EAE mice [54–56]. This apparent discrepancy might reflect, at least in part, the different degree of neurodegeneration in the EAE and cuprizone models. Indeed, neuronal damage and synaptic loss are prominent features in the EAE model, whereas demyelination-associated degeneration of axons is only marginal during cuprizonefeeding [6]. Conversely, the strong increase in CB2 receptor mRNA expression associated to cuprizone challenge is in good agreement with findings in EAE and TMEV mice [20, 43, 56], and consistent with the proposed relevance of this protein in the control of neuroinflammation, as demonstrated during the last decade [57]. Nevertheless, CB2 receptor upregulation at the transcriptional level was not associated to significant increases in protein expression in western blot assays. This discrepancy may be attributed, at least in part, to the limited sensitivity of the western blot technique to detect local upregulation of CB2 receptors in the demyelinated corpus callosum. In addition, the possibility also exists that translational suppression mechanisms limit CB2 receptor protein responses, as recently reported for highly expressed innate immune genes in activated microglia [58]. In this study, we did not address the immunohistochemical localization of CB2 receptors in demyelinated white matter due to the lack of a decisive negative control test allowing for unambiguous detection of the protein in tissue sections. This issue might be relevant to add further information on the biological significance of CB2 receptor function during primary demyelination. Based on the existing literature in the MS field and keeping in mind that demyelination in the cuprizone model takes place without marked infiltration of peripheral immune cells [6, 42], it can be hypothesized that increased CB2 receptor expression by cuprizone feeding most likely occurs in glial cells [49, 57]. With regard to the cell-specificity of CB2 receptor upregulation, we provide direct evidence that activated microglia increase CB2 receptor transcripts in response to cuprizone intoxication. It is noteworthy, however, that the extent of Cnr2 upregulation was much lower in microglia than in whole brain tissue extracts from cuprizone treated mice (Figures 2A and C), suggesting that other cell types, likely astrocytes and OPCs, may upregulate CB2 receptor during primary demyelination. Finally, increased Cnr2 expression in microglial cells has been often correlated to changes in receptor levels and/or function, both in vitro and ex vivo [57, 59, 60], supporting the hypothesis that activated microglia in the cuprizone model translate CB2 receptor transcript upregulation to increased protein levels at the plasma membrane, at least to a certain extent.
On the other hand, in this study we also show increased CB1 levels in demyelinated corpus callosum partly localized to microglial cells. These histological observations suggest that microglia activated during the time-course of toxin-induced demyelination upregulate CB1 receptor expression. Unexpectedly, gene expression analysis in purified microglia showed reduced transcriptional expression of CB1 receptors associated to cuprizone feeding. This striking observation likely reflects a complex regulation of CB1 receptor transcription/translation mechanisms in microglia activated during the demyelination process that may involve accelerated mRNA translation, increased protein half-life and feedback mechanisms leading to attenuated gene transcription, among others. Mechanistic considerations aside, our immunohistochemical results provide novel evidence pointing to a relevant role of CB1 receptors in regulating microglial function during primary demyelination in vivo. Collectively, our observations also support the hypothesis that ABHD6 inactivation modulates microglial reaction in the cuprizone model at least in part through activation of CB1 and CB2 receptors upregulated by these cells. Alternatively, cannabinoid receptorindependent mechanisms can also be suggested, based on the existing literature addressing the effects of 2-AG hydrolysis blockade in models of neuroinflammation [16].
In summary, our data show that demyelination induced by cuprizone feeding is associated to specific changes in the endocannabinoid system that may contribute to and/or regulate damage progression in this animal model. These findings may have significant implications for brain disorders with an important component of primary damage to oligodendrocytes and myelin.
Acknowledgements
We thank Saioa Marcos for expert technical assistance. AM, AB-C, AM-G and MC-A were predoctoral fellows from the Basque Government (AM), the University of the Basque Country (AB-C, AM-G) and the Spanish Ministry of Economy and Competitiveness (MC-A). Dr. JMB is supported by National Institutes of Health grants R01-HL122283 and P50-AA024333. This study was funded by grants from the Spanish Ministry of Economy and Competitiveness MINECO (SAF2013–45084-R and SAF2016–75292-R to CM and SAF2016–78792-R to SOG and MMF), CIBERNED (CB06/0005/0076 to CM), Basque Government (IT702–13 to CM), ARSEP Foundation (to CM and SM) and Fundación WOP (to SM and CM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5. References
- [1].Reich DS, Lucchinetti CF, Calabresi PA, Multiple Sclerosis N Engl J Med 378 (2018) 169–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H, Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination, Ann Neurol 47 (2000) 707–17. [DOI] [PubMed] [Google Scholar]
- [3].Kipp M, Nyamoya S, Hochstrasser T, Amor S, Multiple sclerosis animal models: a clinical and histopathological perspective, Brain Pathol 27(2) (2017) 123–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Molina-Holgado E, Arévalo-Martín A, Ortiz S, Vela JM, Guaza C, Theiler’s virus infection induces the expression of cyclooxygenase-2 in murine astrocytes: inhibition by the anti-inflammatory cytokines interleukin-4 and interleukin-10, Neurosci Lett 324 (2002) 237–41. [DOI] [PubMed] [Google Scholar]
- [5].Kipp M, Clarner T, Dang J, Copray S, Beyer C, The cuprizone animal model: new insights into an old story, Acta Neuropathol 118 (2009) 723–36. [DOI] [PubMed] [Google Scholar]
- [6].Skripuletz T, Gudi V, Hackstette D, Stangel M, De- and remyelination in the CNS white and grey matter induced by cuprizone: the old, the new, and the unexpected, Histol Histopathol 26 (2011) 1585–97. [DOI] [PubMed] [Google Scholar]
- [7].Gudi V, Gingele S, Skripuletz T, Stangel M, Glial response during cuprizone-induced de- and remyelination in the CNS: lessons learned, Front Cell Neurosci 8 (2014) 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chiurchiù V, van der Stelt M, Centonze D, Maccarrone M, The endocannabinoid system and its therapeutic exploitation in multiple sclerosis: Clues for other neuroinflammatory diseases, Prog Neurobiol 160 (2018) 82–100. [DOI] [PubMed] [Google Scholar]
- [9].Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Makriyannis A, Khanolkar A, Layward L, Fezza F, Bisogno T, Di Marzo V, Endocannabinoids control spasticity in a multiple sclerosis model, FASEB J 15 (2001) 300–2. [DOI] [PubMed] [Google Scholar]
- [10].Maresz K, Pryce G, Ponomarev ED, Marsicano G, Croxford JL, Shriver LP, Ledent C, Cheng X, Carrier EJ, Mann MK, Giovannoni G, Pertwee RG, Yamamura T, Buckley NE, Hillard CJ, Lutz B, Baker D, Dittel BN, Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells, Nat Med 13 (2007) 492–7. [DOI] [PubMed] [Google Scholar]
- [11].Ortega-Gutiérrez S, Molina-Holgado E, Arévalo-Martín A, Correa F, Viso A, López-Rodríguez ML, Di Marzo V, Guaza C, Activation of the endocannabinoid system as therapeutic approach in a murine model of multiple sclerosis, FASEB J 19 (2005) 133840. [DOI] [PubMed] [Google Scholar]
- [12].Bernal-Chico A, Canedo M, Manterola A, Victoria Sánchez-Gómez M, Pérez-Samartín A, Rodríguez-Puertas R, Matute C, Mato S, Blockade of monoacylglycerol lipase inhibits oligodendrocyte excitotoxicity and prevents demyelination in vivo, Glia 63 (2015) 163–76. [DOI] [PubMed] [Google Scholar]
- [13].Feliú A, Bonilla Del Río I, Carrillo-Salinas FJ, Hernández-Torres G, Mestre L, Puente N, Ortega-Gutiérrez S, López-Rodríguez ML, Grandes P, Mecha M, Guaza C, 2-Arachidonoylglycerol reduces proteoglycans and enhances remyelination in a progressive model of demyelination, J Neurosci 37 (2017) 8385–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mecha M, Feliú A, Machín I, Cordero C, Carrillo-Salinas F, Mestre L, Hernández-Torres G, Ortega-Gutiérrez S, López-Rodríguez ML, de Castro F, Clemente D, Guaza C, 2-AG limits Theiler’s virus induced acute neuroinflammation by modulating microglia and promoting MDSCs, Glia 66 (2018) 1447–63. [DOI] [PubMed] [Google Scholar]
- [15].Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu QS, Lichtman AH, Cravatt BF, Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system, Nat Neurosci 13 (2010) 1113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Alhouayek M, Masquelier J, Muccioli GG, Controlling 2-arachidonoylglycerol metabolism as an anti-inflammatory strategy, Drug Discov Today 19 (2014) 295–304. [DOI] [PubMed] [Google Scholar]
- [17].Wen J, Ribeiro R, Tanaka M, Zhang Y, Activation of CB2 receptor is required for the therapeutic effect of ABHD6 inhibition in experimental autoimmune encephalomyelitis, Neuropharmacology 99 (2015) 196–209. [DOI] [PubMed] [Google Scholar]
- [18].Naydenov AV, Horne EA, Cheah CS, Swinney K, Hsu KL, Cao JK, Marrs WR, Blankman JL, Tu S, Cherry AE, Fung S, Wen A, Li W, Saporito MS, Selley DE, Cravatt BF, Oakley JC, Stella N, ABHD6 blockade exerts antiepileptic activity in PTZ-induced seizures and in spontaneous seizures in R6/2 mice, Neuron 83 (2014) 361–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tchantchou F, Zhang Y, Selective inhibition of alpha/beta-hydrolase domain 6 attenuates neurodegeneration, alleviates blood brain barrier breakdown, and improves functional recovery in a mouse model of traumatic brain injury, J Neurotrauma 30 (2013) 565–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Loría F, Petrosino S, Mestre L, Spagnolo A, Correa F, Hernangómez M, Guaza C, Di Marzo V, Docagne F, Study of the regulation of the endocannabinoid system in a virus model of multiple sclerosis reveals a therapeutic effect of palmitoylethanolamide, Eur J Neurosci 28 (2008) 633–41. [DOI] [PubMed] [Google Scholar]
- [21].Witting A, Chen L, Cudaback E, Straiker A, Walter L, Rickman B, Möller T, Brosnan C, Stella N, Experimental autoimmune encephalomyelitis disrupts endocannabinoid-mediated neuroprotection, Proc Natl Acad Sci U S A 103 (2006) 6362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Centonze D, Bari M, Rossi S, Prosperetti C, Furlan R, Fezza F, De Chiara V, Battistini L, Bernardi G, Bernardini S, Martino G, Maccarrone M, The endocannabinoid system is dysregulated in multiple sclerosis and in experimental autoimmune encephalomyelitis, Brain 130 (2007) 2543–53. [DOI] [PubMed] [Google Scholar]
- [23].Hsu KL, Tsuboi K, Chang JW, Whitby LR, Speers AE, Pugh H, Cravatt BF, Discovery and optimization of piperidyl-1,2,3-triazole ureas as potent, selective, and in vivo-active inhibitors of α/β-hydrolase domain containing 6 (ABHD6), J Med Chem 56 (2013) 8270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Thomas G, Betters JL, Lord CC, Brown AL, Marshall S, Ferguson D, Sawyer J, Davis MA, Melchior JT, Blume LC, Howlett AC, Ivanova PT, Milne SB, Myers DS, Mrak I, Leber V, Heier C, Taschler U, Blankman JL, Cravatt BF, Lee RG, Crooke RM, Graham MJ, Zimmermann R, Brown HA, Brown JM, The serine hydrolase ABHD6 is a critical regulator of the metabolic syndrome, Cell Rep 5 (2013) 508–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Paxinos G, Franklin K, The mouse brain in stereotaxic coordinates, Academic Press; (2012). [Google Scholar]
- [26].Abiega O, Beccari S, Diaz-Aparicio I, Nadjar A, Layé S, Leyrolle Q, Gómez-Nicola D, Domercq M, Pérez-Samartín A, Sánchez-Zafra V, Paris I, Valero J, Savage JC, Hui CW, Tremblay M, Deudero JJ, Brewster AL, Anderson AE, Zaldumbide L, Galbarriatu L, Marinas A, Vivanco M, Matute C, Maletic-Savatic M, Encinas JM, Sierra A, Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling, PLoS Biol 14 (2016) e1002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, Langmann T, Holtman IR, Wang X, Eggen BJ, Boddeke HW, Hambardzumyan D, Wolf SA, Kettenmann H, Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1, PLoS One 10 (2015) e0116644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT, The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments, Clin Chem 55 (2009) 611–22. [DOI] [PubMed] [Google Scholar]
- [29].den Boon FS, Chameau P, Schaafsma-Zhao Q, van Aken W, Bari M, Oddi S, Kruse CG, Maccarrone M, Wadman WJ, Werkman TR, Excitability of prefrontal cortical pyramidal neurons is modulated by activation of intracellular type-2 cannabinoid receptors, Proc Natl Acad Sci U S A 109 (2012) 3534–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].McCarthy KD, de Vellis J, Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue., J Cell Biol 85 (1980) 890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Domercq M, Etxebarria E, Pérez-Samartín A, Matute C, Excitotoxic oligodendrocyte death and axonal damage induced by glutamate transporter inhibition, Glia 52 (2005) 36–46. [DOI] [PubMed] [Google Scholar]
- [32].Ahn K, McKinney MK, Cravatt BF, Enzymatic pathways that regulate endocannabinoid signaling in the nervous system, Chem Rev 108 (2008) 1687–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hernández-Torres G, Cipriano M, Hedén E, Björklund E, Canales Á, Zian D, Feliú A, Mecha M, Guaza C, Fowler CJ, Ortega-Gutiérrez S, López-Rodríguez ML, A reversible and selective inhibitor of monoacylglycerol lipase ameliorates multiple sclerosis, Angew Chem Int Ed Engl 53 (2014) 13765–70. [DOI] [PubMed] [Google Scholar]
- [34].Gomez O, Arevalo-Martin A, Garcia-Ovejero D, Ortega-Gutierrez S, Cisneros JA, Almazan G, Sánchez-Rodriguez MA, Molina-Holgado F, Molina-Holgado E, The constitutive production of the endocannabinoid 2-arachidonoylglycerol participates in oligodendrocyte differentiation, Glia 58 (2010) 1913–27. [DOI] [PubMed] [Google Scholar]
- [35].Wen J, Jones M, Tanaka M, Selvaraj P, Symes AJ, Cox B, Zhang Y, WWL70 protects against chronic constriction injury-induced neuropathic pain in mice by cannabinoid receptor-independent mechanisms, J Neuroinflammation 15 (2018) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Huffman JW, Layward L, Cannabinoids control spasticity and tremor in a multiple sclerosis model, Nature 404 (2000) 84–7. [DOI] [PubMed] [Google Scholar]
- [37].Arévalo-Martín A, Vela JM, Molina-Holgado E, Borrell J, Guaza C, Therapeutic action of cannabinoids in a murine model of multiple sclerosis, J Neurosci 23 (2003) 2511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Pryce G, Ahmed Z, Hankey DJ, Jackson SJ, Croxford JL, Pocock JM, Ledent C, Petzold A, Thompson AJ, Giovannoni G, Cuzner ML, Baker D, Cannabinoids inhibit neurodegeneration in models of multiple sclerosis, Brain 126 (2003) 2191–202. [DOI] [PubMed] [Google Scholar]
- [39].Mecha M, Carrillo-Salinas FJ, Feliú A, Mestre L, Guaza C, Microglia activation states and cannabinoid system: Therapeutic implications, Pharmacol Ther 166 (2016) 40–55. [DOI] [PubMed] [Google Scholar]
- [40].Stella N, Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas, Glia 58 (2010) 1017–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Metna-Laurent M, Marsicano G, Rising stars: modulation of brain functions by astroglial type-1 cannabinoid receptors, Glia 63 (2015) 353–64. [DOI] [PubMed] [Google Scholar]
- [42].Remington LT, Babcock AA, Zehntner SP, Owens T, Microglial recruitment, activation, and proliferation in response to primary demyelination, Am J Pathol 170 (2007) 1713–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN, Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli, J Neurochem 95 (2005) 437–45. [DOI] [PubMed] [Google Scholar]
- [44].Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, Wolf S, Hoertnagl H, Raine CS, Schneider-Stock R, Nitsch R, Ullrich O, The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells, Neuron 49 (2006) 67–79. [DOI] [PubMed] [Google Scholar]
- [45].Jean-Gilles L, Feng S, Tench CR, Chapman V, Kendall DA, Barrett DA, Constantinescu CS, Plasma endocannabinoid levels in multiple sclerosis, J Neurol Sci 287 (2009) 212–5. [DOI] [PubMed] [Google Scholar]
- [46].Di Filippo M, Pini LA, Pelliccioli GP, Calabresi P, Sarchielli P, Abnormalities in the cerebrospinal fluid levels of endocannabinoids in multiple sclerosis, J Neurol Neurosurg Psychiatry 79 (2008) 1224–9. [DOI] [PubMed] [Google Scholar]
- [47].Cabranes A, Venderova K, de Lago E, Fezza F, Sánchez A, Mestre L, Valenti M, García-Merino A, Ramos JA, Di Marzo V, Fernández-Ruiz J, Decreased endocannabinoid levels in the brain and beneficial effects of agents activating cannabinoid and/or vanilloid receptors in a rat model of multiple sclerosis, Neurobiol Dis 20 (2005) 207–17. [DOI] [PubMed] [Google Scholar]
- [48].Ueda N, Tsuboi K, Uyama T, Metabolism of endocannabinoids and related Nacylethanolamines: canonical and alternative pathways, FEBS J 280(9) (2013) 1874-94. [DOI] [PubMed] [Google Scholar]
- [49].Benito C, Romero JP, Tolón RM, Clemente D, Docagne F, Hillard CJ, Guaza C, Romero J, Cannabinoid CB1 and CB2 receptors and fatty acid amide hydrolase are specific markers of plaque cell subtypes in human multiple sclerosis, J Neurosci 27 (2007) 2396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Palumbo S, Toscano CD, Parente L, Weigert R, Bosetti F, Time-dependent changes in the brain arachidonic acid cascade during cuprizone-induced demyelination and remyelination, Prostaglandins Leukot Essent Fatty Acids 85 (2011) 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Palumbo S, Toscano CD, Parente L, Weigert R, Bosetti F, The cyclooxygenase-2 pathway via the PGE2EP2 receptor contributes to oligodendrocytes apoptosis in cuprizone-induced demyelination, J Neurochem 121 (2012) 418–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Blankman JL, Simon GM, Cravatt BF, A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol, Chem Biol 14 (2007) 1347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Viader A, Blankman JL, Zhong P, Liu X, Schlosburg JE, Joslyn CM, Liu QS, Tomarchio AJ, Lichtman AH, Selley DE, Sim-Selley LJ, Cravatt BF, Metabolic Interplay between astrocytes and neurons regulates endocannabinoid action, Cell Rep 12 (2015) 798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Berrendero F, Sánchez A, Cabranes A, Puerta C, Ramos JA, García-Merino A, Fernández-Ruiz J, Changes in cannabinoid CB(1) receptors in striatal and cortical regions of rats with experimental allergic encephalomyelitis, an animal model of multiple sclerosis, Synapse 41 (2001) 195–202. [DOI] [PubMed] [Google Scholar]
- [55].Cabranes A, Pryce G, Baker D, Fernández-Ruiz J, Changes in CB1 receptors in motorrelated brain structures of chronic relapsing experimental allergic encephalomyelitis mice, Brain Res 1107 (2006) 199–205. [DOI] [PubMed] [Google Scholar]
- [56].Palazuelos J, Davoust N, Julien B, Hatterer E, Aguado T, Mechoulam R, Benito C, Romero J, Silva A, Guzmán M, Nataf S, Galve-Roperh I, The CB(2) cannabinoid receptor controls myeloid progenitor trafficking: involvement in the pathogenesis of an animal model of multiple sclerosis, J Biol Chem 283 (2008) 13320–9. [DOI] [PubMed] [Google Scholar]
- [57].Navarro G, Morales P, Rodríguez-Cueto C, Fernández-Ruiz J, Jagerovic N, Franco R, Targeting cannabinoid CB2 receptors in the central nervous system. Medicinal chemistry approaches with focus on neurodegenerative disorders, Front Neurosci 10 (2016) 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Boutej H, Rahimian R, Thammisetty SS, Béland LC, Lalancette-Hébert M, Kriz J, Diverging mRNA and protein networks in activated microglia reveal SRSF3 suppresses translation of highly upregulated innate immune transcripts, Cell Rep 21 (2017) 3220–33. [DOI] [PubMed] [Google Scholar]
- [59].Mecha M, Feliú A, Carrillo-Salinas FJ, Rueda-Zubiaurre A, Ortega-Gutiérrez S, de Sola RG, Guaza C, Endocannabinoids drive the acquisition of an alternative phenotype in microglia, Brain Behav Immun 49 (2015) 233–45. [DOI] [PubMed] [Google Scholar]
- [60].Navarro G, Borroto-Escuela D, Angelats E, Etayo Í, Reyes-Resina I, Pulido-Salgado M, Rodríguez-Pérez AI, Canela EI, Saura J, Lanciego JL, Labandeira-García JL, Saura CA, Fuxe K, Franco R, Receptor-heteromer mediated regulation of endocannabinoid signaling in activated microglia. Role of CB1 and CB2 receptors and relevance for Alzheimer’s disease and levodopa-induced dyskinesia, Brain Behav Immun 67 (2018) 139–51. [DOI] [PubMed] [Google Scholar]