

Abstract
UBE2N is a K63-specific ubiquitin conjugase linked to various immune disorders and cancer. Here we demonstrate that UBE2N and its partners UBE2V1 and UBE2V2 are highly expressed in malignant melanoma. Silencing of UBE2N and its partners significantly decreased melanoma cell proliferation and subcutaneous tumor growth. This was accompanied by increased expression of E-cadherin, p16, and MC1R and decreased expression of melanoma malignancy markers including SOX10, Nestin, and ABCB5. Mass spectrometry-based phosphoproteomic analysis revealed that UBE2N loss resulted in distinct alterations to the signaling landscape: MEK/ERK signaling was impaired, FRA1 and SOX10 gene regulators were downregulated, and p53 and p16 tumor suppressors were upregulated. Similar to inhibition of UBE2N and MEK, silencing FRA1 decreased SOX10 expression and cell proliferation. Conversely, exogenous expression of active FRA1 increased pMEK and SOX10 expression and restored anchorage-independent cell growth of cells with UBE2N loss. Systemic delivery of NSC697923, a small molecule inhibitor of UBE2N, significantly decreased melanoma xenograft growth. These data indicate that UBE2N is a novel regulator of the MEK/FRA1/SOX10 signaling cascade and is indispensable for malignant melanoma growth. Our findings establish the basis for targeting UBE2N as a potential treatment strategy for melanoma.
Keywords: melanoma, UBE2N, MEK/FRA1/SOX10 signaling
Introduction
Metastatic melanoma is the most aggressive and difficult to treat skin cancer. The incidence of melanoma is on the rise especially among the young population. The NIH SEER program estimated that 87,110 people were diagnosed with melanoma in the United States in 2017, accounting for 5.2% of all new cases of cancer, and that 11% of these patients would succumb to the disease (1). In recent years, immunotherapies and BRAF/MEK oncokinase inhibitors have yielded a high response rate (2–6). However, these treatments fail to produce a long-lasting benefit for the majority of responders due to the rapid development of resistance through cancer cell intrinsic and extrinsic mechanisms (7,8).
The RAS/RAF/MEK/ERK signaling cascade is commonly activated by growth factors and cytokines via an orchestrated cascade of reversible posttranslational modifications, most notably phosphorylation and ubiquitination. In cancer cells, this pathway is often constitutively active as a result of genetic changes. Specifically, BRAF mutation occurs in nearly 70% of cutaneous melanomas and 90% of these mutations are BRAFV600E which is a potent activator of the downstream MEK/ERK kinases (9). The transformation potency of mutant BRAF is subject to further regulation by ubiquitination (10).
Ubiquitination is a rather complex and multifaceted process (11). Poly-ubiquitination involves binding of additional ubiquitin monomers to a lysine (K) or methionine (M) residue (e.g. K48, K63, and M1) of the preceding ubiquitin, forming structurally and functionally distinct ubiquitin polymers (Ub). K48-Ub primarily targets proteins to the 26S proteasome complex for degradation, whereas K63-Ub regulates signal transduction and gene expression (12). Ubiquitination typically requires concerted actions of an ubiquitin activating E1 enzyme, an E2 ubiquitin conjugase and E3 ubiquitin ligases and it is proteolytically cleaved by deubiquitinases (11). While E1 is functionally ubiquitous, E2 and E3 enzymes and deubiquitinases are multifaceted. For example, CYLD is a deubiquitinase specific for M1-Ub and K63-Ub and it inhibits inflammation and tumorigenesis (13)(14). In contrast, UBE2N (also called Ubc13) is a K63-Ub-specific E2 enzyme recently characterized as a crucial growth promoter of several human cancers, such as breast cancer, neuroblastoma, B-cell lymphoma, and colon cancer (15–19). UBE2N interacts with a non-catalytic variant UBE2V1 (UeV1) or UBE2V2 (MMS2) to activate NF-κB and p38 signaling pathways (15) and DNA repair (20), respectively. However, little is understood about the role of UBE2N in melanoma and the mechanisms underlying its function in cancer.
In this study, we demonstrated that UBE2N and variants are essential for melanoma cell proliferation, survival, and malignant progression. Using unbiased proteomic approach, we revealed a global effect of UBE2N on cell signaling and gene regulation, and identified a MEK/FRA1/SOX10 signaling cascade acting downstream of UBE2N. We also verified FRA1 as a key promoter of pMEK and SOX10 expression and melanoma growth. Finally, we showed the feasibility of pharmacologically interfering with UBE2N function to hinder melanoma xenograft growth in mice.
Materials and Methods
Cell culture and gene transduction
Cells were cultured at a 37°C incubator supplemented with 5% CO2 and all culture media and supplements were obtained from (Thermo Fisher Scientific, Waltham, MA). A375, A2058, and Skmel28, and B16-F10 cells were obtained from (ATCC, Manassas, VA) via Duke Cell Culture Facility. DM598, DM733, and DM738 cells were kindly provided by Dr. Hilliard Seigler (Duke University Medical Center). They were derived from primary biopsies of metastatic melanoma obtained under a Duke University Institutional Review Board approved protocol with written informed consent from patients. These studies were conducted in accordance with U.S. Common Rule. A375, A2058, B16, DM598, DM733, and DM738 were maintained in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS). Cell lines were confirmed to express Mart-1 and Nestin, but no further authentication was performed in this study. A2058, A375, and B16 were negative for all pathogens, except mycoplasma sp., as screened by PCR (IDEXX BioResearch, Columbia, MO). Other cell lines were not screened. Normal human melanocytes were isolated from surgically discarded foreskin samples obtained in accordance to an IRB protocol approved by Duke, and cultured in complete melanocyte growth medium 254. The pLKO lenti-viral shRNA and CRISPR constructs specific for UBE2N, UBE2V1, UBE2V2, and FRA1 (Supplementary Table S1) were obtained from (Duke Functional Genomics Shared Resource, Durham, NC). The retroviral expression vector for the constitutively active FRA1DD mutant was generated as previously described (21). For gene transduction, A375, A2058, and DM598 cells were incubated with the viral media supplemented with 8 μg/ml polybrene overnight at a 37°C incubator supplemented with 5% CO2. Transduced cells were selected with 1 μg/ml puromycin for 1–2 weeks.
Cell growth assay and soft agar colony formation
For cell proliferation assay, cells were seeded onto 96-well dishes at 5,000 cells/well and next day, cells were incubated in quadruples for 48 hours with increasing concentrations of NSC697923 (0, 1, 2, 4, and 8 μM) (Selleckchem, Houston, TX), cells were incubated with 5 μL 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (20mg/mL, Sigma-Aldrich, St. Louis, MO) for 2 hours and media were then replaced with DMSO to dissolve the thiazol crystals. Absorbance was measured using a plate reader (Synergy H1, BioTek Winooski, VT). For soft agar colony formation, 1 mL of 0.5% noble agar was transferred to each well of a 6-well plate to form the base layer. Gene transduced A375 cells were mixed with 0.35% Noble agar, and then seeded in triplicates at 5,000 cells/well onto the solidified base. About 15 minutes later, one mL of 10% FBS/DMEM was carefully added to each well. The plates were kept in a 37°C incubator with 5% CO2 and 200 μl of 10% FBS/DMEM fresh media were added to each well every other day. Two weeks later colonies were stained with nitro blue tetrazolium (NBT) dye overnight at 37°C. Colonies were imaged and counted using the Olympus microscopic imaging system.
Immunoblotting
Primary human melanocytes, A375, A2058, DM598, DM733, DM738, and B16 cells were grown to about 90% confluence in either the 10%FBS/DMEM medium or the complete melanocyte medium. Protein lysates were collected with RIPA buffer. Transduced cells were re-seeded 1–2 weeks after selection with puromycin and protein lysates were collected 24 hours later. For Western blotting, proteins (20 μg/sample) were separated by 10–12% polyacrylamide gel electrophoresis, transferred onto nitrocellulose membrane, and then blotted with the following antibodies listed in (Supplementary Table S2). The blots were detected with IRDye-conjugated secondary antibodies (Invitrogen), and imaged with the Odyssey Imagining system (Li-COR, Lincoln, NE).
Flow Cytometry
A375 cells transduced to express shCon, shUBE2N, shUBE2V1 or shUBE2V2 were fixed with 70% ethanol for 30 minutes at 4°C, treated with 50 μL (100 μg/mL) of ribonuclease solution, and stained with 200 μL of Propidium Iodide. Cells were analyzed using the BD LSRFortessa cell analyzer. Cell cycle analysis was performed using FlowJo software.
Histology and immunostaining
Immunofluorescent (IF) staining was performed with frozen tissue sections, as previously described (21). Primary antibodies used for IF are listed in (Supplementary Table S2). Samples were counterstained with DAPI (Thermo Fisher Scientific). Images were taken with the Olympus BX41 microscopic imaging system or the Olympus IX73 imaging system (Center Valley, PA).
Real-time RT-PCR
Total RNA was isolated from cells using the Qiagen RNAeasy kit (Qiagen, Valencia, CA). cDNA was generated by iScript reverse transcriptase supermix and real time PCR was carried out using the SYBR green master mix (BioRad, Hercules, CA) in StepOnePlus Real time systems (Applied Biosystems). PCR primers spanning the exon-exon junctions were designed specifically for SOX10, FRA1, UBE2N, UBE2V1, UBE2V2, p53, p16, and GAPDH (Supplementary Table S3).
Phosphoproteomic analysis
Phosphoproteomic analysis was performed as previously described (Foster MW et al. JPR 2017), and expanded methods are in Supplemental Methods. Briefly, lysates were prepared in 8 M urea buffer, and following reduction/alkylation, proteins were digested overnight with TPCK-trypsin. Following cleanup, phosphopeptides were enriched using titanium dioxide, and enriched peptides were analyzed using a nanoAcquity UPLC system (Waters) interfaced to a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific). Label-free quantitation using accurate-mass and retention time alignment was performed in Rosetta Elucidator, and database searching was performed using Mascot 2.5 (Matrix Science). Raw data have been deposited to the MassIVE repository (MSV000082208).
Animal studies
Animal studies were performed in compliance with Duke Animal Care and Use Committee. Four to six-weeks old NSG SCID mice were obtained from Duke Cancer Institute Animal Facility and animals (n=5/group) were injected subcutaneously in the right and left flank regions with 150,000 A375 cells expressing shCon, shUBE2N, shUBE2V1, or shUBE2V2 suspended in 100 μl (PBS/matrigel mixed at 3:1 vol/vol). Tumors were measured biweekly with a caliper for up to 4 weeks after which the animals were sacrificed. Tumor tissues were fixed in 10% neutral buffered formalin for histology, flash frozen for protein extraction, or embedded in OCT for cryosectioning and immunofluorescence staining. For the pharmacological study, 8 NSG SCID mice were injected subcutaneously with 150,000 A375 cells in both flanks. Four mice were randomly selected the next day, and treated every other day for a total of 18 days with either solvent (0.5% DMSO and 30% Corn oil) or 5 mg/kg NSC697923. Tumors were measured every 4 days and animals were euthanized for tissue collection as described above.
Results
UBE2N is required for melanoma cell proliferation.
To assess the relevance of UBE2N in melanoma, we examined NCBI GEO gene expression datasets, and found that the UBE2N mRNA levels were higher in the metastatic tumors compared to normal skin and benign nevi (Figure 1A). By immunoblotting, we found that UBE2N, UBE2V1 and UBE2V2 were ubiquitously expressed in melanocytes, but UBE2V2 and UBE2N, particularly the mono-ubiquitinated UBE2N (UBE2N-Ub) (22), exhibited increased expression in the metastatic melanoma cell lines, including A2058, A375, DM598, DM733, DM738, and Skmel28, compared to normal melanocytes. The increased expressions of UBE2N, UBE2V1, and UBE2V2 were especially pronounced in melanoma cells grown with the melanocyte culture media (Figure 1B). These data indicate that UBE2N and variants are highly expressed in melanoma cells.
Figure 1. UBE2N is important for melanoma cell proliferation.

(A) Relative UBE2N mRNA expression levels obtained from NCBI GEO data sets GDS3966 (31 primary and 52 metastatic melanomas) and GDS1375 (7 normal skin, 18 nevi and 37 malignant melanomas). Graphs represent averages of UBE2N mRNA normalized to that of primary tumor or normal skin +/− SD. (B) Immunoblotting for UBE2N, UBE2V1 and UBE2V2 in primary human melanocytes and metastatic human melanoma cell lines cultured with either 10% FBS/DMEM or melanocyte culture media. Actin was used as a loading control. (C) Confirmation of shRNA-mediated gene silencing via immunoblotting. (D) Cell proliferation via MTT assay. Graphs represent average percentages of cell proliferation normalized to control A375 cells at 72 hours after seeding +/− SD, p<0.05. (E) Cell cycle analysis of gene transduced A375 cells via flow cytometry. (F) Graph represents average percent of cells in subG0, G0, S and M-phases +/− SD. P-values of ≤0.01 were obtained via unpaired student T-test.
To determine the functional importance of UBE2N, we first performed shRNA-mediated gene silencing of UBE2N, UBE2V1, and UBE2V2 in A375 melanoma cells. The efficiency of gene silencing was as verified by immunoblotting and real time q-PCR (Figure 1C, Supplementary Figure S1A-C). Consistent with UBE2N being the primary E2 enzyme responsible for K63-Ub, gene silencing of UBE2N and variants markedly reduced K63-Ub of the whole cell lysates, as shown by immunoblotting (Supplementary Figure S1D). MTT-based cell growth analysis showed that gene silenced cells grew significantly slower than control cells (Figure 1D). Further cell cycle analysis via flow-cytometry revealed that UBE2N-loss significantly increased the percentage of cells in the subG0 and G0/G1, and inhibited M-phase entry (Figure 1E-F). To verify the observed growth effects, we performed gene silencing with a second shRNA construct targeting UBE2N and CRISPR-constructs targeting UBE2V1 and UBE2V2 in both A375 and A2058 cells. Time-course growth analysis showed that each of these targeting strategies resulted in decreased cell proliferation (Supplementary Figure S2A-B). These data indicate that UBE2N and variants are crucial for melanoma cell proliferation.
UBE2N and variants are required for melanoma growth and progression in vivo.
To determine whether UBE2N and its variants are important for melanoma growth in vivo, we performed subcutaneous tumor growth analysis. We found that cells with gene silencing of UBE2N or the variants formed significantly smaller tumors than those of the control cells (Figure 2A-B). Consistently, these tumors were less proliferative and more apoptotic, as indicated by reduced number of Ki-67-positive cells and increased number of cleaved caspase 3-positive cells, respectively (Figures 2C-D). In addition, the shUBE2N, shUBE2V1 and the shUBE2V2 tumors showed a markedly increased expression of the epithelial cell marker E-cadherin (Figures 2E). Interestingly, gene silencing of UBE2N and UBE2V1, but not UBE2V2, reduced expression of the cell adhesion marker β1-integrin (Figure 2F).
Figure 2. Gene silencing of UBE2N inhibits melanoma growth and progression.

(A) Image of subcutaneous tumors generated in NSG SCID mice with gene transduced A375 cells. (B) Tumor size. Graph depicts average tumor size (n=10/group) +/− SD. P-values of less than 0.05 were obtained via unpaired student T-Test comparing gene silenced groups to the control group. (C-F) Immunofluorescence staining of 4-weeks old subcutaneous tumors for Ki-67, cleaved caspase 3, E-cadherin and β1-integrin [orange]. Nuclei [blue].
Cells with UBE2N loss often appeared bigger and flatter than control cells in culture, which is indicative of differentiation and senescence. By immunostaining, we found that both the melanocyte differentiation marker MC1R and the cell growth inhibitor and senescent marker p16 were significantly increased in the UBE2N, UBE2V1 and UBE2V2 gene silenced tumors compared to the control tumors (Figure 3A-B). In contrast, markers previously linked to melanoma malignancy, including SOX10 (23) and Nestin (24), were significantly decreased in these tumors (Figure 3C-D). These results indicate that UBE2N and its variants are required for melanoma growth and malignant progression.
Figure 3. UBE2N is required for the suppression of melanoma cell differentiation.

(A-D) Immunostaining of subcutaneous tumors for MC1R, p16, SOX10, and Nestin [orange], Nuclei [blue]. Graphs depict average percentages of MC1R, p16, SOX10, and Nestin-positive cells over total cell population quantified from 5–10 images of each group +/− SD. p-values of less than 0.01 were obtained via student T-test.
UBE2N is essential for the expression of a pro-oncogenic protein landscape.
Phosphoproteomics has emerged as an important technique for investigating aberrant signaling in cancer and response to therapy (25)(26). We used mass spectrometry to quantify changes in the phosphoproteomes of A375 cells in response to gene silencing of UBE2N. A total of 7,022 phosphopeptides were quantified (Supplementary Table S4–5), with 2,127 peptides that reached an FDR-corrected p<0.05; of these 258 were upregulated >5-fold, and 509 were downregulated <5-fold in the shUBE2N-expressing vs. control cells (Figure 4A). Contained within these dysregulated phosphopeptides were a number of well-characterized and regulatory sites in a range of pro-growth and pro-invasion molecules (Figure 4B). For example, phosphorylation of ERK1/2 (at sites critical for kinase activation) was markedly reduced in shUBE2N-expressing cells while ERK1/2 protein levels were unchanged (Figure 4C), and this correlated with the reduction of phosphorylation of ERK sensors such as the PXTP site (Thr526) of the transcriptional repressor ERF (Figure 4B), an Ets family member crucial for cell-cycle progression (27). We also found that the phosphorylation of MEK1/2 (Ser217/221), the upstream activator of ERK1/2, was similarly affected (Figure 4C). Interestingly, phosphorylation of MEK2 (Thr394), a modification known to be regulated by CDK5 to inhibit the canonical MEK activity (28), was increased in cells with UBE2N loss (Figure 4B). These effects on the MEK1/2-ERK1/2 pathway were consistent with the anti-proliferative effects of UBE2N knockdown.
Figure 4. UBE2N is essential for the expression of a pro-oncogenic protein landscape.

(A) Phosphopeptide expression in A375 cells expressing shUBE2N versus shCon was visualized by volcano plot. Phoshopeptides in red and blue were ≥5-fold higher and ≥−5-fold lower, respectively, in shUBE2N cells compared to shCon cells (w/ FDR-corrected p<0.05; unpaired T-test). (B) Differentially-expressed phosphopeptides were queried in the phosphosite database (www.phosphosite.org). The expression values of select phosphosites identified in low throughput studies (i.e. validated sites with known target kinases and/or regulatory function) were converted to Z-scores followed by two-dimensional hierarchical clustering using JMP Pro. (C) Protein and phosphoprotein expressions were confirmed by Western blotting. (D) mRNA expression was quantified by real-time RT-PCR. Total RNA samples were isolated from A375 cells expressing shCon and shUBE2N. Graphs represent averages of relative mRNA levels of FRA1, SOX10, p53, and p16 normalized to GAPDH +/− SD. p-values <0.01 were obtained via unpaired student T-test.
While total levels of ERK and MEK1/2 proteins were unchanged, we identified a number of dysregulated phosphoproteins with marked changes in total protein abundance in shUBE2N-expressing cells. These included SOX10 and Fos-related antigen 1 (FRA1), transcription factors important for melanoma progression and therapeutic resistance (23)(29) (Figure 4C). In addition, we observed an upregulation of p53 and p16 tumor suppressors (Figure 4C). The relative mRNA levels of FRA1, SOX10, p16, and p53 in shUBE2N versus control cells correlated well with their protein expression, demonstrating that these proteins were transcriptionally regulated (Figure 4D). To verify the observed molecular changes, we performed immunoblotting with lysates collected from A375 and A2058 cells that had undergone gene targeting with a second shRNA construct for UBE2N and a CRISPR construct for each of the variants. We found that each of these gene targeting approaches resulted in decreased expression of pMEK, pERK, SOX10, and FRA1 in both A375 and A2058 cells (Supplementary Figure S2C-D). Collectively, these data show that UBE2N regulates an oncogenic proteome at both post-translational and transcriptional levels.
A375 and A2058 cells express the BRAF(V600E) mutant. We examined whether the observed growth and molecular effects of UBE2N are specific to BRAF mutant cells. For this, we targeted UBE2N and variants in DM598 cells which harbor an NRAS mutation (30). Interestingly, we found that silencing of UBE2N and variants each reduced cell proliferation of DM598 cells, but the effects were less dramatic than those on A375 and A2058 cells (Supplementary Figure S2A-B and S3A). Immunoblotting showed that pMEK, pERK, SOX10, and FRA1 were not affected by UBE2N loss in DM598 cells (Supplementary Figure S3B). These data implicate that UBE2N regulation of MEK/ERK signaling might be specific to BRAF mutant cells.
FRA1 mediates UBE2N regulation of MEK and SOX10, and plays a pivotal role in melanoma growth.
To verify that the observed molecular changes were due to reduced UBE2N function, we treated cells with increasing doses of NSC697923, a small molecular compound that specifically inhibits K63-Ub via disruption of UBE2N interaction with UBE2V1 and UBE2V2 (31). By immunoblotting, we found that NSC697923 decreased FRA1 and SOX10 expression in a dose-dependent manner in both A375 and A2058 cells, as did treatments of the MEK inhibitor trametinib (Supplementary Figure S4A-B), suggesting that UBE2N acts through MEK to regulate FRA1 and SOX10.
FRA1 is an AP1 family gene regulator and promoter of many malignancies including melanoma (29). We asked whether FRA1 loss contributes to the growth defects and other molecular changes observed in shUBE2N cells. To address this question, we first expressed a constitutively active FRA1 mutant (FRA1DD) in A375 cells expressing shUBE2N. By immunoblotting, we found that exogenous FRA1DD expression restored the expressions of pMEK, pERK, and SOX10 that were otherwise downregulated by UBE2N gene silencing or treatment of NSC697923 (Figure 5A, Supplementary Figure S4C). ATPase-based cell growth analysis showed that FRA1DD expression restored proliferation of cells with UBE2N-targeted gene silencing (Figure 5B). Additionally, FRA1DD decreased sensitivity of A375 cells to NSC697923 treatment at both high and low serum culture conditions as measured for the 2 and 4 μM doses (Supplementary Figure S4D-E). Moreover, FRA1DD expressing cells showed increased subcutaneous tumor growth (Supplementary Figure S4F-G).
Figure 5. FRA1 mediates UBE2N regulation of MEK and SOX10, and plays a pivotal role in melanoma growth.
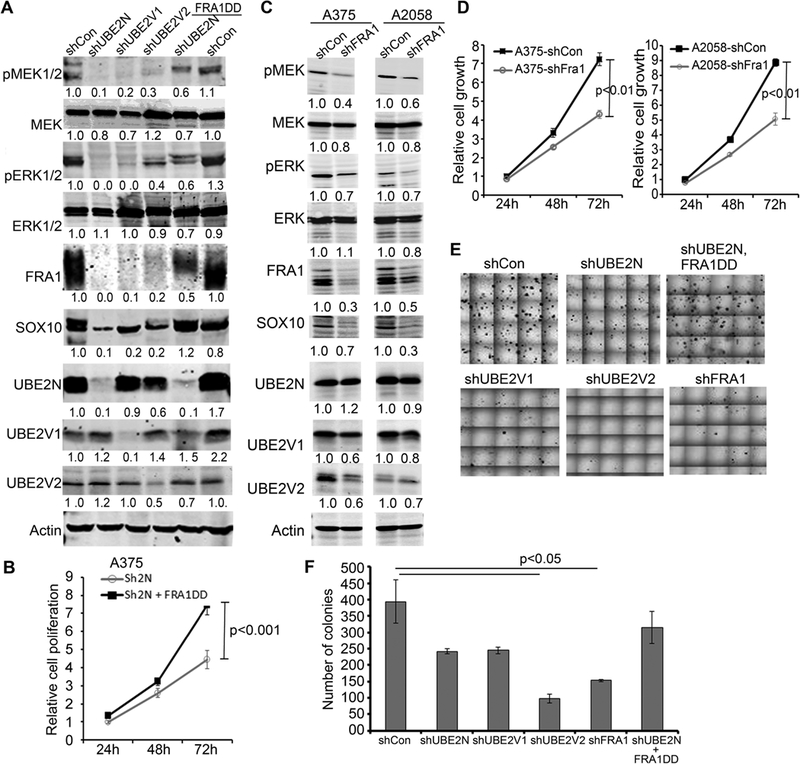
(A) Immunoblotting of protein lysates isolated from A375 cells transduced to express shCon, shUBE2N, shUBE2V1, shUBE2V2, and FRA1DD. (B) Cell growth analysis via MTT assay. Graph depicts growth curve of A375 cells in triplicates expressing shUBE2N alone or with FRA1DD +/− SD. (C) Immunoblotting of lysates isolated from A375 and A2058 cells transduced to express shCon or shFRA1. (D) Cell growth analysis via MTT assay. Graphs depict time-course growth of triplicates of transduced A375 and A2058 cells expressing shFRA1 normalized to control cells +/− SD. (E) Representative images of soft agar colonies of transduced A375 cells. (F) Colony counts. Graph represents average number of soft agar colonies of triplicate dishes +/− SD, p-values<0.05 were obtained via unpaired student T-test.
Next, we performed FRA1-targeted gene silencing with lentiviral shRNA in A375 and A2058 cells. We found that FRA1 gene silencing decreased pMEK, pERK, and SOX10 expressions (Figure 5C), and inhibited proliferation of both cell lines (Figure 5D). Further soft agar colony formation assay showed that gene silencing of FRA1, UBE2N, UBE2V1, or UBE2V2 each resulted in a significant decrease of colony formation, whereas FRA1DD increased it (Figure 5E-F). These data indicated that FRA1 is an important mediator of UBE2N regulation of MEK/SOX10 signaling, and plays a pivotal role in melanoma growth.
Pharmacological inhibition of UBE2N mitigates melanoma growth in vivo.
Consistent with gene silencing effects, treatment of NSC697923 inhibited the proliferation of A375, A2058, and B16 melanoma cell lines in a dose response manner, and sensitized cells to death under serum-starved conditions (Figure 6A-C). In addition, we observed that NSC697923 significantly slowed cell migration, as demonstrated by scratch-wounding assay (Supplementary Figure S5A-B).
Figure 6. Systemic treatment of NSC697923 inhibits melanoma xenograft growth, and decreases melanoma tumor growth and malignancy.

(A) Cell growth analysis via MTT assay. A375, A2058, and B16 were treated with varying concentrations of NSC697923 in the presence of 1% or 10% FBS. Graphs represent averages of tetrad wells +/− SD, p-values<0.05 were obtained via unpaired student T-test. (B) Subcutaneous tumor growth kinetics. A375 cells (1.5×105/injection) were injected into both flanks of NSG mice. Animals (n=4/group) were then treated every other day with intraperitoneal dose of 100 μl solvent (5% DMSO in 30% corn oil) or NSC697923 (5 mg/Kg body weight). (C) Images of the subcutaneous tumors collected 22 days after injection. (D) Immunoblotting of protein lysates isolated from subcutaneous tumors for K63-Ub and Actin. (E-J) Immunostaining of the frozen sections of the subcutaneous tumors for Ki-67, cleaved caspase 3, SOX10, Nestin, ABCB5, and p16 [orange]. Nuclei [blue]. Graphs depict quantification of relative percentages or intensities of cells positively stained for the above markers. P-values less than 0.05 were obtained via unpaired student T-test.
To establish the utility of NSC696723 for in vivo applications, we examined its effect on the A375 xenograft model. We found that intraperitoneal injections of NSC697923 (5 mg/Kg) administered every other day significantly reduced subcutaneous tumor growth of A375 cells in immunodeficient NSG mice (Figure 6B-C). Immunoblotting of tumor protein lysates confirmed that total K63-Ub was reduced by about 2-fold in tissues treated with NSC697923 (Figure 6D). By immunostaining, we found that tumors treated with NSC697923 were less proliferative and more apoptotic than the control tumors, as indicated by the reduced number of Ki-67 cells (Figure 6E) and the increased number of cleaved caspase 3-positive cells, respectively (Figure 6F). In addition, we found that NSC697923 treated tumors expressed markedly reduced levels of SOX10, Nestin and ABCB5 (Figure 6G-I). In contrast, p16 was increased in the drug-treated tissues (Figure 6J). During the course of treatment, none of the treated animals showed any superficial signs of toxicity or significant weight loss (Supplementary Figure S6A-C). These results indicate that systemic delivery of NSC697923 impedes melanoma growth by decreasing the expression of melanoma malignancy markers and increasing the expression of tumor suppressors.
Discussion
Our study demonstrates a critical role of UBE2N and its variant partners in melanoma growth and progression. We characterized UBE2N as a negative regulator of tumor suppressors and a positive regulator of oncogenic proteins. Specifically, our data support a working model where FRA1 as a crucial effector molecule acting downstream of UBE2N to maintain the MEK/FRA1/SOX10 signaling cascade and melanoma malignancy (Figure 7). Most importantly, we established the preclinical feasibility of applying a small molecular inhibitor of UBE2N to attenuate melanoma growth in vivo. Our findings underscore UBE2N as a new promising therapeutic target for melanoma. Given that UBE2N is relevant to many other cancers (15–19), the mechanistic insights obtained from this study have broad implications.
Figure 7. Working model for UBE2N function in melanoma.

UBE2N acts in part through a MEK/FRA1/SOX10 signaling cascade to promote melanoma growth, survival, and malignant progression.
Previous studies have linked UBE2N to NF-κB, p38, and p53 signaling pathways in several cancer models (15–19). Our studies have for the first time identified a novel MEK/FRA1/SOX10 signaling cascade downstream of UBE2N. It is rather intriguing that UBE2N regulation appears to be specific to BRAF but not NRAS mutant cells. BRAF(V600E) is subject to K63-Ub which is required for BRAF(V600E)-driven transformation (10). While our attempts to detect BRAF-K63-Ub were not successful, we observed that UBE2N loss increased MEK2 phosphorylation at a non-canonical Thr394 residue, a modification previously shown to be responsible for CDK5-mediated MEK/ERK inactivation and control of insulin resistance (28). Further studies are needed to determine whether this is the main mechanism underlying MEK inactivation by UBE2N-loss. It is also interesting to observe that knockdown of UBE2V2 appears to induce a more potent effect on cell proliferation than that of other subunits. Like any other gene targeting approach, there is a risk of off-target effects. RT-PCR and immunoblotting data show that UBE2N and UBE2V1 are highly expressed in shUBE2V2 cells, suggesting that non-specific targeting of UBE2N and UBE2V1 is unlikely the main cause of the dramatic phenotype. While the high efficiency of UBE2V2 knockdown and other unknown non-specific effects might contribute to the observed phenotype, it is conceivable that UBE2V2 has UBE2N-independent functions, as were recently reported for UBE2N (32,33). It is also clear that UBE2V2 has MEK-independent functions (34). This is shown by our data demonstrating that knockdown of UBE2V2 significantly decreased cell proliferation of DM598 cells but it has a negligible effect on MEK activation. Further studies are required to better understand the distinct mechanisms mediating the functions of each UBE2N subunit and how these proteins crosstalk with BRAF oncogene.
As an AP1 family transcription factor, FRA1 regulates expression of genes involved in cell cycle progression, tumor cell migration, and angiogenesis (29). Additionally, FRA1 regulates membrane lipid synthesis independent of its transcriptional activity and subsequently AKT activation (21) (35). FRA1-tageted gene silencing decreased MEK/ERK activation, and MEK-inhibition decreased FRA1 expression, supporting the existence of a positive feedback signaling loop. FRA1 silencing also slightly downregulated the expression of UBE2V2 (Figure 5C). FRA1 is known to be induced by MEK (36) and interestingly, in turn activates MEK/ERK signaling through AP1 target genes such as HBEGF, thereby maintaining a positive feedback signaling loop (37). Since the effects of UBE2N loss on MEK and FRA1 both appear to be specific to BRAF mutant cells, it is conceivable that they are regulated through a BRAF-related common target of UBE2N.
SOX10 is a neuronal cell marker expressed at a basal level in normal human melanocytes, but its expression is significantly upregulated in malignant melanomas (38). SOX10 regulates the expression of Nestin, another neural progenitor cell marker associated with melanoma growth and migration (39). ABCB5 belongs to a family of membrane transporters that regulate membrane potential and chemoresistance (40). The downregulation of SOX10, Nestin, and ABCB5 by genetic and/or pharmacological inhibition of UBE2N supports that UBE2N is essential for malignant growth of melanoma cells. Further, we established that in melanoma, SOX10 is regulated by UBE2N via FRA1. In line with our findings, UBE2V1 and UBE2V2 are overexpressed in human embryonic stem cells and cancer cell lines compared to their normal adult counterparts (41). It is highly conceivable that UBE2N and variants play a role in cancer stem cell phenotype.
Lastly, UBE2N may also be involved in modulation of a tumor microenvironment that favors tumor progression and resistance to therapy. In this regard, UBE2N loss altered expression of a plethora of molecules relevant to extracellular matrix and immune function (Supplementary Table S5). Further studies are needed to understand how UBE2N regulates the metabolic landscape of the tumor microenvironment which by itself provides a goldmine of opportunities for new cancer therapeutics (42). UBE2N also inhibits the conversion of regulatory T-cells into effector T-cells (43), suggesting that treatment of UBE2N inhibitors such as NSC697923 could improve the anti-tumor immune response stimulated by immune checkpoint inhibitors.
Supplementary Material
Acknowledgement
This work was funded by NCI (RCA188619) to J.Y. Zhang and Department of Dermatology of Duke School of Medicine. We thank So Young Kim (Duke Functional Genomics Shared Resource) for providing the CRISPR and shRNA constructs and Arthur Moseley (Duke Proteomic Core) for guidance in proteome analysis.
Financial support
NCI (RCA188619) to J.Y. Zhang, Department of Dermatology, Duke School of Medicine.
Footnotes
Conflict of interest disclosure
None
References
- 1.NA Howlader N, Krapcho M, Miller D, Bishop K, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (eds). SEER Cancer Statistics Review, 1975–2014. https://seercancergov//1975_2014/ 2017 [Google Scholar]
- 2.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robert C, Thomas L, Bondarenko I, O’Day S, M DJ, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364:2517–26 [DOI] [PubMed] [Google Scholar]
- 4.Luke JJ, Ott PA. PD-1 pathway inhibitors: the next generation of immunotherapy for advanced melanoma. Oncotarget 2015;6:3479–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14 [DOI] [PubMed] [Google Scholar]
- 7.Zhu J, Powis de Tenbossche CG, Cane S, Colau D, van Baren N, Lurquin C, et al. Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes. Nat Commun 2017;8:1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer treatment reviews 2017;52:71–81 [DOI] [PubMed] [Google Scholar]
- 9.Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, et al. High frequency of BRAF mutations in nevi. Nat Genet 2003;33:19–20 [DOI] [PubMed] [Google Scholar]
- 10.An L, Jia W, Yu Y, Zou N, Liang L, Zhao Y, et al. Lys63-linked polyubiquitination of BRAF at lysine 578 is required for BRAF-mediated signaling. Sci Rep 2013;3:2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol 2016;18:579–86 [DOI] [PubMed] [Google Scholar]
- 12.Gallo LH, Ko J, Donoghue DJ. The importance of regulatory ubiquitination in cancer and metastasis. Cell Cycle 2017;16:634–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mathis BJ, Lai Y, Qu C, Janicki JS, Cui T. CYLD-mediated signaling and diseases. Curr Drug Targets 2015;16:284–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ke H, Augustine CK, Gandham VD, Jin JY, Tyler DS, Akiyama SK, et al. CYLD inhibits melanoma growth and progression through suppression of the JNK/AP-1 and beta1-integrin signaling pathways. J Invest Dermatol 2013;133:221–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Z, Shen S, Zhang Z, Zhang W, Xiao W. Ubiquitin-conjugating enzyme complex Uev1A-Ubc13 promotes breast cancer metastasis through nuclear factor-кB mediated matrix metalloproteinase-1 gene regulation. Breast cancer research : BCR 2014;16:R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pulvino M, Liang Y, Oleksyn D, DeRan M, Van Pelt E, Shapiro J, et al. Inhibition of proliferation and survival of diffuse large B-cell lymphoma cells by a small-molecule inhibitor of the ubiquitin-conjugating enzyme Ubc13-Uev1A. Blood 2012;120:1668–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng J, Fan YH, Xu X, Zhang H, Dou J, Tang Y, et al. A small-molecule inhibitor of UBE2N induces neuroblastoma cell death via activation of p53 and JNK pathways. Cell Death Dis 2014;5:e1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gombodorj N, Yokobori T, Yoshiyama S, Kawabata-Iwakawa R, Rokudai S, Horikoshi I, et al. Inhibition of Ubiquitin-conjugating Enzyme E2 May Activate the Degradation of Hypoxia-inducible Factors and, thus, Overcome Cellular Resistance to Radiation in Colorectal Cancer. Anticancer Res 2017;37:2425–36 [DOI] [PubMed] [Google Scholar]
- 19.Wu X, Zhang W, Font-Burgada J, Palmer T, Hamil AS, Biswas SK, et al. Ubiquitin-conjugating enzyme Ubc13 controls breast cancer metastasis through a TAK1-p38 MAP kinase cascade. Proc Natl Acad Sci U S A 2014;111:13870–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersen PL, Zhou H, Pastushok L, Moraes T, McKenna S, Ziola B, et al. Distinct regulation of Ubc13 functions by the two ubiquitin-conjugating enzyme variants Mms2 and Uev1A. J Cell Biol 2005;170:745–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, Wu J, Luo S, Lechler T, Zhang JY. FRA1 promotes squamous cell carcinoma growth and metastasis through distinct AKT and c-Jun dependent mechanisms. Oncotarget 2016;7:34371–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bekes M, Okamoto K, Crist SB, Jones MJ, Chapman JR, Brasher BB, et al. DUB-resistant ubiquitin to survey ubiquitination switches in mammalian cells. Cell reports 2013;5:826–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graf SA, Busch C, Bosserhoff A-K, Besch R, Berking C. SOX10 promotes melanoma cell invasion by regulating melanoma inhibitory activity. The Journal of investigative dermatology 2014;134:2212–20 [DOI] [PubMed] [Google Scholar]
- 24.Akiyama M, Matsuda Y, Ishiwata T, Naito Z, Kawana S. Inhibition of the stem cell marker nestin reduces tumor growth and invasion of malignant melanoma. J Invest Dermatol 2013;133:1384–7 [DOI] [PubMed] [Google Scholar]
- 25.Basken J, Stuart SA, Kavran AJ, Lee T, Ebmeier CC, Old W, et al. Specificity of phosphorylation responses to MAP kinase pathway inhibitors in melanoma cells. Mol Cell Proteomics 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stuart SA, Houel S, Lee T, Wang N, Old WM, Ahn NG. A Phosphoproteomic Comparison of B-RAFV600E and MKK1/2 Inhibitors in Melanoma Cells. Mol Cell Proteomics 2015;14:1599–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le Gallic L, Sgouras D, Beal G Jr., Mavrothalassitis G. Transcriptional repressor ERF is a Ras/mitogen-activated protein kinase target that regulates cellular proliferation. Mol Cell Biol 1999;19:4121–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik-Bogoslavski D, et al. An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature 2015;517:391–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maurus K, Hufnagel A, Geiger F, Graf S, Berking C, Heinemann A, et al. The AP-1 transcription factor FOSL1 causes melanocyte reprogramming and transformation. Oncogene 2017;36:5110–21 [DOI] [PubMed] [Google Scholar]
- 30.Augustine CK, Toshimitsu H, Jung S-H, Zipfel PA, Yoo JS, Yoshimoto Y, et al. Sorafenib, a multikinase inhibitor, enhances the response of melanoma to regional chemotherapy. Molecular cancer therapeutics 2010;9:2090–101 [DOI] [PubMed] [Google Scholar]
- 31.Hodge CD, Edwards RA, Markin CJ, McDonald D, Pulvino M, Huen MS, et al. Covalent Inhibition of Ubc13 Affects Ubiquitin Signaling and Reveals Active Site Elements Important for Targeting. ACS Chem Biol 2015;10:1718–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huen MSY, Huang J, Yuan J, Yamamoto M, Akira S, Ashley C, et al. Noncanonical E2 variant-independent function of UBC13 in promoting checkpoint protein assembly. Mol Cell Biol 2008;28:6104–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brun J, Chiu R, Lockhart K, Xiao W, Wouters BG, Gray DA. hMMS2 serves a redundant role in human PCNA polyubiquitination. BMC Mol Biol 2008;9:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andersen PL, Zhou H, Pastushok L, Moraes T, McKenna S, Ziola B, et al. Distinct regulation of Ubc13 functions by the two ubiquitin-conjugating enzyme variants Mms2 and Uev1A. The Journal of cell biology 2005;170:745–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Motrich RD, Castro GM, Caputto BL. Old players with a newly defined function: Fra-1 and c-Fos support growth of human malignant breast tumors by activating membrane biogenesis at the cytoplasm. PLoS One 2013;8:e53211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verde P, Casalino L, Talotta F, Yaniv M, Weitzman JB. Deciphering AP-1 function in tumorigenesis: fra-ternizing on target promoters. Cell cycle 2007;6:2633–9 [DOI] [PubMed] [Google Scholar]
- 37.Mullenbrock S, Shah J, Cooper GM. Global expression analysis identified a preferentially nerve growth factor-induced transcriptional program regulated by sustained mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) and AP-1 protein activation during PC12 cell differentiation. J Biol Chem 2011;286:45131–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohamed A, Gonzalez RS, Lawson D, Wang J, Cohen C. SOX10 expression in malignant melanoma, carcinoma, and normal tissues. Applied Immunohistochemistry & Molecular Morphology 2013;21:506–10 [DOI] [PubMed] [Google Scholar]
- 39.Klein WM, Wu BP, Zhao S, Wu H, Klein-Szanto AJ, Tahan SR. Increased expression of stem cell markers in malignant melanoma. Modern pathology 2007;20:102–7 [DOI] [PubMed] [Google Scholar]
- 40.Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, et al. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer research 2005;65:4320–33 [DOI] [PubMed] [Google Scholar]
- 41.Ma L, Broomfield S, Lavery C, Lin SL, Xiao W, Bacchetti S. Up-regulation of CIR1/CROC1 expression upon cell immortalization and in tumor-derived human cell lines. Oncogene 1998;17:1321–6 [DOI] [PubMed] [Google Scholar]
- 42.Sugiura A, Rathmell JC. Metabolic Barriers to T Cell Function in Tumors. J Immunol 2018;200:400–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang JH, Xiao Y, Hu H, Jin J, Yu J, Zhou X, et al. Ubc13 maintains the suppressive function of regulatory T cells and prevents their conversion into effector-like T cells. Nat Immunol 2012;13:481–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.