

Summary
Studying metabolic activities in living cells is crucial for understanding human metabolism, but facile methods for profiling metabolic activities in an unbiased, hypothesis-free manner are still lacking. To address this need, we here introduce the deep labeling method, which combines a custom 13C medium with high-resolution mass spectrometry. A proof-of-principle study on human cancer cells demonstrates that deep labeling can identify hundreds of endogenous metabolites as well as active and inactive pathways. For example, protein and nucleic acids were almost exclusively de novo synthesized, while lipids were partly derived from serum; synthesis of cysteine, carnitine and creatine was absent, suggesting metabolic dependencies; and branched-chain keto acids (BCKA) were formed and metabolized to short-chain acylcarnitines, but did not enter the TCA cycle. Remarkably, BCKA could substitute for essential amino acids to support growth. The deep labeling method may prove useful to map metabolic phenotypes across a range of cell types and conditions.
Graphical abstract
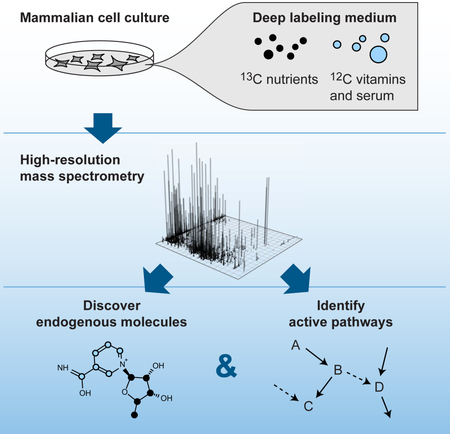
eTOC blurb
Grankvist et al. describe a simple, high-throughput method to catalogue endogenous metabolites and detect metabolic pathway activity in mammalian cells, using isotope tracing with a custom-designed 13C-labeled medium. This technique can be used to survey the metabolic state of a range of cell types and tissues in various experimental conditions.
Introduction
To understand the role of cellular metabolism in human health and disease, measuring the activity of enzymes or pathways in living human cells and tissues is central. Metabolism is highly cell type- and cell state-specific (Jain et al. 2012; Shlomi et al. 2008; Agren et al. 2012), suggesting opportunities for diagnosis and treatment of human disorders. Besides the well-known metabolic disorders of obesity, diabetes, and the metabolic syndrome, mutations in metabolic enzymes underlie a variety of genetic diseases, and metabolism is increasingly appreciated as a key mechanism in disorders of cell proliferation as well. In cancer biology, for example, metabolic enzymes associated with transformed cells may present new targets for drugs that aim to reduce cell proliferation (Vander Heiden 2011), and similarly, metabolic traits of specific immune cell types are being explored in the context of autoimmune disorders (Freitag et al. 2016). Systematic profiling of metabolic activities in human cells is therefore of fundamental importance for understanding the physiology of normal cells and their metabolic derangements in human disease (Hoppe 2012).
While there has been much progress in advanced measurement techniques for detailed studies of specific metabolic pathways (Buescher et al. 2015), a straightforward, high-throughput methodology for systematic, hypothesis-free assessment of metabolic activities in human cells is still lacking. Mass spectrometry (MS) or nucleic magnetic resonance (NMR) based metabolomics methods measure (relative) abundance of cellular metabolites, which can help detect differences in metabolic state, but such abundance data does not by itself inform on metabolic activities (Buescher et al. 2015). Measuring metabolite uptake and release can be done with reasonably high throughput, but provides only limited information on cellular metabolism (Allen et al. 2003; Jain et al. 2012). For a detailed assessment of intracellular metabolic activity in living cells, metabolic tracing with isotope-labeled nutrients is required. However, in mammalian cells, isotope labeling has mainly been used in a targeted, hypothesis-driven fashion, where selected pathways of interest are studied in depth using specific isotopic tracers in combination with sophisticated mathematical modeling (Niedenführ et al. 2015). Such tracer experiments are the gold standard for quantifying metabolic fluxes, but are challenging to perform and require detailed knowledge of the pathway of interest beforehand. On the other hand, several studies have used untargeted metabolomics methods to detect products of specific tracers, notably glucose and glutamine (Kluger et al. 2014; Huang et al. 2014; Capellades et al. 2016; Hiller et al. 2010); but in such cases, products of other medium nutrients are not detected, and only a subset of metabolic activities can be investigated. Indeed, the complement of metabolites synthesized by human cells is still not known (Viant et al. 2017). Comprehensive identification of endogenously synthesized compounds have to our knowledge been reported only in microorganisms that can grow on a single carbon source, where fully 13C cells can been generated, allowing global detection of endogenous metabolites by the IROA method (Qiu et al. 2016). However, since mammalian cells require a complex mixture of nutrients, including chemically undefined serum (Niklas et al. 2010), generating fully 13C mammalian cells is not feasible. In addition, full 13C labeling affords no information on the pathways used to synthesize the measured metabolites.
In this paper, we report a facile method, “deep labeling”, for hypothesis-free discovery of endogenous metabolites and metabolic activities in human cells from a single isotope tracing experiment. By culturing cells in a custom growth medium where the basic precursors of metabolic pathways are 13C, while vitamins and serum components are 12C, endogenously synthesized metabolites can be identified by incorporation of 13C atoms (Figure 1A). Simultaneously, presence of 12C atoms in a metabolite indicates incorporation of serum fat, protein or other exogenous compounds, and also serves to identify inactive metabolic pathways. This method is simple to perform, requires no computational modeling or a priori knowledge about metabolism, and is applicable to any cell type that can be maintained in culture conditions.
Figure 1. Cataloguing endogenously synthesized metabolites using deep labeling.

(A) Schematic of the deep labeling experiment strategy. (B) 13C enrichment of 1,319 LC-HRMS peaks with a unique predicted formula, in extract of cells grown in 12C medium (blue) or 13C medium (black). Individual enrichment values for three replicates are shown. (C-E) MIDs of pyruvate, acetyl-CoA and a novel pentose acid (ribonate; see text) in cells grown in 12C medium (blue) or 13C medium (black). (F-H) MIDs of pentose acid (ribonate), gulonate and ribose-5-phosphate/ribulose-5-phosphate in cells cultured with 1-13C1-glucose. Ribose-5-phosphate and ribulose-5-phosphate could not be separated by chromatography. Error bars denote standard deviation. MI fractions not shown were negligible. See also Figure S1 and Figure S2.
Results
Cataloguing endogenously synthesized metabolites using deep labeling
To obtain wide coverage of endogenous metabolites and metabolic activities in human cells, we synthesized a growth medium where glucose and all amino acids were fully 13C, while choline and the various vitamins were 12C (Figure 1A). An atom-level computational analysis of a large human metabolic network model (Nilsson & Jain 2016) covering 342 metabolites and 659 reactions indicated that in these conditions, 13C isotopes should reach at least one atom in every metabolite (except the vitamins and choline itself), suggesting that the design is feasible. As a proof-of-principle study, we chose to characterize the HCT116 colorectal carcinoma cell line, a near-diploid, p53-positive epithelial-like cell type widely used as a model system (Brattain et al. 1981). We cultured these cells in the 13C medium for at least 6 population doublings to ensure that > 98 % of cellular material was newly synthesized, and analyzed polar cell extracts with liquid chromatography-high resolution mass spectrometry (LC-HRMS). We detected 3,299 metabolite ions that were reproducibly observed across replicate cultures but absent in blanks, which likely represent cellular (but not necessarily endogenously synthesized) metabolites. Since we were interested in mass isotopomer distributions, which can only be calculated when the number of carbons is known, we focused on 1,319 metabolite ions for which we could assign a unique sum formula based on exact mass (Table S1; STAR Methods text). About 70 % of these ions acquired some 13C atoms in the 13C cultures, but not in 12C control cultures, demonstrating that they represent endogenous metabolites (Figure 1B). We were able to annotate 958 ions (73 %) with putative HMDB identifiers (Wishart et al. 2009) based on accurate mass, and we confirmed 105 out of 112 selected annotations (94%) against pure standards, suggesting that error rates are quite low. The majority of metabolites were small (64 % had 10 carbons or less), but we also detected larger species of up to 64 carbons, most of which were likely phospholipids (Figure S1A). Most endogenous metabolites (68 %) contained nitrogen, 18 % were phosphorylated and 17 % contained sulfur, while 22 % were pure carbohydrate (CxHyOz) compounds (Figure S1B). Endogenous metabolites exhibited a wide range of 13C enrichment (Figure 1B), with small metabolites such as pyruvate containing only nutrient-derived 13C atoms (Figure 1C), while more complex metabolites such as coenzyme A were mosaics of 12C vitamin moieties and nutrient-derived 13C atoms (Figure 1D), in agreement with known synthesis pathways in human cells.
One important application of the deep labeling methodology is identification of novel metabolites synthesized by various human cell types, given that only a fraction of human metabolites is known (Viant et al. 2017). As an example of discovery of endogenous metabolites, we observed a 13C5-labeled ion in cell extracts (Figure 1E) with formula C5H10O6, consistent with a pentose acid. This ion was not present in the medium (Figures S2A–S2C) and had no co-eluting compound (Figure S2D), indicating it was not an in-source fragment. The known pentose acids of biological origin are arabinonate and ribonate, which derive from the pentose sugars arabinose and ribose, respectively; and xylonate and lyxonate, which derive from the hexose acid gulonate. Culturing the HCT116 cells and two additional cell lines with a 1-13C1-glucose (Figures1F–H) or 2-13C1-glucose (Figures S2E–G) tracer resulted in clear 13C labeling of the metabolite ion (Figures 1F and S2E), demonstrating that it derives from glucose. Moreover, the mass isotopomer distribution (MID) of gulonate (Figures 1G and S2F) differed markedly from that of the pentose acid, rendering xylonate/lyxonate unlikely candidates, while the MID of ribose-5-phosphate/ribulose-5-phosphate (Figures 1H and S2G) was similar that of the pentose acid, suggesting that the compound is ribonate. A pure standard for ribonate eluted at the same retention time (Figure S2H) and gave an identical MS/MS spectrum (Figure S2I), supporting this conclusion. Ribonate has previously been observed in human plasma and was recently associated with risk for high-grade glioma (J. et al. 2017), but has to our knowledge not previously been shown to be synthesized by human cells, and its function in humans is unknown. These data exemplify that deep labeling experiments can uncover novel metabolites in human cells.
Profiling active and inactive metabolic pathways
Because deep labeling results in 13C incorporation into virtually any endogenously synthesized metabolite, it allows determining active and inactive metabolite pathways based on the observed mass isotopomers (Figure 1A). Metabolites in glycolysis, the pentose phosphate pathway, the TCA cycle, amino acid metabolism as well as de novo purine and pyrimidine synthesis all showed the expected 13C mass isotopomers, indicating that these central metabolic pathways are active (Table S1). Nucleobases were mostly 13C4, indicating active turnover of nucleotide and deoxynucleotide pools, but little salvage of medium (13C0) nucleotides (Figure 2A). S-adenosylmethionine (SAM)-dependent methylation was active, indicated by 13C14 SAM and 13C13 SAH (Figure 2B), while cystathionine was 13C0, indicating that the “transsulfuration” pathway that converts methionine into cysteine and taurine was inactive (Figure 2B). Polyamines were both synthesized and degraded, indicated by 13C10 methylthioadenosine and 13C4 acetylputrescine, respectively (Figure 2B), consistent with the notion that polyamine metabolism oscillates over the cell cycle (Bettuzzi et al. 1999).
Figure 2. Profiling active and inactive metabolic pathways.

(A-E) MIDs for selected metabolites with carbon number in parentheses (left) are shown as stacked bar charts (middle) with numbers indicating mass isotopomers, together with simplified schemas of the corresponding pathways (right). White regions of bars charts denote the 13C0 MI. Gray arrows denote inactive pathways. Numbers at the right side of the bars indicate 13C enrichment. Metabolites in parentheses were not measureable. Nonstandard abbreviation are hxan, hypoxanthine; thym, thymine; 5mta, 5-methiothioadenosine; aptrc, acetyl-putrescine; cyst, cystathionine; taur, taurine; tmabutn, trimethylammoniobutanoate; crn, carnitine; acrn, acetylcarnitine; orn, ornithine; creat, creatine; pcreat, phosphocreatine, gudac, guanidinoacetate; 5htrp, 5-hydroxytryptophan; kynr, kynurenine; ncam, nicotinamide. For enzymes indicated, see text.
Identifying inactive pathways in human cells is of great interest to predict dependency on metabolites provided by the cells’ environment, which could be exploited to prevent cell growth. In the HCT116 cells, we found that the fatty acid carrier carnitine was 13C0 and hence not synthesized, although the initial steps of this pathway leading to 13C7 trimethylammoniobutanoate were found to be active, suggesting that the carnitine-synthesizing BBOX1 enzyme was absent. Yet, acetylcarnitine was 13C2 (Figure 2C), indicating that the cells utilize 12C carnitine from the serum for carrying 13C acyl groups, and hence may be dependent on extracellular carnitine. Similarly, creatine and its downstream metabolites phosphocreatine and creatinine were present but 13C0, indicating that HCT116 cells lack the creatine synthesis pathway (Figure 2D).
We also observed synthesis of signaling molecules, bridging between metabolism and regulation. For example, the tryptophan-derived signaling metabolite kynurenine was de novo synthesized (13C10) but also acquired from the medium (13C0) (Figure 2E). Kynurenine is generated by some cancers and is thought to suppress immune cell responses (Platten et al. 2012). In agreement with a signaling role of this metabolite, kynurenine was not used for downstream synthesis of NAD, since a 13C20 mass isotopomer of NAD was not observed; instead, cells incorporated 12C nicotinamide from the medium to form 13C14 NAD (Figure 2E). We also detected synthesis of 13C11 5-hydroxytryptophan (Figure 2E), the initial metabolite in the branch of tryptophan metabolism that gives rise to serotonin and related hormones; as well as 13C4 gamma-aminobutyrate (GABA; Table S1), a signaling metabolite that can inhibit proliferation in colon cancer cells (Ortega 2003). Taken together, these data show that deep labeling can reveal activity of a variety of metabolic pathways from a single experiment.
Carbon sources for oxidation and biomass synthesis
Deep labeling experiments can also to some extent identify the carbon sources used by cells for oxidation and biomass synthesis. In the HCT116 cells, the acetyl group of acetyl-CoA was 97 % 13C (Figure 3A), indicating that the contribution of medium-derived fatty acids or other unlabeled material to acetyl-CoA was very small. Alpha-ketoglutarate, the other major carbon source for the tricarboxylic acid (TCA) cycle, was also nearly completely 13C, indicating that most oxidized carbon was derived from the medium nutrients. Yet, citrate/isocitrate showed a prominent 12C1mass isotopomer (Figure 3A), suggesting substantial incorporation of 12CO2 into TCA carbon by isocitrate dehydrogenase (IDH), similar to previous reports (Metallo et al. 2011). Fumarate, malate and aspartate also exhibited notable 12C1 mass isotopomers, indicating that 12CO2 propagates to these metabolites as well, possibly via citrate lyase (Figure 3A). We note that these 12C isotopomers may be due to exchange fluxes at IDH, and do not demonstrate net carbon fixation into biomass. In addition, succinate exhibited a prominent 13C0 isotopomer that was not present in other TCA cycle metabolites, suggesting a substantial, separate succinate pool derived from an unlabeled substrate. A possible candidate is peroxisomal oxidation of fatty acids, which may form succinate via the ACOT4 thioesterase (Westin et al. 2005).
Figure 3. Carbon sources for oxidation and biomass synthesis.

(A) MIDs for TCA cycle metabolites and a simplified schema of the TCA cycle, as in Figure 2. For enzymes indicated, see text. (B) MID for lysophosphatidylcholine 16:0. (C) Plot of 13C enrichment vs. number of carbons for phospholipids, for extracts of HCT116 cells grown in the 13C medium. (D) MID for phosphophatidylcholine 34:1 Notable mass isotopomers are indicated. Percentages denote sums of indicated MI fractions, representing labeled/unlabeled acyl chains (see text). The fraction of heavy chains for PC 34:1 is given by (0.55 + 2 × 0.28) / 2 = 0.55. (E) MID for glycerol-3-phosphocholine. Error bars denote standard deviation. See also Figure S3.
The source of protein and lipids, the major constituents of cell biomass, remains an open question for most human cell types (Hosios et al. 2016). In the present data set, cellular amino acid 13C enrichment was very similar to that of the 13C medium (Table S2), indicating that amino acids are fully derived from medium nutrients in HCT116 cells, and suggesting that proteins are endogenously synthesized rather than obtained from the serum. A proteomics analysis of 13C-labeled cells confirmed this prediction (Figure S3). In contrast, the observed MIDs of phospholipids indicated a mix of endogenous synthesis and consumption. For example, for LPC 16:0, we estimated from the observed MID that 63 % of the fatty acyl chains were de novo synthesized (Figure 3B); the remaining acyl chains are presumable obtained from serum fat. Interestingly, larger lipids generally had lower 13C enrichment, suggesting that longer acyl chains are preferentially obtained from the serum (Figure 3C). Moreover, the MID of phosphatidylcholine exhibited two distinct modes corresponding to either one (C16/C18) or two (C34) de novo synthesized acyl chains (Figure 3D), indicating that consumed lipids are actively remodeled by cells. Consistent with this, the phospholipid catabolism product glycerol-3-phosphocholine was 30 % 13C0 (Figure 3E), indicating turnover of the glycerol backbone from serum-derived lipids. It is clear from this data that de novo fatty synthesis contributes substantially to cellular lipids in cultured HCT116 cells, consistent with studies showing that knockdown of fatty acid synthase (FASN) reduces cell growth (Currie et al. 2013), but exogenous lipids are an important source as well.
Branched-chain keto acid metabolism
Branched-chain amino acid (BCAA) catabolism has recently been identified in several cancer types (Hattori et al. 2017; Mayers et al. 2014), but its function remains unclear. In HCT116 cells, we observed several metabolite ions with accurate mass and labeling pattern matching products of the branched-chain amino acids (BCAAs), including the corresponding branched-chain keto acids (BCKAs) and several branched-chain acylcarnitine species (Figure 4A), indicating that BCAA catabolism is active in HCT116 cells (Figure 4B). We confirmed the identity of the BCKA using pure standards (Figure S4), and further pursued these observations with individual isotope tracing experiments using either 13C6-isoleucine, 13C6-leucine, or 13C5-valine. In each case, the amino acid was taken up by cells and transaminated to its corresponding keto acid, confirming that a branched-chain amino acid transaminase is active in HCT116 cells (Figure 4C). A total of 169 ions acquired at least 5% 13C enrichment from either of the BCAA tracers (Table S3). All three branched-chain acylcarnitine species were formed, and isoleucine additionally gave rise to 13C3 propanoyl-carnitine, demonstrating that the complete isoleucine degradation pathway is active in these cells (Figure 4D). However, we observed no 13C labeling of TCA cycle metabolites, suggesting that the function of BCAA metabolism in these cells is unrelated to oxidation for energy. The branched-chain acylcarnitines have recently been shown to be precursors of fatty acid synthesis in human preadipocytes (Green et al. 2016; Crown et al. 2015; Hirosuke et al. 1994), and are associated with growth phenotypes in C.Elegans (Han et al. 2016), but have not previously been observed in cancer cells.
Figure 4. Branched-chain keto acid metabolism in HCT116 cells.

(A) MI fractions of BCKA and branched-chain acylcarnitine species from deep labeling tracing experiment. (B) Simplified schematic of the relevant BCAA catabolism pathways. Numbers in parentheses denote number of carbons, excluding the CoA or carnitine moieties. (C) MI fractions from isotope tracing experiments with either 13C6-isoleucine (black), 13C6-leucine (blue), or 13C5-valine (red) for 48 h. The isoleucine and leucine peaks have similar retention times, resulting in some overlapping labeling pattern regarding both the amino acids and keto acids. (D) MIDs of isobutyryl-, propanoyl-, methylbutyryl- and isovaleryl-carnitine in extracts of cells cultured in (left to right) unlabeled medium, with 13C-isoleucine, 13C-leucine, or 13C-valine. Methylbutyryl- and isovaleryl-carnitine were not separable by LC-HRMS, and are therefore observed as a single peak that acquires labeling from both 13C-isoleucine and 13C-leucine. (E) MI fractions from cells labeled with 13C-isoleucine, 13C-leucine and 13C-valine simultaneously for 48 h, in presence (gray) or absence (black) of unlabeled BCKA. Error bars denote standard deviation. (F) Cell growth curves from cells cultured the absence or presence of the BCAA and BCKA for 72 h and 120 h. Data represent mean±SD. See also Figure S4.
Branched-chain keto acids are present in human plasma at 30—50 uM, comparable to the BCAA levels (Wishart et al. 2013), suggesting that utilization of BCKA could be advantageous to proliferating cancer cells as an alternative source of BCAA for protein synthesis, particularly in poorly vascularized tumors where nutrients are scarce. To investigate whether BCKA can be utilized by HCT116 cells, we performed isotope tracing using 13C BCAA in medium supplemented with 12C BCKA. We found that the fraction of labeled BCAA declined in the presence of unlabeled BCKA (Figure 4E), demonstrating that BCKA were indeed taken up by the cells and transaminated into BCAA. Finally, we found that BCKA could fully substitute for BCAA to support growth of HCT116 cells: at equal nutrient concentrations, cells grew at the same rate on keto acids as on the corresponding amino acids, demonstrating that BCKA are efficiently utilized (Figure 4F). Taken together, these data show that deep labeling experiments can reveal novel metabolic phenotypes of human cells.
Discussion
In this paper, we have introduced deep labeling as a simple experimental strategy for profiling cellular metabolism, and applied it to chart the metabolism of a human cancer cell line, as an example. The resulting data shows that a single deep labeling experiment allows detecting hundreds of metabolites, assessing the activity of metabolic pathways, and determining the source of biomass and metabolic fuels. We therefore anticipate that deep labeling could be broadly applicable to systematically study the metabolism of cultured human cells in a variety of experimental conditions.
In isotope tracing experiments, the precise choice of tracer is a key factor determining the information gained about pathway activity. Our medium design, where glucose and the amino acids are 13C while serum-derived nutrients are 12C, allows us to detect de novo synthesis of most cellular metabolites (Figure 1A). A drawback is that, since the resulting MIDs depend on the metabolic state of the cells, metabolite formulas must be identified from other sources, such as the HMDB database. In contrast, the Isotope Ratio Outlier Analysis (IROA) methodology (Qiu et al. 2016) allows identifying the number carbon number directly from mass isotopomer distributions; however, this requires complete 13C labeling of all biomass, which is only feasible in microorganisms that can grow on a single carbon source, and the resulting data yields no information about pathway activity. It must also be noted that our method cannot distinguish between usages of alternative pathways leading from a labeled nutrient to a labeled product, unless there is at least one unlabeled intermediate. To resolve such cases, metabolic flux analysis experiments are needed. Also, deep labeling requires that the cell culture reaches isotopic steady state, which means that cells need to be grown in the deep labeling for several cell doublings so that biomass is fully renewed (here we ensured at least 6 doubling, so that 1 – 2−6 = 98% of biomass is newly synthesized). Hence, our methodology is not suitable for studying rapid metabolic responses to acute stimuli.
In the HCT116 cells, we find that both endogenous fatty acid synthesis and serum fat contributes to lipids in cultured cancer cells, consistent with previous evidence that fatty acids can be obtained from serum (Kamphorst et al. 2013). The observation that endogenous fatty acyls are more common in shorter acyl chains could reflect de novo synthesis of palmitate (C16) by fatty acid synthase (FASN), but lack of further fatty acid elongation; or may be related to acyl chain specificity of lipid remodeling enzymes. We note that, since culture medium has lower lipid content than plasma, cancer cells in vivo would be expected to have better access to exogenous lipids. On the other hand, our data indicates that cell protein is fully derived from medium nutrients in HCT116 cells, and not from serum protein, in contrast to previous studies in pancreatic and bladder carcinoma cells (Commisso et al. 2013; Kamphorst et al. 2015). Hence, protein consumption from the medium appears to be cell type-dependent.
A notable finding was that HCT116 cells transaminate branched-chain amino acids (BCAA) to BCKA, which were further metabolized by the mitochondrial oxidative pathway into branched-chain carnitines, but did not enter the TCA cycle. The BCKA are similar to (branched) short chain fatty acids, and their metabolism involves enzymes also utilized for fatty acid metabolism. BCKA metabolism has recently been shown to be important for adipocyte differentiation (Green et al. 2016), where BCKA carbon is incorporated into other fatty acids (Crown et al. 2015). We found no incorporation of BCKA into phospholipids, which indicates that BCKA are not a major contributor to lipid synthesis overall. In addition, adipocytes metabolized BCAA into citrate, while the HCT116 cells did not. While the ultimate function of BCKA metabolism remains unclear, the finding that BCKA can fully substitute for the corresponding amino acids suggests that this is a major metabolic pathway in HCT116 cells. In line with this, another recent study highlighted BCKA as a possible alternative nutrient to branched-chain amino acids in leukemia cells (Hattori et al. 2003). Also, glioma (Tönjes et al. 2013) and non-small lung carcinoma cells (Mayers et al. 2016) are thought to utilize BCAA as a nitrogen source, while pancreatic carcinoma cells did not (Mayers et al. 2016). Overall, it appears that BCAA/BCKA metabolism is a flexible system that can satisfy several metabolic needs of proliferating cancer cells. The diversity of metabolic modes seen in these cell types further underscores the need for facile methods for systematic profiling of metabolism in human cells. We hope that the deep labeling strategy will prove useful for this purpose.
STAR Methods text
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Roland Nilsson (roland.nilsson@ki.se).
Experimental Model and Subject Details
Cell culture conditions
HCT116 (human colorectal cancer, male), HeLa (human cervix carcinoma, female) and MCF7 (human breast adenocarcinoma, female) cells were cultured in custom made RPMI 1640 medium (Table S2) supplemented with 5 % dialyzed heat inactivated fetal bovine serum (FBS), 100U/ml penicillin-streptomycin (Life Technologies) and 1000x phenol red (Sigma-Aldrich) at 37 °C and 5 % CO2. The FBS was dialyzed in SnakeSkin 10K MWCO dialysis tubing (Thermo Fisher Scientific #88245).
Method Details
Stable isotope tracing experiments
For deep labeling, cells were cultured in 10 cm tissue culture dishes for 120 h in a custom made RPMI 1640 labeled medium containing the following stable isotopes (Cambridge Isotope Laboratories, Inc): 13C6-glucose (CLM-1396-0), 13C6-arginine:HCl (CLM-2265-H), 13C4-asparagine:H2O (CLM-8699-H), 13C4-aspartic acid (CLM-1801-H), 13C3-cysteine (CLM-4320-H), 13C5-glutamic acid (CLM-1800-H), 13C5-glutamine (CLM-1822-H), 13C2-glycine (CLM-1017-0), 13C6-histidine:HCl:H2O (CLM-2264-0), 13C6-isoleucine (CLM-2248-H), 13C6-leucine (CLM-2262-H), 13C6-lysine:2HCl (CLM-2247-H), 13C5-methionine (CLM-893-H), 13C9-phenylalanine (CLM-2250-H), 13C5-proline (CLM-2260-H), 13C3-serine (CLM-1574-H), 13C4-threonine (CLM-2261-0), 13C11-tryptophan (CLM-4290-H), 13C9-tyrosine (CLM-2263-H) and 13C5-valine (CLM-2249-H).
To evaluate the labeling capability of the three BCAA and effects of the BCKA, cells were cultured in 6-well plates with custom made RPMI 1640 medium with the tracers; 13C6-isoleucine, 13C6-leucine and 13C5-valine, individually or simultaneously w/wo co-culture of 0.3 mM per BCKA for 48 h.
For glucose isotope tracing experiment, HCT116, MCF7 and HeLa cells were cultured in 6-well plates with custom made RPMI 1640 media with the stabled isotope 1-13C1-glucose or 2-13C1-glucose for 48 h.
For proteomics analysis, HCT116 cells were cultured in custom made RPMI 1640 medium with the tracers 13C6-arginine:HCl and 13C6-lysine:2HCl for 120 h. Cells were collected by trypsinization and the pellet were washed with phosphate buffered saline prior analysis.
Common for all tracing experiments; cells were seeded at a density to achieve ~85 % confluence at the time of extraction. Separate wells with identical plating were utilized for confirmation of cell numbers. Three independent biological replicates were used for all cell culture experiments and conditions. As controls, additional cells were cultured in an unlabeled custom made RPMI 1640 medium (unlabeled nutrients from Sigma-Aldrich).
Metabolite extraction and mass spectrometry
For metabolomics studies, HCT116 cells were analyzed by liquid chromatography-high resolution mass spectrometry (LC-HRMS), as previously described. At the time of extraction, spent medium was collected and cells were rapidly washed twice with cold phosphate buffered saline to remove contaminating medium. We estimate that two rounds of washing reduces medium concentrations by a factor ~106, bringing glucose (the most severe contaminant) from 11 mM to 11 uM. Metabolites were then extracted using 4.5 ml (10 cm tissue culture dishes) or 1 ml (6-well plates) of −80 °C pre-cooled HPLC grade methanol. The microplates were kept on dry ice and cell material was scraped in the cold methanol, collected, vortexed for 1 minute and stored in −80 °C until analysis.
For LC-HRMS analysis, cellular material was centrifuged at 10000 RPM for 10 min at 4 °C and the cell pellets were resuspended in solution followed by three freeze-thaw cycles, with alternating 37 °C and −80 °C liquid baths in 60 second intervals. To ensure complete cell lysis, samples were vortexed for 30 seconds and sonicated for two minutes. Samples were then dried down in vacuo using a vacuum concentrator and re-suspended in 25 μL of 80:20 methanol:water. Resuspended samples were mixed by aspiration, sonicated for 2 minutes, vortexed for 30 seconds, centrifuged at 14,000 g × 10 minutes at 4 °C and supernatants were transferred to LC-HRMS vials containing a 200 μl glass insert. An injection volume of 5 μL, which contained metabolic material from ~80,000 cells, was used for all cell samples. LC-MS/MS based metabolomics analysis was performed using a Thermo QExactive orbitrap mass spectrometer coupled to a Thermo Vanquish UPLC system. Chromatographic separation of metabolites was achieved using a Millipore (Sequant) Zic-pHILIC 2.1×150 mm 5 um column maintained at 25 °C. Compounds were eluted via a 19 minute linear gradient starting from 90:10 acetonitrile:20 mM ammonium bicarbonate to 45:55 acetonitrile:20 mM ammonium bicarbonate. A Thermo Q-Exactive orbitrap mass spectrometer was operated in positive and negative ion modes using a heated electrospray ionization (HESI) source at 35,000 resolution, 100 ms ion trap time for MS1 and 17,500 resolution, 50 ms ion trap time for MS2 collection. Data was collected over a mass range of m/z 67-1000, using a sheath gas flow rate of 40 units, auxillary gas flow rate of 20 units, sweep gas flow rate of 2 units, spray voltage of 3.5 and 2.5 kV for positive and negative ion modes (respectively), capillary inlet temperature of 275 °C, auxillary gas heater temperature of 350 °C and an S-lens RF level of 45. For MS2 collection, MS1 ions were isolated using a 1.0 m/z window and fragmented using a normalized collision energy of 35. Fragmented ions were placed on dynamic exclusion for 30 seconds before being allowed to be fragmented again.
LC-HRMS data processing
Primary Thermo .raw data files were processed using in-house software to detect all LC-HRMS peaks with an apex intensity of at least 70,000. Peak detection was done using a variation of the centWave algorithm (Tautenhahn et al. 2008), with the following modifications: (1) a chromatogram was defined by initializing with the highest intensity m/z peak, and iteratively adding any m/z peaks located within 10ppm and 3 scans from the peaks already included in the chromatogram; (2) after wavelet decomposition of a chromatogram, each wavelet component was scaled so as to not exceed the original chromatogram intensity at any point in the time domain, in order to reduce false positives from low-frequency components. Only metabolite ions (LC-MS peaks) that were reproducible detected in both unlabeled and labeled cells and reach > 5x the blank intensity were retained. Metabolites ions were matched to (putative) identifiers in the Human Metabolome Database (HMDB) 1 by m/z with a tolerance of 5 ppm and considering H+, H−, Na+, K+, Cl− and NH4+ adducts, while for ions with no matches in HMDB, any molecular formula resulting in a theoretical m/z within 5 ppm of the measured m/z was considered. Where possible, final metabolite identification was made based on agreement with the observed retention time of pure standards, as well as appearance of the expected isotopomers in 13C-labeled samples. For concentration estimation, the LC peak shape of the observed endogenous (labeled) compound was also verified in this manner.
Ions with a unique predicted formula were considered for mass isotopomer analysis (given the predicted number of carbons), resulting in a total of 16,949 isotopic peaks. We restricted ourselves to ions of with a unique formula to ensure that the carbon number is known, which is required to account for all possible mass isotopomers peaks and fully determine the MIDs (although it is possible to obtain relative mass isotopomer abundances without knowing the number of carbons). To avoid false isotopic peaks (due to contamination by other ions of similar m/z), we discarded mass isotopomers whose intensity ratio over the base (13C0) mass isotopomers exceeded 3% of the expected ratio (from natural MI distribution). For metabolite of interest, false isotopomers were also excluded by examining the chromatographic shape of isotopic peaks, which must closely match that of the base isotopomer. The 13C enrichment E was defined for a compound with n carbons and MI fraction x0,x1,…xn as
Protein extraction and digestion for mass spectrometry analysis
Cell pellets were suspended in 8 M urea (Sigma-Aldrich), 50 mM ammonium bicarbonate and 10 % acetonitrile (ACN) resulting in protein yields 516-528 μg. The samples were sonicated using VibraCell probe (Sonics & Materials, inc) for 1 min with pulse 2/2, at 20 % amplitude, and sonicated in bath for 10 minutes. Ten μg of each sample were subjected to tryptic digestion following protein reduction in 5 mM DTT at 37°C for 45 min and alkylation in 15 mM iodoacetamide for 30 min at room temperature in the dark. Trypsin was added in an enzyme to protein ratio of 1:30 and digestion was carried out over night at 37°C.
Liquid chromatography tandem mass spectrometry
Tryptic peptides were cleaned on C18 StageTips (Thremo Scientific) and 25 % of the resulting peptide mixture was injected into a nano LC-1000 system on-line coupled to a QExactive Plus mass spectrometer (Thermo Scientific, Bremen, Germany). The chromatographic separation of the peptides was achieved using a 50 cm long C18 EASY spray column (Thermo Scientific, Bremen, Germany), with the following gradient: 4-26 % ACN in 180 min, 26-80 % ACN in 5 min and 80 % ACN for 8 min all at a flow rate of 300 nl/min. The MS acquisition method was comprised of one survey full scan ranging from m/z 300 to 1650, acquired with a resolution of R= 140,000 (at m/z 200), followed by data-dependent HCD fragmentations from maximum 16 most intense precursor ions with a charge state ≥ 2. The tandem mass scans were acquired with a resolution of R=17,500, targeting 2e5 ions, setting isolation width to m/z 4 and normalized collision energy to 26.
Cell growth experiments
Cells were cultured in custom made RPMI 1640 medium w/wo the BCAA (Sigma-Aldrich #I7403, #L8912, and #V0513) and w/wo the BCKA (Toronto Research Chemicals, #M326420, #M326425 and #K193500), both individually and in combination, for 72 h and 120 h. After exposure, cells were washed with phosphate buffered saline, trypsinized with Trypsin-EDTA (0.05 %) (Thermo Fisher Scientific) and suspended in growth medium. Cell numbers were measured in the cellsuspension with TC20 automated cell counter (Bio-Rad Laboratories, Inc.) and/or Scepter cell counter (Merck Millipore).
Quantification and Statistical Analysis
Mathematica scripts were used for calculations of peak areas and building plots. GraphPad Prism version 7 software was used for bar charts.
Data and Software Availability
Mass spectrometry data presented in this study is available from www.nilssonlab.se.
Additional Resources
N/A
Supplementary Material
Related to Figure 1. Peak list of 1,319 metabolites from deep labeling tracing experiment
Related to Figure 1 and STAR methods text. Custom made RPMI 1640 medium formulation
Related to Figure 4. Peak list from untargeted analysis
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Methanol (JT Baker), HPLC grade | VWR | Cat#BAKR8402.250 0 |
| Ammonium Bicarbonate | Honeywell | Cat#40867-50G |
| LCMS Grade Water | Optima | Cat#W64 |
| LCMS Grade Methanol | Honeywell | Cat#BJLC230-4 |
| LCMS Grade Acetonitrile | Honeywell | Cat#BJLC015-4 |
| 8M Urea | Sigma-Aldrich | Cat#U4883 |
| RPMI 1640 vitamins solution 100x | Sigma-Aldrich | Cat#R7256 |
| Trypsin-EDTA (0.05%), phenol red | Gibco | Cat#25300062 |
| PBS, pH 7.4 | Gibco | Cat#10010056 |
| Fetal bovine serum, qualified, heat inactivated US origin | Gibco | Cat#16140071 |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | Cat#15140122 |
| Phenol red solution | Sigma-Aldrich | Cat#P0290 |
| D-Glucose (dextrose) | Sigma-Aldrich | Cat#D9434 |
| Glycine | Sigma-Aldrich | Cat#50046 |
| L-Arginine | Sigma-Aldrich | Cat#A8094 |
| L-Asparagine | Sigma-Aldrich | Cat#A4159 |
| L-Aspartic acid | Sigma-Aldrich | Cat#A7219 |
| L-Cysteine | Sigma-Aldrich | Cat#C7352 |
| L-Glutamic acid | Sigma-Aldrich | Cat#G8415 |
| L-Glutamine | Sigma-Aldrich | Cat#G8540 |
| L-Histidine | Sigma-Aldrich | Cat#H6034 |
| trans-4-Hydroxy-L-proline | Sigma-Aldrich | Cat#H54409 |
| L-Isoleucine | Sigma-Aldrich | Cat#I7403 |
| L-Leucine | Sigma-Aldrich | Cat#L8912 |
| L-Lysine monohydrochloride | Sigma-Aldrich | Cat#L8662 |
| L-Methionine | Sigma-Aldrich | Cat#M3508 |
| L-Phenylalanine | Sigma-Aldrich | Cat#P5482 |
| L-Proline | Sigma-Aldrich | Cat#P5607 |
| L-Serine | Sigma-Aldrich | Cat#S4311 |
| L-Threonine | Sigma-Aldrich | Cat#T8441 |
| L-Tryptophan | Sigma-Aldrich | Cat#T8941 |
| L-Tyrosine | Sigma-Aldrich | Cat#T8566 |
| L-Valine | Sigma-Aldrich | Cat#V0513 |
| Calcium nitrate tetrahydrate | Sigma-Aldrich | Cat#C2786 |
| Sodium phosphate dibasic | Sigma-Aldrich | Cat#S0876 |
| Magnesium sulfate | Sigma-Aldrich | Cat#M2643 |
| Potassium chloride | Sigma-Aldrich | Cat#P5405 |
| Sodium bicarbonate | Sigma-Aldrich | Cat#S5761 |
| Sodium chloride | Sigma-Aldrich | Cat#S7653 |
| L-Glutathione reduced | Sigma-Aldrich | Cat#G6013 |
| 4-Methyl-2-oxovaleric Acid Sodium Salt (4mop) | Toronto Research Chemicals | Cat#M326425 |
| α-Keto Isovaleric Acid Sodium salt (3mob) | Toronto Research Chemicals | Cat#K193500 |
| 3-Methyl-2-oxovaleric Acid Sodium Salt (3mop) | Toronto Research Chemicals | Cat#M326420 |
| D-Glucose (U-13C6, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-1396-0 |
| Glycine (U-13C2, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-1017-0 |
| L-Arginine (U-13C6, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-2265-H |
| L-Asparagine (U-13C4, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-8699-H |
| L-Aspartic acid (U-13C4, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-1801-H |
| L-Cysteine (U-13C3, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-4320-H |
| L-Glutamic acid (U-13C5, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-1800-H |
| L-Glutamine (U-13C6, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-1822-H |
| L-Histidine (U-13C6, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-2264-0 |
| L-Isoleucine (U-13C6, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-2248-H |
| L-Leucine (U-13C6, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-2262-H |
| L-Lysine hydrochloride (U-13C6, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-2247-H |
| L-Methionine (U-13C5, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-893-H |
| L-Phenylalanine (U-13C9, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-2250-H |
| L-Proline (U-13C5, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-2260-H |
| L-Serine (U-13C3, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-1574-H |
| L-Threonine (U-13C4, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-2261-0 |
| L-Tryptophan (U-13C11,99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-4290-H |
| L-Tyrosine (U-13C9, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-2263-H |
| L-Valine (U-13C5, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-2249-H |
| D-Glucose (2-13C, 99%) | Cambridge Isotope Laboratories, Inc | Cat#CLM-746-PK |
| D-Glucose (1-13C) | Omicron Biochem. Inc. | Cat#GLC-018 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| HCT116 | ATCC | Cat#ATCC® CCL-247™ |
| MCF7 | ATCC | Cat#ATCC® HTB-22™ |
| HeLa | ECACC | Cat#93021013 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and Algorithms | ||
| mzAccess (http://www.mzaccess.org/) | (Lyutvinskiy et al. 2017) |
N/A |
| Mathematica v10 | Wolfram Research | N/A |
| GraphPad Prism version 7 software | GraphPad Software | N/A |
| Other | ||
| SnakeSkin™ Dialysis Tubing, 3.5K MWCO, 35 mm | Thermo Fisher Scientific |
Cat#88244 |
| Cell scraper with two-position blade, scraper length: 25 cm | Sarstedt | Cat#83.1830 |
| Cell culture dish 10 cm | Sarstedt | Cat#83.1830 |
| Cell culture plates, 6-well | Sarstedt | Cat#83.3920 |
Highlights.
Rich data on endogenous metabolites and active pathways from a single cell culture
In HCT116 cancer cells, hundreds of synthesized metabolites were unknown
Cells scavenge nutrients from serum and lack central synthesis pathways
Branched-chain keto acids were metabolized and could substitute for amino acids
Significance.
As cellular metabolism is increasingly recognized as a key factor for a range of human disorders, charting the metabolic activity of various human cell types is of great importance. Measuring activity of metabolic enzymes and pathways in living cells or tissues is challenging, and there is currently no systematic method for unbiased profiling of metabolism in mammalian cells. In this paper, we propose such a method, based on a custom-designed 13C-labeled growth medium, and demonstrate that a single experiment using less than 106 cells provides rich data on endogenous metabolites, including hundreds of still unknown species, and in many cases also allows identifying active / inactive metabolic pathways. We believe that this method will be valuable for profiling the metabolism of a variety of cell systems in an unbiased manner. Such experiments are needed to catalogue and understand the metabolic phenotypes of human cells in normal and pathological conditions.
Acknowledgements
This work was supported by grants from the Swedish Foundation for Strategic Research (FFL12-0220.006), the Strategic Programme in Cancer Research and Karolinska Institutet to R.N., and the National Institutes of Health 5R03HL133720, 1R01ES027595 and 1S10OD020025 to M.J. Mass spectrometric analysis for proteomics analysis were carried out at the Proteomics Biomedicum core facility, Karolinska Institutet, Stockholm.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agren R et al. , (2012). Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS computational biology, 8(5), p.e1002518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen J et al. , (2003). High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nature Biotechnology, 21(6), 692–696. [DOI] [PubMed] [Google Scholar]
- Bettuzzi S et al. , (1999). Coordinate changes of polyamine metabolism regulatory proteins during the cell cycle of normal human dermal fibroblasts. FEBS letters, 446(1), 18–22. [DOI] [PubMed] [Google Scholar]
- Brattain MG et al. , (1981). Heterogeneity of Malignant Cells from a Human Colonic Carcinoma. Cancer research, 41, 1751–1756. [PubMed] [Google Scholar]
- Buescher JM et al. , (2015). A roadmap for interpreting 13C metabolite labeling patterns from cells. Current Opinion in Biotechnology, 34, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capellades J et al. , (2016). GeoRge: A Computational Tool to Detect the Presence of Stable Isotope Labeling in LC/MS-Based Untargeted Metabolomics. Analytical Chemistry, 88(1), 621–628. [DOI] [PubMed] [Google Scholar]
- Commisso C et al. , (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature, 497(7451), 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crown SB, Marze N & Antoniewicz MR, (2015). Catabolism of branched chain amino acids contributes significantly to synthesis of odd-chain and even-chain fatty acids in 3T3-L1 adipocytes. PLoS ONE, 10(12), 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie E et al. , (2013). Cellular Fatty Acid Metabolism and Cancer. Cell Metabolism, 18(2), 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag J et al. , (2016). Immunometabolism and autoimmunity. Immunology and Cell Biology, 94(10), 925–934. [DOI] [PubMed] [Google Scholar]
- Green CR et al. , (2016). Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nature chemical biology, 12(1), 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M et al. , (2016). Developmental defects of Caenorhabditis elegans lacking branched-chain α-ketoacid dehydrogenase are mainly caused by monomethyl branched-chain fatty acid deficiency. Journal of Biological Chemistry, 291(6), 2967–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori A et al. , (2017). Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M et al. , (2003). Development of a chemical structure comparison method for integrated analysis of chemical and genomic information in the metabolic pathways. Journal of the American Chemical Society, 125(39), 11853–65. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, (2011). Targeting cancer metabolism: a therapeutic window opens. Nature reviews. Drug discovery, 10(9), 671–84. [DOI] [PubMed] [Google Scholar]
- Hiller K et al. , (2010). Nontargeted elucidation of metabolic pathways using stable-isotope tracers and mass spectrometry. Analytical chemistry, 82(15), 6621–8. [DOI] [PubMed] [Google Scholar]
- Hirosuke O et al. , (1994). Precursor role of branched-chain amino acids in the biosynthesis of iso and anteiso fatty acids in rat skin. Biochimica et Biophysica Acta (BBA)/Lipids and Lipid Metabolism, 1214(3), 279–287. [PubMed] [Google Scholar]
- Hoppe A, (2012). What mRNA Abundances Can Tell us about Metabolism. Metabolites, 2, 614–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosios AM et al. , (2016). Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Developmental Cell, 36(5), 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X et al. , (2014). X13CMS: global tracking of isotopic labels in untargeted metabolomics. Analytical chemistry, 86(3), 1632–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- J. H et al. , (2017). A prospective study of serum metabolites and glioma risk. Oncotarget, 8(41), 70366–70377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M et al. , (2012). Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science, 336(6084), 1040–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ et al. , (2015). Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Research, 75(3), 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ et al. , (2013). Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proceedings of the National Academy of Sciences of the United States of America, 110(22), 8882–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluger B et al. , (2014). Untargeted profiling of tracer-derived metabolites using stable isotopic labeling and fast polarity-switching LC-ESI-HRMS. Analytical Chemistry, 86(23), 11533–11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayers JR et al. , (2014). Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nature medicine, 20(10), 1193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayers JR et al. , (2016). Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science, 3453(6304), 1161–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo C et al. , (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature, 481, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedenführ S, Wiechert W & Nöh K, (2015). How to measure metabolic fluxes: A taxonomic guide for 13C fluxomics. Current Opinion in Biotechnology, 34, 82–90. [DOI] [PubMed] [Google Scholar]
- Niklas J, Schneider K & Heinzle E, (2010). Metabolic flux analysis in eukaryotes. Current opinion in biotechnology, 21(1), 63–9. [DOI] [PubMed] [Google Scholar]
- Nilsson R & Jain M, (2016). Simultaneous tracing of carbon and nitrogen isotopes in human cells. Mol. BioSyst, 12(C), 1929–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega A, (2003). A new role for GABA: Inhibition of tumor cell migration. Trends in Pharmacological Sciences, 24(4), 151–154. [DOI] [PubMed] [Google Scholar]
- Platten M, Wick W & Van Den Eynde BJ, (2012). Tryptophan Catabolism in Cancer: Beyond IDO and Tryptophan Depletion. Cancer research, 72(21), 5435–5440. [DOI] [PubMed] [Google Scholar]
- Qiu Y et al. , (2016). Isotopic Ratio Outlier Analysis of the S. cerevisiae Metabolome Using Accurate Mass Gas Chromatography/Time-of-Flight Mass Spectrometry: A New Method for Discovery. Analytical Chemistry, 88, 2747–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomi T et al. , (2008). Network-based prediction of human tissue-specific metabolism. Nature biotechnology, 26(9), 1003–1010. [DOI] [PubMed] [Google Scholar]
- Tautenhahn R, Böttcher C & Neumann S, (2008). Highly sensitive feature detection for high resolution LC/MS. BMC bioinformatics, 9, p.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tönjes M et al. , (2013). BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nature medicine, 19(7), 901–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viant MR et al. , (2017). How close are we to complete annotation of metabolomes? Current Opinion in Chemical Biology, 36, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westin MAK, Hunt MC & Alexson SEH, (2005). The identification of a succinyl-CoA thioesterase suggests a novel pathway for succinate production in peroxisomes. Journal of Biological Chemistry, 280(46), 38125–38132. [DOI] [PubMed] [Google Scholar]
- Wishart DS et al. , (2009). HMDB: a knowledgebase for the human metabolome. Nucleic acids research, 37(Database issue), D603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS et al. , (2013). HMDB 3.0-The Human Metabolome Database in 2013. Nucleic Acids Research, 41(D1). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Related to Figure 1. Peak list of 1,319 metabolites from deep labeling tracing experiment
Related to Figure 1 and STAR methods text. Custom made RPMI 1640 medium formulation
Related to Figure 4. Peak list from untargeted analysis