

Abstract
Antibody-based immune therapies targeting the T cell checkpoint molecules CTLA-4 and PD-1 have impacted cancer therapy. However, this immune therapy requires complex manufacturing and frequent dosing, limiting the global use of this treatment. Here we focused on the development of a DNA-encoded monoclonal antibody (DMAb) approach for delivery of anti-CTLA-4 monoclonal antibodies in vivo. With this technology, engineered and formulated DMAb plasmids encoding IgG inserts were directly injected into muscle and delivered intracellularly by electroporation, leading to in vivo expression and secretion of the encoded IgG. DMAb expression from a single dose can continue for several months without the need for repeated administration. Delivery of an optimized DMAb encoding anti-mouse CTLA-4 IgG resulted in high serum levels of the antibody as well as tumor regression in Sa1N and CT26 tumor models. DNA-delivery of the anti-human CTLA-4 antibodies ipilimumab and tremelimumab in mice achieved potent peak levels of approximately 85μg/mL and 58μg/mL, respectively. These DMAb exhibited prolonged expression, with maintenance of serum levels at or above 15μg/mL for over a year. Anti-human CTLA-4 DMAbs produced in vivo bound to human CTLA-4 protein expressed on stimulated human PBMC and induced T cell activation in a functional assay ex vivo. In summary, direct in vivo expression of DMAb encoding checkpoint inhibitors serve as a novel tool for immunotherapy that could significantly improve availability and provide broader access to such therapies.
Introduction
Immune modulatory monoclonal antibodies (mAbs), in particular antibodies targeting the immune checkpoint molecules CTLA-4 and PD-1, have shown unprecedented impact in the clinic for patients with multiple types of solid tumors(1). Antibodies targeting CTLA-4, ipilimumab and tremelimumab, were the first of this class to enter clinical studies in patients with solid tumors in 2000. Ipilimumab, was the first therapy to improve both progression free and overall survival in patients with melanoma, and was FDA approved for treatment of unresectable or metastatic melanoma in 2011(1). Additional clinical trials are ongoing for both ipilimumab and tremelimumab as well as next generation CTLA-4 blocking antibodies alone or in combination therapies for a variety of different malignancies(1).
Despite the success of these therapies in the clinic, the price tag may limit the availability of these life-saving drugs for underserved populations(2). The high price is due in part to the complexity of manufacturing mAbs, and the high doses at which they are required in patients(3). Approaches that could allow for less frequent delivery and more simple formulations might be very valuable.
The use of gene delivery technologies has been proposed for delivery of prophylactic or therapeutic mAbs for infectious disease and cancer(4–7). The major delivery methods investigated have been viral vectors, DNA plasmids and in vitro transcribed RNA. Due to a host of likely limitations, to date none of these platforms have been used to encode antibodies targeting the immune checkpoint inhibitors PD-1 or CTLA-4.
Here, we report the design and development of DNA encoded mAbs (DMAbs) expressing antibodies targeting CTLA-4 directly in vivo. We show that synthetic sequence optimized DMAbs targeting mouse CTLA-4 protein can be robustly expressed in vivo, have a reasonable and unique half-life and can drive protective anti-tumor immune responses in vivo. We further show that optimized DMAbs encoding ipilimumab and tremelimumab are potently expressed in vivo, bind to human regulatory T cells and activate human effector T cells. This strategy potentially provides a novel approach to immune checkpoint therapy, allowing for more novel and widely useful options for this technology in the management and treatment of cancer.
Materials and Methods
Cell Culture and Transfection
HEK293T cells, CT26 and Sa1N tumor cells were obtained from ATCC, which performs thorough testing and authentication of their cell lines using morphology, karyotyping and PCR based approaches. They were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). They were both routinely tested for Mycoplasma contamination, and maintained at low passage (<20 passages) in cell culture. Only Sa1N or CT26 cells at lower than passage 5 were implanted into mice. HEK293T cells were transfected with GeneJammer transfection reagent according to the manufacturer’s recommendations (Agilent). Cells and conditioned media were harvested 48 hours after transfection using RIPA lysis buffer (Cell Signaling Technology) containing EDTA-free protease inhibitor (Roche) for analysis by western blot.
DNA plasmid construction
The amino acid sequences for 9D9, ipilimumab and tremelimumab were obtained from published patents or available DrugBank sequences (US9868961B2 for 9D9). The nucleotide sequence for the mouse IgG2b (9D9) was codon optimized for mouse to enhance mammalian expression, and the nucleotide sequences for the human IgG1 (ipilimumab) and IgG2 (tremelimumab) were optimized for both mouse and human codon biases. All sequences were also RNA optimized and included a Kozak sequence. Plasmids were cloned into the modified pVax1 plasmid with a human cytomegalovirus promoter and bovine growth hormone polyA sequence (GenScript). Both heavy and light chains were encoded in the same plasmid, separated by a furin cleavage site (RGRKRRS) and a P2A peptide to ensure cleavage. Additional sequence modifications for 9D9 were made based on sequence alignment to the mouse germline IGHV1–19*01 sequence, and are indicated in Supplementary Figure 1.
DMAb injection and mouse tumor studies
C57Bl/6, Balb/c and A/J mice were purchased from Jackson laboratories. DNA plasmids were formulated with 12 Units of hyaluronidase enzyme (Sigma-Aldrich) in 30μL total injection volume. Formulated DNA plasmid was injected at one site (100μg) in the tibialis anterior (TA) muscle, or at 4 sites (100μg per site) in both TA muscles and quadriceps muscles. Following plasmid injection, the muscles were pulsed with two 0.1 Amp electric constant current square-wave pulses using the CELLECTRA®−3P device (Inovio Pharmaceuticals). For tumor challenge studies, A/J or Balb/c mice were implanted subcutaneously with 10 million Sa1N tumor cells or 500,000 CT26 tumor cells, respectively, in PBS on the right flank. As human antibodies are immunogenic in immune competent mice, we studied their expression in Balb/c mice that were depleted of CD4+ and CD8+ T cells transiently at the time of DMAb injection (using a 200μg injection of clone GK1.5 and clone YTS 169.4, BioXCell). For tumor studies, mice were euthanized when tumors reached 1.5cm in diameter. All mice still alive at the end of study cleared their tumors completely. All animal studies were performed in accordance with guidelines from the National Institute of Health, and were approved by the Wistar Institutional Animal Care and Use Committee.
Human patient samples
Human blood was obtained from consenting adult healthy volunteers through the Wistar Phlebotomy core under Institutional Review Board (IRB) approved protocol #21801304. Written informed consent was obtained from all patients, and studies were conducted in accordance with recognized ethical guidelines (Delaration of Helsinki). Whole blood was collected in heparinized tubes and subsequently layered on top of an equal volume of histopaque 1083 (Sigma-Aldrich).
CTLA-4 blockade luciferase assay
T cell activation after CTLA-4 blockade was assessed using the CTLA-4 Blockade Bioassay (Promega), according to manufacturer’s instructions. Ipilimumab and tremelimumab DMAb was purified from individual mice for this assay (n=3 mice for each DMAb), using the Nab Protein A/G Spin Kit (ThermoFisher), and was concentrated using Amicon Ultra Centrifugal Filters (Millipore Sigma). Luciferase activity was measured using the Synergy2 plate reader (Biotek).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism software. Error bars represent the mean ± the standard error of the mean (SEM) or the mean ± the standard deviation (SD), as indicated in the figure legend. Statistical significance was determined by a Student’s t-test for experiments containing two experimental groups, and by a one-way ANOVA followed by Tukey’s post-hoc HSD test for experiments with more than two groups. For tumor growth over time, multiple t-tests were performed for each time point. For mouse survival analysis, significance was determined using a Gehan-Brelow-Wilcoxon test. IC50 values were calculated using a non-linear regression of serum concentration versus OD450 value. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Detailed methods related to western blot, ELISA, immunofluorescence staining and human PBMC stimulation and staining are included in the Supplementary Methods.
Results
Design, expression and binding of mouse anti-mouse CTLA-4 DMAbs
We used the mouse anti-mouse CTLA-4 9D9 clone to encode in our optimized DNA expression system, based on its previously described anti-tumor activity(8,9). The design for this DMAb plasmid was built off prior DMAb work from our group in the infectious disease space, and is described in detail in the methods section(4,5).
Transfected HEK293T cells were able to produce and secrete 9D9 DMAb antibody in vitro, detected by ELISA and western blot (Figure 1A,B). However, expression of this DMAb was low (~660ng/mL) compared to other previously examined DMAbs(4,5). We therefore engineered several modifications into the DMAb to improve expression, including modification of the beginning and end of the heavy chain sequence (Supplementary Figure 1A,B). While modification of the end sequence alone (mod #2) only slightly improved antibody production in vitro, modification of the beginning sequence or both sequences significantly improved antibody production, with nearly a 10-fold improvement in antibody secretion to the media for mod #4 (Figure 1B). These framework modifications did not alter the binding to mouse CTLA-4 protein by ELISA, with similar IC50 values compared to recombinant 9D9 (range 36.105–44.25ng/mL) (Figure 1C).
Figure 1. Expression and binding of mouse anti-mouse CTLA-4 DMAbs.

A) Secreted mouse IgG levels for the indicated DMAb from transfected HEK293T cells. B) Western blot of mouse IgG from lysates (left) and supernatants (right). Red bands indicate the ladder, green bands indicate mouse IgG. C) Binding of purified 9D9 or supernatants from transfected cells to mouse CTLA-4 protein. IC50 is indicated in the figure legend. Individual curves from biological replicates are shown. D) Serum concentration of anti-CTLA-4 mouse IgG from C57Bl/6 mice injected with 100μg of the indicated DMAb. Error bars indicate mean ±SD for in vitro studies, and mean ±SEM for in vivo studies. A and C, n= at least 2 biological replicates. D, n=5 mice per group.
We next tested expression of these DMAbs in C57Bl/6 mice through delivery by IM-EP (100μg) (Figure 1D). Similar to the in vitro results, the original 9D9 DMAb produced antibody in the serum at relatively low levels (~1.2μg/mL of serum) (Figure 1D). All three modified DMAbs expressed at higher levels, with the mod #4 producing levels of ~7.9μg/mL, over 6-fold higher than the original DMAb sequence (Figure 1D). These important framework modifications therefore greatly improved both in vitro and in vivo expression of this DMAb without altering binding to mouse CTLA-4 protein.
Anti-tumor activity of anti-mouse CTLA-4 DMAb in multiple tumor models
We next studied the highest expressing 9D9 DMAb (9D9 DMAb mod #4) in mouse tumor challenge models. We first utilized the Sa1N fibrosarcoma model, which is one of the first models used to demonstrate anti-tumor immunity from CTLA-4 blockade(10). We compared anti-tumor activity of the 9D9 DMAb to that of the recombinant 9D9 antibody (Figure 2A). Because DMAbs take a few days to be secreted from the muscle tissue, we started DMAb delivery 4 days earlier than recombinant 9D9. We compared one injection of DNA (400μg) to three injections of recombinant 9D9 antibody, delivered three days apart (10μg per injection), and observed similar kinetics of expression (Figure 2A, Supplementary Figure 2A,B), indicating prolonged duration of expression of the DMAb. Upon challenge with Sa1N tumor cells, both the 9D9 DMAb and the recombinant 9D9 were effective at inducing tumor clearance compared to control groups (Figure 2B, Supplementary Figure 2C). Tumors grew in all mice initially upon implantation; however, upon DMAb delivery, 8/10 mice cleared their tumors (Figure 2B). Upon recombinant 9D9 delivery, 9/10 mice completely cleared their tumors (Supplementary Figure 2D). Due to the immunogenic nature of this tumor, 3/10 mice in the mouse IgG control group also cleared their tumors spontaneously (Supplementary Figure 2D). To test for immunologic memory after DMAb exposure, we re-challenged the mice that cleared their tumors 6 months after the initial treatment (Supplementary Figure 3). 100% of the mice that were previously treated with either recombinant 9D9 antibody or 9D9 DMAb cleared the re-implanted tumors (Supplementary Figure 4). We also demonstrated that earlier DMAb administration (7 days prior to tumor implantation) was also effective at inducing tumor clearance in 6/10 mice (Supplementary Figure 4A-C). In summary, anti-CTLA4 DMAbs exhibit prolonged serum antibody levels exhibiting an injection sparing effect with similar anti-tumor activity compared to recombinant mAb.
Figure 2. Anti-tumor activity of anti-CTLA-4 DMAb in Sa1N and CT26 tumor models.

A) Tumor study outline for DMAb delivery using prophylactic Sa1N tumor model in A/J mice (top), and serum levels of anti-CTLA-4 mouse IgG from these mice (bottom). 400μg DMAb was delivered by IM-EP 4 days prior to implantation of tumor cells. B) Tumor volume measurements and survival analysis of the mice described in A. C) Tumor study outline for DMAb delivery using therapeutic CT26 tumor model in Balb/c mice (top), and serum levels of anti-CTLA-4 mouse IgG from these mice (bottom). 400μg DMAb was delivered by IM-EP 3 days after implantation of CT26 tumor cells. D) Tumor volume measurements and survival analysis of the mice described in C. Error bars indicate mean ±SEM. N=10 mice per group. Shown is a representative of two independent experiments.
We next tested the impact of 9D9 DMAb on the tumor microenvironment prior to tumor clearance at day 10 (Supplementary Figure 5A). At this early time point, tumors from both groups were similar sizes. The 9D9 DMAb induced higher levels of global lymphocyte infiltration (CD3+ cells) as well as specifically CD8+ T cell infiltration, compared to isotype control mice, indicating potent immune stimulatory capacity driven by the DMAb (Supplementary Figure 5B,C). In addition, the CD8+ T cells infiltrating the 9D9 DMAb-treated tumors expressed higher levels of activation markers, including CD44, CD69 and PD1 (Supplementary Figure 5D). Importantly, tumors treated with the 9D9 DMAb had a significantly lower proportion of regulatory T cells (CD4+/CD25+/FoxP3+) (Supplementary Figure 5E).
We next tested the efficacy of this DMAb in a therapeutic setting in the CT26 tumor model. For this model, we began DMAb administration 3 days after tumor implantation (Figure 2C). The 9D9 DMAb exhibited high expression in this mouse strain (Figure 2C), and was effective at controlling tumor growth in this therapeutic setting, inducing tumor clearance in 8/10 mice (Figure 2D). These results support the versatility of this DMAb platform across multiple mouse strains and tumor models.
Expression and binding of human anti-human CTLA-4 DMAbs
We next studied both in vitro and in vivo production of clinically relevant ipilimumab and tremelimumab DMAbs (ipi-DMAb and treme-DMAb) (Figure 3). Both of these DMAbs were expressed and secreted at very high levels into the media of transfected cells in vitro (~14.3μg/mL for ipi-DMAb and ~5.8μg/mL for treme-DMAb, Figure 3A). In addition, both heavy and light chains were clearly visible in both lysate and media by western blot (Figure 3B).
Figure 3. Expression and binding of human anti-human CTLA-4 DMAbs.
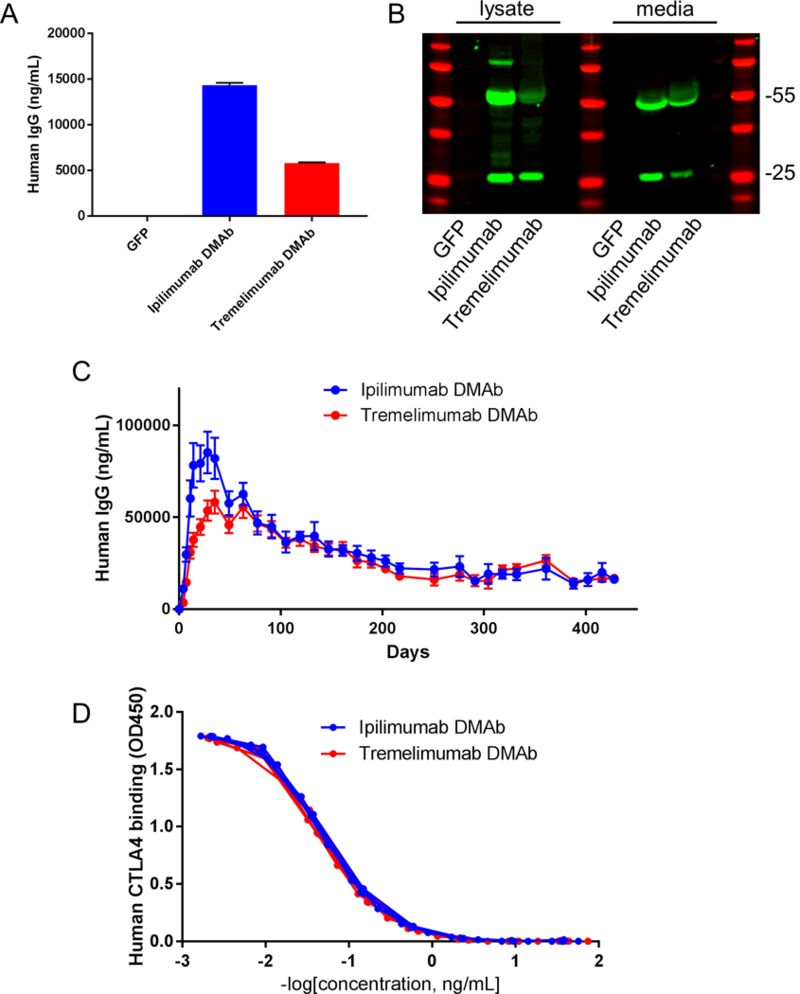
A) Secreted human IgG levels for the indicated DMAb from transfected HEK293T cells. B) Western blot of human IgG from lysates (left) and supernatants (right). Red bands indicate the ladder, green bands indicate human IgG. C) Serum concentration of human IgG over time in Balb/c mice injected with 400μg of the indicated DMAb by IM-EP. D) Binding of ipi-DMAb and treme-DMAb purified from mouse serum to human CTLA-4 protein by ELISA. Curves from individual mice are shown. For in vitro experiments, error bars indicate mean ±SD. For in vivo experiments, error bars indicate mean ±SEM. A, n= 2 biological replicates. C, n=5 mice per group. D, n=3 mice per group.
Dosing of 400μg of formulated DNA in the tibialis anterior and quadriceps muscles of Balb/c mice demonstrated robust expression of both DMAbs, with potent peak expression levels of ~85μg/mL for ipi-DMAb and ~58 μg/mL for treme-DMAb (Figure 3C). These studies were done in mice depleted of CD4 and CD8 T cells to eliminate the anti-human immune response (Supplementary Figure 6). Both DMAbs produced mAb for prolonged periods of over one year (Figure 3C). Importantly, the DMAb harbored in the serum of the treated animals bound robustly to human CTLA-4 by ELISA (Figure 3D).
Functionality of human anti-human CTLA-4 DMAbs
Functionality of the ipi-DMAb and treme-DMAbs was assessed using in vitro human T cell assays (Figure 4). Peripheral blood mononuclear cells (PBMCs) were isolated from three healthy donors, and stimulated with PMA/ionomycin to induce CTLA-4 surface expression on regulatory T cells (Figure 4A)(11). Because CD4 surface expression is down-regulated upon stimulation with PMA/ionomycin, regulatory T cells (Tregs) were classified as CD3+, CD8- and CD25+ PBMCs. Similar to the positive control anti-human CTLA-4 antibody, in vivo produced ipi-DMAb and treme-DMAb efficiently stained stimulated Tregs, but not unstimulated Tregs (Figure 4A,B).
Figure 4. Functionality of human anti-human CTLA-4 DMAbs.

A) Flow cytometric staining of CD3+CD8-CD25+ human PBMCs for CTLA-4 with the indicated antibodies, with or without PMA/ionomycin stimulation. B) Quantification of the staining depicted in A, for 3 individual donors. C) Illustration of CTLA-4 blockade bioassay. D) Results from bioassay described in C. The Relative Luciferase Units (RLU) are graphed relative to the RLU from no antibody control wells. Ipi-DMAb and treme-DMAb were purified from mouse serum. Error bars indicate ±SD. For Figure D, curves indicate 4-parameter nonlinear fit.
A functional T cell activation assay was utilized to test the ability of the DMAbs to induce T cell activation in vitro. For this assay, aAPC/Raji cells were coincubated with Jurkat cells that were transduced with a construct expressing luciferase off of the IL-2 promoter (Figure 4C). Upon efficient blockade of the CTLA-4/CD80/CD86 interaction, these Jurkat cells can be efficiently activated and express luciferase (Figure 4C). We found that ipi-DMAb, treme-DMAb and the positive control αCTLA-4 antibody induced luciferase expression in a dose-dependent manner (Figure 4D). As expected, the negative control antibody (9D9) did not induce luciferase expression (Figure 4D). Interestingly, the treme-DMAb induced luciferase expression at lower concentrations compared to the ipi-DMAb, potentially indicating more potent blocking function (Figure 4D). Together, these results demonstrate that anti-CTLA-4 antibodies produced by DNA plasmids in vivo are functional.
Discussion
Here we have described and validated a novel platform for the administration of immune checkpoint blockade antibodies through the use of DNA plasmids encoding IgG. The CELLECTRA electroporation approach described here has been widely used in clinical DNA vaccine trials, has a favorable safety and tolerability profile, and would be more rapid and cost efficient for mAb delivery compared to intravenous injection, which may broaden the applications that can be used for checkpoint antibodies (12,13). In these pre-clinical studies, engineered DMAbs were efficient at driving in vivo expression of anti-CTLA-4 mAbs, and exhibited properties of IgG encoded CTLA-4 mAb. The DMAbs were capable of inducing potent anti-tumor immunity and CD8 T cell infiltration while decreasing Treg infiltration. These results suggest that this technology could be used for novel therapeutic approaches that are currently limited for biologic mAbs, such as maintenance therapies.
Both DNA plasmid and viral delivery approaches have been used in pre-clinical models to deliver therapeutic mAbs for cancer therapy(14–16). However, these approaches thus far have focused on antibodies targeting cancer surface antigens or angiogenic factors. While viral vectors can drive high expression, their use is limited to seronegative individuals, they can genetically mark patients, and they are difficult to re-administer due to seroconversion (7). Here, we report that the DMAb approach for immune checkpoint delivery can result in significant and prolonged in vivo expression from as little as a single dose.
Immune checkpoint blockade combination therapies are showing synergy in the clinic for certain indications(1). While combination therapy between ipilimumab and nivolumab is highly effective in melanoma patients, it also results in even more toxicity compared to monotherapy(17). Unfortunately, the full scope of this toxicity was difficult to predict using pre-clinical mouse or non-human primate models(18,19). Due to this toxicity concern, next generation versions of ipilimumab that can be selectively activated within tumors are currently being developed and tested in clinical trials(9,20). Additional designs are being developed to enhance the effector function induced by these antibodies, including Fc mutations that enhance binding to the human FcγRIIIa as well as non-fucosylated versions with enhanced antibody-dependent cell-mediated cytotoxicity activity(9,21). These important antibody improvements may provide expanded uses for CTLA-4 targeted antibodies in the future.
Additional areas for further development of this technology could include exploration of different isotypes to control function or expression in vivo, as well as new approaches for in vivo regulation of DMAb expression such as gene switch platforms that utilize small molecule regulation as well as allosteric ribozymes(22–24). These interesting approaches could allow for self-regulation of therapy.
Using the DMAb platform, we have successfully delivered complex bi-specific antibodies, in addition to multiple different antibodies simultaneously within the same animal in the infectious disease arena(4,5). This technology could therefore be adapted to include combination therapy with anti-PD1 DMAbs or with vaccines to open up additional therapeutic avenues. Further study of this novel approach is likely to provide valuable additions to the cancer therapy toolbox.
Supplementary Material
Statement of Significance.
DNA-encoded monoclonal antibodies represent a novel technology for delivery and expression of immune checkpoint blockade antibodies, thus expanding patient access to, and possible clinical applications of, these therapies.
Acknowledgments
Financial Support:
This work was supported by an NIH/NCI NRSA Individual Fellowship (F32 CA213795 to E.K. Duperret), a Penn/Wistar Institute NIH SPORE (P50CA174523 to D.B. Weiner), the Wistar National Cancer Institute Cancer Center (P30 CA010815), the W.W. Smith Family Trust (to D.B. Weiner), funding from the Basser Foundation (to D.B. Weiner) and a grant from Inovio Pharmaceuticals (to D.B. Weiner).
Footnotes
Conflicts of Interest:
M. C. Wise, T. Smith, K. Broderick, E. Masteller, J. J. Kim and L. Humeau are employees of Inovio Pharmaceuticals and as such receive salary and benefits, including ownership of stock and stock options. K.M. receives a commercial grant from Inovio Pharmaceuticals, and has served as a consultant/advisor for Inovio Pharmaceuticals. D.B. Weiner receives a commercial research grant from Inovio Pharmaceuticals, has received honoraria from Inovio Pharmaceuticals, GeneOne, AstraZeneca, has ownership interest (including patents) in Inovio Pharmaceuticals and is a consultant/advisory board member for Inovio Pharmaceuticals. The other authors declare no competing financial interests.
References:
- 1.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade Science. American Association for the Advancement of Science; 2018;359:1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrews A Treating with Checkpoint Inhibitors-Figure $1 Million per Patient Am Heal drug benefits. Engage Healthcare Communications, LLC; 2015;8:9. [PMC free article] [PubMed] [Google Scholar]
- 3.Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs. Taylor & Francis; 2015;7:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elliott STC, Kallewaard NL, Benjamin E, Wachter-Rosati L, McAuliffe JM, Patel A, et al. DMAb inoculation of synthetic cross reactive antibodies protects against lethal influenza A and B infections. npj Vaccines. 2017;2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel A, DiGiandomenico A, Keller AE, Smith TRF, Park DH, Ramos S, et al. An engineered bispecific DNA-encoded IgG antibody protects against Pseudomonas aeruginosa in a pneumonia challenge model. Nat Commun. 2017;8:637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muthumani K, Marnin L, Kudchodkar SB, Perales-Puchalt A, Choi H, Agarwal S, et al. Novel prostate cancer immunotherapy with a DNA-encoded anti-prostate-specific membrane antigen monoclonal antibody. Cancer Immunol Immunother. 2017;66:1577–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hollevoet K, Declerck PJ. State of play and clinical prospects of antibody gene transfer J Transl Med. BioMed Central; 2017;15:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1:32–42. [DOI] [PubMed] [Google Scholar]
- 9.Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies Cancer Cell Cell Press; 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–6. [DOI] [PubMed] [Google Scholar]
- 11.Jago CB, Yates J, Câmara NOS, Lechler RI, Lombardi G. Differential expression of CTLA-4 among T cell subsets Clin Exp Immunol. Wiley-Blackwell; 2004;136:463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trimble CL, Morrow MP, Kraynyak KA, Shen X, Dallas M, Yan J, et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: a randomised, double-blind, placebo-controlled phase 2b trial. Lancet (London, England). 2015;386:2078–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tebas P, Roberts CC, Muthumani K, Reuschel EL, Kudchodkar SB, Zaidi FI, et al. Safety and Immunogenicity of an Anti-Zika Virus DNA Vaccine - Preliminary Report. N Engl J Med. 2017;NEJMoa1708120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang M, Shi W, Zhang Q, Wang X, Guo M, Cui Z, et al. Gene Therapy Using Adenovirus-Mediated Full-length Anti-HER-2 Antibody for HER-2 Overexpression Cancers. Clin Cancer Res. 2006;12:6179–85. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe M, Boyer JL, Crystal RG. AAVrh.10-mediated genetic delivery of bevacizumab to the pleura to provide local anti-VEGF to suppress growth of metastatic lung tumors Gene Ther. NIH Public Access; 2010;17:1042–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi J, Liu Y, Zheng Y, Guo Y, Zhang J, Cheung P -t., et al. Therapeutic Expression of an Anti-Death Receptor 5 Single-Chain Fixed-Variable Region Prevents Tumor Growth in Mice. Cancer Res. 2006;66:11946–53. [DOI] [PubMed] [Google Scholar]
- 17.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma N Engl J Med. Massachusetts Medical Society; 2017;377:1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keler T, Halk E, Vitale L, O’Neill T, Blanset D, Lee S, et al. Activity and safety of CTLA-4 blockade combined with vaccines in cynomolgus macaques J Immunol. American Association of Immunologists; 2003;171:6251–9. [DOI] [PubMed] [Google Scholar]
- 19.Selby MJ, Engelhardt JJ, Johnston RJ, Lu L-S, Han M, Thudium K, et al. Preclinical Development of Ipilimumab and Nivolumab Combination Immunotherapy: Mouse Tumor Models, In Vitro Functional Studies, and Cynomolgus Macaque Toxicology. Ahmad A, editor PLoS One. Public Library of Science; 2016;11:e0161779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korman AJ, Engelhardt J, Loffredo J, Valle J, Akter R, Vuyyuru R, et al. Abstract SY09–01: Next-generation anti-CTLA-4 antibodies Cancer Res. American Association for Cancer Research; 2017;77:SY09–01-SY09–01. [Google Scholar]
- 21.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function Proc Natl Acad Sci U S A. National Academy of Sciences; 2006;103:4005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nomura Y, Zhou L, Miu A, Yokobayashi Y. Controlling mammalian gene expression by allosteric hepatitis delta virus ribozymes ACS Synth Biol. American Chemical Society; 2013;2:684–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DRAGHIA-AKLI R MALONE PB, HILL LA, ELLIS KM, SCHWARTZ RJ, NORDSTROM JL. Enhanced animal growth via ligand-regulated GHRH myogenic-injectable vectors FASEB J. Federation of American Societies for Experimental Biology; 2002;16:426–8. [DOI] [PubMed] [Google Scholar]
- 24.Burnside ER, De Winter F, Didangelos A, James ND, Andreica E-C, Layard-Horsfall H, et al. Immune-evasive gene switch enables regulated delivery of chondroitinase after spinal cord injury Brain. Oxford University Press; 2018;141:2362–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.